

Abstract
Dementia associated with the Alzheimer's disease is thought to be correlated with the conversion of the β − Amyloid (Aβ) peptides from soluble monomers to aggregated oligomers and insoluble fibrils. We present a discrete-time mathematical model for the aggregation of Aβ monomers into oligomers using concepts from chemical kinetics and population dynamics. Conditions for the stability and instability of the equilibria of the model are established. A formula for the number of monomers that is required for producing oligomers is also given. This may provide compound designers a mechanism to inhibit the Aβ aggregation.
Introduction
Alzheimer's disease (AD) is the most cause of dementia and the most prevalent neurodegenerative disorder [1, 2, 3]. It is a progressive degenerative disorder that is age-related and is characterized by the loss of synapses and neurons from the brain and by the presence of extracellular protein-containing deposits (senile plaques) and intracellular neurofibrillary tangles [4, 2]. Β − Amyloid is the principal protein component of the extracellular plaques. Aβ is a 39- to 43- residue proteolytic product of a parental amyloid precursor protein (APP) that localizes to the plasma membrane, trans-Golgi network, endoplasmic reticulum (ER) and endosomal, lysosomal and mitochondrial membranes [5, 6]. Aβ contains sequences from extracellular and transmembrane regions of the parent protein [7, 8]. The spontaneous conversion of Aβ monomers into febrillar aggregates is found to be associated with the development of AD [9]. In fact, the neurodegenerative effects of AD are hypothesized to arise from Aβ. This is commonly known as the amyloid hypothesis, and is the dominant model of AD pathogenesis [10, 11, 5, 12, 13, 14, 15]. Albeit, amounting support from biochemical, genetic, and transgenic animal studies that supports the amyloid hypothesis, [16, 17, 18, 19, 20], debate over the amyloid hypothesis remains controversial.
In general, protein aggregation from soluble to non-soluble structures has been linked to be a causative factor of several diseases, including AD, Parkinson's disease, Huntington's disease, Prion disease, among others [21, 15]. For the case of Aβ linkage to AD, it has been found that Aβ becomes indeed toxic once aggregated [22, 23, 24, 25, 26]. Furthermore, numerous studies show a strong correlation between soluble Aβ oligomer levels and the extent of synaptic loss [27, 28, 29, 30, 31, 32], further suggesting that the soluble oligomers are the causative agents of AD [33, 34, 35, 36]. This in turn has motivated several studies aimed at exploiting Aβ aggregation mechanisms and kinetics of Aβ conversion. Especially that synthetic Aβ was found to spontaneously aggregates into sheet-rich fibrils, resembling those in plaques [5].
Naiki and Nakakuki [37] proposed a simple mathematical model in which fibril elongation is postulated to occur by reversible addition of monomers to preexisting fibrils. The model, however, does not explain the generation of new fibrils neither it simulates fibril length. Lomakin [38] proposed a detailed kinetic model in which they postulated that rapid reversible equilibrium between monomers and micelles occurs, followed by spontaneous generation of nuclei from micelles, in an irreversible process. Fibrils then grew by adding monomers to the fibril tip or the nucleous. The model accounts for the co-existence of monomers and fibrils and is capable of predicting fibril evolution (mass and length) in time. However, the experiments leading to the model development were performed in non-physiological conditions (pH 1). Pallitto [3] developed a kinetic model that qualitatively de-scribed Aβ self-association kinetics from the unfolded state. The model incorporated information about mass distribution and length changes of Aβ and accounted for the co-existence of monomer, dimer, and aggregated species. The model provided mechanisms for both generation and elongation of fibrils, and was able to capture all the essential features of the experimental data.
The structure of the Aβ monomer is difficult to characterize due to its tendency to aggregate. Experiments focusing on the understanding of physical structure of Aβ showed that they are not necessarily homogenous in shape. Filament (3–4 nm in diameter) and fibril (8–10 nm in diameter) structures have been observed in several electron microscopy and atomic force microscopy experiments [39, 40, 26]. Furthermore, Malinchik et al. [41] suggested that fibers are made of three to five laterally associated filaments. Fraser [42] reported on observing amyloid fibers made of five to six globular units, each with 2.5–3 nm in diameter. Finally, Reixach et al. [43] showed that oligomers are formed from the aggregation of at most six monomers.
