

Abstract
Both schizophrenia (SZ) and substance abuse (SA) exhibit significant heritability. Moreover, N-methyl-D-aspartate receptors (NMDARs) have been implicated in the pathophysiology of both SZ and SA. We hypothesize that the high prevalence of co-morbid SA in SZ is due to dysfunction of NMDARs caused by shared risk genes. We used transgenic mice with a null mutation of the gene encoding serine racemase (SR), the enzyme that synthesizes the NMDAR co-agonist D-serine, to mimic an established genetic risk factor for SZ. We determined the effect of NMDAR hypofunction resulting from the absence of D-serine on motivated behavior using intracranial self-stimulation (ICSS) and neurotransmitter release in the nucleus accumbens using in vivo microdialysis. Compared to wild-type mice (WT), SR−/− mice exhibited similar baseline ICSS thresholds but were less sensitive to the threshold-lowering (rewarding) and the performance-elevating (stimulant) effects of cocaine. While basal dopamine (DA) and glutamate release were elevated in the nucleus accumbens of SR−/− mice, cocaine-induced increases in DA and glutamate release were blunted. γ-Amino-butyric acid (GABA) efflux was unaffected in the SR−/− mice. Together, these findings suggest that the impaired NMDAR function and a consequent decrease in sensitivity to cocaine effects on behavior are mediated by blunted DA and glutamate responses normally triggered by the drug. Projected to humans, NMDAR hypofunction due to mutations in SR or other genes impacting glutamatergic function in SZ may render abused substances less potent and effective, thus requiring higher doses to achieve a hedonic response, resulting in elevated drug exposure and increased dependence/addiction.
Keywords: D-serine, N-methyl-D-aspartate receptor, schizophrenia, serine racemase, addiction
INTRODUCTION
Substance abuse (SA) disorders are highly prevalent in individuals with schizophrenia (SZ), a disabling mental disorder that exhibits high heritability (∼80%) and affects 1% of the population worldwide (Perälä et al. 2007). Excluding caffeine dependence, studies have reported that people with SZ are 3-6 times more likely to have a SA disorder than the general public (for review, Coyle 2006). For example, nearly 90% of individuals with SZ smoke cigarettes heavily, more than 3-times the rate in the general population. Nearly 50% are addicted to alcohol, followed by cannabis and cocaine as the most abused agents (Green 2007). Thus, SA susceptibility in SZ appears to comprise all classes of abused agents including nicotine, stimulants, depressants (alcohol), and cannabis. Critically, clinical data show that SA is associated with poor outcomes in SZ, most likely because of poor compliance in treatment, exacerbation of psychosis, homelessness, more frequent hospitalizations, violence (Erkiran et al. 2006), and SA increases the cost of care (Green 2007). These adverse consequences point to the pressing need for a better understanding of factors that may contribute to comorbid SA in SZ, which will facilitate the identification of more effective treatments.
Mounting evidence from genetic, post-mortem and pharmacologic studies strongly indicate that glutamatergic dysfunction, especially decreased activity of N-methyl-D-aspartate receptors (NMDARs), is a core pathophysiologic feature of SZ (Balu & Coyle 2015; Coyle, Tsai & Goff 2002; Javitt 2007). NMDAR hypofunction is thought to be due, at least in part, to decreased availability of the forebrain NMDAR co-agonist, D-serine and the effects of other risk genes whose proteins cluster around the glutamatergic synapse (Balu and Coyle 2015). In fact, patients with SZ exhibit decreased levels of D-serine in cerebrospinal fluid and serum (Hashimoto et al. 2005) and adjunct treatment with D-serine has been shown to reduce the positive symptoms, negative symptoms, and cognitive impairments of the disorder (Coyle, 2006; Tsai & Lin 2010). There is also evidence of down-regulation of the cortical fast-firing, parvalbumin-positive γ-aminobutyric acid (GABA) interneurons, resulting in the disinhibition of glutamatergic pyramidal neurons (Benes & Berretta 2001) and a hyperglutamatergic state (Lisman et al. 2008), is a consequence of NMDAR hypofunction, and likely contributes to the negative symptoms and cognitive impairments that are hallmarks of the disease.
The NMDARs also have been implicated in neuroplastic changes underlying addictive behaviors (Hopf 2017). Specifically, modulation of the mesocorticolimbic dopamine (DA) system (the circuit in the brain responsible for reward processing) by glutamatergic inputs from cortical and subcortical regions appears to regulate both rewarding and aversive mood states, which may drive maladaptive drug-seeking behaviors (see Carlezon & Thomas 2009 for a review). Furthermore, extinction of both cocaine-induced conditioned place preference (CPP) and cocaine self-administration have been shown to be NMDAR-dependent and can be enhanced by treatment with the partial glycine modulatory site (GMS) agonist D-cycloserine (Thanos et al. 2011) or D-serine (Kelamangalath, Seymour & Wagner 2009). Of interest, several clinical studies have demonstrate that the atypical antipsychotic clozapine reduces cigarette smoking, cocaine abuse, and alcohol consumption in patients with chronic SZ, effects that are unique to clozapine (Green, 2007). Both clinical and preclinical research strongly suggests that the ‘je ne sais quoi’ of clozapine’s effects on negative symptoms and, likely on SA, is through enhancement of NMDAR function (Coyle & Tsai 2004). Furthermore, clozapine exhibits a pattern of cortical cfos expression that mirrors that of the NMDAR glycine modulatory site (GMS) partial agonist D-cycloserine (Kinney et al. 2003). Taken together, such findings have led to the view that NMDAR function has a broad influence on the development and maintenance of addictive behaviors.
Our laboratory has focused on the serine racemase (SR) gene SRR, now an established risk gene associated with SZ, (Ripke et al. 2014) as a plausible mediator of NMDAR hypofunction in cortico-limbic regions of the brain. SR converts L-serine to D-serine. Genetically silencing srr in mice (SR−/−) results in an ~85% reduction in brain D-serine levels (Basu et al. 2009) and produces a phenotype sharing many features with SZ including structural, neurochemical, and cognitive abnormalities that closely resemble those observed in humans with SZ (Balu et al. 2012; 2013; Puhl et al. 2015; Steullet et al. 2017). Notably, most of these deficits can be normalized by treatment with exogenous D-serine or with an mGluR5 positive allosteric modulator (Balu et al. 2013; 2016). Also, an immunohistochemistry study demonstrated that more than half of the D-serine positive neurons in the neocortex and hippocampus of SR−/− mice are GABAergic interneurons (Balu et al. 2014). The SR−/− mice also exhibit a down-regulation of PV-expression of the cortical GABAergic neurons that is reversed by D-serine (Steullet et al. 2017). Together these studies have established the genetic construct validity of the SR−/− model for SZ.
With regard to addiction, SR−/−mice exhibit a reduced ability to extinguish conditioned responses to amphetamine-associated stimuli and a failure to reverse amphetamine-induced synaptic changes in nAcc (Benneyworth & Coyle 2012). Moreover, alteration of NMDAR co-agonist availability either via genetic manipulation or pharmacologic treatment affects cocaine-induced CPP (i.e., extinction learning and cocaine-induced reinstatement) and locomotor sensitization (Puhl et al. 2015). Such results provide evidence for the idea that the SR−/−mouse model for NMDAR hypofunction may provide a novel and useful means for delineating neurobiological and behavioral factors that play a key role in SZ and co-morbid SA.
