

Abstract
Introduction
Sepsis is the third leading cause of morbidity and mortality in neonates. Sepsis in neonates is characterized as the systemic inflammation due to infection within the first 28 days after birth. The molecular mechanism causing the exaggerated inflammation phenotype in neonates has not been completely elucidated. Receptor interacting protein kinase 3 (RIPK3) is a protein identified as a mediator in programmed necrosis or necroptosis. We hypothesize that RIPK3, could be responsible for the inflammatory response in neonates and that deficiency in the RIPK3 protein attenuates inflammation and organ injury in neonatal sepsis.
Methods
Male and female C57BL6 wild-type (WT) and RIPK3 knock-out (KO) newborn mice aged 5–7 days (3–4 g body weight) were injected intraperitoneally with 0.9 mg/g cecal slurry (CS). At 10 h after injection, the newborns were euthanized and blood, the lungs and gut tissues were collected.
Results
At 10 h after CS injection, serum cytokines IL-6 and IL-1β in the WT mice were increased by 511- and 43-fold whereas in KO mice, these levels were increased by 166-fold and 22-fold, respectively. Lung IL-1β in the WT mice increased by 7-fold after CS injection whereas only a 4-fold increase was seen in the KO mice. In the lungs of CS injected KO mice, the injury score, MIP-2 mRNA, myeloperoxidase (MPO) activity and TUNEL staining were significantly reduced by 76%, 70%, 26% and 74%, respectively compared to the CS WT mice. Gut TUNEL staining was also reduced by 80%.
Conclusion
The deficiency in RIPK3 attenuated serum and lung cytokines, lung injury and neutrophil infiltration and lung and gut apoptosis. These data suggest that RIPK3, in part, is responsible for the systemic inflammatory response in neonatal sepsis.
Keywords: RIPK3, neonatal sepsis, lung injury, cytokines, apoptosis
Introduction
Neonatal sepsis poses a significant health burden and is associated with high morbidity and mortality [1]. Low birth weight infants are especially vulnerable to sepsis and appropriate interventions are needed as early as possible in improving survival outcome [2]. The pathogenesis of neonatal sepsis is characterized by an exaggerated inflammatory response leading to single or multi-organ dysfunctions. Increase in pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 are frequently observed in both animal models and in infants suffering from neonatal sepsis [3–7]. While there is much improvement in diagnosis of neonatal sepsis, little is known about the molecular factors responsible for the immune response leading to end organ damage and cell death during neonatal sepsis.
Receptor interacting protein kinase 3 (RIPK3) is a member of the RIP kinase family and is expressed in multiple tissues including the tissues of the hemopoietic cell lineages. Receptor interacting protein belongs to a family of serine/threonine kinases that are sensors of cellular stress [8]. Two members of the RIPK family, RIPK1 and RIPK3, interact together and mediate programmed necrosis or necroptosis [9–11]. Necroptosis is a type of cell death that is controlled by the kinase activity of RIPK1 [12]. The crucial role of RIPK1 in cellular models of necrosis in tumor necrosis factor (TNF) receptor 1, TNF-related apoptosis inducing ligand receptor (TRAIL), Fas, Toll-Like Receptor (TLR) 3 and 4 has been shown in a number of studies [13,14]. Allosteric inhibition of the kinase activity of RIPK1 by necrostatin blocks necrosis without affecting the activation of NF-kB or MAPK pathways. Interestingly, in the presence of caspase inhibitor, zVAD-fmk, cells were protected from TNF-induced apoptosis whereas in other cells, zVAD treatment caused TNF-induced necrosis [15–17]. At first, RIPK3 was regarded as a protein associated with RIPK1. However, several studies implicated RIPK3 in the regulation of apoptosis and NF-kB signaling [18–20]. Mutagenesis studies showed that phosphorylation of RIPK1 by RIPK3 is crucial in inhibiting either RIPK1 or TNF-induced NF-kB activation [21]. Therefore, although RIPK3 was first identified as a protein associated with RIPK1, more recent work identified RIPK3 as a mediator of necroptosis which is caspase-independent. A combination of genome wide siRNA analysis and biochemical studies revealed that RIPK3 is the key determinant of necrotic cell death downstream to RIPK1 [10]. Later it was confirmed that RIPK3 expression determines the cells to undergo necroptosis implicating RIPK3 as the molecular switch which determines whether cells undergo apoptosis or necroptosis [9–12].