Mathematical models of the Aβ kinetics provide a clearer mechanistic understanding of the amyloid fibril growth, improve our ability to design compounds that alter and modulate fibril formation, and provide therapeutic potential venues for the AD.
This paper develops a discrete mathematical model for the aggregation of monomers to oligomers and discusses a mechanism to reduce the production of oligomers. The model is based on the assumption that soluble Aβ oligomers are the causative agents of AD. First, we provide a brief review of chemical kinetics and then proceed to develop the discrete model. A stability analysis follows which determines the aggregation condition. Finally, A formula for the number of monomers that is required for producing oligomers is provided. In [44], Puri and Li developed a continuous-time (differential equation) model focusing on the network cross talk among microglia, neuron, and astroglia, and the corresponding pathological consequence. However, in this work, we take a different approach (discrete) in modeling the aggregation of β − Amyloid into diamers, triamers, etc., and finally into oligomers. The novelty of our approach is the utilization of ideas from chemical kinetics [45] and population dynamics [46], to develop a discrete-time model describing this process.
A brief review of chemical kinetics
In chemical kinetics [45], if A is the reactant and B is the product, so that A→B, then the average rate of the reaction describes the change in the concentration of either A or B, and is given by,
| (1) |
and
| (2) |
moreover,
| (3) |
Letting Δt = 1, we obtain from (3),
| (4) |
In general, for the reaction,
| (5) |
where a, b, c, d are the number of molecules of A, B, C, D respectively, we have,
| (6) |
The reaction rate law expression relates the rate of an elementary reaction to the concentration of each reactant, that is
| (7) |
where K is the reaction constant, to be determined experimentally.
The construction of the model: Making oligomers
In its simplest forms, Amyloid formation can be described by protein aggregation, involving the misfolding of Aβ into soluble and insoluble assemblies [47, 48]. Kinetic studies have suggested that the misfolding of monomeric Aβ has been shown to precede the formation of oligomers, which then serve as seeds for accelerated fibril growth, [49], as illustrated in Fig 1.
Fig 1. The aggregation of β − Amyloid: From monomers to oligomers.
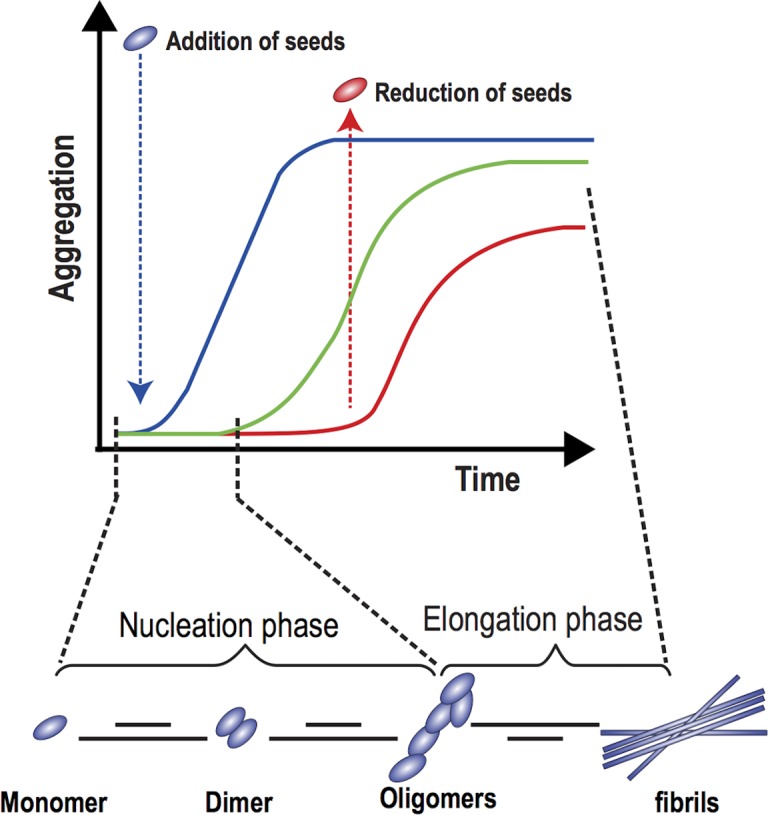
See text for details. Adapted from [48].