The present research was conducted to further evaluate the neurobiological and behavioral effects of the psychomotor stimulant cocaine in SR−/− and wild-type (WT) mice to determine how this SZ risk genotype influences cocaine’s actions on brain reward mechanisms and addiction liability. The mutations affecting the NMDAR in schizophrenia like SR might increase the hedonic effects of cocaine, thereby increasing the risk for substance abuse. Or, counter-intuitively, the mutations could reduce the hedonic response, thereby requiring higher doses to achieve a hedonic response, thus increasing the risk of dependency.
First, we employed intracranial self-stimulation (ICSS) in SR homozygous knockout (SR−/−) and WT mice to determine the cocaine-induced changes in the reward threshold measured as current and/or frequency and maximum rate of responding. ICSS can serve as a highly sensitive assay of hedonic state in rodents (Markou et al. 1991; Carlezon & Chartoff 2007) and, has been successfully used to examine how molecular and genetic manipulations can alter reward-related effects of brain stimulation and drugs of abuse (Donahue et al. 2014). Next, to determine whether changes in rewarding behavior may be accompanied by alterations in neurotransmitter release, we used in vivo microdialysis to measure DA, glutamate, and GABA efflux within the addiction-related brain region nucleus accumbens in freely-moving SR−/− and WT mice at baseline and following administration of cocaine.
MATERIALS AND METHODS
Transgenic mice
SR−/− mice (backcrossed onto a C57Bl/6J background for > 10 generations) were bred in-house by crossing heterozygous (SR+/-) males and females. Homozygous (SR−/−) offspring expressed the SR gene with exon 1 (encoding the catalytic domain of the enzyme) deleted (Basu et al. 2009). WT littermates also were used for the SR−/− strain. Prior to behavioral and neurochemical experiments, animals were group-housed with littermates in polycarbonate cages and maintained on a 12:12 h light/dark cycle in a temperature- (22 °C) and humidity-controlled vivarium. Food and water were available ad libitum. All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, 2011) and were approved by the McLean Hospital Institutional Animal Care and Use Committee.
ICSS Procedures
Mice (approximately 2 months old) were anesthetized (ketamine 80 mg/kg and xylazine 10 mg/kg; i.p.), and monopolar stimulating electrodes were stereotactically implanted in the medial forebrain bundle (MFB; in mm from bregma, anterior-posterior: −1.9, mediolateral: −1.0, dorsal-ventral: −5.0 below dura, according to the atlas of Paxinos & Franklin [2001]). Following ICSS electrode implantation surgery, animals were individually housed and allowed a 1-week recovery period. Thereafter, all mice were trained to respond for brain stimulation by spinning an operant wheel during daily 1-hour sessions over the course of 2–4 weeks (Muschamp et al. 2012). Stimulation frequency was fixed at 141 Hz, and stimulation current was adjusted to the lowest value that would support stable responding (i.e., one quarter wheel turn, which translated to one brain stimulation reward, per second). After identifying this minimum current for each mouse, the current was held constant during subsequent training sessions, where mice were allowed to respond to 15 stimulation frequencies presented in descending order (0.05 log10 unit steps) during fifteen 50-second trials. Each trial was preceded by a 5-second priming period, during which non-contingent stimulation was given at the frequency used for that trial. Also, each trial was followed by a 5-second timeout period, during which responding was not reinforced. Responding was recorded during each 50-second trial in each set of 15 trials (termed a “pass”). The range of frequencies was adjusted over the course of training for 4–6 weeks, so that mice responded through the highest 6–7 frequencies stably over six passes (90 minutes of training). The lowest frequency that supported responding (ICSS threshold or T0) was computed with least-squares line of best-fit analysis (Carlezon & Chartoff 2007). When animals were observed to have stable mean ICSS thresholds (<10% variability over three consecutive days), the effects of vehicle and different doses of cocaine (3–12 mg/kg) were determined on ICSS threshold. Mice responded through three passes immediately before drug treatment, and thresholds from the second and third passes were averaged to obtain the baseline (ICSS threshold and maximal response rate) parameters. Each mouse then received either vehicle or cocaine (3–12 mg/kg) and was tested for three passes. Mice were treated with vehicle or each dose of cocaine twice, in ascending then descending order, with a minimum of two days between treatments.
In Vivo Microdialysis Procedures
WT and SR−/− mice were anesthetized (120 mg/kg ketamine and 16 mg/kg xylazine; i.p.) and placed into a mouse stereotaxic apparatus (Stoelting Co., Wood Dale, IL, USA). An incision was made to expose the skull, and a small (2-mm in diameter) hole was drilled in the skull either on the left or right side of the mouse brain to expose the dura. Mice were then implanted with a concentric dialysis probe (see below) that was continuously perfused with saline, while secured in a CMA/10 clip (CMA/Microdialysis AB, Solna, Sweden) on a stereotaxic holder. Based on previous studies and the Paxinos & Franklin (2001) mouse brain atlas, the dialysis probe was aimed at the nucleus accumbens (shell/core co-ordinates: Anterior: +1.3–1.5; Lateral: ± 0.6–1.3; Vertical: −4.9–5.1; A and L millimeters from bregma; V millimeters from dura; Mereu et al. 2015) and, once implanted, secured to the skull using dental cement (GlasIonomer Cement CX-Plus, Henry Schein, Melville, NY). Following surgery, mice recovered overnight in hemispherical CMA-120 cages (CMA/Microdialysis AB, Solna, Sweden) equipped with fluid swivels (375/D/22QM; Instech Laboratories Inc, Plymouth, PA) for connection to the dialysis probes.
Concentric dialysis probes were assembled with AN69 dialyzing membranes (Hospal Dasco, Lyon, France) as described previously (Desai et al. 2010). Briefly, two 4-cm pieces of silica-fused capillary tubes (serving as an inlet and outlet tubing of the probe) were inserted into a 22-gauge stainless steel needle and fixed into place using glue. The inlet and outlet tubing were, respectively, set at 6- and 5-mm from the tip of the needle, and inserted into a 4-mm capillary (0.25-mm external diameter) AN69 dialyzing membrane that was enclosed with a drop of glue; the inlet tubing was set at approximately 0.1 mm from the closed end of the dialyzing fiber. Next, the dialyzing membrane was fixed to the inlet and outlet tubing with glue that limited the exposed dialyzing surface of membrane to the lower 1-mm of probe, i.e., the space between the inlet and outlet not covered by glue.
Microdialysis studies were performed on freely moving mice in the same CMA-120 cages in which they recovered overnight following surgery. Approximately 20 hr after surgery, dialysis probes were connected to fluid swivels, and Ringer’s solution (147.0 mM NaCl, 2.2 mM CaCl2, and 4.0 mM KCl) was delivered at a constant flow rate of 2.5 μl/min, through the probe using a 2.5-ml syringe (Hamilton Co., Reno, NV) controlled by CMA 402 Syringe Pump (CMA/Microdialysis AB). Dialysate sample collection began approximately 1 hr after the pump was started, and 50-μl samples were collected every 20 minutes thereafter. DA, glutamate, and GABA efflux were analyzed as described below. Cocaine or saline was administered after 6 baseline samples were taken approximately 3 hr after initiating delivery of Ringer solution) and DA levels had stabilized, i.e., less than 10% variability between the last three samples. Beginning 30-min after the saline injection, incremental cumulative doses of 3.2 to 56 mg/kg cocaine were administered every 30-min, permitting the evaluations of up to four cumulative doses during a single test session to attain complete within-subject dose-effect functions. Sampling continued every 20-min for two hours after the final cocaine injection. Mice were used only once.
