

Abstract
A fitness landscape (FL) describes the genotype-fitness relationship in a given environment. To explain and predict evolution, it is imperative to measure the FL in multiple environments because the natural environment changes frequently. Using a high-throughput method that combines precise gene replacement with next-generation sequencing, we determine the in vivo FL of a yeast tRNA gene comprising over 23,000 genotypes in four environments. Although genotype-by-environment interaction (G×E) is abundantly detected, its pattern is so simple that we can transform an existing FL to that in a new environment with fitness measures of only a few genotypes in the new environment. Under each environment, we observe prevalent, negatively biased epistasis between mutations (G×G). Epistasis-by-environment interaction (G×G×E) is also prevalent, but trends in epistasis difference between environments are predictable. Our study thus reveals simple rules underlying seemingly complex FLs, opening the door to understanding and predicting FLs in general.
The fitness landscape (FL) of a gene in a given environment maps each mutational variant of the gene to the fitness of the organism carrying the variant. FLs allow explaining as well as predicting evolutionary trajectories, and are therefore of fundamental biological importance1. Because fitness is expected to vary by environment, which frequently changes in nature, characterizing FLs in multiple environments offers not only more relevant and accurate evolutionary explanations and predictions but under certain circumstances also insights that are otherwise impossible to gain. For example, a gene that seems harmful to an organism in one environment is nonetheless retained in its genome, because it is beneficial in other environments2. Two alleles at a locus may be respectively favored in two environments, resulting in a stable genetic polymorphism if the population switches sufficiently frequently between these environments3,4. A population trapped at a suboptimal local fitness peak under one environment may escape upon an environmental shift, even if the environment later shifts back5,6. Differences in FL between two environments may also lead to ecological speciation7. Notwithstanding, due to the huge genotype space (4n genotypes for a gene with n nucleotides), even the characterization of a small fraction of the FL of a gene under one environment had been a formidable challenge until recently8-13. As a result, past quantifications of FLs typically probe only one environment. Adopting a recently developed high-throughput method9 with minor modifications, we map the in vivo FL of a tRNA gene in four environments in order to study the environmental impact on FLs. Of particular interest are genotype-by-environment interactions (G×E) and epistasis-by-environment interactions (G×G×E). In humans, G×E has been implicated in numerous diseases such as cancer and mental disorders14,15. Epistasis, or interaction between mutations (G×G), has multiple important functional and evolutionary implications16 and is known to vary among environments17-22. Because the method employed can measure the fitness of tens of thousands of genotypes9, the collected data permit the quantification of G×E and G×G×E at an unprecedentedly large scale, providing opportunities for identifying general principles of these interactions.
Results
Mapping multi-environment FLs of a tRNA gene
is a transfer RNA that uses its anticodon 5′-CCU-3′ to bind to the arginine codon AGG in translation. It is encoded by a single-copy gene (HSX1) in the budding yeast Saccharomyces cerevisiae. Deleting this gene is nonlethal, because , another tRNA for arginine, can wobble-pair with AGG. We synthesized the 72-nucleotide gene with a 3% per-site mutation rate (1% to each alternative nucleotide) at 69 sites; the remaining three sites were kept invariant for technical reasons9 (Fig. 1; see Methods). This mutation rate was chosen to maximize the representation of genotypes carrying two mutations for the purpose of studying epistasis between mutations (see Methods). We constructed a library of more than 100,000 yeast strains, each carrying a gene variant at its native genomic location. Three to five parallel competitions of this strain pool were conducted in each of four environments: 30°C in the rich medium YPD (hereafter Env30), 23°C in YPD (Env23), 37°C in YPD (Env37), and 30°C in YPD with 3% dimethyl sulfoxide (DMSO) added (EnvDMSO) (Fig. 1). Among them, Env30 is the optimal growth condition for the wild-type yeast strain used, with the other environments imposing cold, heat, and oxidative stresses23, respectively. The selection of these four environments was based on previous studies showing that tRNA folding and decay are temperature-dependent24-26 and RNA structure and function are affected by DMSO27.
Figure 1. Determining the fitness landscape of the yeast gene in multiple environments.
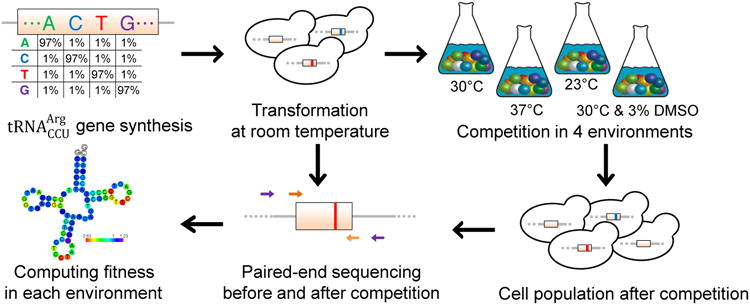
Chemically synthesized gene variants are incorporated at the native genomic locus of the gene. Transformation is conducted at room temperature to ensure comparatively equal representation of mutant strains. The tRNA variant-carrying cells compete for ∼13 generations in each environment at log phase, and the tRNA gene variants are sequenced before and after the competition. The fitness of each genotype relative to wild-type is calculated from the relative frequency change of paired-end sequencing reads covering the tRNA gene variant during competition.