The two phases of Amyloid formation are shown: (i) nucleation phase, in which monomers undergo misfolding and associate to form oligomeric nuclei, and (ii) elongation phase, in which the oligomeric nuclei rapidly grow by further addition of monomers, forming larger fibrils. As explained in [48], the nucleation phase occurs gradually and at a slower rate than the elongation phase which proceeds faster being more favorable. A sigmoidal curve can thus describe the process. Addition of more monomers (seeds) speeds up the process and induces faster aggregate formation (blue curve). In contrast, the lack of monomers introduces lag time and slows down the aggregation process.
We now proceed to the modeling part. We assume that (i) monomers of Aβ aggregate to form diamers (2 monomers), triamers (3 monomers), …, etc. (ii) only monomers aggregate with diamers, triamers, …. (iii) this process of aggregation is irreversible., and finally, (iv) oligomers are formed from the aggregation of six monomers.
Let M1, M2, M3,…,Mn−1, denote the number of monomers, diamers, triamers,:…, respectively. It is assumed that n monomers aggregate to make an oligomer as shown in Fig 1. A mathematical scenario is illustrated in Table 1.
Table 1. Kinetics of monomers aggregation as a function of time.
| Reaction | Reaction rate | Change in ΔM1 |
|---|---|---|
| M1 + M1 → M2 | ΔM1(t) = −2K1M1(t)M1(t + 1) | |
| M1 + M2 → M3 | −ΔM1 = −ΔM2 = −ΔM3 | ΔM1(t) = −K2M2(t)M1(t + 1) |
| M1 + M3 → M4 | −ΔM1 = −ΔM3 = −ΔM4 | ΔM1(t) = −K3M3(t)M1(t + 1) |
| ⋮ | ⋮ | ⋮ |
| M1 + Mn−2 → Mn−1 | −ΔM1 = −ΔMn−2 = −ΔMn−1 | ΔM1(t) = −Kn−2Mn−2(t)M1(t + 1) |
It is important to note that the change ΔM1 for each reaction are different. In Table 1 column 3, we determine the change in this concentration for each reaction. Adding ΔM1 for all reactions, we get, the overall change for M1:
Which can be written as,
| (8) |
Similarly, for i >1 at the ith reaction, we have,
therefore, the overall change for Mi, i>1:
| (9) |
which can be expanded as,
Since monomers are produced by the body, we assume a source function that this is represented by f(M1). Hence the discrete model is finally given by
We now take . In case of no interaction, the first equation becomes, , which is the popular Beverton-Holt model and is illustrated in Fig 2.
Fig 2. The cobweb diagram of the Beverton-Holt model of monomer's production in the absence of aggregation.
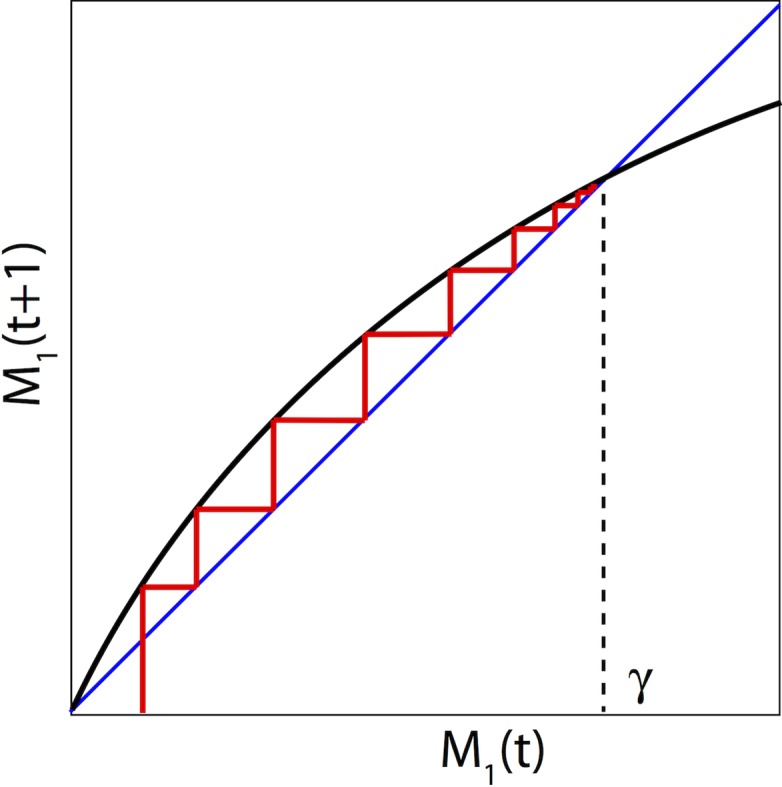
The size of monomers increases over time and reaches its carrying capacity γ.
