

Abstract
The focus of cancer treatment has recently shifted towards targeted therapies, including immunotherapy, which allow better individualization of care and are hoped to increase the probability of success for patients. Specifically, T cells genetically modified to express chimeric antigen receptors (CARs; CAR-T cells) have generated exciting results. Recent clinical successes with this cutting-edge therapy have helped to push CAR-T cells towards approval for wider use. However, several limitations need to be addressed before the widespread use of CAR-T cells as a standard treatment. Here, we will give a succinct background on adoptive T-cell therapy, followed by a brief overview of the structure of CARs, how they are introduced into T cells, and how CAR-T cell expansion and selection is achieved in vitro. We will discuss some of the challenges in CAR design, as well as the difficulties that arise in large-scale CAR-T cell manufacture that will need to be addressed to achieve successful commercialization of this type of cell therapy. Finally, we will discuss developments already on the horizon.
Keywords: Chimeric Antigen Receptors, Genetic engineering, Immunotherapy, Manufacturing, T cells
1. Introduction
Surgery, chemotherapy and radiation therapy, alone or in combination, have been the mainstay of cancer treatment. Together with newer and more accurate diagnostic tools, these approaches have contributed to substantially improved outcomes. However, the prognosis of most malignancies remains poor. Given their complexity, most cancers will ultimately require more personalized management to achieve cure or control. Recently, the focus of cancer treatment has shifted towards targeted therapies, including immunotherapy, which allow better individualization of care and are hoped to increase the probability of success for patients.
Adoptive T-cell therapy (ATCT) is a form of immunotherapy that involves the isolation of lymphocytes with the intent to stimulate and expand ex vivo potent antigen-specific T cells that are subsequently infused into a patient to treat a disease. Adoptively transferred T cells are a “living” drug that has potential advantages over conventional therapies: T cells recognize tumor antigens through their antigen receptor (T cell receptor, or TCR) allowing them to mount a strong specific immune response potentially capable of eliminating tumors in a short period of time and, in addition, are able to proliferate and potentially survive in vivo for years [1], possibly granting them the ability to control tumors that relapse. The most exciting results with ATCT have been achieved by the genetic modification of T cells with chimeric antigen receptors (CARs; CAR-T cells). Recent clinical successes with this cutting-edge technology have helped to push T cell therapy towards approval for wider use. However, several limitations need to be addressed before the widespread use of CAR-T cells as a standard treatment.
Here, we will give a succinct overview of the structure of CARs, how they are introduced into T cells, and how CAR-T cell expansion and selection is achieved in vitro. We will provide a basic description of CAR design and clinical applications. We will mention some of the challenges in CAR design, and some of the difficulties that arise in large-scale CAR-T cell manufacture that will need to be overcome to achieve successful commercialization of this type of cell therapy. Finally, we will discuss developments already on the horizon.
2. Chimeric antigen receptors (CARs)
Some of the limitations seen with earlier types of ATCT can be overcome by redirecting T cell activity towards an antigen using a recognition system that relies on the antigen binding abilities of an antibody molecule. A chimeric antigen receptor (CAR) is a synthetic molecule that does just that. In brief, a CAR is a protein that fuses an extracellular, antibody-derived antigen recognition domain with intracellular TCR-derived, activating domains [2] (Fig. 1). The extracellular binding moiety provides the antigen specificity and is commonly a single-chain fragment variable (scFv) originating from a monoclonal antibody. When T cells are engineered to express a CAR, after binding the target antigen via the scFv, they get activated through the signaling components included in the CAR.
Figure 1. Schematic representation of a Chimeric Antigen Receptor (CAR).
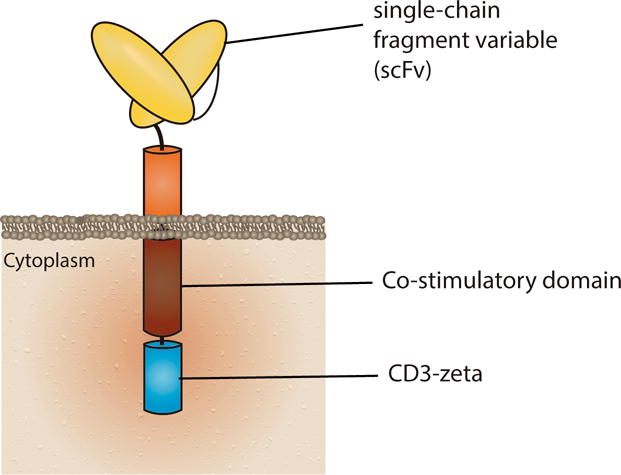
CARs currently in clinical use have an extracellular antigen recognition domain (here represented by an antibody-derived single-chain variable fragment, scFv), one or more co-stimulatory domains (signal 2), and CD3ζ (signal 1).
Since CARs provide MHC-independent antigen recognition, some of the mechanisms used by tumor cells for immune escape, such as downregulation of MHC molecules, can be mitigated [3]; in addition, a CAR specific for a certain antigen could be used in patients regardless of their specific HLA type (a common limitation of other engineered T cells). Another advantage of CAR-mediated antigen recognition is that antigens other than proteins, such as carbohydrates and lipids, can be recognized [4,5].