The wild-type yeast growth rates (GR) in these four environments show GR(Env30) > GR(EnvDMSO) > GR(Env37) > GR(Env23), while the gene importance (GI) to growth, measured by the fractional reduction in growth rate caused by deleting the tRNA gene, shows GI(Env37) > GI(EnvDMSO) > GI(Env30) > GI(Env23) (Supplementary Fig. S1). The gene is more important to cell growth at higher temperatures probably because the probability that is correctly folded decreases with temperature, reducing its ability to compensate the loss of the gene.
After the competition, the gene amplicons from each replicate competition (T1) under each environment, as well as those from the common starting population (T0; with two technical repeats), were sequenced with 125-nucleotide paired-end Illumina sequencing (Fig. 1). The sequences provide the genotype as well as genotype frequency information. A total of 881 million read pairs were generated. Read numbers per genotype are highly correlated between technical repeats (Pearson's correlation r =0.994; Supplementary Fig. S2a) and between biological replicates (r =0.9911-0.9999; Supplementary Fig. S2b). The technical repeats are combined in subsequent analysis. The fitness of each genotype relative to the wild-type (fitness hereafter) in an environment was estimated using the genotype frequencies at T0 and T1 as well as the number of wild-type generations in the competition (see Methods). We analyzed 23,284 genotypes with read counts ≥100 at T0. Because the gene is nonessential, mutations in the gene are unlikely to be lethal. When an estimated fitness is < 0.5 (likely due to stochasticity), we set it at 0.5, meaning that the genotype has no growth9.
G×E is pervasive
We previously reported the FL of the yeast gene in Env37 and its reliability, consistency with evolutionary data, and potential mechanistic basis9. With the FL data in four environments, we here focus on the impact of the environment on the FL rather than describing the FL under each environment. Specifically, we study whether the same mutation has different fitness effects in different environments, formally known as genotype-by-environment interaction28 (G×E) in fitness (Fig. 2a). Under the null hypothesis of no G×E, the fitness effect of a mutation is environment-independent, and hence the mutant fitness distribution should be identical across environments. In stark contrast to this null expectation, the mutant fitness distribution differs drastically among each pair of the four environments (P < 10-300, Kolmogorov-Smirnov test; Fig. 2b), suggesting pervasive G×E in our data. Mean mutant fitness (F̄) shows F̄(Env23) > F̄(Env30) > F̄(EnvDMSO) > F̄(Env37), exactly opposite to the aforementioned order of GI in the four environments. That is, mean mutant fitness decreases as gene importance in the environment rises. Although testing G×E for individual genotypes has a much-reduced statistical power, a substantial fraction showed significantly different fitness values between environments (nominal P < 0.05, t-test; lower left triangle in Fig. 2c). This fraction generally increases with the extent of environmental difference that can be gauged (e.g., temperature difference). It also increases for genotypes carrying a single point mutation (N1 mutants; upper right triangle in Fig. 2c), owing to their relatively high genotype frequencies in the strain pool that lead to relatively high numbers of sequencing reads, which empower the statistical test. Because only 5% of genotypes are expected by chance to show significantly different fitness values between any pair of environments under the null hypothesis of no G×E, our findings establish the prevalence of G×E. In some studies, G×E is defined by a between-environment variation in the growth rate difference between a genotype and the wild-type28-31. Pervasive G×E is also found when growth rate difference instead of relative fitness is used in the definition of G×E (Supplementary Fig. S3; see Methods).
Figure 2. The yeast gene fitness landscape in each of the four environments examined.

a, Definition of G×E based on mutant fitness relative to the wild-type in two environments. b, Frequency distribution of the fitness of all 23,284 genotypes in each environment. Note the different scales on the Y axis for different environments. c, Fraction of mutants exhibiting significant G×E. The lower left triangle shows the fraction of all genotypes whose fitness values differ significantly between the two environments compared (nominal P < 0.05, t-test), whereas the upper right triangle shows the corresponding fraction of N1 mutants.
Quantitative relationship in genotype fitness between environments
Despite the abundance of G×E, the fitness of a genotype in one environment exhibits a strong linear correlation with that in another environment (r = 0.78 - 0.94; lower left triangle in Fig. 3a), especially for N1 mutants (r = 0.94 - 0.99; upper right triangle in Fig. 3a), which generally have more precise fitness estimates than mutants carrying two or more mutations. The correlation is confirmed by individually measuring the fitness of 55 mutants across the four environments on the basis of growth curves (r = 0.85 - 0.96; Supplementary Fig. S4).
Figure 3. A piecewise linear model predicts fitness landscape in one environment from that in another environment.