The Beverton-Holt model [50] is a discrete-time population model which gives the population size (density) M1(t + 1) as a function of the size (density) of the previous generation M1(t). Note that (δ + 1) represents the average growth of monomer production with δ>0 and γ is the carrying capacity. Fig 3 illustrates the growth rate behavior [50, 46].
Fig 3. The figure shows the function (on the vertical axis) versus M1(t) (on the horizontal axis).
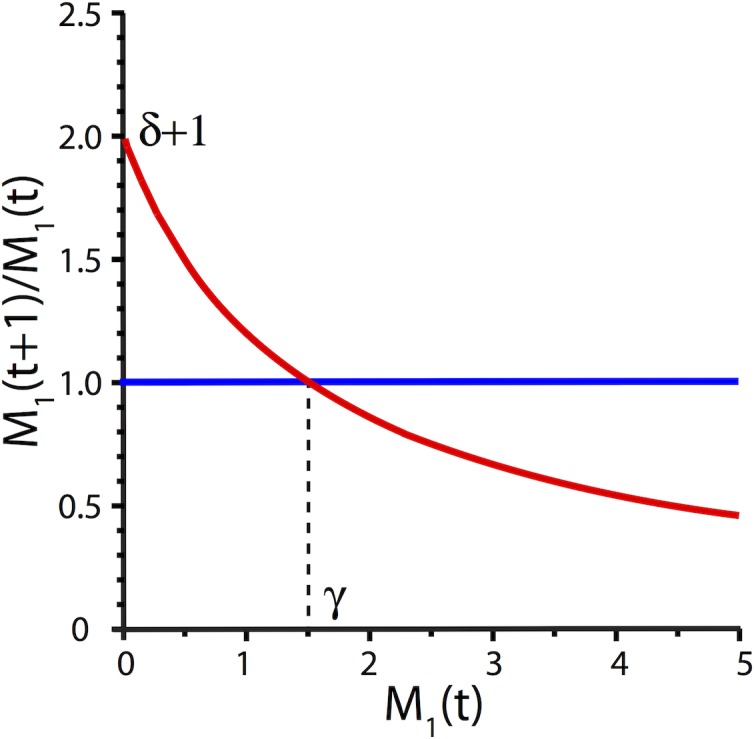
The growth rate decreases from δ + 1 when M1 is small, to γ and eventually close to zero.
Using this, we obtain,
| (10) |
The basic equilibria are the extinction point E* = (0,0,…,0) and the coexistence point , where .
Moreover, all the points on the surface are equilibrium points that are of no practical interest since in this case, no chemical reaction is present.
Stability analysis
As mentioned earlier, [43], it is accepted that 6 monomers are needed to aggregate to make one oligomer. In model (10), let us assume that n = 6. Hence we have the two equilibria E* = (0,0,0,0,0), and . The Jacobian matrix of (10) is now given by
Thus E* is unstable, since δ > 0. Next we compute J(M*).
To determine the stability of M*, we are going to use the following result due to Gerschgorin [51].
Theorem (Gerschgorin)
Let A = (aij) be a k × k matrix. Let Si be the disk in the complex plane with center at aij, and radius . Then all eigenvalues of A lie in .