RIPK3 mediated necrotic cell death accelerates systemic inflammation and mortality [22]. In addition, in animal models of cerulean-induced pancreatitis, in adult severe sepsis models and in high dose endotoxemia, the expression of RIPK3 was dramatically increased [23–25]. To delineate the importance of RIPK3 signaling, mice deficient in RIPK3 was generated by gene targeting. Mice lacking RIPK3 were healthy and fertile and showed no signs of impairment in either TNF receptor or Toll like receptor-induced NF-kB activation [26]. When embryonic fibroblasts from wild type mice experienced cell death, those cells from RIPK3 knockout mice were resistant to necrosis following treatment with various necrosis inducing agents [10]. Neonatal immune response is considered immature and the cytokine responses are polarized to Thelper (Th) 2 and Th-17 with impairment in Th-1 cytokines. As such, the immunological profiles of the neonates are distinct from adults [27]. In this study, we hypothesized that the deficiency in RIPK3 attenuates inflammation and tissue injury in neonatal sepsis. To test this, sepsis was induced in the wild-type (WT) and RIPK3 deficient newborn mice (RIPK3KO) and inflammatory response leading to end-organ damage and cell death was examined in neonatal sepsis.
1. Materials and Methods
1.1. Experimental animals
Breeder pairs of RIPK3KO (Genentech, San Francisco, CA) and C57BL6 (WT) mice (Charles River Laboratories, Wilmington, MA) were purchased and bred at our animal facility. All mice were kept in temperature-controlled room under 12 h light/dark cycle and fed a standard Purina rodent chow diet. Newborn mice from each strain aged 5–7 days (3–4 g body weight) were used for all studies. Pups were not identified as either male or female and they remained with their mothers throughout. Experimental procedures were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. This project was approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research.
1.2. Mouse model of neonatal sepsis
Neonatal sepsis was induced by a cecal slurry (CS) method adapted from Wynn et al., [28] with some modifications [29]. Cecal slurry was prepared from six adult house-bred mice (3 males and 3 females) aged 11–14 weeks. Briefly, mice were euthanized via CO2 and cecal contents were collected and suspended in 5% dextrose for a dilution of 84.7 mg/ml. The cecal slurry was filtered through a 70 μm filter, separated as 1ml aliquots, and stored at −80°C. To administer CS, newborn mice were removed from the mothers, separated into two groups, placed on a 37°C heating pad and anesthetized using 2.5% inhalational isoflurane. One group was injected intraperitoneally with 0.9 mg/g body weight CS with a 29-gauge needle and the other group was left untreated and served as controls for the experiment. Subsequently, they were returned to cages with their respective mothers for recovery from anesthesia. A new aliquot of CS was used within 2 h of thawing for each experiment. At 10 h after CS injection, the pups were re-anesthetized and blood, lungs and gut samples were harvested. Blood was centrifuged at 7000 g for 10 min and serum collected. Lungs and gut samples were either flash-frozen in liquid nitrogen or fixed in formalin for histology. Collected serum and frozen tissue samples were stored at −70°C until analysis. The 10 h time point was chosen based on a pilot study in the wild type mice (n=6) where we observed no mortality at 10 h after induction while 100% mortality was seen at 24 h after induction.
1.3. Experimental groups
For each experiment, four groups of newborn mice (WT control, WT CS, KO control and KO CS) aged 5–7 days were used. In general, we obtain 6–8 pups from each mother for each strain. Pups used were randomly separated for controls and CS injection for both WT and KO strains. All assays performed were conducted at the same time for all four groups. To achieve statistical power, 6–8 pups per group were used and a maximum of two litters from each strain were included.
