

Abstract
Epigenetic regulation is emerging as a key feature in the molecular characteristics of various human diseases. Epigenetic aberrations can occur from mutations in genes associated with epigenetic regulation, improper deposition, removal or reading of histone modifications, DNA methylation/demethylation, and impaired non-coding RNA interactions in chromatin. Menin, the protein product of the gene causative for the multiple endocrine neoplasia type 1 (MEN1) syndrome, interacts with chromatin-associated protein complexes and also regulates some non-coding RNAs, thus participating in epigenetic control mechanisms. Germline inactivating mutations in the MEN1 gene that encodes menin predispose patients to develop endocrine tumors of the parathyroids, anterior pituitary and the duodenopancreatic neuroendocrine tissues. Therefore, functional loss of menin in the various MEN1-associated endocrine cell types can result in epigenetic changes that promote tumorigenesis. Because epigenetic changes are reversible, they can be targeted to develop therapeutics for restoring the tumor epigenome to the normal state. Irrespective of whether epigenetic alterations are the cause or consequence of the tumorigenesis process, targeting the endocrine tumor-associated epigenome offers opportunities for exploring therapeutic options. This review presents epigenetic control mechanisms relevant to the interactions and targets of menin, and the contribution of epigenetics in the tumorigenesis of endocrine cell types from menin loss.
Keywords: Neuroendocrine, Pancreas, Parathyroid, Pituitary
Introduction
Epigenetic changes observed in various human diseases have been shown to play important roles as disease drivers, biomarkers and therapeutic targets. Epigenetic changes can occur due to mutations in factors that control epigenetic modifications or in response to metabolic or environmental signals, that lead to improper deposition, removal or reading of histone modifications, DNA methylation/demethylation, and impaired non-coding RNA interactions in chromatin (Allis et al. 2015). The evidence for the role of epigenetic regulation in tumor etiology is highlighted by the discovery of mutations in genes encoding factors that participate in epigenetic regulatory mechanisms in various cancers (Koschmann et al. 2017). Menin, the protein encoded by the gene responsible for causing the multiple endocrine neoplasia type 1 (MEN1) syndrome participates in epigenetic regulation (Shilatifard 2012, van Nuland et al. 2013). Patients with the MEN1 syndrome carry germline heterozygous inactivating mutations in the MEN1 gene located at chromosome 11q13, that encodes menin, which predisposes to the development of tumors in multiple endocrine organs (Chandrasekharappa et al. 1997, Lemmens et al. 1997). The main MEN1-associated endocrine cell types that form tumors are located in the parathyroids, anterior pituitary and the duodenopancreatic neuroendocrine tissues (Thakker et al. 2012). Other MEN1-associated endocrine tumors include foregut carcinoids that arise from neuroendocrine cells of the lung, thymus or gastrointestinal (GI) tract. The tumors are classified as functioning when they hypersecrete the hormones characteristic of the endocrine cell types affected, or non-functioning when they do not hypersecrete any hormones. Parathyroid tumors are functioning causing primary hyperparathyroidism due to excessive parathyroid hormone (PTH) secretion. Pituitary and duodenopancreatic neuroendocrine tumors in MEN1 patients are observed to be either functioning (prolactinoma, 20%; somatotropinoma, 10%; gastrinoma, 40%; insulinoma,10%), or non-functioning (pituitary, 5%; PNETs, 20–55%) (Marini et al. 2017, Schussheim et al. 2001, Thakker et al. 2012). In the context of these endocrine tumors, MEN1 acts as a classic tumor suppressor gene because human tumors show loss of heterozygosity (LOH) at 11q13 resulting in biallelic loss/inactivation of MEN1. (Chandrasekharappa et al. 1997, Lemmens et al. 1997). Similarly, in genetically engineered mouse models, germline targeted deletion of both copies of the Men1 gene causes embryonic lethality, while germline targeted deletion of one copy of the Men1 gene generates live mice that later in life develop endocrine tumors similar to those seen in the human MEN1 syndrome with LOH at the Men1 locus (Agarwal 2014, Agarwal 2017b).