To apply this result, let us determine the disks ,
We need to show that,
and
Hence
| (11) |
and
| (12) |
substituting condition (11) into (12) and letting , we obtain,
| (13) |
which is true. Hence, the first condition for the eigenvalue λ1 to be inside the unit disk is
| (14) |
For and we need
| (15) |
or,
or,
| (16) |
and
| (17) |
| (18) |
resulting in,
| (19) |
which is true assuming (16).
Hence the condition that all eigenvalues λi, 2 ≤ i ≤ n − 1 lie inside the unit disk is Ki−1 ≤ Ki, 2 ≤ i ≤ n − 1
The following theorem summarizes the above stability analysis.
Theorem
The following statements hold true
(i) E* is always unstable
(ii) M* is stable if
| (20) |
and Ki−1 ≤ Ki for 2 ≤ i ≤ n − 1
Now to prevent the aggregation of monomers to oligomers and reduce the toxicity level of the neuron cells, one should make the system unstable.
This can be accomplished by either (i) using a catalyst that would reduce K1 so that
| (21) |
and, consequently, the equilibrium point M* is unstable, or (ii) using a suppressant to limit the production of monomers to the effect that the net reproduction rate δ is reduced.
Another interesting problem is to determine exactly how many monomers are needed to make an oligomer. As mentioned earlier, we followed the literature and assumed that this number is 6.
However, one may use the formula to find the number of monomers needed to make an oligomer, as
| (22) |
Fig 4 shows the behavior of the number of monomers (n) needed to create an oligomer as a function of K1 (with gamma fixed; blue curve) and as a function of gamma (with K1 fixed; red curve).
Fig 4. The variation of n, the number of monomer that aggregates to an oligomer, relative to K1 and γ increases or K1 decreases.

As can be seen, to increase n, we can either increase γ or lower K1. By increasing n, one may reduce the number of oligomers, and consequently, may decrease the likelihood of developing the Alzheimer's disease.
Conclusion
AD is an irreversible progressive degenerative disorder that is characterized by the loss of synapses and neurons from the brain and by the presence of extracellular protein containing deposits, with β − Amyloid as the principal protein component. The presence of β − Amyloid is strongly suggested to be a causative of neural degeneration as celebrated by the celebrated Aβ hypothesis. β − Amyloid monomers aggregate to oligomers and in turn oligomers aggregate to fibrils. In this paper, we have developed a discrete mathematical model for the aggregation of monomers to oligomers. The model is based on the assumption that oligomers are the toxic stage of the aggregation, and is built using concepts from chemical kinetics and population dynamics. Based on the model, we propose a mechanism to slow down the aggregation from monomers to oligomers,
Here, γ is the carrying capacity of the β − Amyloid, δ is its growth rate, and Ki is the reaction constant of i monomers (for instance, K1 is for a monomer, K2 is for a diamer,…etc).
Furthermore, we develop an equation for the number of monomers needed to form an oligomer
where is the equilibrium state of monomers.
In this paper, we present a formula for the reduction of the aggregation of monomers to the toxic oligomers, a process thought to contribute to the development of Alzheimer Disease. To our knowledge, this is the first discrete mathematical modeling study that combines population dynamics and kinetics principles to develop a prevention mechanism that would potentially reduce the risk of Alzheimer Disease. We note that this is a mathematical model that has not been tested or implemented in a clinical trial. We derive formulations for critical parameters thought to affect the progression of the AD. Validating the model is beyond the scope of this paper and is a possible venue for future follow-up work.
Acknowledgments
This work was supported by a grant from King Abdul Aziz University, SAU.
Data Availability
No particular data set has been used. This is a mathematical modelling paper. All equations are detailed and the results can be fully derived by following these equations.