Nonetheless, CAR-T cells have some drawbacks. Whereas traditional T cell responses through TCR and MHC interaction can recognize intracellular proteins (which account for more than 90% of potential TAAs) that are processed and presented by the MHC molecule, binding of the CAR is limited to molecules that are present on the surface of tumor cells. Another potential disadvantage is that, because scFv sequences are usually derived from murine proteins, humoral and cellular immune responses can occur against this portion of the molecule and lead to elimination of CAR-T cells from circulation. This problem, which has been previously documented in some patients, may be minimized by using scFv whose amino acid sequences have been fully humanized [6].
Although early (first generation) CARs contained only one activating domain (usually a portion of the ζ chain in the TCR complex) and thus mimicked only TCR activation (signal 1), all clinically effective CARs include extra activating domains that trigger additional stimulatory pathways (signal 2). Most commonly, the costimulatory domains included are CD28 or 4-1BB (CD137). These second (with one additional activating domain) and third (with two or more additional domains) generation CARs are associated with increased CAR-T cell proliferation and cytokine production both in preclinical models and in clinical trials [7].
While CAR-T cells can in theory be redirected against any surface-expressed target of choice, most clinical trials so far have been directed at CD19, an antigen that is expressed in B-cell malignancies (reviewed elsewhere [8,9]). For example, phase I and II clinical trials of CD19 CAR-T cells have consistently shown high antitumor efficacy, with complete response rates of 70 to 90% in pediatric and adult patients with relapsed or refractory, chemotherapy-resistant acute B-cell leukemia. These encouraging results have attracted the interest of pharmaceutical and biotech companies and a CD19 CAR-T cell product was recently approved by the FDA for commercial use outside clinical trials by Novartis Pharmaceuticals Corp. (Kymriah™), and more companies are likely to see their products approved in the next months. Meanwhile, CAR-T cells directed against other disease targets, both in hematological (such as BCMA [10], CD20 [11], CD33[12] or CD123[13]) and solid tumors (such EFGRvIII, GD2[14], and Her2[15]), are now being tested in clinical trials.
3. CAR optimization
Despite encouraging clinical results with CD19 CAR-T cells, there is considerable heterogeneity in the structure of CARs (reviewed elsewhere [16]) used across trials in different institutions. Yet, it is unclear whether one type of CAR has advantages over another, even when targeting the same antigen. CARs have been designed empirically; distinct backbones, types and number of signaling endodomains, and methods of CAR delivery into T cells likely affect antitumor activity. For instance, the spatial interaction between a cancer cell and a T cell depends on the exact location of the epitope being recognized on the target molecule and the structure of the extracellular domain of the CAR. Differences in T cell physiology and activation owing to the use of distinct scFvs recognizing the same antigen, different co-stimulatory endodomains [17] or different lengths and sequences of the CAR extracellular domain [18] have been documented. While it is conceivable that there is an optimal distance and angle of interaction between CAR-T cells and target cells for proper T-cell activation, these properties are often impossible to predict.
Therefore, although the backbone of the CAR and the number and type of its endodomains are an absolute determinant of the overall antitumor effect observed in patients, their optimization is currently a laborious and trial-and-error procedure, with multiple iterations of “bench to bedside and back again” approaches. This process makes translation to the clinic difficult and time consuming. It is clear, however, that the CAR design process does not follow a “one size fits all” model and that each CAR must be designed, optimized and tested for the specific target molecule in order to generate the ideal CAR for a particular tumor.
4. Engineering the T cell to express a CAR
After a CAR is designed, its gene needs to be permanently introduced into T cells. The most commonly used vectors for this purpose are replication-defective retroviruses of two types: γ-retrovirus and lentivirus. Both have an RNA genome that is reverse-transcribed into DNA after T-cell transduction, which then gets integrated into the host genome. This integration process raises concerns about potential insertional mutagenesis [19], to which lentivirus may be less prone by favoring insertion sites away from active genes [20,21]. Factors such as the transgene and its promoter, and the cell type being transduced also seem to influence the risk of mutagenesis [22]. T-cell transduction appears to be safe especially considering that retroviruses have been used for this purpose for the last 30 years. Nevertheless, both types of integrative viral vectors are considered potentially oncogenic and need to be carefully tested for the absence of replication-competent particles and require extensive patient follow-up when used for clinical applications.
Instead of viral vectors, which require extensive and expensive biosafety testing, transposon/transposase systems, which are usually delivered into T cells by electroporation with plasmids, can be used. These systems have lower costs, simpler manufacturing procedures, and are potentially safer since insertions have a nearly random distribution [23]. However, these systems have disadvantages, including lower transduction efficiency, which may require prolonged periods of CAR-T cell expansion that may potentially reduce their antitumor effect in vivo. The efficacy of CAR-T cells generated using non-viral systems remains to be demonstrated, as there has been only a small number of clinical trials using them, but modest antitumor activity was observed in a clinical trial using CD19 CAR-T cells transfected with a Sleeping Beauty (a form of transposon/transposase) system [24].
Another option for T-cell transduction is the electroporation of mRNA encoding the CAR into to their cytoplasm [24]. With this strategy, there is no integration of an exogenous gene into the genome, mitigating concerns regarding genotoxicity. This system has already been tested in clinic [25] and CAR expression was detected on the T cell surface only for up to one week. Since expression of the CAR is transient, this method may be optimal in instances where persistent targeting of an antigen may be associated with life-threatening toxicities. Clinical trials of mesothelin-specific CAR-T cells transduced using this method are now in progress (NCT01355965) to investigate if CAR expression will persist long enough to elicit antitumor effects but short enough to prevent side effects.