DA was quantified by injecting 10-μl of the dialysate samples, without purification, into a high-performance liquid chromatography system consisting of a 150 × 3.2 mm chromatographic column (particle size 3.0 μm; ESA Inc., Chelmsford, MA), and a coulometric detector (Coulochem III; ESA, Dionex, Sunnyvale, CA). Oxidation and reduction electrode potentials of the analytical cell (5014B; ESA Inc.), respectively, were set at +125 mV and −175 mV. The mobile phase consisting of 100 mM NaH2PO4, 0.1 mM Na2EDTA, 0.5 mM n-octyl sulfate, and 18% (v/v) methanol (pH adjusted to 5.5 with Na2HPO4) was pumped at a flow rate of 0.6 ml/min by an ESA 584 solvent delivery module. Assay sensitivity for DA was 2 fmol/sample. The remaining 40 μl dialysate sample was mixed with 10 μl of 10% trichloracetic acid (TCA), and then processed through three vials, each containing 200 μl diethyl ether to neutralize the acid. Samples were then divided into 20 or 25 μl samples to, respectively, quantify glutamate (samples frozen immediately on dry ice and stored in −80 freezer until ready to be analyzed) or GABA according to Donazanti & Yamamoto (1988). Briefly, stock solutions of glutamate and GABA were prepared in Ringer solution (see above), and 40 μL of each standard was mixed with 10 μL of 10% TCA. A stock solution of o-phthaldialdehyde (OPA)/2-mercaptoethanol (βME) derivatizing reagent was prepared by dissolving 27 mg OPA in 1 ml of a 1:1 mixture of methanol and HPLC grade water (27 mg/ml), and 5 μL of βME and 9 mL of tetraborate buffer (0.1M sodium tetraborate; pH 9.3) were then added. The working derivatizing reagent was freshly prepared by diluting 2.5 ml of OPA/βME derivatizing reagent with 7.5 ml of 0.1M sodium tetraborate buffer. Both glutamate (20 μl) and GABA (25 μl) samples and standards (before, during, and after each mouse) were mixed with 15 μl of derivatizing reagent and analyzed using a Waters Xterra MS column (2.5 μm; 50 × 3.0 mm; Waters, Milford, MA, USA), an UltiMate 3000 WPS autosampler, and a coulometric detector (Dionex Coulochem III; Thermo Scientific). Oxidation and reduction electrode potentials of the analytical cell (5014B; ESA Inc.), respectively, were set at +150 mV and +550 mV. The mobile phase consisted of 100 mM Na2HPO4, 20% methanol (v/v) [adjusted to 13% methanol (v/v) to quantify glutamate], and 3.5% acetonitrile (pH adjusted to 6.7 with phosphoric acid). Assay sensitivity for glutamate and GABA was 0.6 pmol/sample and 0.2 pmol/sample, respectively.
After completion of the experiment, mice were euthanized with pentobarbital (130 mg/kg, i.p.) overdose. Brains were removed and immersed in 4% formaldehyde in saline solution. After a minimum of one week, fixed brains were sliced by microtome (Microm HM 650 V; Microm International, Thermo Scientific, Waldorf, Germany) into 30 μm serial coronal sections (orientation according to Paxinos & Franklin 2001) to identify the location of the probes. In all subjects, the location of the probes was verified by microscope; data from mice with probe placements outside of the nucleus accumbens were excluded from analysis. Figure 4 shows a schematic representation of the typical locations of the dialyzing portion of the probes implanted in the nucleus accumbens.
Figure 4.
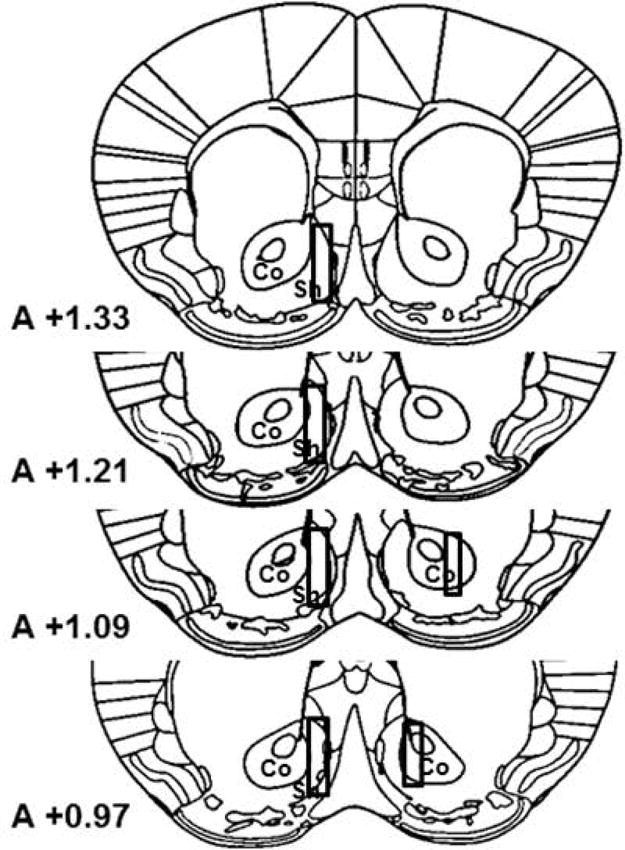
Drawings of the forebrain sections based on Paxinos and Franklin (2001) with superimposed rectangles that show the confines within which the microdialysis probe tracks were considered to be in the nucleus accumbens shell or core. Data were only included from all subjects with probe tracts within the rectangles. The anterior coordinate (measured from the bregma) is located on each section.
Drugs
Cocaine hydrochloride was obtained from the National Institute on Drug Abuse (Bethesda, MD). Cocaine was dissolved in 0.9% bacteriostatic saline and injected intra-peritoneally in a volume of 10 mL/kg. Doses of cocaine were selected on the basis of preliminary dose-ranging experiments and/or published reports on the behavioral and neurochemical effects of cocaine (Puhl et al. 2015; Desai et al. 2010; Mereu et al. 2015). Doses are expressed in terms of the free base.
Statistical Analyses
Mixed factorial analysis of variance (ANOVA) and unpaired t tests were conducted using GraphPad Prism software (Version 7.03, GraphPad Software, Inc., La Jolla, CA) to analyze ICSS data. Fisher’s least significant difference (LSD) post-hoc tests were conducted using Statistica software (Version 12, StatSoft, Tulsa, OK) when appropriate.
Time course data from in vivo microdialysis experiments are expressed as a percentage of basal DA, glutamate and GABA values, which were calculated from the basal sample that was taken immediately before the first injection. The effects of each cumulative dose of cocaine were determined as the mean of at least two dialysate samples taken from all WT and SR−/− subjects immediately after each injection. All results are presented as group means (+SEM). Time course data and effects of cumulative dose were analyzed using a two-way ANOVA (genotype and time and genotype and drug dose as factors, respectively) for repeated measures over time and dose; overall changes from basal levels were subjected to Dunnett’s t-test. Differences between basal levels of extracellular DA, glutamate, and GABA in dialysates from nucleus accumbens of WT and SR−/− mice were calculated as the mean of the last four consecutive samples from all WT and SR−/− subjects immediately preceding the first injection (Table 1), and were analyzed using an unpaired t-test. All data were examined using SigmaPlot 13.0 (Systat Software, Inc. San Jose, CA) or GraphPad Prism 5.02 (GraphPad Software, Inc. La Jolla, CA). Results were considered significant at p<0.05.