Interestingly, MEN1 mutations are also detected in 30–40% of sporadic tumors of MEN1-associated endocrine cell types - parathyroid adenomas, gastrinomas, pancreatic neuroendocrine tumors (PNETs), and lung neuroendocrine tumors (Cromer et al. 2012, Debelenko et al. 1997, Jiao et al. 2011, Newey et al. 2012, Scarpa et al. 2017, Simbolo et al. 2017, Zhuang et al. 1997). Among sporadic PNETs, MEN1 mutations are detected in the non-functioning PNETs, but rarely in the functioning PNETs (insulinomas) (Cao et al. 2013, Chou et al. 2016, Jiao et al. 2011, Lichtenauer et al. 2015, Scarpa et al. 2017, Wang et al. 2017). Also, MEN1 mutations are rarely detected in sporadic pituitary tumors (Bi et al. 2017, Marques and Korbonits 2017, Newey et al. 2013, Poncin et al. 1999). Because menin participates in epigenetic regulation, functional loss of menin in the various MEN1-associated endocrine cell types would lead to epigenetic changes that could potentially promote tumorigenesis. Furthermore, evidence from genome and exome sequencing efforts of sporadic non-functioning and functioning PNETs, parathyroid adenoma, and lung neuroendocrine tumors have shown additional mutations in other genes associated with epigenetic control mechanisms that predict epigenetic changes to promote tumorigenesis in these endocrine cell types (Chou et al. 2016, Cromer et al. 2012, Jiao et al. 2011, Romano et al. 2017, Scarpa et al. 2017, Simbolo et al. 2017, Wang et al. 2017).
Menin is a 67 kDa ubiquitously expressed predominantly nuclear protein, with no intrinsic enzymatic activity. As stated above, menin is a tumor suppressor in the context of MEN1-associated endocrine cell types; however, menin has been shown to act as a pro-oncogenic co-factor in mixed lineage leukemia (Muntean and Hess 2012, Yokoyama et al. 2005). The crystal structure of menin shows a deep pocket that can act as a binding site for interacting proteins (Protein Data Bank No. 3RE2 and 3U84) (Huang et al. 2012, Murai et al. 2011). Evidence from protein-protein interaction studies have shown that menin is involved in epigenetic regulation and gene transcription through interaction with proteins in chromatin-associated protein complexes and transcription factors, affecting the expression of target genes such as genes that control cell proliferation (Karnik et al. 2005, Wu and Hua 2011). Interacting partners of menin also include proteins that participate in cell adhesion, cell division, cell motility, cell signaling, cytoskeletal structure, DNA repair in response to DNA damage, and studies conducted in Drosophila melanogaster have shown a role of menin in the maintenance of genomic stability (Agarwal 2017a, Busygina et al. 2004, Feng et al. 2017a, Hendy et al. 2009).
Investigations to elucidate the biological function of menin through protein-protein interaction studies have significantly advanced our knowledge about the potential roles of menin in various cellular pathways, but they have been mainly conducted using cell lines (HEK293, HeLa, HepG2, K562, and Mouse Embryo Fibroblasts (MEFs)) that are unrelated to MEN1-associated endocrine cell types. Follow-up functional characterization of some interactions of menin have been performed in endocrine/endocrine-related cell lines (βlox5, BON1, INS1, MIN6, NIH-H720, NIH-727, and QGP1) albeit not in endocrine cell lines with MEN1 mutations or that lack menin because no such cell lines are currently available. The focus of this review will be on studies conducted using endocrine/endocrine-related cell lines, or menin-null endocrine tumors from mouse models. This review excludes the discussion of menin’s interaction with transcription factors because they were recently well-covered elsewhere (Agarwal 2017a, Dreijerink et al. 2017, Feng et al. 2017a). In this review, we present a survey of the published literature to describe epigenetic control mechanisms that are relevant to the interactions and targets of menin, the contribution of epigenetics in the tumorigenesis of endocrine cell types from menin loss, and experimental therapeutic strategies to potentially correct the tumor-specific epigenetic changes.