The most recent approach to genetically modify T cells uses gene editing tools such as CRISPR or TALEN to create a double break in a particular DNA site, with the CAR gene being provided as a DNA template that is introduced into that selected genomic site. This system minimizes the risk of unrestrained genomic integration and has demonstrated good antitumor effect in a preclinical model [26]. It is, however, more complex than other approaches. Nonetheless, the use of this engineering system is under intense study.
Regardless of the actual method used for T cell transduction, from a biotechnology point of view, all these vectors can be made in large quantities and frozen for several years. When T cells are available for transduction, an aliquot can be thawed and used as needed.
5. CAR-T cell manufacturing
The first step in the generation of autologous CAR-T cells is the collection of peripheral mononuclear blood cells (PBMC) from a patient, usually through leukapheresis, whereby blood is removed from the donor and separated into different components, with the immune cells being collected while all other components returned to the donor’s circulation. T cells are then cultured in the presence of a nonspecific T cell stimulus, which is required not only for expansion but also for efficient genetic modification, since most of the transduction/transfection systems require actively dividing cells.
Because activation of T cells occurs physiologically through their interaction with myeloid cells, such as dendritic cells, these primary cells have been tested as a system for T-cell activation in vitro [27]. However, the manufacture of these natural antigen presenting cells (APCs) is time-consuming, expensive, and has variable success from donor to donor, which hampers their use as a reliable source for T-cell activation. To overcome this limitation, artificial APCs, such as the chronic myeloid leukemia cell line K562, have been used as feeder cells, allowing consistent T cell activation [28]. These artificial APCs can be modified to express costimulatory molecules (such as CD40, CD80 and CD86) that further help with T-cell stimulation. Additionally, artificial APCs can be further engineered to express the antigen targeted by the CAR, leading to selective expansion of CAR+ T cells once the artificial receptor gets expressed (shown, for example, with a PSCA-specific CAR [29]). Most often, however, simpler alternatives employing T-cell activating CD3 and CD28 monoclonal antibodies are used for T-cell stimulation [30]; these antibodies coat plasticware or are attached to suspension beads present during T-cell culture. Any of these methods leads to the generation of T cells with high replicative capacity.
T cells are then transduced or electroporated to introduce the CAR gene and expanded in culture for days to weeks (Fig 2). The primary goal of this step is to generate sufficient cells (up to 1011) to inject back into the patient to produce adequate antitumor effects. Although commonly used in research labs, conventional static cell culture systems (i.e., plates and flat flasks) are expensive, labor intensive, and difficult to scale up. CAR-T cell production for clinical trials has thus relied mostly on bioreactor culture systems, which are able to hold large volumes of culture medium and have documented advantages (reviewed elsewhere [31]). Systems such as WAVE Bioreactors™ (GE Healthcare Bio-Sciences, USA) or the more recent G-Rex™ (Wilson Wolf, USA) have been used in several forms of ATCT and in clinical trials of CAR-T cells [32]. The former consist of single-use cell culture bags that are placed on top of a rocking base that maintains a constant temperature and enables gas transfer and mixing; the latter use tall flasks that have a gas-permeable membrane on their base that allows gas exchange through convection, permitting maximum cell growth with uninterrupted access to nutrients. Both systems allow fast expansion, with capacity for scaling up. With recent improvements, CAR-T expansion requires usually 7 to 14 days until an adequate number of cells is attained.
Figure 2. Schematic representation of the manufacture process of CAR-T cells used in clinical trials.
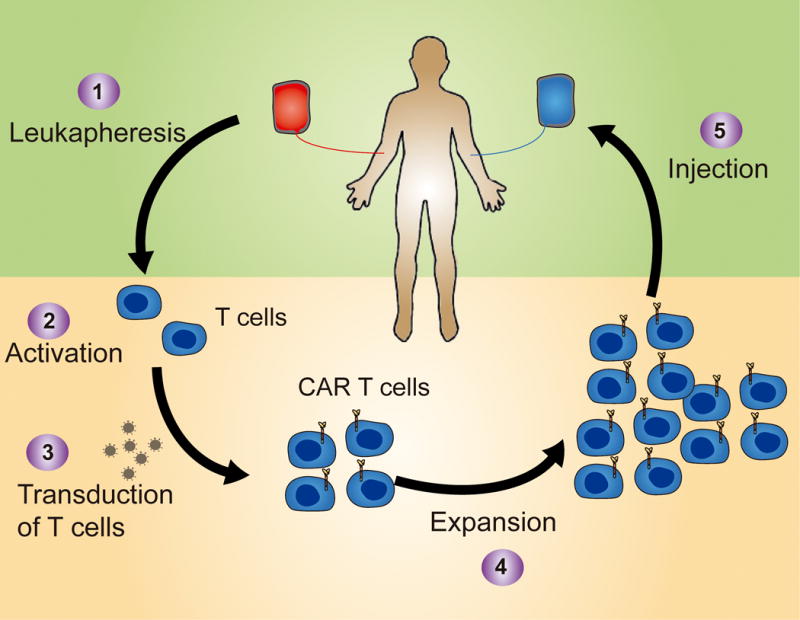
T cells are collected from the patient usually through leukapheresis (1) and then activated (2) and transduced with a retroviral vector (3). CAR-T cells are then expanded (4) to obtain sufficient numbers to infuse back into the patient a few days or weeks later (5).