1.4. Measurement of serum and lung cytokines
Frozen lung tissues were powdered in liquid nitrogen and homogenized with a sonic dismembrator in lysis buffer (10mM TBS pH 7.5, 1% Triton-X 100, 1mM ethylenediaminetetraacetic acid, 1mM ethlyleneglycoltetraacetic acid) with protease inhibitor cocktail (Thermo Scientific, Rockford, IL). Protein concentrations were measured using DC Protein Assay kit (Bio-Rad Laboratories) and a standard amount of protein was loaded onto ELISA plates. Serum and lung cytokines were measured using commercial ELISA kits specific for IL-6 and IL-1β (BD Biosciences). The assays were conducted according to manufacturer’s instructions.
1.5. Histological examination
Lung tissues fixed in 10% formalin were paraffin-embedded, cut into 5μm sections, collected on glass slides and stained with hematoxylin and eosin (H&E). Stained slides were examined under a light microscope and scored for tissue injury as reported previously [30]. The injury was scored from 0 to 2 based on the presence of neutrophils in the alveolar space, neutrophils in the interstitial space, hyaline membranes, proteinaceous debris filling the airspaces, and alveolar septal thickening. The sum of the scores was weighted as a maximum grade of 100 per visual field.
1.6. MIP-2 mRNA expression
Lung tissues were homogenized in TRIzol reagent (Invitrogen, Carlsbad, CA) and total RNA extracted. The RNA was reverse transcribed and subjected to quantitative real time polymerase chain reaction (qPCR). The PCR was carried out in 25 μl reaction containing 0.08 μmol each forward and reverse primer, complementary DNA, and 12.5 μl 2X SYBR Green PCR Master Mix. The amplification was performed in a 7300 real time PCR machine (Applied Biosystems, Foster City, CA) under the thermal profile of 50°C for 2 min, 95°C for 10 min, followed by 45 cycles of 95°C for 15 s, 60°C for 1 min and 72°C for 1 min. Mouse β-actin expression was used to normalize each sample and the relative expression was quantified using the 2−ΔΔCt method. The sequences of primers used are the follows: mouse MIP-2 5′-CCC TGG TTC AGA AAA TCA TCC A-3′ (forward) and 5′-GCT CCT CCT TTC CAG GTC AGT-3′ (reverse); mouse β-actin, 5′-CGT GAA AAG ATG ACC CAG ATC A-3′ (forward) and 5′-TGG TAC GAC CAG AGG CAT ACA G-3′ (reverse).
1.7. Myeloperoxidase (MPO) activity assay
Lung tissues were homogenized in potassium phosphate (KPO4) buffer containing 0.5% hexadecyltrimethylammonium bromide. Samples were centrifuged and the supernatant was diluted in reaction solution (o-dianisidine hydrochloride and hydrogen peroxide). For MPO activity, the rate of change in optical density was measured at 460 nm and calculated as units per mg tissue.
1.8. TUNEL assay
Lung and gut tissue sections were deparffinized and processed with Proteinase K (20 μg/ml) for 20 min at room temperature. Sections were then stained with green fluorescent TUNEL kit (Roche Diagnostics, Indianopolis, IN) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). TUNEL-positive cells per field per section in the lungs were counted at 200 X magnification under a fluorescence microscope.
1.9. Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). Data were compared by one way analysis of variance (ANOVA) for multiple groups and Student Newman Keuls (SNK) test was used for between comparisons. Differences in values were considered significant if P<0.05.
2. Results
2.1. RIPK3 deficiency decreased serum and lung cytokines after neonatal sepsis
At 10 h after CS injection, serum IL-6 and IL-1β were increased by 511-fold and 43-fold in WT mice, respectively. In the KO mice after CS injection, serum IL-6 and IL-1β were increased by 166-fold and 22-fold, respectively (Figure 1A–B). In WT mice after CS injection, lung IL-1β was increased by 7.0-fold whereas in the KO mice there was only a 4-fold increase in these levels (Figure 1C). Serum IL-6, IL-1β and lung IL-1β in WT sham and KO sham were 0.09 ± 0.01 vs. 0.10 ± 0.01; 8.9 ± 4.8 vs. 0.05 ± 0.01; 7.0 ± 0.7 vs. 5.5 ± 0.03, respectively. There was no significant difference between WT sham and KO sham in either serum or lung cytokines measured.