Epigenetic Mechanisms
Eukaryotic DNA with core histone proteins (H2A, H2B, H3, and H4) is packaged into nucleosomes to form chromatin fibers (Olins and Olins 2003). This packaging regulates access to DNA for various biological processes such as DNA replication, DNA repair and transcription into RNA. Modifications to chromatin are essential to permit or restrict access, for example, to the transcriptional machinery including transcription factors for gene expression (Flavahan et al. 2017). Chromatin modifiers and remodelers constitute ‘epigenetic factors’ that function in multi-protein complexes that may also include non-coding RNAs, to modify the DNA or histones contained in nucleosomes (Allis and Jenuwein 2016). The multi-protein complexes contain enzymes and proteins referred to as the ‘writers’, ‘readers’ and ‘erasers’ of chromatin modifications (Allis and Jenuwein 2016). Modifications to DNA include methylation, hydroxymethylation and further oxidation. Histones can undergo a variety of post-translational modifications (PTMs) such as methylation, acetylation, phosphorylation and ubiquitination to generate an active, repressive or poised state to control gene expression; and the array of PTMs that govern a specific chromatin state is referred to as the ‘histone code’ (Huang et al. 2014). Also, chromatin interactions can occur over short and long distances thereby generating conditions that enhance gene transcription or insulate the regions of gene transcription (Rowley and Corces 2016). ‘Epigenetic changes’ through alterations in DNA and histone modifications, chromatin remodeling, and alterations in non-coding RNA interactions, create a positive or negative environment affecting the outcome of gene transcription and chromatin structure (Allis et al. 2015). The ‘epigenetic marks’ on DNA and histones that are generated as a result of the enzymatic activity of various chromatin modifying enzymes can survive successive cell divisions, and they are reversible, which presents an opportunity for correction to the normal state, for example when such changes promote disease processes (Zoghbi and Beaudet 2016). Technological advances in chromatin immunoprecipitation and next-generation sequencing have allowed comprehensive characterization of genome-wide epigenetic marks to reveal the status of the epigenome in normal and pathological conditions that can help to identify epigenomic changes that are disease drivers, or to serve as biomarkers and therapeutic targets. Below we describe the various epigenetic mechanisms and chromatin modifications that are relevant to MEN1-associated endocrine cell types (Figure 1).
Figure 1. Overview of menin-dependent epigenetic regulation in MEN1-associated endocrine cell types.
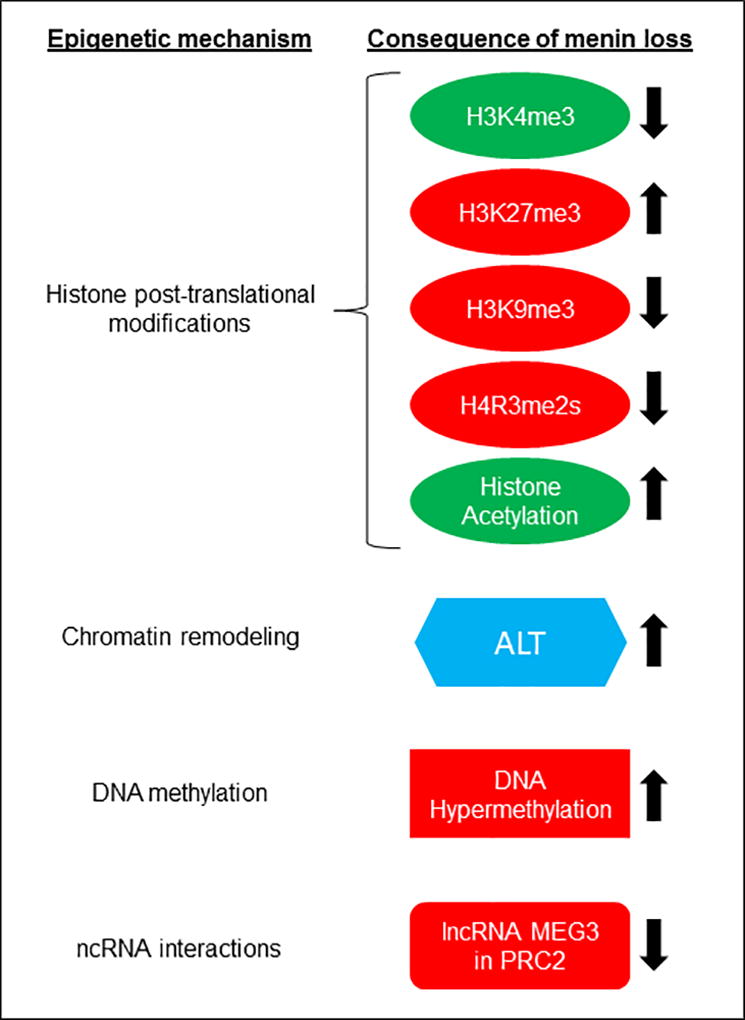