Although T cells expand well in vitro, a balance is also required between maximizing T cell numbers during expansion and preventing their differentiation and exhaustion, because the latter leads to T cell senescence and limited persistence in vivo [33]. During expansion, T cells are exposed to a combination of proinflamatory cytokines that promote their growth but can also prevent their full differentiation, improving their persistence and reactivity when injected back into patients [34,35]. Different institutions use a combination of different cytokines such as IL-2, IL-7 and IL-15, or IL-15 and IL-21, but whether any particular cocktail has advantages over the others is an area of ongoing research. Nevertheless, the dual role of IL-2 in expanding conventional and regulatory T cells [33] may be a drawback to using it in this context.
To date, CAR-T cells used in most clinical trials have been expanded from a heterogeneous PBMC population without any form of selection before manufacture and administration, leading to a final product whose characteristics depend mostly on the intrinsic attributes of each patient’s circulating PBMCs. However, recent studies have suggested that certain subsets of T cells, such as naïve [36] and central memory [36] populations, may possess functional advantages, such as longer-term persistence, better proliferative capacity in vivo, and prolonged survival after adoptive cell transfer compared to more differentiated effector T cells [37,38]. Injection of less differentiated T cells has been positively correlated with antitumor response in clinical trials [39].
The reductionist approach of selecting a particular T cell subset might, however, exclude populations with lower cytotoxic effect but with complementary function. For instance, although the ratio of CD4+ to CD8+ T cells at the beginning of culture reflects that of peripheral blood, ex vivo expansion of T cells often favors the CD8+ subset. Although a pure CD8+ population is expected to have better cytotoxic activity, the use of 1:1 ratio of CD4+:CD8+ CAR-T cells has improved antitumor effect in preclinical models and showed impressive activity in a group of patients with acute lymphoblastic leukemia [40]. CD4+ T cells do not merely enhance CD8+ T cell function by release of cytokines, but they can also efficiently promote tumor elimination through their direct role in tumor rejection, as recently demonstrated [41].
Most CAR clinical trials conducted to date have not selected for a particular subset of T cells. However, if adjustment of CD4+:CD8+ ratios or enrichment for naïve/central memory populations ultimately demonstrate superior antitumor effects, manufacturing protocols may need to be altered to include extra purification steps. Clinical grade magnetic sorting for these T cell subsets has been developed [42] and can be easily adopted for the generation of CAR-T cells if required. These sorting procedures may even be helpful for isolating bulk T cells from the rest of the PBMCs prior to starting expansion, as some populations of monocytes (which are collected concomitantly during leukapheresis) can inhibit CAR-T cell expansion [43], a finding that may explain failure of CAR-T cell production from some patients.
6. Scalability of autologous CAR-T cell therapy
To date, the cost and complexity associated with manufacturing restricts CAR-T cell production to specialized centers that have the capacity to deliver the therapy to the small number of patients required for the early phase clinical trials. A jump from “proof-of-concept trials” to commercialization will require a paradigm shift from the way conventional drugs and other biologic therapies are generated.
Regardless of the specific CAR being used, CAR-T cell production needs to occur according to a set of rules that are defined under Good Manufacturing Practice (GMP) guidelines. Thus far, CAR-T cell generation in GMP facilities is performed using protocols adapted from basic research, often in open systems that require constant manipulation by highly skilled technicians. These requirements preclude production of several CAR-T cell products in parallel without exponentially increasing the number of technicians and space needed, associated costs, and risk of failure and contamination. They also complicate the design of automated systems due to the complex nature of the protocols, which entail many different steps performed over a short period of time. While some of the bioreactors described earlier allow fast expansion of T cells, CAR-T cell production depends on complex and integrated steps, especially during genetic modification, which are usually carried in independent, uncoupled systems. Under this circumstances, personalized cell therapies such as CAR-T cells cannot easily become widely available. If CAR-T cell processing became more automated, cell products could be produced for a greater number of patients more efficiently.
Conversion of these protocols, which rely on multiple manual manipulations during activation, transduction, feeding, expansion, washing, and harvesting, to fully sealed robotic procedures has proven difficult but will likely be required to reduce the risk of contamination and the overall costs associated with labor, laboratory usage, and materials. Closed and automated systems are now being developed, such as the CliniMACS Prodigy™ designed by Miltenyi Biotec, which combines a cell washer that can purify immune cells directly from blood, a magnetic cell separation system, and a cell cultivation device that also allows viral transduction of T cells [44], all of which may reduce the need for large sterile rooms. Still one limitation of this type of all-in-one machines is that during one run of T-cell expansion, the system cannot be used to produce CAR-T cells from another donor, and thus its scalability is directly dependent on the number of machines available to run in parallel.
On the other hand, patient-specific cell products can only be used for one patient per batch, and thus one of the biggest limitations of current CAR-T cell production lies in its personalized nature. Irrespective of the system used to produce CAR-T cells, large-scale production of a genetically engineered autologous cell product poses a number of challenges regarding manufacture timing and cost. Scaling up the manufacturing process of patient-specific cells requires the capacity to generate multiple independent expansions of T cells in parallel. While the conventional biopharmaceutical economic model follows the principle that costs are reduced by scaling up production, the cost per batch of CAR-T cells (specific for each patient) cannot be reduced by producing a larger batch. Thus, since it is personalized for each individual, CAR-T cell manufacturing may not have an economy of scale. Nonetheless, improvements in engineering and manufacturing technology, the use of closed systems, and automation of complex, labor-intensive steps should improve scalability to some degree, which in turn should reduce the costs [45].