Fig. 1. Serum and lung cytokines were attenuated in KO neonates.
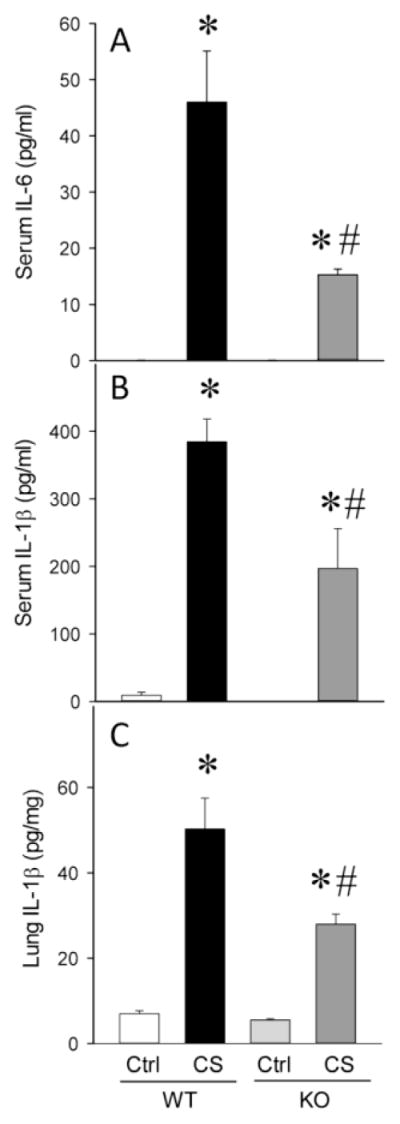
Serum IL-6 (A), serum IL-1β (B) and Lung IL-1β (C) from wild type (WT) and RIPK3 knockout (KO) mice at 10 h after CS injection. Data are expressed as mean ± SEM and compared by one-way ANOVA and SNK test. *P<0.05 vs. WT Ctrl; #P<0.05 vs. WT CS.
2.2. RIPK3 deficiency decreased lung injury after neonatal sepsis
Tissue histology evaluated by H&E staining showed that in CS injected KO mice, decreased presence of neutrophils in the alveolar space, and reductions in the hyaline membranes and alveolar thickening were observed as compared to CS injected WT mice (Figure 2A). While the lung injury score in the WT mice was increased by 9.0-fold after CS injection, there were only a 2.0-fold increase in the lung injury score in the KO mice (Figure 2B).
Fig. 2. Lung architecture was improved in KO neonates.

H&E stain of lungs from WT and KO newborn mice. A. Representative photographs at 200 X magnification. B. Histological score calculated as described in Materials and Methods. Data are expressed as mean ± SEM and compared by one-way ANOVA and SNK test. *P<0.05 vs. WT Ctrl; #P<0.05 vs. WT CS.
2.3. RIPK3 deficiency decreased lung neutrophil infiltration after neonatal sepsis
Lung tissues were evaluated for MIP-2 mRNA expression and MPO activity to assess neutrophil infiltration in CS injected WT and KO mice. In WT mice after CS injection, lung MIP-2 mRNA increased by 84-fold whereas in the KO mice the expression was increased by only 25-fold (Figure 3A). Likewise, the lung MPO activity was increased by a significant 50% in the WT mice while only a 10% increase was observed in the KO mice (Figure 3B). No significant difference was observed in sham newborns between the strains in any of the above measurements.
Fig. 3. Lung neutrophil infiltration was attenuated in KO neonates.