Table 1.
Basal levels of extracellular DA, glutamate, and GABA in dialysates from nucleus accumbens of WT and SR−/− mice. Data were calculated as the mean of the last four consecutive samples from all subjects immediately preceding the first injection. Significant differences between genotypes
| Genotype | DA (fmols/10μl sample) | Glutamate (pmols/20μl sample) | GABA (pmols/25μl sample) |
|---|---|---|---|
| WT | 6.42 (±0.88) | 3.92 (±0.42) | 1.16 (±0.17) |
| SR −/− | 10.79 (±1.79)* | 9.01 (±1.45)* | 1.33 (±0.12) |
= P<0.05.
RESULTS
Effects of cocaine on ICSS responding in WT and SR−/− mice
Twenty-one mice were implanted with ICSS electrodes and trained to respond for electrical stimulation (see Methods section). Upon reaching stability criteria for ICSS responding, the mice received cocaine treatment, and were included in the statistical analyses: 16 WT and 5 SR−/−. An unpaired t test showed that there was no difference in the minimum current required to maintain ICSS responding between the two genotypes, p>0.05 (see Figure 1), suggesting that the mutation did not affect baseline sensitivity to the rewarding brain stimulation. ICSS thresholds and maximum response rates were analyzed using repeated measures ANOVAs varying genotype (WT or SR−/−) and cocaine dose (0, 3, 6, or 12 mg/kg). For ICSS thresholds, significant main effects of genotype, F(1, 124)=13.80, P<0.01, and cocaine dose, F(3, 372)=29.49, P<0.01, as well as a significant genotype × cocaine dose interaction, F(3, 372)=18.50, P<0.01, were found. The main effect of cocaine dose indicates that, overall, ICSS thresholds decreased (i.e., shifted leftward) in a dose-dependent manner when cocaine was administered. Post hoc analysis showed that WT, mice exhibited significant decreases in ICSS thresholds when cocaine was administered (ps<0.01) when comparing ICSS threshold following saline versus 3, 6, and 12 mg/kg cocaine (see Figure 2). In contrast, SR−/− mice exhibited no change in ICSS threshold following cocaine administration (ps>0.05) when comparing ICSS threshold following saline versus 3, 6, and 12 mg/kg cocaine (see Figure 2).
Figure 1.
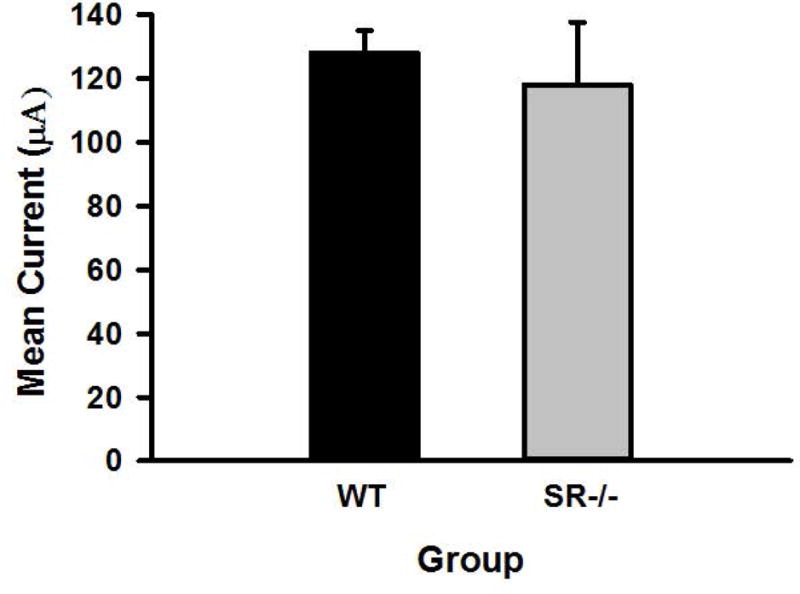
Mean minimum current (μA) required to maintain ICSS responding in WT (n=16) and SR−/− (n=5), mice.
Figure 2.
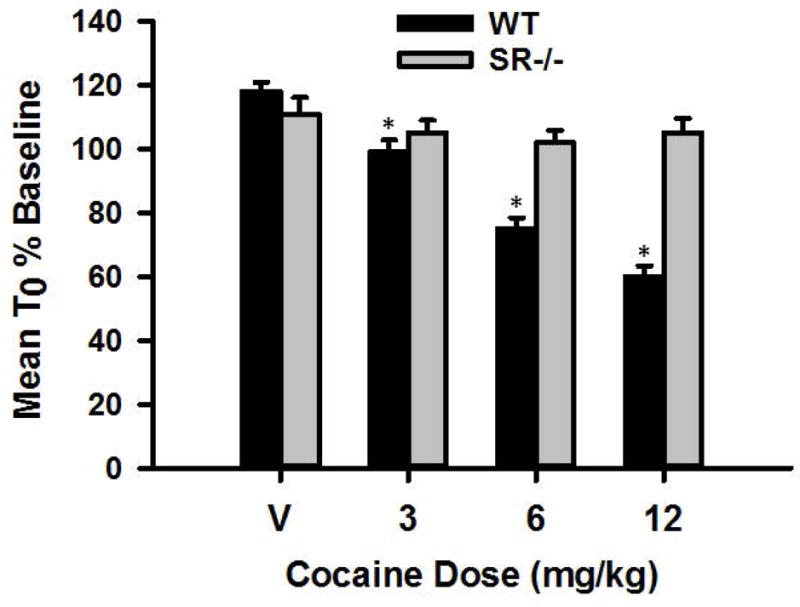
Mean ICSS threshold (T0) expressed as a percentage of baseline for WT (n=16) and SR−/− (n=5) mice following treatment with 0, 3, 6, and 12 mg/kg cocaine. * indicates statistical significance (p<0.01) compared to saline (0 mg/kg) treatment.
For maximum response rates, significant main effects of genotype, F(1, 124)=6.29, P<0.05, and cocaine dose, F(3, 372)=4.38, P<0.01 were found. The main effect of cocaine dose indicates that, overall, maximum response rate increased in a dose-dependent manner when cocaine was administered. Post hoc analysis showed that WT mice exhibited significant increases in maximum response rates when cocaine was administered (ps<0.01), i.e., when comparing maximum response rates following saline versus 3, 6, and 12 mg/kg cocaine (see Figure 3). Conversely, SR−/− mice exhibited no change in maximum response rates (ps>0.05) when comparing ICSS threshold following saline versus 3, 6, and 12 mg/kg cocaine (see Figure 3).
Figure 3.