Epigenetic mechanisms, and the observed or predicted consequence of menin loss are shown that were investigated in various studies of human or mouse endocrine tumors, or studies conducted in endocrine/endocrine-related cell lines. Green or Red, indicates the normal effect of the epigenetic event resulting in the activation or repression of gene expression, respectively. Up or Down arrow, indicates increase or decrease, respectively, of the epigenetic event in a cell line or tumor with menin-deficiency. ALT (alternative lengthening of telomeres) is shown in blue (and not in red or green) because it is an event associated with chromatin structure at telomeres.
Histone methylation and demethylation
Methylation of histones at lysine or arginine residues in the chain of amino acids that protrude from the nucleosome, referred to as the ‘histone tail’, is achieved by histone methyltransferases (KMTs) that exist in multi-subunit protein complexes (Cheng 2014). KMTs methylate histone H3 on lysine residues at positions 4, 9, 27, 36, and 79 in the in mono-, di-, or tri-methylated form depending on whether one, two, or three methyl groups are added, respectively (Chi et al. 2010). In general, the histone H3 lysine-4 trimethyl mark (H3K4me3) specifies a state of transcriptional activation, that is located at the transcriptional start site (TSS) of actively transcribed genes. On the other hand, H3K9me3 and H3K27me3 are associated with transcriptional silencing. When both H3K4me3 and H3K27me3 occur together, the gene is said to be bivalently poised - a state maintained, for example, by embryonic stem cells during lineage differentiation and development, to enable rapid switching between active and repressive gene transcription states (Voigt et al. 2013). Eraser KDMs such as lysine specific demethylase 1 (LSD1/KDM1A), lysine specific demethylase 2 (LSD2/KDM1B), and Jumonji AT-rich interacting domain 1A (JARID1A/KDM5A/RBP2), remove mono- and di-methylation of H3K4 and H3K9 (Cheng 2014). Depending on the specific amino acid residue methylated or demethylated, and the level of methylation, the process of histone methylation and demethylation generates active or repressive chromatin states.
H3K4me3 and H3K27me3
There are at least six different protein complexes that contain KMTs for catalyzing the gene activating histone modification H3K4me3 (Dreijerink et al. 2017). Menin has been shown to interact with KMTs, MLL1 (KMT2A) and MLL2 (KMT2B), in two of those multi-subunit protein complexes, and studies using chromatin immunoprecipitation have shown that menin is present at the TSS with H3K4me3 (Hughes et al. 2004, Scacheri et al. 2006, Yokoyama et al. 2004). Several lines of evidence have established the importance of this interaction of menin involving H3K4me3 and the target genes affected by using MEN1-associated endocrine cell types, mainly pancreatic islets.