Funding Statement
This work was supported by a grant from King Abdul Aziz University, SAU. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Wang X, Ding H. Alzheimer's disease: Epidemiology, genetics, and beyond. Neurosc. Bull. 2008;24: 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hao W, Friedman A. Mathematical model on Alzheimers disease, BMC Systems Biology. 2016;10:108 doi: 10.1186/s12918-016-0348-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pallitto MM, Murphy RM. A Mathematical Model of the Kinetics of β -Amyloid Fibril Growth from the Denatured State. Biophysical Journal. 2001;81, 1805:1822 doi: 10.1016/S0006-3495(01)75831-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purves D, Augustine G, Fitzpatrick D, William WC, Anthony-Samuel L, White LE, et al. Neuroscience (5th ed.). 2012;Sunderland, MA: Sinauer Associates; p. 713. [Google Scholar]
- 5.Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Ab oligomers. FEBS. 2010;277, 1348:1358. [DOI] [PubMed] [Google Scholar]
- 6.Livadiotis G, Assas L, Dayeh MA, Elaydi S, Phea C, Roberts JL, et al. Experimental analysis of interacting plasma membrane cholesterol & ß-Amyloid. Advances in Alzheimer’s Disease. 2017;6, 75–96. [Google Scholar]
- 7.Kang J, Lemaire H-G, Unterbeck A, Salbaum JM, Masters CL, Grzeschik K-H, et al. The precursor of Alzheimers disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325, 733:736. doi: 10.1038/325733a0 [DOI] [PubMed] [Google Scholar]
- 8.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core proteins in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 1985;82, 4245:4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joachim CL, Selkoe DJ. The seminal role of β -amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1992;6, 7:34. [DOI] [PubMed] [Google Scholar]
- 10.Beyreuther K, Masters CL. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer's disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1991;1: 241–251. [DOI] [PubMed] [Google Scholar]
- 11.Hardy JA, Allisop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease Trends in Pharmac. 1991;12: 383–388. [DOI] [PubMed] [Google Scholar]
- 12.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis Science. 1992;256: 184–185. [DOI] [PubMed] [Google Scholar]
- 13.Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6: 487–498. [DOI] [PubMed] [Google Scholar]
- 14.Selkoe DJ, Hardy J. The Amyloid hypothesis of Alzheimer's disease at 25 years, EMBO Molecular Medicine. 2016;8: 595–608. doi: 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pryor NE, Moss MA, Hestekin CN. Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size Int. J. Mol. Sci. 2012;13, 3038:3072. doi: 10.3390/ijms13033038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid b-protein: reversal by tachykinin neuropeptides. Science 1990;250, 279:282. [DOI] [PubMed] [Google Scholar]
- 17.Mattson MP, Cheng B, David D, Bryant K, Lieberburg I, Rydel RE. β - Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992;12, 376:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F b-amyloid precursor protein. Nature. 1995;373, 523:527. doi: 10.1038/373523a0 [DOI] [PubMed] [Google Scholar]
- 19.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996;274:99–102.. [DOI] [PubMed] [Google Scholar]
- 20.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Nature Med. 1998;4:97–100.. [DOI] [PubMed] [Google Scholar]
- 21.Kagan B, Jang H, Capone R, Arce FT, Ramachandran S, Lal R, et al. Antimicrobial properties of amyloid peptides. Mol. Pharm. 2012. April 2;9(4):708–17. doi: 10.1021/mp200419b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 1993;13:1676 1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, et al. Secondary structure of amyloid β peptide correlates with neurotoxic activity in vitro. Mol. Pharmacol. 1994;45:373–379.. [PubMed] [Google Scholar]
- 24.Seilheimer B, Bohrmann B, Bondolfi L, Muller F, Stuber D, Dobeli H. The toxicity of the Alzheimers β -amyloid peptide correlates with a distinct fiber morphology. J. Struct. Biol. 1997;119:59 71. doi: 10.1006/jsbi.1997.3859 [DOI] [PubMed] [Google Scholar]
- 25.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, et al. Protofibrillar intermediates of amyloid β -protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 1999;19:8876–8884.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward RV, Jennings KH, Jepras R, Neville W, Owen DE, Hawkins J, et al. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of β -amyloid peptide. Biochem. J. 2000;348:137–144.. [PMC free article] [PubMed] [Google Scholar]