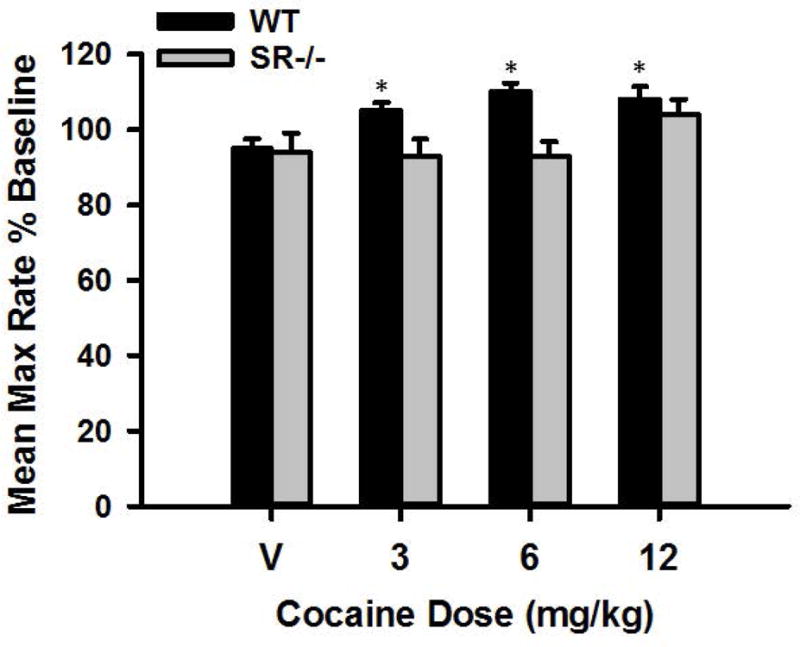
Mean ICSS maximum response rate expressed as a percentage of baseline for WT (n=16) and SR−/− (n=5) mice following treatment with 0, 3, 6, and 12 mg/kg cocaine. * indicates statistical significance (p<0.01) compared to saline (0 mg/kg) treatment.
Effects of cocaine on nucleus accumbens DA, glutamate, and GABA efflux in WT and SR−/− mice
We used in vivo microdialysis to measure basal and cocaine-induced changes in extracellular levels of DA, glutamate, and GABA within the nucleus accumbens in SR−/− and WT mice. Table 1 shows a significant difference between the baseline DA [t(42)=2.1, p<0.05] and glutamate [t(42)=3.12, p<0.05] values in dialysates of the nucleus accumbens for the two genotypes. Mean basal levels of extracellular DA and glutamate were, respectively, 1.7- and 2.3-fold higher in SR−/− mice compared to WT mice (Table 1). In contrast, no significant difference was observed between the baseline GABA dialysate values within the nucleus accumbens for the two genotypes [t(42)=0.83, p>0.05; Table 1].
Figure 5 (panels A-F) shows the effects of cumulative doses of cocaine on extracellular levels of DA, glutamate, and GABA in the nucleus accumbens in SR−/− and WT mice. The effects of vehicle injections on levels of extracellular DA, glutamate, and GABA at the outset of the experimental session were not significantly different between the two genotypes (Dunnett’s t-test: p>0.05; Figure 5, panels A-F); some modest non-significant increases in glutamate levels were observed in WT mice (Dunnett’s t-test: p>0.05; Figure 5, panels B and E).
Figure 5.
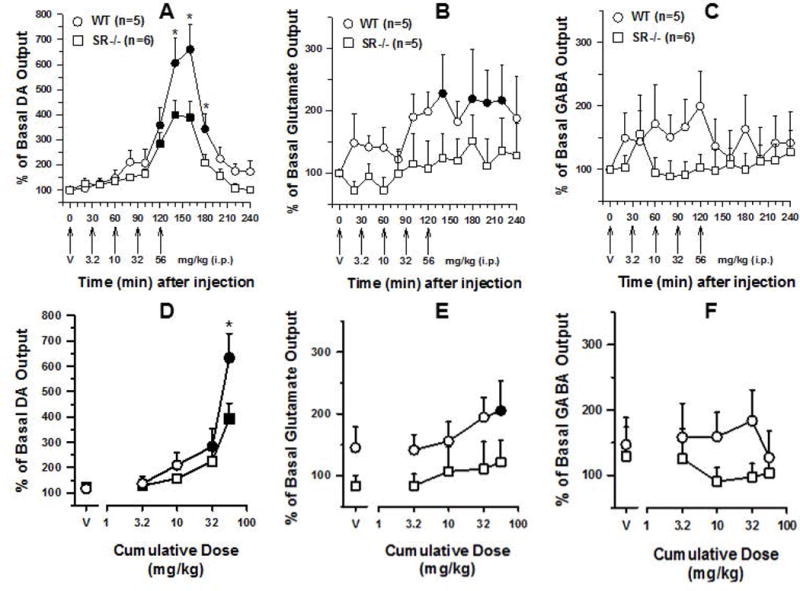
Top Panels: Time course of the effects of cumulative administration of cocaine (3.2–56 mg/kg) on the extracellular levels of DA (A), glutamate (B), and GABA (C) in the nucleus accumbens of WT (n = 5) and SR−/− (n=5/6) mice. Dialysate samples were taken from the nucleus accumbens every twenty minutes. Each arrow indicates time points at which incremental cumulative injections of cocaine were administered. Ordinates; percentage of basal DA, glutamate, and GABA level; abscissae, time in minutes after injection. Each point indicates the mean (±SEM) effect shown as of percentage of basal DA, glutamate, or GABA levels; levels were uncorrected for probe recovery. Bottom Panels: Cumulative dose-response. Changes in extracellular levels of DA (D), glutamate (E), and GABA (F) in the nucleus accumbens of WT (n = 5) and SR−/− (n=5/6) mice after administration of cocaine are shown. Ordinates, percentage of basal DA, glutamate, and GABA levels; abscissae cumulative drug dose in milligrams per kilogram. Each data point represents the mean (±SEM) of at least two dialysate samples taken from all WT and SR−/− subjects immediately following administration of each cumulative dose. Filled symbols represents values that are significantly different from basal or vehicle values (i.e., at time point “0”). Significant differences between genotypes, * = P<0.05.
Cumulative administration of cocaine (3.2–56 mg/kg) produced a significant dose- and time-dependent increase in DA levels above baseline and vehicle values in the dialysate samples from the nucleus accumbens in both genotypes (Figure 5, panels A and D). The two-way ANOVA indicated significant main effects of time [(F(12,108)=44.0, P<0.05], dose [F(4,36)=59.7, P<0.05], genotype × time [F(12,108)=3.81, P<0.05], and genotype × dose [F(4,36)=5.53, P<0.05] interactions, with non-significant main effects of genotype [Fs(1,36–108)≤2.80, Ps>0.05). Data from Figure 5 (panels A and D) show that the lower cumulative doses of cocaine (3.2 and 10 mg/kg) did not significantly increase levels of DA above baseline or vehicle values in either SR−/− or WT mice (ps>0.05), whereas higher doses of cumulatively administered cocaine (32 and 56 mg/kg) produced significant increases in levels of extracellular DA in both SR−/− and WT mice after each injection (i.e., from approximately 120 to 180 min of the observation period; ps<0.05). However, significant differences in extracellular DA levels emerged between the two groups as the cumulative dose of cocaine increased (Figure 5, panels A and D). Thus, the highest cumulative dose of 56 mg/kg cocaine produced significantly greater increases in extracellular DA levels in WT mice compared to SR−/− mice (633 ±95% versus 394 ±60%; Figure 5, panel D) for approximately 20 to 60 minutes after administration [i.e., 140 minutes WT mice = 606 ±98% versus SR−/− mice = 400 ±59%; 160 minutes WT mice = 660 ±101% and SR−/− mice = 389 ±66%; and 180 minutes WT mice = 343 ±60% vs SR−/− mice = 208 ±33%; ps<0.05; Figure 5, panels A and D). Levels of DA returned to near baseline or vehicle values approximately 90–120 minutes after administration of 56 mg/kg cocaine (Figure 5, panel A).