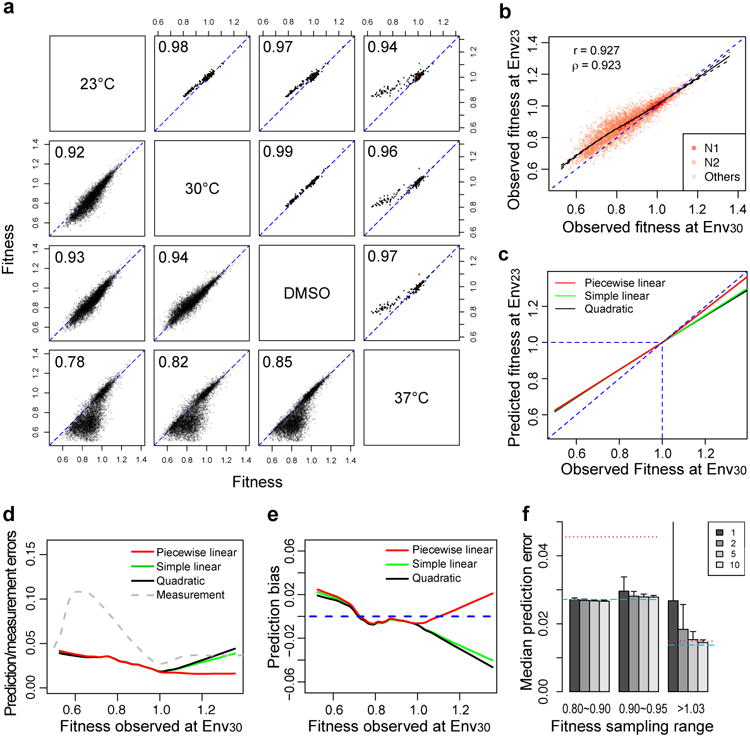
a, Fitness values from two environments are highly correlated among all mutants (lower left triangle), especially N1 mutants (upper right triangle). Pearson's correlation coefficient is presented for each environment pair. b, LOESS curve (black solid curve) describing the relationship between genotype fitness in Env30 and Env23, with 95% confidence intervals indicated (black dashed curves). In a and b, each dot is a genotype, and the diagonal is indicated by a blue dashed line. c, A piecewise linear model, a simple linear model, and a quadratic model are respectively plotted to capture the relationship between genotype fitness in Env30 and Env23. The blue dashed lines show Y=X, X=1, and Y=1, respectively. d, Prediction error and measurement error of fitness at Env23. The three solid curves respectively show prediction errors under three different prediction models when fitness at Env23 is predicted from that in Env30, whereas the grey dashed curve shows the measurement error based on one measurement at Env23. e, Prediction bias of fitness at Env23. The three solid curves respectively show prediction bias under three different models when predicting fitness at Env23 from that in Env30. The blue dashed line shows zero bias. f, Measuring fitness of only a few mutants in Env23 is sufficient for deriving the (piecewise) linear model used in predicting the fitness landscape in Env23 from that in Env30. When 1, 2, 5, or 10 N1 mutants (indicated by different shades of grey) chosen for fitness measurement at Env23 have fitness in Env30 (f30) < 1, median prediction errors shown are for all mutants with f30 < 1. When N1 mutants chosen for fitness measurement at Env23 have f30 > 1, median prediction errors shown are for all mutants with f30 > 1. The x-axis shows the f30 range in which mutants are randomly picked for fitness measurement at Env23. Blue dashed lines show median prediction errors when all genotypes are measured for fitness at Env23. Red dotted lines show median prediction errors assuming no G×E.
To more accurately describe the relationship of genotype fitness in two environments, we plotted a local regression (LOESS) of the fitness in Env30 (f30) and the fitness in Env23 (f23) across all genotypes (Fig. 3b). As expected, the regression goes near the point of (f30 = 1, f23 = 1) because of a large number of genotypes showing similar fitness as the wild-type. The regression suggests that the data can be described approximately by a piecewise linear model of f23-1 = k(f30-1), with one slope (ka) for genotypes of f30 < 1 and a different slope (kb) for genotypes of f30 >1 (see Supplementary Table S1 for other environment pairs). In other words, the fitness effect of a mutation in Env23 (i.e., f23-1) is proportional to that in Env30 (i.e., f30-1). We observed a general trend that kb is closer to 1 than is ka (i.e., |kb-1| < |ka-1|) across all environment pairs (Fig. 3b; Supplementary Table S1). This pattern may reflect different properties of deleterious and beneficial mutations other than their effect size differences, because the same trend holds even for deleterious and beneficial mutations of comparable fitness effect sizes. We also respectively tested a simple linear model with only one parameter and a quadratic model in describing the between-environment fitness relationships. Although the quadratic model generally outperforms the simple and piecewise linear models, the coefficient of the quadratic term is small. Also, the advantage of the quadratic model over the piecewise linear model, measured by the difference in the fraction of variance explained, is small (Supplementary Table S2). Furthermore, this advantage arises from the fact that the vast majority of genotypes are less fit than the wild-type (Fig. 2b) and that the quadratic model allows one more parameter than the piecewise linear model to describe these genotypes. When equal numbers of genotypes that are fitter and less fit than the wild-type are considered, the piecewise linear model generally performs the best (Supplementary Table S3). Because of the distinct biological meanings and evolutionary roles of deleterious and beneficial mutations, the piecewise linear model, which has equal weights for these two types of mutations in fitting, appears more appropriate.