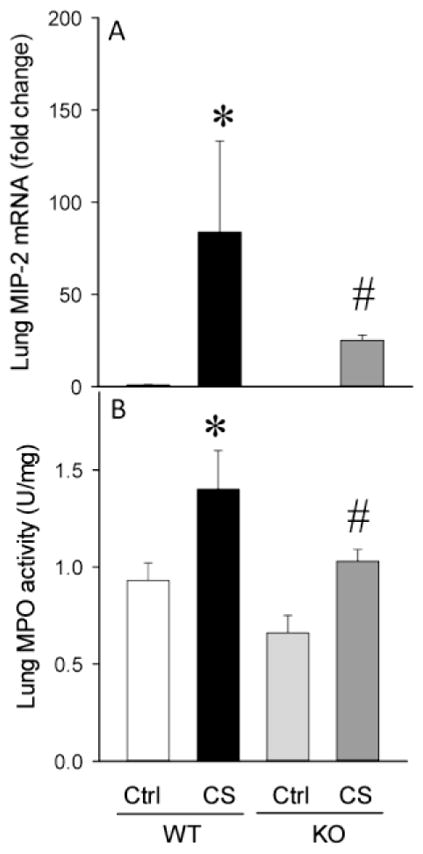
Lung MIP-2 mRNA expression (A) by real time quantitative polymerase chain reaction (qPCR) and Lung myeloperoxidase (MPO) activity (B). Data are expressed as mean ± SEM and compared by one-way ANOVA and SNK test. *P<0.05 vs. WT Ctrl; #P<0.05 vs. WT CS.
2.4. RIPK3 deficiency decreased lung and gut apoptosis after neonatal sepsis
Lung and gut tissues were evaluated by TUNEL assay to assess apoptosis (Figure 4A). In WT mice after CS injection, TUNEL positive cells in the lungs were increased by 7.8-fold whereas in the KO mice there was only a 2-fold increase in TUNEL staining (Figure 4B). TUNEL positive cells in the gut were increased by 10-fold in the WT mice after CS injection whereas in the KO mice, TUNEL staining was increased by 2.0-fold (Figure 5A–B).
Fig. 4. Lung apoptosis was reduced in KO neonates.

A. Representative photographs of TUNEL staining of lung tissues. B. TUNEL-positive cells per field calculated from A. Data are expressed as mean ± SEM and compared by one-way ANOVA and SNK test. *P<0.05 vs. WT Ctrl; #P<0.05 vs. WT CS.
Fig. 5. Intestinal apoptosis was reduced in KO neonates.

A. Representative photographs of TUNEL staining of gut tissues. B. Gut TUNEL positive cells per field calculated from A. Data are expressed as mean ± SEM and compared by one-way ANOVA and SNK test. *P<0.05 vs. WT Ctrl; #P<0.05 vs. WT CS.
3. Discussion
Neonatal sepsis is a systemic inflammatory response occurring in neonates during the first 28 days of their birth. It is the third leading cause of neonatal death and low birth weight infants are more prone to sepsis than term infants. This increased susceptibility has been attributed to their immature or underdeveloped immune system [31]. The pathogenesis of neonatal sepsis is characterized as the exaggerated inflammatory response due to infection with the presence of high levels of proinflammatory cytokines, i.e., TNF-α, IL-1β and IL-6 [7]. While there is much improvement in diagnosis, the molecular factors mediating such inflammatory conditions were not completely elucidated. In the current study using an established CS model in newborn mice, serum IL-6 and IL-1β were significantly elevated in the WT mice whereas in the KO mice these levels were markedly attenuated. While lung injury score, lung neutrophil infiltration and lung and gut apoptosis were significantly increased in the WT mice, the KO mice showed a more attenuated phenotype in all these parameters. These data suggest that RIPK3 could be involved in the uncontrolled inflammatory response leading to end organ damage and cell death observed during neonatal sepsis.