Unlike the effects of cocaine on extracellular DA levels, cumulatively administered cocaine (3.2–56 mg/kg) produced a modest but significant dose- and time-dependent increase in glutamate levels above baseline and vehicle values in the dialysate samples from the nucleus accumbens in WT mice (ps<0.05), but not SR−/− mice (Figure 5, panels B and E). Although the two-way ANOVA indicated significant main effects of time (F(12,108)=2.66, p<0.05) and dose [F(4,36)=4.11, P<0.05), variability among individual subjects did not permit statistical confirmation in grouped data, leading to non-significant main effects of genotype [Fs(1,36–108)≤3.02, Ps>0.05], genotype × time [F(12,108)=0.64, P>0.05), and genotype × dose [F(4,36)=0.48, P>0.05] interactions. Data from Figure 5 (panels B and E) show that the cumulative doses of 3.2 to 10 mg/kg cocaine did not produce statistically significant increases in extracellular glutamate levels above baseline or vehicle values and were generally comparable in SR−/− and WT mice (ps>0.05). In contrast, higher doses of cumulatively administered cocaine (32 and 56 mg/kg) produced some modest but significant increases in levels of extracellular glutamate above baseline or vehicle values in WT mice after each injection (from 140 to 220 minutes after first injection; ps<0.05), whereas glutamate levels remained near baseline or vehicle levels in SR−/− mice throughout the observation period (ps>0.05; Figure 5, panels B and E).
In both genotypes, cumulative administration of cocaine (3.2–56 mg/kg) produced no significant changes from baseline and vehicle values for GABA levels in the dialysate samples from the nucleus accumbens (Figure 5, panels C and F). The two-way ANOVA confirmed non-significant main effects of time [F(12,108)=0.86, P>0.05), dose [F(4,36)=1.24, P>0.05), genotype [Fs(1,36–108)≤0.89, Ps>0.05), genotype × time [F(12,108)=1.25, P>0.05), and genotype × dose [F(4,36)=2.10, P>0.05) interactions. Data from Figure 5 (panels C and F) show that the cumulative doses of cocaine (3.2–56 mg/kg) did not significantly modify levels of GABA from baseline or vehicle values and were generally comparable in SR−/− and WT mice throughout the observation period (ps>0.05).
Discussion
The present studies were conducted to determine whether a null mutation of SR, an established risk gene for SZ that synthesizes the forebrain NMDAR co-agonist D-serine, affects the behavioral and neurochemical effects of cocaine. Consistent with previous ICSS studies (Gilliss et al. 2002; Roybal et al. 2007; DiNieri et al. 2009; Donahue et al. 2014), cocaine produced a dose-dependent decrease in ICSS thresholds and a corresponding increase in maximum ICSS response rates in WT mice. In contrast, impairing NMDAR function in constitutive SR−/− mice by reducing (>85%) D-serine levels prevented cocaine-induced augmentation of ICSS responding without altering maximum ICSS rates of responding. Thus, doses of cocaine up to 12 mg/kg did not alter ICSS thresholds in SR−/− mice, whereas doses of cocaine tested (3.0, 6.0, or 12 mg/kg) produced significant decreases in ICSS thresholds in WT mice, reflecting a reward-related effect (Fig. 2). The failure of cocaine to induce a characteristic leftward shift in ICSS responding in homozygous SR knockout that exhibit a global disruption of NMDAR signaling not only highlights the importance of this receptor in mediating cocaine’s behavioral actions but also suggests that SR−/− mice are much less sensitive to cocaine’s rewarding and stimulant effects compared to WTs. This interpretation is consistent with data from our and other laboratories showing a diminished behavioral (e.g., conditioned place preference, locomotor sensitization) response to cocaine or other psychomotor stimulants (e.g., amphetamine, methamphetamine) in SR−/− mice compared to WT mice (Puhl et al. 2015; Benneyworth & Coyle 2012; Horio et al. 2012). Taken together, our results from ICSS studies designed to assess the effects of genotype on the reward-related behavioral response to cocaine strongly suggest that hypofunction of forebrain NMDARs leads to a substantial decrease in the hedonic effects of this highly addictive substance.
What might account for the diminished hedonic effects of cocaine in the SR−/− mice? Previous studies have established that the behavioral effects of cocaine, including the rewarding and reinforcing effects that are thought to underlie abuse liability, are primarily mediated by the inhibition of DA transport and, a subsequent increase in extracellular DA concentrations (e.g., Madras et al. 1989). Recently, modulation of the mesocorticolimbic DA system, the circuit in the brain responsible for reward processing, by glutamatergic inputs from cortical and subcortical regions has also been implicated in regulating both rewarding and aversive mood states, which may drive maladaptive drug-seeking behaviors (see Carlezon & Thomas 2009; Hopf 2017 for a review). Thus, it is conceivable that differences in the interplay between DA and glutamate neurotransmission may provide an explanation for the blunted hedonic effects of cocaine observed in SR−/− mice relative to WTs. In this regard, our in vivo microdialysis experiments indicate that baseline levels of extracellular DA in the nucleus accumbens are nearly 70% greater in SR−/− mice compared to WT mice (Table 1). This elevated DA output is accompanied by a significant ~3-fold increase in basal glutamate efflux without any alteration in extracellular GABA release. Interestingly, the effect of SR−/− induced NMDAR hypofunction on basal levels of accumbens DA and glutamate efflux mirrors previous observations after pharmacologically inhibiting NMDAR function with phencyclidine (Takahata & Moghaddam 2003) and following inhibition of NMDA receptors in the prefrontal cortex (Del Arco & Mora 2014). Of interest, elevated subcortical DA release is also consistent with the moderate spontaneous hyperactivity observed in the male SR−/− mice (Basu et al. 2009).
Consistent with previous studies in rodents (e.g., Mereu et al. 2015), cocaine produced a dose- and time-dependent increase in extracellular levels of DA in the nucleus accumbens of WT and SR−/− mice. However, the relative peak of extracellular DA concentrations after injection of the highest cumulative dose of cocaine (56 mg/kg) was substantially blunted in SR−/− mice by nearly 50% compared to WT mice, no differences were observed at lower cumulative doses of cocaine. In addition, cocaine failed to produce increases in glutamate efflux in the nucleus accumbens of SR−/− mice compared to WTs. These results are in line with a previous report by Horino et al. (2012) showing reduced methamphetamine-induced increases in extracellular DA in the nucleus accumbens in SR−/− mice that have been previously treated with methamphetamine compared to methamphetamine-treated WT mice. Together, our findings raise the possibility that NMDAR hypofunction and a consequent decrease in sensitivity to the stimulant and rewarding effects of cocaine are mediated by blunted DA and glutamate responses normally triggered by the drug. Another example of a link between receptor hypofunction and drug addiction stems from recent research showing that the α5 nicotinic acetylcholine receptor allelic variant that is hypofunctional is most robustly associated with tobacco smoking/nicotine addiction (Wen et al. 2016).
Of interest, the elevated basal and blunted cocaine-induced increases in extracellular DA in SR−/− mice are consistent with the likelihood that the DA receptors have been desensitized by the chronically elevated synaptic DA. Such an interpretation is supported by data from previous research showing desensitization of the calcium signal associated with agonist activiation of DA D1 and D2 receptor hetro-oligomers (So et al. 2007). Of course, other adaptive changes secondary to chronically elevated extracellular DA in nucleus accumbens may also contribute to the reduced hedonic effects of acute cocaine-induced increases in DA, such as up-regulation of dynorphin expression (Berke & Hyman 2000).