Predicting the FL under new environments
To evaluate how well the piecewise linear model (Fig. 3c) predicts the f23 of a genotype from its f30 estimate, we focused on 6319 genotypes that have relatively precise experimental estimates of f30 and f23 (with ≥ 500 reads at T0 and ≥ 1 read at T1 in each of the five replicates under each of the two environments). Such restriction is necessary because of the difficulty in judging whether a prediction is good or not by comparing it with an actual measurement that is imprecise. We treated the f23 of a genotype estimated from the mean of five biological replicates as the observed value. The predicted and observed f23 values are highly correlated (Spearman's ρ = 0.908, Pearson's r = 0.915, P < 10-300 in both tests). We define prediction error by the absolute difference between the observed and predicted f23. By contrast, measurement error is the mean absolute difference between the observed f23 and that estimated from one biological replicate. Remarkably, the prediction error is no greater than the measurement error across the entire range of f30 in our data (Fig. 3d), and this pattern holds for four of the 12 environment pairs (Supplementary Fig. S5). In addition, we quantified the prediction bias by the median difference between the predicted and observed f23, and found it to be minimal except when f30 < 0.6 (Fig. 3e), which is expected (see Methods). For comparison, we respectively tested a linear model with one k and a quadratic model (Fig. 3c). These two models have larger biases and errors when f30 > 1 but are otherwise similar in performance to the piecewise linear regression (Fig. 3d, e). These observations hold in all 12 pairwise predictions from one environment to another (Supplementary Fig. S5). Another performance indicator of our prediction is the fraction of predictions that deviate significantly from the observation. Only 0.51% of the 6319 predictions of f23 from f30 belong to this category (FDR = 0.05).
In the above exercise, the prediction model is determined by comparing the FLs respectively measured in Env30 and Env23. In practice, we would measure the FL in Env30 and then predict it in Env23. Because the piecewise linear model involves only two parameters (ka and kb), in principle we need to measure the fitness of only two genotypes, respectively fitter and less fit than the wild-type, in Env23 to parameterize the model. This is indeed the case, as long as the f30 of the genotypes chosen for fitness measurement in Env23 is not too close to 1 (Fig. 3f). This pattern holds for most of the 12 environment-to-environment fitness predictions (Supplementary Fig. S6).
Epistasis between mutations is negatively biased
We estimated epistasis between mutations in the tRNA gene using the fitness estimates of all 207 N1 mutants and 8,101 double mutants (N2 mutants) (see Methods). In each environment, a large fraction of mutation pairs show significant epistasis and the epistasis is strongly biased toward negative values (Fig. 4a; Supplementary Fig. S7) except at paired sites (Supplementary Fig. S8), similar to observations in other molecules8,12,32. Furthermore, the average fitness of all genotypes carrying the same number (≥ 2) of mutations is lower than expected under the multiplicative model, again indicating pervasive negative epistasis (Fig. 4b).
Figure 4. Epistasis-by-environment interaction is prevalent.
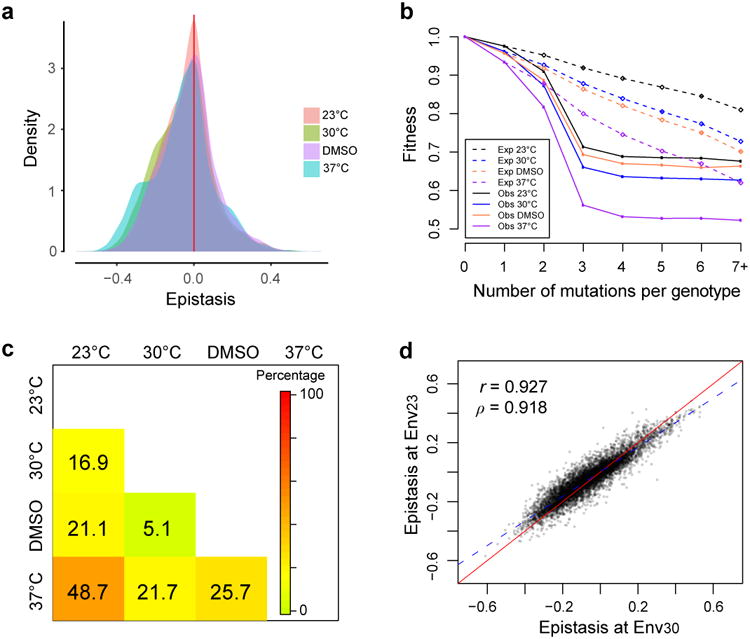
a, Frequency distributions of 8,101 epistasis between mutations in four environments. b, Observed mean fitness of mutants carrying a given number of mutations (solid dots) is lower than expected under no epistasis (open diamonds) in each environment. c, Fractions of mutation pairs that exhibit significant G×G×E for each environment pair (nominal P < 0.05, t-test). d, Epistasis measured in Env23 and that measured in Env30 are highly correlated. Pearson's correlation (r) and Spearman's correlation (ρ) are indicated. The red solid line shows the diagonal, whereas the blue dashed line shows the linear regression.