Cells may die through multiple means and the two predominant ways of death are apoptosis and necrosis [32]. Apoptosis involves activation of caspases which manifests as condensed chromatin with the plasma membrane being intact. For decades unlike apoptosis, necrosis has been considered as merely fortuitous and unregulated form of cell death caused by permeabilization of the plasma membrane and subsequent cytoplasmic swelling with no signs of nuclear shrinkage [32]. However, recent evidence suggests that as in the case of apoptosis, necrosis can also be regulated. At the forefront of such regulation is the receptor interacting protein kinase (RIPK). Although RIPK3 was first identified as a protein associated with RIPK1, more recent work established RIPK3 as the molecular switch that determines the cell to undergo apoptosis or necrosis [10,11]. In an animal model of cerulein-induced pancreatitis, the expression of RIPK3 was dramatically increased in the pancreas and 67% of the mice showed pancreas acinar cell loss and necrosis whereas these effects were prevented in the KO mice [10]. In an adult severe sepsis model and in high dose endotoxemia, RIPK3 protein was increased significantly and RIPK3 deficiency attenuated sepsis-induced increases in cytokines and neutrophil infiltration to the lung tissues [23–25]. The current study in newborn mice, sepsis-induced increases in cytokines, neutrophil infiltration to the lungs, and lung and gut apoptosis were attenuated in the KO mice. It can be speculated that the effect could be due to attenuation of neonatal sepsis associated necrosis in the lungs and the gut.
How RIPK3 deficiency attenuates necrosis in the lungs and gut in neonatal sepsis has not been determined. Alarmins or damage associated molecular patterns (DAMPs) released from the stressed cells undergoing necrosis act as endogenous danger signals to exacerbate inflammatory response [33]. Increased levels of these DAMPs have been associated with inflammatory conditions such as adult sepsis, arthritis, lupus, Crohn’s disease and cancer [34,35]. In our CS model of the newborn mice, IL-1β and IL-6 were significantly elevated at 10 h after sepsis induction. These cytokines are pro inflammatory cytokines which are crucial and sensitive biomarkers shown to be elevated in neonatal sepsis [36]. In addition, RIPK3 has shown to be a stimulator of IL-1 activation [37]. However, studies in TNF-α induced mouse adult sepsis model, RIPK3 is not essential for cytokine transcription but it sustains cytokine secretion presumably by DAMP release induced by cell necrosis [24]. Lung integrity was severely compromised in the WT mice 10 h after cecal slurry injection whereas no substantial tissue damage was observed in the gut [29]. The circulating levels of IL6 and IL-1β could come from other organs such as the liver or the circulating immune cells. Neutrophils in the alveolar space, expression of the lung MIP-2 mRNA and lung MPO activity were attenuated in the CS injected RIPK3 deficient mice as compared to the WT counterpart. Presence of neutrophils in the alveolar space, MIP-2 mRNA expression and MPO activity are measures of neutrophil infiltration to the tissues [38]. Increased neutrophil infiltration to the tissues, particularly the lungs, has been implicated in systemic inflammatory response [39]. Several studies also support that necroptosis caused by DAMPs released from cells promote neutrophil infiltration [40,41]. Therefore, it is possible that the deficiency in RIPK3 attenuates cellular necrosis thereby prevents the release of these DAMPs and decreases inflammation in neonatal sepsis.
There are several limitations for our study. The major limitation is that we have not addressed the mechanism of action of the RIPK3 depletion to inflammatory response in neonatal sepsis. However, the purpose of this study was to determine whether deficiency in RIPK3 has any effect in inflammation and injury during neonatal sepsis. Our study clearly established an association between RIPK3 depletion and end organ damage in neonatal sepsis. However, the mechanism by which RIPK3 facilitates necrosis in neonatal sepsis has not been elucidated. In a high dose adult LPS-induced acute respiratory distress syndrome (ARDS) model, RIPK3 protein, p-RIPK3, mixed lineage kinase domain like (MLKL) protein and cleaved caspase-3 were increased whereas in the KO mice, the MLKL protein was decreased and cleaved caspase-3 was unaltered suggesting that RIPK3-mediated inflammation and lung injury as the major mechanism in this model [23]. In the low dose adult LPS model, LPS increased apoptosis whereas in the presence of high dose LPS, apoptosis was inhibited presumably leading to increased necroptosis and subsequent inflammation and lung injury. This study suggested that inhibiting RIPK3 could protect mice against LPS-induced acute lung injury by reducing lung necrosis and inflammation [23]. In our studies, CS injection to RIPK3 deficient mice significantly decreased TUNEL positive cells in the lungs and the gut. It has also been reported that mice lacking RIPK3 do not exhibit any defects in cells undergoing apoptosis or NF-kB activation [26]. Thus, the potential interplay between apoptosis and necrosis in the lungs and gut during neonatal sepsis requires further investigation.