7. Cost of CAR T cell technology
Although it is too soon to assess real world amounts, it has been estimated that production of CAR-T cells for a single patient may reach up to $40,000 [46]. Such costs are due to the need for sophisticated manufacturing facilities, highly trained personnel, expensive materials, and assurance of biosafety and quality. The commercial costs of CAR-T cell therapy are, however, expected to be much higher than just that of CAR-T cell production [47], likely around 10 times or more that amount. In addition, when estimating the total cost of CAR T cell therapy, other expenses, such as that of hospitalization and co-administration of other agents or drugs with the CAR-T cells, must also be considered. Nevertheless, although an expensive therapeutic approach, its potential economic benefits for the patient and society also need to be considered in any financial analyses.
8. Towards universal CAR-T cells
Ideally, any commercial cellular therapy product should allow prefabrication, be collected from a healthy third party, be easy to store, and be quickly delivered as an off-the-shelf product. Several solutions are already being tested to overcome the limitations of using autologous T cells.
One approach is to insert the CAR into other types of cells that can also be adoptively transferred. Natural Killer (NK), NK-T, and γδ T cells collected from a healthy donor may be able to replace autologous conventional T cells for CAR-cell generation and be used in cancer patients without causing graft-versus-host disease (GvHD). A potential advantage of NK cells over T cells is their innate potential antitumor activity: even if antigen loss renders tumor cells invisible to the CAR, CAR-NK cells will still express an array of receptors that may recognize aberrant expression of TAA in cancer cells [48–50]. However, whether any of these other immune cells has similar antitumor response is still unknown because they have never been tested with a CAR in a clinical trial. In addition, these cells may have some limitations as they can still be recognized and eliminated by the host immune system; they do not seem to survive as long as T cells in vivo, at least in part because of the lack of a memory population; and some of them (e.g. NK cells) may not be easily frozen without decreasing their antitumor activity [51].
The use of an immortalized cell line, which is easily expanded and scalable, is a potential solution to prevent variability among batches. Thus far, there is no immortalized T-cell line available for clinical use, but a continuously growing NK cell line (NK-92) is available [52]. This solution would be limited to a few countries since, for example, the European regulatory agency did not approve their use. Nonetheless, clinical trials using NK-92 cells are ongoing in China (NCT02944162, NCT02892695). If these current clinical trials show successful results and demonstrate safety, increased attention to this technology is expected.
The generation of allogeneic “universal”, off-the-shelf T cells is also under intense investigation. These platforms use genome editing tools to inactivate the endogenous TCR, preventing modified T cells from producing GvHD [53,54]. Disrupting the TCR at the same time that the CAR is introduced allows the T cell to be activated only through the CAR. Off-the-shelf CAR-T cells have shown efficacy in lymphoma xenograft models and in two infant patients who were treated for B-cell leukemia with CD19 CAR-T cells that had their endogenous TCRs ablated by TALEN technology and were magnetically depleted of TCR+ cells (<1% TCR+ T cells remaining) [55]. Of note, GvHD was still observed in these patients, although mild and limited only to skin, in contrast to the more widespread manifestations observed in typical GvHD; and there was evidence of alloreactivity in the marrow. In any case, this experience demonstrates that sorting of any particular subset may not be enough to totally prevent potential complications from allogeneic T cell injection.
Production of off-the-shelf CAR-T cells faces problems similar to that of autologous CAR-T cells, since it requires similar expansion protocols, in addition to the extra steps of TCR knockout and selection of TCR-negative cells (Fig. 3). Nonetheless, each batch could be used to treat more than one patient, thus decreasing dramatically the cost of production per patient, which may push adoptive cell therapy towards an industrialization and standardization production model with consistent pharmaceutical release criteria from batch to batch. Although the idea of universal CAR-T cells is attractive, their safety and efficacy has not yet been proven, and further research is required before this approach could be implemented.
Figure 3. Workflow of CAR-T cell manufacturing.