Epistasis by environment interaction is prevalent
Except for the pair of Env30 and EnvDMSO in which mutant fitness values are similar (Fig. 2c), a substantial fraction of epistasis varies between environments (Fig. 4c), revealing prevalent epistasis-by-environment interactions (G×G×E). G×G×E is also abundantly observed under a commonly used alternative definition of epistasis (Supplementary Fig. S9).
Given that the fitness of a genotype in one environment can be predicted from that in another environment and that epistasis is estimated from the fitness values of three relevant genotypes, it is possible to predict epistasis in one environment from the fitness data in another environment (see Methods). Nevertheless, epistasis prediction is expected to be less precise than fitness prediction, because each epistasis prediction relies on three fitness predictions. Not with standing, predicting the general trend in epistasis changes upon an environmental shift may be sufficiently reliable, because errors in individual epistasis predictions are random and do not alter the general trend. Our prediction of epistasis at Env23 from the fitness data at Env30 suggests a strong, positive correlation in epistasis between the two environments (Supplementary Fig. S10), which is confirmed by a comparison of epistasis respectively measured in the two environments (Fig. 4d; Supplementary Fig. S11). We also predict that epistasis holds the same direction across environments for most albeit not all mutation pairs (Supplementary Fig. S10), as is observed (Supplementary Table S4). We further predict that, when the mean fitness effect of mutation enlarges upon an environmental shift (e.g., from Env30 to Env37), switches from positive to negative epistasis outnumber the opposite switches, which is confirmed by our data (Supplementary Table S4). Nevertheless, none of the predicted epistasis sign switches show significant epistasis in each of the two environments concerned and exhibit an epistasis sign switch, likely due to the relatively large error in predicting the epistasis of specific mutation pairs.
Discussion
In summary, we detected widespread G×E in the FLs of the yeast gene across four environments, echoing accumulating reports of G×E in humans and model organisms14,15,29,33 and supporting the importance of mapping FLs in multiple environments. Our discovery of a simple rule of G×E provides a convenient and reliable computational means to transform the FL from one environment to another with minimal additional experiments. Our observation that the fitness effect of a mutation can be linearly transformed between environments is equivalent to the statement that the ratio between the fitness effects of any two deleterious (or beneficial) mutations (say A and B) in a gene is roughly constant irrespective of the specific environment. When mutation B is a null mutation, the ratio measures the fraction of the gene's fitness contribution impacted by mutation A. The constancy of this ratio among environments leads to the interpretation that, while a gene's fitness contribution may vary among environments2,34, a mutation in the gene influences the gene's fitness contribution by a fixed proportion that is independent of the environment. The above interpretation does not depend on the specific mechanism by which a mutation affects fitness. One critical question is whether our discovery in a yeast tRNA gene applies to other genes (especially protein-coding genes) and other organisms. To address this question, we analyzed the local FLs of a segment of the yeast heat shock protein Hsp90 gene in four environments35 and found that the observed patterns (Supplementary Fig. S12) resemble those in the tRNA gene. Further verifications, especially in other organisms, are needed when more multi-environment FLs become available in the future. Note that because the fitness contribution of one gene may rise while that of another may decrease upon an environmental shift, the between-environment transformation of FL is expected to be gene-specific.
We also detected widespread G×G×E, which were previously reported at much smaller scales17-22. Because of the broad functional and evolutionary implications of epistasis, between-environment differences in epistasis cannot be ignored, especially in those rare cases where an environmental shift reverses the sign of epistasis.
In previous studies, we and others provided evidence that FLs of RNA genes under a given environment can be explained to a significant extent by RNA folding8,9. In the present work, our FL mapping across environments suggests that a mutation in the gene influences the gene's fitness contribution by a fixed proportion that is independent of the environment. We also explained mechanistically why the tRNA gene has a larger fitness contribution at a higher temperature, at least qualitatively. Together, they suggest that FLs of RNA genes can be understood from a molecular mechanistic perspective and hence predicted from relevant information without exhaustive or extensive experimental mapping. Further endeavors toward this goal for both RNA and protein genes are likely to be highly rewarding.
Methods
Measuring genotype fitness in multiple environments
The chemical synthesis of the yeast gene with random sequence variation was previously described9. Briefly, because IDT (https://www.idtdna.com/site) could not synthesize oligonucleotides longer than 100 nucleotides that require manual mixing of nucleotides and because there is a need for constant regions at the two ends of the oligonucleotides for polymerase chain reaction (PCR), we could only allow 69 variable sites in the tRNA gene. Hence, the first nucleotide and last two nucleotides (counting from the 5′ end) of the 72-nucleotide tRNA gene were invariant and synthesized according to the wild-type sequence. At each variable site, the probability of incorporating each of the three non-wild-type nucleotides was set at 0.01. This “mutation” rate was chosen to maximize the fraction of variants carrying two mutations in order to study pairwise epistasis. In the pool of tRNA gene variants synthesized, the fractions of molecules with 0, 1, 2, 3, 4 and >4 mutations are expected to be 12%, 26%, 27%, 19%, 10%, and 6%, respectively, while the possible numbers of variants with 0, 1, 2, 3, and 4 mutations are 1, 207, 2.1×104, 1.4×106, and 7.0×107, respectively. Hence, the expected genotype frequency for a given genotypes with 0, 1, 2, 3, and 4 mutations is 0.12, 1.26×10-3, 1.29×10-5, 1.36×10-7, and 1.43×10-9, respectively. Previous sequencing data verified these expectations9.