Anhedonia is a core negative symptom of SZ (Blanchard & Cohen 2006), and individuals with SZ less frequently engage in motivated behaviors aimed at obtaining rewards (Strauss, Waltz & Gold 2014). Likewise, anhedonia is frequently reported in substance-dependent individuals, especially during and after withdrawal (Garfield, Lubman & Yücel 2014). Anhedonia also increases the likelihood of relapse to drug abuse (Cook et al. 2010; Hatzigiakoumis et al., 2011) and is correlated with length of abstinence (Garfield et al. 2014). Together, this evidence suggests an increased susceptibility to substance abuse among individuals with SZ that may be due to shared neural substrates, including glutamatergic dysregulation (Coyle 2006).
Although there has been some debate over whether negative symptoms in SZ contribute to stimulant abuse, Bennett et al. (2009) found more severe positive and negative symptoms in individuals with SZ who were abusing cocaine as compared to those who were cocaine free. As SR−/− mice reproduce the cellular pathology, cortical atrophy and cognitive impairments associated with negative symptoms in SZ (Balu et al. 2014) and since the SR gene, SRR, and several genes expressing proteins down-stream to SR are risk genes for SZ (Balu & Coyle 2015), the SR−/− mouse is a genetic model of SZ, especially the negative and cognitive symptoms, with construct validity.
Based upon the evidence of a reduced hedonic response to cocaine in the SR−/− mice and their persistently elevated extracellular DA levels in the nucleus accumbens, we propose that NMDAR hypofunction creates a state in which cocaine becomes less potent and effective and, therefore, considerably higher doses of cocaine are required to achieve a hedonic response in individuals with SZ than in unaffected individuals. As a consequence, individuals with SZ who abuse cocaine are at markedly higher risk of developing severe physiologic dependency and withdrawal. Thus, contrary to our expectation that the high prevalence of substance abuse in individuals with SZ was due to greater hedonic responses to abused substances, our results strongly suggest that the high prevalence of substance abuse in these individuals may be related to their need for considerably higher doses of the drug to obtain a hedonic response, thereby increasing overall drug exposure and the likelihood of physical dependence and addiction (Koob & Volkow 2010).
Acknowledgments
The authors thank Abigail Alexander and Emery Mokler for technical assistance with the ICSS experiments and Dr. Scott Lukas for helpful analytical discussions.
FUNDING AND DISCLOSURES: This research was supported by NIH grant RO1MH51290-18 to JTC, R01MH063266-17 to WC, DA031231 K01 NIH/NIDA grant to RID, and a NARSAD Young Investigator Award to MDP. WC has numerous patents related to the use of kappa- opioid receptor ligands to treat psychiatric illness, which is outside the scope of the present work. He has served as a consultant for Cerecor over the past 3 years. JTC reports a patent on the clinical use of D-serine that is owned by Partners Health Care and consulting with Novartis and Forum Pharm over the last 3 years. Since completing this study, MDP has taken a position with Alkermes. No other conflicts are reported.
Footnotes
AUTHORSHIP CONTRIBUTIONS
M.D. Puhl and R.I. Desai equally contributed to this manuscript and should be considered first authors. M.D. Puhl, R.I. Desai, and JT Coyle participated in research design for behavioral and neurochemical experiments. M.D. Puhl conducted all ICSS studies and R.I. Desai conducted all microdialysis experiments. M.D. Puhl and R.I. Desai performed behavioral and neurochemical data analysis, respectively. M.D. Puhl, R.I. Desai and JT Coyle wrote or contributed to the writing of the manuscript. J.T. Coyle acquired funding for the research.
References
- Balu DT, Coyle JT. The NMDA receptor ‘glycine modulatory site’ in schizophrenia: D-serine, glycine, and beyond. Curr Opin Pharmacol. 2015;20:109–115. doi: 10.1016/j.coph.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Basu AC, Corradi JP, Cacace AM, Coyle JT. The NMDA receptor co- agonists, D-serine and glycine, regulate neuronal dendritic architecture in the somatosensory cortex. Neurobiol Dis. 2012;45:671–82. doi: 10.1016/j.nbd.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Li Y, Puhl MD, Benneyworth MA, Basu AC, Takagi S, Bolshakov VY, Coyle JT. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci U S A. 2013;110(26):E2400–9. doi: 10.1073/pnas.1304308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Takagi S, Puhl MD, Benneyworth MA, Coyle JT. D-serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell Mol Neurobiol. 2014;34:419–435. doi: 10.1007/s10571-014-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Li Y, Takagi S, Presti KT, Ramikie TS, Rook JM, Jones CK, Lindsley CW, Conn PJ, Bolshakov VY, Coyle JT. An mGlu5-Positive Allosteric Modulator Rescues the Neuroplasticity Deficits in a Genetic Model of NMDA Receptor Hypofunction in Schizophrenia. Neuropsychopharmacology. 2016;41(8):2052–61. doi: 10.1038/npp.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, Jiang ZI, Benneyworth MA, Froimowitz MP, Lange N, Snyder SH, Bergeron R, Coyle JT. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry. 2009;14:719–27. doi: 10.1038/mp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Bennett ME, Bellack AS, Brown CH, DiClemente C. Substance dependence and remission in schizophrenia: A comparison of schizophrenia and affective disorders. Addict Behav. 2009;34:806–14. doi: 10.1016/j.addbeh.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benneyworth MA, Coyle JT. Altered acquisition and extinction of amphetamnine-paired context conditioning in genetic mouse models of altered NMDA receptor function. Neuropsychopharmacology. 2012;37(11):2496–2504. doi: 10.1038/npp.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–32. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Blanchard JJ, Cohen AS. The structure of negative symptom within schizophrenia: Implications for assessment. Schizophr Bull. 2006;32:238–245. doi: 10.1093/schbul/sbj013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protoc. 2007;2:2987–2995. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Thomas MJ. Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology. 2009;56(Suppl 1):122–32. doi: 10.1016/j.neuropharm.2008.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook J, Spring B, McChargue D, Doran N. Effects of anhedonia on days to relapse among smokers with a history of depression: A brief report. Nicotine Tob Res. 2010;12:978–982. doi: 10.1093/ntr/ntq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT. Substance use disorders and schizophrenia: A question of shared glutamatergic mechanisms. Neurotox Res. 2006;10:221–233. doi: 10.1007/BF03033359. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology (Berl) 2004;174(1):32–8. doi: 10.1007/s00213-003-1709-2. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G, Goff DC. Ionotropic glutamate receptors as therapeutic targets in schizophrenia. Curr Drug Targets CNS Neurol Disord. 2002;1:183–189. doi: 10.2174/1568007024606212. [DOI] [PubMed] [Google Scholar]