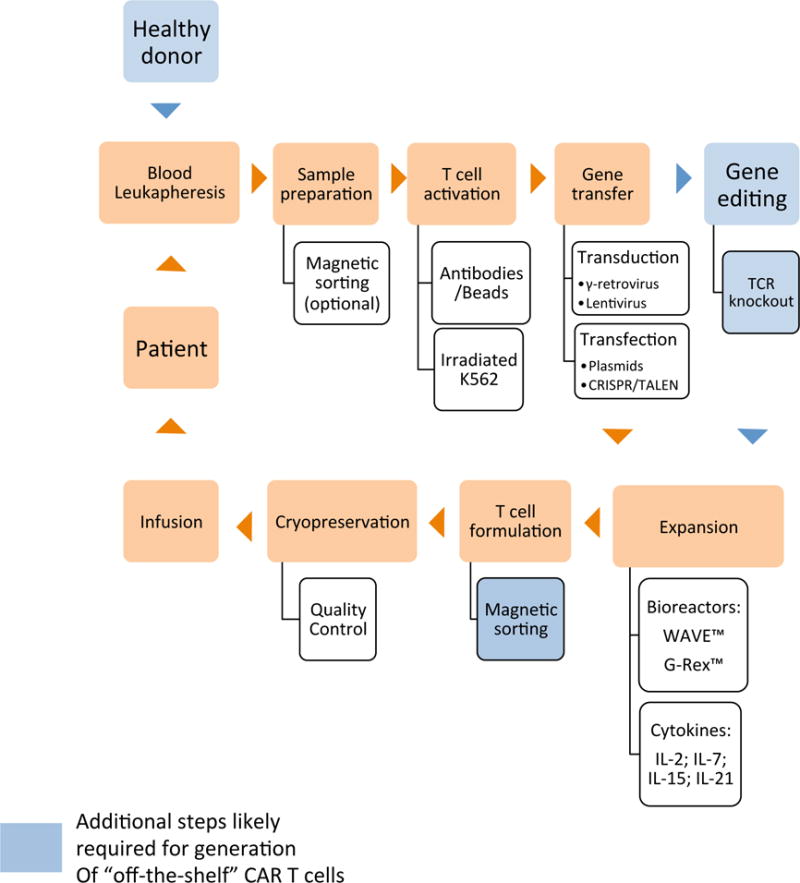
Starting with blood collection, T cells undergo a complex process evolving multiple sequential steps until infusion into the patient. Each step is performed in GMP conditions with several alternatives from institute to institute. Steps in blue are usually required for generation of “off-the-shelf” CAR T cells
Finally, the search for a “universal” effector is also being done with the CAR itself. While most CARs currently being tested in the clinic have been designed for a specific target according to the scFv in their antigen binding domains, a new line of research is developing a universal CAR that recognizes a soluble molecule that acts as a bridge between the CAR itself and the target molecule. This bridging molecule is recognized by the CAR after it binds to the target molecule on the tumor cell surface, thus directing the CAR-T cells to specific antigens [56,57]. This technology would allow the generation of one T cell product for all types of cancers. Since it would be possible to target multiple antigens simultaneously with different bridging molecules, this approach may also reduce the risk of tumor antigen escape variants. Clinical trials using this approach are in progress (NCT02776813) [58].
9. Future directions
Although CARs allow targeting of tumor cells by T cells, most cancers and their supporting stroma have evolved a multitude of ways to evade recognition by immune effectors and thus prevent tumor killing by T cells. Further T cell engineering may thus be required to extend the success observed in hematologic malignancies to solid tumors, which so far have been much more difficult to address. Examples of these additional genetic modifications include forcing the expression of chemokine receptors to increase trafficking and survival to specific tumor sites such as the brain [59]; increasing cytokine production to modulate the tumor microenvironment [60,61]; making T cells resistant to inhibitory signals [62]; or inserting artificial receptors that change how T cells perceive inhibitory signals from the tumor microenvironment [63,64]. Alternatively, targeting simultaneously immunoinhibitory pathways, including, for example, combining checkpoint blockade with CAR-T cells [65], may provide additional benefits. All these approaches are the focus of current research.
10. Conclusions
Adoptive cell therapy has entered a new phase in which genetic transfer of tumor-specific receptors into T cells can convert them into effective cancer killers. CARs allow antigen recognition without MHC restriction and thus have removed one of the obstacles to more widespread application of T cell therapy. CAR-T cells are highly targeted, like monoclonal antibodies, but possess the additional benefits of active trafficking to cancer sites, in vivo expansion and long-term persistence. Moreover, further genetic modifications can serve as countermeasures to tumor immune evasion strategies. Optimized CAR design, better understanding of the processes leading to T cell activation and memory, and improved gene transfer methods have allowed this strategy to be successfully brought to the clinic. In addition, technical improvements in cell processing, such as automation and better culture systems, will likely lead to simpler and faster CAR-T cell production, hopefully driving down its costs. Challenges regarding scalability and changes in manufacturing models to allow mass production remain hurdles to the widespread use of CAR therapy. Nonetheless, the striking results obtained with CD19-specific CAR-T cells support the idea that this therapy will soon join the mainstream of oncologic treatment, a noteworthy instance of a concept taken from the research bench to the assembly line.
Acknowledgments
The authors thank Catherine Gillespie for editing the manuscript. Diogo Gomes-Silva acknowledges Fundação para a Ciência e Tecnologia (FCT, Portugal) for financial support (SFRH/BD/52479/2014). This work was supported in part by grants from the Leukemia and Lymphoma Society Specialized Center of Research (grant 7018) and the National Institutes of Health National Cancer Institute (grant 3P50CA126752).