Strain construction, competition assay, and library preparation were conducted as previously described9 except that KAPA HiFi HotStart DNA Polymerase instead of AccuPrime™ Pfx DNA polymerase was used for PCR amplification to further increase the accuracy of genotyping. In short, we collected over 100,000 BY4742 yeast strains carrying gene variants at its native genomic location and mixed them as the starting strain pool. Cells derived from the same starting population were cultured in four separate environments and were transferred every few hours to grow in log phase for ∼13 generations. The exact number of generations for the whole population was computed by tracking optical density at 660 nm (OD660) over time, and the number of generations for the wild-type was then inferred from its frequency change in the population. Five parallel competitions were conducted in Env23 and Env30, respectively, whereas three parallel competitions were conducted in EnvDMSO and Env37, respectively. Cells were then harvested and lysed to extract DNA. Two rounds of PCR amplification were conducted to amplify the gene incorporated at the correct genomic location and to add adaptors for sequencing, respectively9. We sequenced the common population before the competition in two technical repeats and the population after each competition.
Only perfectly matched fully overlapping paired-end reads were used in estimating genotype frequencies. The change in relative genotype frequency during the competition was used to estimate the fitness of each genotype relative to the wild-type. The fitness of a genotype is calculated by averaging the fitness among biological replicates. To ensure relatively accurate fitness estimation, we focused on 23,284 genotypes with read counts ≥ 100 before the competition unless otherwise mentioned. The reliability of genotype fitness estimation using en masse competition followed by sequencing was previously demonstrated by comparing with three other methods9. In the present study, the biological replicates allow the assessment of estimation error.
Quantifying growth rates and fitness of selected genotypes
The growth rates of selected yeast strains were measured using Bioscreen C OD reader in the specific environment considered. Cells were first grown overnight until saturation, and then diluted by a factor of 50 to roughly OD600 = 0.1. OD measurements at the wide band (450-580 nm) were taken every 20 minutes for 48 hours. The maximum growth rate was calculated following standard procedures36. At least two biological replications in growth measurement were performed per genotype per environment. The fitness (f) of a genotype relative to the wild-type was calculated by , where R and R0 are the maximum growth rates of the genotype and wild-type, respectively. The above formula was derived the following way. After one generation of exponential growth of a population of wild-type cells, we have eR0T = 2, where T is the generation time of the wild-type. Similarly, we have eRT = 2f for the genotype concerned. Combining the above two equations, we obtain .
Quantifying G×E and G×G×E
Based on fitness estimates in multiple biological replicates, a t-test is used to examine if the fitness of a genotype (relative to the wild-type) is significantly different between two environments at a nominal P-value of 0.05, which constitutes G×E. In addition to the G×E definition used in the main text (i.e., the fitness of a genotype relative to the wild-type differs between environments), we also used an alternative definition of G×E that the growth rate difference between a genotype and the wild-type varies between environments28,29.
Epistasis is defined by ε = fAB – fAfB, where fA and fB are the fitness values of two single mutants and fAB is the fitness of the corresponding double mutant. We similarly tested G×G×E by examining if epistasis is significantly different between two environments, using a t-test at a nominal P-value of 0.05. We also tested G×G×E using an alternative definition of epistasis: ε′ = ln fAB –ln fA –ln fB37.
Piecewise robust linear regression between fitness values in two environments
Local regression (LOESS) with a span of 0.5 was used to visualize the general trend in the relationship between mutant fitness measured in two environments. Using the rlm function of the MASS package in R, we conducted a piecewise robust linear regression between the fitness at one environment and that at another for genotypes with ≥ 500 reads at T0 and ≥ 1 read at T1 in each biological replicate of the two environments concerned. Compared with the ordinary least squares linear regression, the robust linear regression reduces the potential impact of outliers. The model is fitted separately for genotypes fitter than the wild-type and those less fit than the wild-type. The regression is forced to go through (1, 1), because the relative fitness is defined as 1 for the wild-type in each environment. Specifically, our regression model is
where f1 and f2 are the fitness values of a genotype in environment 1 and 2, respectively.
In addition, we tried two alternative models to regress the fitness estimates from two environments. The first alternative model is a simple robust linear model that goes through (1, 1) without separate slopes for genotypes fitter than the wild-type and those less fit than the wild-type. The second alternative model is a quadratic model that goes through (1, 1). We calculated adjusted R2 to represent the fraction of variance in fitness explainable by each model. None of the above models would be accepted if correlations between observations and predictions are weak, prediction errors are large, or genotypes with significant prediction errors are numerous.