- So CH, Verma V, O’Dowd BF, George SR. Desensitization of the Dopamine D1 and D2 Receptor Hetero-Oligomer Mediated Calcium Signal by Agonist Occupancy of Either Receptor. Mol Pharmacol. 2007;72:450–462. doi: 10.1124/mol.107.034884. [DOI] [PubMed] [Google Scholar]
- Del Arco A, Mora F. Prefrontal cortex-nucleus accumbens interaction: in vivo modulation by dopamine and glutamate in the prefrontal cortex. Pharmacol Biochem Behav. 2008;90:226–35. doi: 10.1016/j.pbb.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Desai RI, Paronis CA, Martin J, Desai R, Bergman J. Monoaminergic psychomotor stimulants: discriminative-stimulus effects and dopamine efflux. J Pharmacol Exp Ther. 2010;333:834–843. doi: 10.1124/jpet.110.165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue RJ, Muschamp JW, Russo SJ, Nestler EJ, Carlezon WA., Jr Effects of striatal ∆FosB overexpression and ketamine on social defeat stress-induced anhedonia. Biological Psychiatry. 2014;76:550–558. doi: 10.1016/j.biopsych.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donazanti BA, Yamamoto BK. An improved and rapid HPLC-EC method for the isocratic separation of amino acid neurotransmitters from brain tissue and microdialysis perfusates. Life Sci. 1988;43:913–22. doi: 10.1016/0024-3205(88)90267-6. [DOI] [PubMed] [Google Scholar]
- Erkiran M, Ozünalan H, Evren C, Aytaçlar S, Kirisci L, Tarter R. Substance abuse amplifies the risk for violence in schizophrenia spectrum disorder. Addict Behav. 2006;31(10):1797–805. doi: 10.1016/j.addbeh.2005.12.024. [DOI] [PubMed] [Google Scholar]
- Garfield JB, Lubman DI, Yücel M. Anhedonia in substance use disorders: A systematic review of its nature, course and clinical correlates. Aust N Z J Psychiatry. 2014;48:36–51. doi: 10.1177/0004867413508455. [DOI] [PubMed] [Google Scholar]
- Gilliss B, Malanga CJ, Pieper JO, Carlezon WA., Jr Cocaine and SKF-82958 potentiate brain stimulation reward in Swiss-Webster mice. Psychopharmacology (Berl) 2002;163(2):238–248. doi: 10.1007/s00213-002-1153-8. [DOI] [PubMed] [Google Scholar]
- Green AI. Pharmacotherapy for schizophrenia and co-occurring substance use disorders. Neurotox Res. 2007;11(1):33–40. doi: 10.1007/BF03033480. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindström LH, Iyo M. Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:767–9. doi: 10.1016/j.pnpbp.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hatzigiakoumis DS, Martinotti G, Giannantonio MD, Janiri L. Anhedonia and substance dependence: Clinical correlate and treatment options. Front Psychiatry. 2011;2:10. doi: 10.3389/fpsyt.2011.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW. Do specific NMDA receptor subunits act as gateways for addictive behaviors? Genes Brain Behav. 2017;16(1):118–138. doi: 10.1111/gbb.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horio M, Kohno M, Fujita Y, Ishima T, Inoue R, Mori H, Hashimoto K. Role of serine racemase in behavioral sensitization in mice after repeated administration of methamphetamine. PLoS ONE. 2012;7(4):e35494. doi: 10.1371/journal.pone.0035494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. Glutamate and schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- Kelamangalath L, Wagner JJ. D-serine treatment reduces cocaine-primed reinstatement in rats following extended access to cocaine self-administration. Neuroscience. 2010;169:1127–35. doi: 10.1016/j.neuroscience.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney GG, Sur C, Burno M, Mallorga PJ, Williams JB, Figueroa DJ, Wittmann M, Lemaire W, Conn PJ. The glycine transporter type 1 inhibitor N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine potentiates NMDA receptor-mediated responses in vivo and produces an antipsychotic profile in rodent behavior. J Neurosci. 2003;23(20):7586–91. doi: 10.1523/JNEUROSCI.23-20-07586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madras BK, Fahey MA, Bergman J, Canfield DR, Spealman RD. Effects of cocaine and related drugs in nonhuman primates. I. [3H]cocaine binding sites in caudate-putamen. J Pharmacol Exp Ther. 1989;251:131–141. [PubMed] [Google Scholar]
- Markou A, Koob GF. Postcocaine anhedonia. An animal model of cocaine withdrawal. Neuropsychopharmacology. 1991;4:17–26. [PubMed] [Google Scholar]
- Martinotti G, Nicola MD, Reina D, Andreoli S, Focà F, Cunniff A, Tonioni F, Bria P, Janiri L. Alcohol protracted withdrawal syndrome: The role of anhedonia. Subst Use Misuse. 2008;43:271–284. doi: 10.1080/10826080701202429. [DOI] [PubMed] [Google Scholar]
- Muschamp JW, Nemeth CL, Robison AJ, Nestler EJ, Carlezon WA., Jr ∆FosB enhances the rewarding effects of cocaine while reducing the pro-depressive effects of the kappa opioid receptor agonist U50488. Biol Psychiatry. 2012;71:44–50. doi: 10.1016/j.biopsych.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council. Guide for the care and use of laboratory animals (Eighth Edition) The national academies press; Washington, D.C.: 2011. www.nap.edu. [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd. Academic Press; San Diego, CA: 2001. p. 2001. [Google Scholar]
- Perälä J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsä E, Pirkola S, Partonen T, Tuulio-Henriksson A, Hintikka J, Kieseppä T, Härkänen T, Koskinen S, Lönnqvist J. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch Gen Psychiatry. 2007;64:19–28. doi: 10.1001/archpsyc.64.1.19. [DOI] [PubMed] [Google Scholar]
- Puhl MD, Berg AR, Bechtholt AJ, Coyle JT. Availability of N-methyl-D- aspartate receptor co-agonists affects cocaine-induced conditioned place preference and locomotor sensitization: Implications for co-morbid schizophrenia and substance abuse. Journal of Pharmacology & Experimental Therapeutics. 2015;353:465–470. doi: 10.1124/jpet.115.223099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JT, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steullet P1, Cabungcal JH, Coyle J, Didriksen M, Gill K, Grace AA, Hensch TK, LaMantia AS, Lindemann L, Maynard TM, Meyer U, Morishita H, O’Donnell P, Puhl M, Cuenod M, Do KQ. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol Psychiatry. 2017;22(7):936–943. doi: 10.1038/mp.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss GP, Waltz JA, Gold JM. A review of reward processing and motivational impairment in schizophrenia. Schizophr Bull. 2014;40(Suppl 2):S107–16. doi: 10.1093/schbul/sbt197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata R, Moghaddam B. Activation of glutamate neurotransmission in the prefrontal cortex sustains the motoric and dopaminergic effects of phencyclidine. Neuropsychopharmacology. 2003;28:1117–24. doi: 10.1038/sj.npp.1300127. [DOI] [PubMed] [Google Scholar]
- Thanos PK, Subrize M, Lui W, Puca Z, Ananth M, Michaelides M, Wang GJ, Volkow ND. D-cycloserine facilitates extinction of cocaine self-administration in C57 mice. Synapse. 2011;65:1099–105. doi: 10.1002/syn.20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522–37. doi: 10.2174/138161210790361452. 2010. [DOI] [PubMed] [Google Scholar]
- Wen L, Jiang K, Yuan W, Cui W, Li MD. Contribution of Variants in CHRNA5/A3/B4 Gene Cluster on Chromosome 15 to Tobacco Smoking: From Genetic Association to Mechanism. Mol Neurobiol. 2016;53(1):472–484. doi: 10.1007/s12035-014-8997-x. [DOI] [PubMed] [Google Scholar]