Abbreviations
- ATCT
adoptive T-cell therapy
- CAR
chimeric antigen receptor
- GMP
good manufacturing practices
- GvHD
graft-versus-host disease
- HLA
human leucocyte antigen
- MHC
major histocompatibility complex
- scFv
single-chain fragment variable
- PBMC
peripheral blood mononuclear cells
- TAA
tumor associated antigen
- TCR
T cell receptor
- TILs
tumor infiltrating lymphocytes
Footnotes
Conflict of interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, Ambrose D, Grupp SA, Chew A, Zheng Z, Milone MC, Levine BL, Melenhorst JJ, June CH. Science Translational Medicine. 2015;7:303. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gross G, Waks T, Eshhar Z. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:10024. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seliger B. Journal of Immunotoxicology. 2008;5:361. doi: 10.1080/15476910802482870. [DOI] [PubMed] [Google Scholar]
- 4.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Blood. 2011;118:6050. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, Chen K, Shin M, Wall DM, Hönemann D, Gambell P, Westerman DA, Haurat J, Westwood JA, Scott AM, Kravets L, Dickinson M, Trapani JA, Smyth MJ, Darcy PK, Kershaw MH, Prince HM. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:2122. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Barrett DM, Ambrose DE, Rheingold SR, Aplenc R, Teachey DT, Callahan C, Barker CS, Mudambi M, Shaw PA, Brogdon J, Young RM, Scholler J, Loew A, Marcucci KT, Finklestein J, Kulikovskaya I, Nazimuddin F, Zheng Z, Levine BL, Porter DL, Lacey SF, Melenhorst JJ, June CH, Grupp SA. Blood. 2015;126:683. [Google Scholar]
- 7.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu M, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Blood. 2011;118:6050. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turtle C, Riddell S, Maloney D. Clinical Pharmacology & Therapeutics. 2016;100:252. doi: 10.1002/cpt.392. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Geyer MB, Brentjens RJ. Blood. 2016;127:3312. doi: 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, Brudno JN, Stetler-Stevenson M, Feldman SA, Hansen BG, Fellowes VS, Hakim FT, Gress RE, Kochenderfer JN. Blood. 2016;128:1688. doi: 10.1182/blood-2016-04-711903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, Raubitschek A, Forman SJ, Greenberg PD, Riddell SR, Press OW. Blood. 2012;119:3940. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q, Wang Y, Lv H, Han Q, Fan H, Guo B, Wang L, Han W. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23:184. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo Y, Chang LJ, Hu Y, Dong L, Wei G, Huang H. Blood. 2015;126:3778. [Google Scholar]
- 14.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Blood. 2011;118:6050. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, Gray T, Wu MF, Liu H, Hicks J, Rainusso N, Dotti G, Mei Z, Grilley B, Gee A, Rooney CM, Brenner MK, Heslop HE, Wels WS, Wang LL, Anderson P, Gottschalk S. Journal of Clinical Oncology. 2015;33:1688. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srivastava S, Riddell SR. Trends in immunology. 2015;36:494. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL. Nature Medicine. 2015;21:581. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR. Cancer Immunology Research. 2015;3:125. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen-Ong G, Potter M, Mushinski J, Lavu S, Reddy E. Science. 1984;226:1077. doi: 10.1126/science.6093260. [DOI] [PubMed] [Google Scholar]
- 20.McGarrity GJ, Hoyah G, Winemiller A, Andre K, Stein D, Blick G, Greenberg RN, Kinder C, Zolopa A, Binder-Scholl G, Tebas P, June CH, Humeau LM, Rebello T. The Journal of Gene Medicine. 2013;15:78. doi: 10.1002/jgm.2691. [DOI] [PubMed] [Google Scholar]
- 21.Schambach A, Zychlinski D, Ehrnstroem B, Baum C. Human Gene Therapy. 2013;24:132. doi: 10.1089/hum.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, Mitsuyasu RT, Bernstein WB, Aronson NE, Levine BL, Bushman FD, June CH. Science Translational Medicine. 2012;4:132. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, Ivics Z, Hudecek M. Leukemia. 2017;31:186. doi: 10.1038/leu.2016.180. [DOI] [PubMed] [Google Scholar]
- 24.Kebriaei P, Huls H, Jena B, Munsell M, Jackson R, Lee DA, Hackett PB, Rondon G, Shpall E, Champlin RE, Cooper LJN. Human Gene Therapy. 2012;23:444. doi: 10.1089/hum.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon SH, Lee JM, Cho HI, Kim EK, Kim HS, Park MY, Kim TG. Cancer Gene Therapy. 2009;16:489. doi: 10.1038/cgt.2008.98. [DOI] [PubMed] [Google Scholar]
- 26.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M. Nature. 2017;543:113. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasegawa K, Noguchi Y, Koizumi F, Uenaka A, Tanaka M, Shimono M, Nakamura H, Shiku H, Gnjatic S, Murphy R, Hiramatsu Y, Old LJ, Nakayama E. Clinical Cancer Research. 2006;12:1921. doi: 10.1158/1078-0432.CCR-05-1900. [DOI] [PubMed] [Google Scholar]
- 28.Maus MV, Thomas AK, Leonard DGB, Allman D, Addya K, Schlienger K, Riley JL, June CH. Nature Biotechnology. 2002;20:143. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 29.Bajgain P, Mucharla R, Anurathapan U, Lapteva N, Leen AM, Heslop HE, Rooney CM, Vera JF. Biology of Blood and Marrow Transplantation. 2013;19:S194. [Google Scholar]
- 30.Orchard PJ, Blazar BR, Burger S, Levine B, Basso L, Nelson DMK, Gordon K, McIvor RS, Wagner JE, Miller JS. Human Gene Therapy. 2002;13:979. doi: 10.1089/10430340252939087. [DOI] [PubMed] [Google Scholar]