Evaluating fitness predictions
We can treat the regression line between fitness values in two environments as predictions of fitness in environment 2 from those in environment 1. Two metrics were used to evaluate these predictions. The first metric is prediction error, which is the absolute difference between the predicted and observed fitness in environment 2. Here the observed fitness is the mean of the measured fitness across all biological replicates. As a comparison, we compute measurement error, which is the absolute difference between the fitness estimated from one biological replicate and that averaged across all biological replicates in environment 2. The second metric is prediction bias, which is the difference between the predicted and observed fitness in environment 2. A LOWESS (local polynomial regression) curve fitting is used to acquire a general trend of the prediction error, measurement error, and prediction bias across different fitness ranges (with the smoother span =0.2). As shown in Fig. 3e, we observed a relatively large positive bias for the prediction of f23 at the low fitness end (f30< 0.6). This bias is expected for the following reason. When f30 < 1, f23 is generally higher than f30 (Fig. 3a). Because here we analyze only those genotypes that have ≥1 read at T1 in both Env23 and Env30, f30 is more likely to be overestimated than f23, which led to a positive prediction bias of f23. In identifying outliers, fitness at Env23 was individually predicted from the fitness values of five biological replicates at Env30 using the piecewise linear model. A t-test was then used to compare the five predicted and five observed fitness values at Env23, followed by a correction for multiple testing with a cutoff of FDR = 0.05.
To predict the FL in a new environment using FL in an old environment and the fitness measures of a few genotypes in the new environment, we randomly picked (without replacement) various numbers of N1 mutants whose fitness values in the old environment are within designated ranges. We used the fitness measures of these mutants in both environments to construct a robust linear model, which was then used to predict the FL in the new environment. We repeated this process 1000 times and computed the prediction bias and error as aforementioned from each repeat. Note that a mutant may be used in multiple replicates.
Expected fitness under no epistasis
Under the assumption of no epistasis, the expected fitness of a genotype carrying n mutations is the product of the fitness of the n constituent N1 mutants or 0.5 whichever is larger.
Predicting epistasis in a new environment
For two N1 mutants and the corresponding double mutant, we first predict their fitness in environment 2 from their measured fitness in environment 1. We then use the definition of epistasis to predict the epistasis between the two mutations in environment 2 from the three predicted fitness values in environment 2. This predicted epistasis in environment 2 is compared with the corresponding epistasis in environment 1.
Let the fitness of two N1 mutants and the corresponding N2 mutant be fA, fB, and fAB in environment 1 and f'A, f'B, and f'AB in environment 2, respectively. Because the vast majority of N1 and N2 mutants are less fit than the wild-type, we assume that formula (f′-1) = k(f-1) holds for all three genotypes considered here. It can be shown that the difference in epistasis between the two environments is
When the mean fitness effect of mutations enlarges upon an environmental shift, k > 1. Thus, when ε1 < 0, which occurs to most pairs of mutations, ε2 – ε1 is negative. This result explains why negative epistasis rarely becomes positive upon an environmental shift that enlarges the mean fitness effect of mutations.
Analysis of a previously published fitness landscape
For most FL mapping in the past, the exact number of generations in the competition was not reported, and hence the data cannot be used to estimate the fitness per generation. Here, we focused on a dataset that allowed estimation of fitness per generation. The dataset contains 189 mutants, representing all 20 amino acids and a stop codon at each of nine examined positions of yeast Hsp90 35. The fitness data were collected in four environments: low salinity in 30°C (30L), high salinity in 30°C (30H), low salinity in 36°C (36L), and high salinity in 36°C (36H). Because no mutant showed a fitness > 1.01 at 30L or 36L, we fitted a simple linear model with a single slope k for prediction of FL in one environment from that in another. If the predicted fitness was < 0.5, the lower bound value of 0.5 was assigned.
Data availability
The NCBI accession number for the sequencing data is GSE111508. Other data are available upon request.
Code availability
The computer code is available on github.com/lichuan199010/tRNAMultiEnv.
Supplementary Material
Acknowledgments
We thank W.-C. Ho, W. Qian, X. Wei, and J.-R. Yang for valuable comments. This work was supported by NSF DDIG (DEB-1501788) to J.Z. and C.L. and by NIH (R01GM103232) to J.Z.
Footnotes
Author contributions: J.Z. conceived the project; C.L. and J.Z. designed the experiment; C.L. performed the experiment and analyzed the data; C.L. and J.Z. wrote the paper.
Competing interests: The authors declare no competing financial interests.