Another limitation is that the inflammatory mediators were measured only at a single time point. The 10 h time point after CS injection was chosen based on the pilot study in WT mice where we observed no mortality whereas at 24 h later, mortality increased to 100%. We have not extended the time point beyond 10 h in the KO mice. Future studies will address the potential role of this deletion at different time points and long term survival outcome during neonatal sepsis. We acknowledge that the utilization of naïve control mice as opposed to control mice receiving the vehicle (5% dextrose) as another limitation for the study. It is highly unlikely that the vehicle could cause any inflammatory response in neonates. However, it is possible that intraperitoneal (IP) injection could trigger inflammation. Since both WT and KO underwent IP injections, the concern that the observed changes between the WT and KO could be due to IP injections can be ruled out. Nevertheless, the use of the naïve control mice is a limitation of this study. Another limitation is the choice of CS model to induce neonatal sepsis. One caveat to the CS model is the variability of the cecal contents among different batches of preparation. In order to rule out both intra- and inter-variations, the cecal contents were made in batches and stored as frozen aliquots prior to the start of experiment. On the day of the experiment, the aliquots were thawed and used within 2 h of thawing. In addition, we recognize that the cecal perforation causing peritonitis as in the adult mice would have been more clinically relevant than the CS model. However, this model is rather technically challenging to establish in a newborn mouse. Future studies using clinical conditions such as necrotizing colitis and/or microbial organisms, i.e., E. coli are necessary to confirm our findings.
In conclusion, the current study showed that the deficiency in RIPK3 attenuated inflammatory response leading to end organ damage and cell death in a mouse model of neonatal sepsis. Further experiments addressing the potential mechanism of action of the RIPK3 depletion to inflammatory response in neonatal sepsis are warranted to determine whether RIPK3 could be a potential target for therapeutics in neonatal sepsis.
Acknowledgments
This work was in part supported by the National Institutes of Health grants, R35GM118337 and R01GM053008 (PW).
Footnotes
This study was in part published as an abstract for the 102nd Annual Conference of the American College of Surgeons, October 20, 2016, Washington DC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, et al. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. 2015;385:430–40. doi: 10.1016/S0140-6736(14)61698-6. [DOI] [PubMed] [Google Scholar]
- 2.Wynn JL, Levy O. Role of innate host defenses in susceptibility to early-onset neonatal sepsis. Clin Perinatol. 2010;37:307–37. doi: 10.1016/j.clp.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leviton A, O’Shea TM, Bednarek FJ, Allred EN, Fichorova RN, Dammann O, et al. Systemic responses of preterm newborns with presumed or documented bacteraemia. Acta Paediatr. 2012;101:355–9. doi: 10.1111/j.1651-2227.2011.02527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reinhart K, Bauer M, Riedemann NC, Hartog CS. New approaches to sepsis: molecular diagnostics and biomarkers. Clin Microbiol Rev. 2012;25:609–34. doi: 10.1128/CMR.00016-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Remick DG. Pathophysiology of sepsis. Am J Pathol. 2007;170:1435–44. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y, Du WX, Jiang HY, Ai Q, Feng J, Liu Z, et al. Multiplex Cytokine Profiling Identifies Interleukin-27 as a Novel Biomarker For Neonatal Early Onset Sepsis. Shock. 2017;47:140–7. doi: 10.1097/SHK.0000000000000753. [DOI] [PubMed] [Google Scholar]
- 7.Khaertynov KS, Boichuk SV, Khaiboullina SF, Anokhin VA, Andreeva AA, Lombardi VC, et al. Comparative Assessment of Cytokine Pattern in Early and Late Onset of Neonatal Sepsis. J Immunol Res. 2017;2017:8601063. doi: 10.1155/2017/8601063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–32. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 11.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 12.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 13.Schutze S, Tchikov V, Schneider-Brachert W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat Rev Mol Cell Biol. 2008;9:655–62. doi: 10.1038/nrm2430. [DOI] [PubMed] [Google Scholar]