- 31.Somerville RPT, Dudley ME. Oncoimmunology. 2012;1:1435. doi: 10.4161/onci.21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vera JF, Brenner LJ, Gerdemann U, Ngo MC, Sili U, Liu H, Wilson J, Dotti G, Heslop HE, Leen AM, Rooney CM. Journal of Immunotherapy. 2010;33:305. doi: 10.1097/CJI.0b013e3181c0c3cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maloy KJ, Powrie F. Nature Immunology. 2005;6:1071. doi: 10.1038/ni1105-1071. [DOI] [PubMed] [Google Scholar]
- 34.Pouw N, Treffers-Westerlaken E, Kraan J, Wittink F, Hagen T ten, Verweij J, Debets R. Cancer Immunology, Immunotherapy. 2010;59:921. doi: 10.1007/s00262-010-0818-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaneko S, Mastaglio S, Bondanza A, Ponzoni M, Sanvito F, Aldrighetti L, Radrizzani M, La Seta-Catamancio S, Provasi E, Mondino A, Nagasawa T, Fleischhauer K, Russo V, Traversari C, Ciceri F, Bordignon C, Bonini C. Blood. 2008;113:1006. doi: 10.1182/blood-2008-05-156059. [DOI] [PubMed] [Google Scholar]
- 36.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, Johnson LA, Kerkar SP, Yang S, Muranski P, Palmer DC, Scott CD, Morgan RA, Robbins PF, Rosenberg SA, Restifo NP. Blood. 2011;117:808. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, Verschoor A, Schiemann M, Hofer T, Busch DH. Science. 2013;340:630. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 38.Gerlach C, Rohr JC, Perié L, van Rooij N, van Heijst JWJ, Velds A, Urbanus J, Naik SH, Jacobs H, Beltman JB, de Boer RJ, Schumacher TNM. Science. 2013;340:635. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME. Clinical Cancer Research. 2011;17:4550. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG. Journal of Clinical Investigation. 2016;126:2123. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA. Science. 2014;344:641. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riddell SR, Sommermeyer D, Berger C, Steven Liu L, Balakrishnan A, Salter A, Hudecek M, Maloney DG, Turtle CJ. The Cancer Journal. 2014;20:141. doi: 10.1097/PPO.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, Khuu H, Merchant MS, Mackall CL. Cytotherapy. 2016;18:893. doi: 10.1016/j.jcyt.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mock* U, Nickolay* L, Cheung GWK, Zhan H, Peggs K, Johnston IC, Kaiser A, Pule M, Thrasher A, Qasim W. Blood. 2015:126. doi: 10.1016/j.jcyt.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Hourd P, Chandra A, Alvey D, Ginty P, McCall M, Ratcliffe E, Rayment E, Williams DJ. Regenerative Medicine. 2014;9:799. doi: 10.2217/rme.14.47. [DOI] [PubMed] [Google Scholar]
- 46.Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S, Debets R. Frontiers in immunology. 2013;4:363. doi: 10.3389/fimmu.2013.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abou-El-Enein M, Bauer G, Reinke P. Nature Biotechnology. 2014;32:1192. doi: 10.1038/nbt.3084. [DOI] [PubMed] [Google Scholar]
- 48.Lim O, Jung MY, Hwang YK, Shin EC. Frontiers in immunology. 2015;6:286. doi: 10.3389/fimmu.2015.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, Gagea M, Banerjee P, Cai R, Bdaiwi MH, Basar R, Muftuoglu M, Li L, Marin D, Wierda W, Keating M, Champlin R, Shpall E, Rezvani K. Leukemia. 2017 doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rezvani K, Rouce R, Liu E, Shpall E. Molecular Therapy. 2017;25:1769. doi: 10.1016/j.ymthe.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mata MM, Mahmood F, Sowell RT, Baum LL. Journal of Immunological Methods. 2014;406:1. doi: 10.1016/j.jim.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, Ottmann OG, Tonn T. Journal of Cellular and Molecular Medicine. 2016;20:1287. doi: 10.1111/jcmm.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R, Lebuhotel C, Eyquem J, Cheung GWK, Duclert A, Gouble A, Arnould S, Peggs K, Pule M, Scharenberg AM, Smith J. Cancer Research. 2015;75:3853. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 54.Gouble A, Philip B, Poirot L, Schiffer-Mannioui C, Galetto R, Derniame S, Cheung G Weng-Kit, Arnould S, Desseaux C, Pule M, Smith J. Blood. 2014;124:4689. [Google Scholar]
- 55.Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, Ghorashian S, Pinner D, Ahsan G, Gilmour K, Lucchini G, Inglott S, Mifsud W, Chiesa R, Peggs KS, Chan L, Farzaneh F, Thrasher AJ, Vora A, Pule M, Veys P. Science Translational Medicine. 2017;9:2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 56.Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, Campana D. Cancer Research. 2014;74:93. doi: 10.1158/0008-5472.CAN-13-1365. [DOI] [PubMed] [Google Scholar]
- 57.Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP. Blood Cancer Journal. 2016;6:e458. doi: 10.1038/bcj.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poon M, Linn YC, Shimasaki N, Tan LK, Koh LP, Coustan-Smith E, Campana D. Blood. 2016;128:3031. [Google Scholar]
- 59.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Journal of Immunotherapy. 2010;33:780. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Blood. 2012;119:4133. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, Balyasnikova IV, Gottschalk S. Cancer Immunology Research. 2017 doi: 10.1158/2326-6066.CIR-16-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Su S, Hu B, Shao J, Shen B, Du J, Du Y, Zhou J, Yu L, Zhang L, Chen F, Sha H, Cheng L, Meng F, Zou Z, Huang X, Liu B. Scientific Reports. 2016;6:20070. doi: 10.1038/srep20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, Moon EK. Cancer Research. 2016;76:1578. doi: 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, Anurathapan U, Rendon D, Heslop HE, Rooney CM, Brenner MK, Vera JF. Molecular Therapy. 2014;22:1211. doi: 10.1038/mt.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kenderian SS, Ruella M, Shestova O, Klichinsky M, Kim MY, Porter DL, June CH, Gill SI. Blood. 2015;126:852. [Google Scholar]