References
- 1.de Visser JA, Krug J. Empirical fitness landscapes and the predictability of evolution. Nat Rev Genet. 2014;15:480–490. doi: 10.1038/nrg3744. [DOI] [PubMed] [Google Scholar]
- 2.Qian W, Ma D, Xiao C, Wang Z, Zhang J. The genomic landscape and evolutionary resolution of antagonistic pleiotropy in yeast. Cell Rep. 2012;2:1399–1410. doi: 10.1016/j.celrep.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergland AO, Behrman EL, O'Brien KR, Schmidt PS, Petrov DA. Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila. PLoS Genet. 2014;10:e1004775. doi: 10.1371/journal.pgen.1004775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell-Olds T, Willis JH, Goldstein DB. Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat Rev Genet. 2007;8:845–856. doi: 10.1038/nrg2207. [DOI] [PubMed] [Google Scholar]
- 5.de Vos MG, Dawid A, Sunderlikova V, Tans SJ. Breaking evolutionary constraint with a tradeoff ratchet. Proc Natl Acad Sci U S A. 2015;112:14906–14911. doi: 10.1073/pnas.1510282112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinberg B, Ostermeier M. Environmental changes bridge evolutionary valleys. Sci Adv. 2016;2:e1500921. doi: 10.1126/sciadv.1500921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schluter D. Ecology and the origin of species. Trends Ecol Evol. 2001;16:372–380. doi: 10.1016/s0169-5347(01)02198-x. [DOI] [PubMed] [Google Scholar]
- 8.Puchta O, et al. Network of epistatic interactions within a yeast snoRNA. Science. 2016;352:840–844. doi: 10.1126/science.aaf0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Qian W, Maclean M, Zhang J. The fitness landscape of a tRNA gene. Science. 2016;352:837–840. doi: 10.1126/science.aae0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hietpas RT, Jensen JD, Bolon DN. Experimental illumination of a fitness landscape. Proc Natl Acad Sci U S A. 2011;108:7896–7901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014;513:120–123. doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bank C, Hietpas RT, Jensen JD, Bolon DN. A systematic survey of an intragenic epistatic landscape. Mol Biol Evol. 2015;32:229–238. doi: 10.1093/molbev/msu301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melnikov A, Rogov P, Wang L, Gnirke A, Mikkelsen TS. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014;42:e112. doi: 10.1093/nar/gku511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Risch N, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301:2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorgeirsson TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips PC. Epistasis--the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu CT, Ingelmo P, Rand DM. G×G×E for lifespan in Drosophila: mitochondrial, nuclear, and dietary interactions that modify longevity. PLoS Genet. 2014;10:e1004354. doi: 10.1371/journal.pgen.1004354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wendling CC, Fabritzek AG, Wegner KM. Population-specific genotype × genotype × environment interactions in bacterial disease of early life stages of Pacific oyster larvae. Evol Appl. 2017;10:338–347. doi: 10.1111/eva.12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filteau M, et al. Evolutionary rescue by compensatory mutations is constrained by genomic and environmental backgrounds. Mol Syst Biol. 2015;11:832. doi: 10.15252/msb.20156444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lalic J, Elena SF. Epistasis between mutations is host-dependent for an RNA virus. Biol Lett. 2013;9:20120396. doi: 10.1098/rsbl.2012.0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Vos MG, Poelwijk FJ, Battich N, Ndika JD, Tans SJ. Environmental dependence of genetic constraint. PLoS Genet. 2013;9:e1003580. doi: 10.1371/journal.pgen.1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flynn KM, Cooper TF, Moore FB, Cooper VS. The environment affects epistatic interactions to alter the topology of an empirical fitness landscape. PLoS Genet. 2013;9:e1003426. doi: 10.1371/journal.pgen.1003426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadowska-Bartosz I, Paczka A, Molon M, Bartosz G. Dimethyl sulfoxide induces oxidative stress in the yeast Saccharomyces cerevisiae. Fems Yeast Res. 2013;13:820–830. doi: 10.1111/1567-1364.12091. [DOI] [PubMed] [Google Scholar]
- 24.Bhaskaran H, Rodriguez-Hernandez A, Perona JJ. Kinetics of tRNA folding monitored by aminoacylation. RNA. 2012;18:569–580. doi: 10.1261/rna.030080.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexandrov A, et al. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell. 2006;21:87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 26.Alexandrov A, Grayhack EJ, Phizicky EM. tRNA m7G methyltransferase Trm8p/Trm82p: evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA. 2005;11:821–830. doi: 10.1261/rna.2030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, Vogt CE, McBrairty M, Al-Hashimi HM. Influence of dimethylsulfoxide on RNA structure and ligand binding. Anal Chem. 2013;85:9692–9698. doi: 10.1021/ac402038t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ottman R. Gene-environment interaction: definitions and study designs. Prev Med. 1996;25:764–770. doi: 10.1006/pmed.1996.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei X, Zhang J. The genomic architecture of interactions between natural genetic polymorphisms and environments in yeast growth. Genetics. 2017;205:925–937. doi: 10.1534/genetics.116.195487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlborg O, et al. A global search reveals epistatic interaction between QTL for early growth in the chicken. Genome Res. 2003;13:413–421. doi: 10.1101/gr.528003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sackman AM, Rokyta DR. Additive phenotypes underlie epistasis of fitness effects. Genetics. 2018;208:339–348. doi: 10.1534/genetics.117.300451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. doi: 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- 33.Smith EN, Kruglyak L. Gene-environment interaction in yeast gene expression. PLoS Biol. 2008;6:e83. doi: 10.1371/journal.pbio.0060083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hillenmeyer ME, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hietpas RT, Bank C, Jensen JD, Bolon DNA. Shifting fitness landscapes in response to altered environments. Evolution. 2013;67:3512–3522. doi: 10.1111/evo.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warringer J, et al. Trait variation in yeast is defined by population history. PLoS Genet. 2011;7:e1002111. doi: 10.1371/journal.pgen.1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mani R, St Onge RP, Hartman JLt, Giaever G, Roth FP. Defining genetic interaction. Proc Natl Acad Sci U S A. 2008;105:3461–3466. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NCBI accession number for the sequencing data is GSE111508. Other data are available upon request.