- 14.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–55. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 15.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14:400–10. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 17.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–23. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kasof GM, Prosser JC, Liu D, Lorenzi MV, Gomes BC. The RIP-like kinase, RIP3, induces apoptosis and NF-kappaB nuclear translocation and localizes to mitochondria. FEBS Lett. 2000;473:285–91. doi: 10.1016/s0014-5793(00)01473-3. [DOI] [PubMed] [Google Scholar]
- 19.Pazdernik NJ, Donner DB, Goebl MG, Harrington MA. Mouse receptor interacting protein 3 does not contain a caspase-recruiting or a death domain but induces apoptosis and activates NF-kappaB. Mol Cell Biol. 1999;19:6500–8. doi: 10.1128/mcb.19.10.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM. RIP3, a novel apoptosis-inducing kinase. J Biol Chem. 1999;274:16871–5. doi: 10.1074/jbc.274.24.16871. [DOI] [PubMed] [Google Scholar]
- 21.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–11. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 22.Meng L, Jin W, Wang X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci U S A. 2015;112:11007–12. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie W, et al. Receptor Interacting Protein 3-Mediated Necroptosis Promotes Lipopolysaccharide-Induced Inflammation and Acute Respiratory Distress Syndrome in Mice. PLoS One. 2016;11:e0155723. doi: 10.1371/journal.pone.0155723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 25.Sharma A, Matsuo S, Yang WL, Wang Z, Wang P. Receptor-interacting protein kinase 3 deficiency inhibits immune cell infiltration and attenuates organ injury in sepsis. Crit Care. 2014;18:R142. doi: 10.1186/cc13970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–9. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuenca AG, Wynn JL, Moldawer LL, Levy O. Role of innate immunity in neonatal infection. Am J Perinatol. 2013;30:105–12. doi: 10.1055/s-0032-1333412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wynn JL, Scumpia PO, Delano MJ, O’Malley KA, Ungaro R, Abouhamze A, et al. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007;28:675–83. doi: 10.1097/SHK.0b013e3180556d09. [DOI] [PubMed] [Google Scholar]
- 29.Hansen LW, Khader A, Yang WL, Jacob A, Chen T, Nicastro JM, et al. Deficiency in milk fat globule-epidermal growth factor-factor 8 exacerbates organ injury and mortality in neonatal sepsis. J Pediatr Surg. 2017;52:1520–7. doi: 10.1016/j.jpedsurg.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–38. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugitharini V, Prema A, Berla Thangam E. Inflammatory mediators of systemic inflammation in neonatal sepsis. Inflamm Res. 2013;62:1025–34. doi: 10.1007/s00011-013-0661-9. [DOI] [PubMed] [Google Scholar]
- 32.Galluzzi L, Kepp O, Kroemer G. RIP kinases initiate programmed necrosis. J Mol Cell Biol. 2009;1:8–10. doi: 10.1093/jmcb/mjp007. [DOI] [PubMed] [Google Scholar]
- 33.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 34.Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest. 2012;122:2711–9. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19:1489–95. doi: 10.1038/nm.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santana Reyes C, Garcia-Munoz F, Reyes D, Gonzalez G, Dominguez C, Domenech E. Role of cytokines (interleukin-1beta, 6, 8, tumour necrosis factor-alpha, and soluble receptor of interleukin-2) and C-reactive protein in the diagnosis of neonatal sepsis. Acta Paediatr. 2003;92:221–7. doi: 10.1111/j.1651-2227.2003.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 37.Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–27. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 39.Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 2013;93:329–42. doi: 10.1189/jlb.0912437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–89. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–73. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]