- 27.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 2003;26, 267–298.. doi: 10.1146/annurev.neuro.26.010302.081142 [DOI] [PubMed] [Google Scholar]
- 28.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimers amyloid beta-peptide, Nat Rev Mol Cell Biol 2007;8, 101–112.. doi: 10.1038/nrm2101 [DOI] [PubMed] [Google Scholar]
- 29.Laferla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimers disease. Nat Rev Neurosci 2007;8, 499–509.. doi: 10.1038/nrn2168 [DOI] [PubMed] [Google Scholar]
- 30.Klein WL, Krafft GA and Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimers disease conundrum, Trends Neurosci. 2001;24, 219–224.. [DOI] [PubMed] [Google Scholar]
- 31.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006;75, 333–366.. doi: 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- 32.Ferreira ST, Vieira MN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimers and other amyloid diseases. IUBMB Life. 2007;59, 332–345.. doi: 10.1080/15216540701283882 [DOI] [PubMed] [Google Scholar]
- 33.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95, 6448–6453.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006;440, 352–357.. doi: 10.1038/nature04533 [DOI] [PubMed] [Google Scholar]
- 35.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416, 535–539.. doi: 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- 36.Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924, 133–140.. [DOI] [PubMed] [Google Scholar]
- 37.Naiki H, Nakakuki K. First-order kinetic model of Alzheimers β -amyloid fibril extension in vitro. Lab. Invest. 1996;74:374–383.. [PubMed] [Google Scholar]
- 38.Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic theory of fibrillogenesis of amyloid β -protein. Proc. Natl. Acad. Sci. USA. 1997;94:7942–7947.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stine WB, Snyder SW, Ladror US, Wade WS, Miller MF, Perun TJ, et al. The nanometer-scale structure of amyloid-b visualized by atomic force microscopy. J. Protein Chem. 1996;15:193–203.. [DOI] [PubMed] [Google Scholar]
- 40.Harper JD, Wong SS, Lieber CM, Lansbury PT Jr. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997;4:119 125. [DOI] [PubMed] [Google Scholar]
- 41.Malinchik SB, Inouye H, Szumowski KE, Kirschner DA. Structural analysis of Alzheimers b(1–40) amyloid: protofilament assembly of tubular fibrils. Biophys. J. 1998;74:537–545.. doi: 10.1016/S0006-3495(98)77812-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraser PE, Duffy LK, OMalley MB, Nguyen J, Inouye H, Kirschner DA. Morphology and antibody recognition of synthetic beta-amyloid peptides. J. Neurosci. Res. 1991;28:474–485.. doi: 10.1002/jnr.490280404 [DOI] [PubMed] [Google Scholar]
- 43.Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. PNAS101. 2004;no. 4: 2817 42822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puri IK, Li L. Mathematical modeling for the pathogensis of Alzheimer's disease, PLoS One. 2010;5 12: c15176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hasegawa K, Yamach M, Naiki H. Kinetic modeling and determination of reaction constants of Alzheimer's beta amyloid fibril extension and dissociation using surface plasma resonance, Biochemistry. 2002;41:13489–98. [DOI] [PubMed] [Google Scholar]
- 46.Brauer F, Castillo-Chavez C. Mathematical Models in Population Biology and Epidemiology. Springer; 2012;Volume 40. [Google Scholar]
- 47.Yoshiike Y, Minai R, Matsuo Y, Chen YR, Kimura T, Takashima A. Amyloid oligomer conformation in a group of natively folded proteins. PLoS One 2008;3:e3235 doi: 10.1371/journal.pone.0003235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar S, Walter J. Phosphorylation of amyloid beta (Aβ) peptides A trigger for formation of toxic aggregates in Alzheimers disease. AGING. 2011;Vol. 3 No 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni CL, Shi HP, Yu HM, Chang YC, Chen YR. Folding stability of amyloid-beta 40 monomer is an important determinant of the nucleation kinetics in fibrillization. FASEB J. 2011;25:1390–1401. doi: 10.1096/fj.10-175539 [DOI] [PubMed] [Google Scholar]
- 50.Beverton RJH, Holt SJ. On the Dynamics of Exploited Fish Populations, Fishery Investigations Series II Volume XIX, Ministry of Agriculture, Fisheries and Food; 1957. [Google Scholar]
- 51.Ortega JM. Matrix Theory: A Second Course. Springer. University Series in Mathematics; 1987;XII,262p. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No particular data set has been used. This is a mathematical modelling paper. All equations are detailed and the results can be fully derived by following these equations.