Mice with targeted deletion of Men1 in the germline (Men1+/−) or specifically in the pancreatic islet beta-cells (Men1f/f;RIP-Cre) develop insulin-secreting pancreatic islet tumors (insulinomas) (Agarwal 2014, Agarwal 2017b). Interestingly, both KMT2A and MEN1 co-localize at human chromosome 11q and mouse chromosome 19. LOH at this locus in MEN1 mutation-positive human PNETs and mouse PNETs (insulinomas) in Men1+/− mice contributes to the tumorigenesis process. Targeted conditional deletion of both Kmt2a and Men1 in mice (Kmt2af/f;Men1f/f;RIP-Cre) led to the acceleration of pancreatic islet tumor formation, and also shortened the average life span of these mice (Lin et al. 2016). This study showed that MLL1 can cooperate with menin to prevent tumor formation. In another study, targeted conditional deletion in mice of Men1 and Rbp2 which encodes a H3K4me3 demethylase (Rbp2f/f;Men1f/f;RIP-Cre) decreased the rate of tumor formation and prolonged survival (Lin et al. 2011). These data showed that the histone demethylase activity of RBP2 is linked to islet tumorigenesis downstream of menin loss.
To investigate the contribution of menin-MLL1 mediated epigenetic regulation in tumor formation, genome-wide distribution of the gene activation mark H3K4me3 and its counterpart recessive mark H3K27me3 was examined in menin-deficient pancreatic islets of 2-month-old Men1f/f;RIP-Cre and control wild-type mice (Lin et al. 2015). This study found that within the same set of genes, loss of H3K4me3 correlated with increased H3K27me3 in menin-deficient islets, and the expression of such genes was downregulated. The most significantly downregulated gene in menin-deficient islets was Igfbp2, that also displayed loss of H3K4me3 together with gain of H3K27me3. Interestingly, the decline in Igfbp2 expression in menin-deficient islets could be reversed with simultaneous deletion of Rbp2 (Lin et al. 2015). This observation further confirms that the histone demethylase activity of RBP2 is linked to islet tumorigenesis downstream of menin loss.
Genome-wide occupancy patterns of menin, MLL1 and another component of the MLL-protein-complex (RBBP5) in human pancreatic islet cells, combined with gene expression analysis in menin-null islets from Men1f/f;RIP-Cre mice showed upregulation of the pancreatic islet β-cell differentiation factor HLXB9 (Scacheri et al. 2006). Candidate gene analysis using menin-null islets from Men1f/f;RIP-Cre mice showed downregulation of cell cycle inhibitory genes in menin-null islet tumors compared to normal islets, mainly the cyclin-dependent kinase inhibitor (CDKI) genes CDKN2C/p18 and CDKN1B/p27, with concomitant loss of H3K4me3 in their promoter regions (Karnik et al. 2005). In another study, a long non-coding RNA, MEG3 was identified as an epigenetic target of menin (Agarwal and Jothi 2012). This study used genome-wide assessment of H3K4me3 and gene expression from wildtype mouse embryonic stem cells (mESCs) and menin-null mESCs. Loss of H3K4me3 was observed in menin-null mESCs at the Meg3 gene, together with downregulation of the Meg3 transcript. Further studies showed decreased MEG3 levels in mouse and human islet tumors (Modali et al. 2015). Also, loss of MEG3 expression was reported in sporadic non-functioning pituitary tumors (Zhao et al. 2005). When mESCs were in vitro differentiated into pancreatic-islet like endocrine cells (PILECs), reduced H3K4me3 and reduced gene expression was observed of the Hox genes in PILECs derived from menin-null mESCs (Agarwal and Jothi 2012). HOX genes have been identified as the direct target of the MLL1-complex in many different cell types, particularly in hematopoetic cells where the menin-MLL association partner as an oncogenic complex in MLL-fusion leukemia (Muntean and Hess 2012). It is important to note that the PILECs were heterogeneous in terms of the cell population as assessed by the expression of pancreatic marker genes, and not entirely consisted of pancreatic endocrine cells (Agarwal and Jothi 2012). Whether HOX genes play a role in MEN1-associated endocrine tumorigenesis remains to be determined.
H3K9me3
Another histone lysine modification regulated by menin is the repressive H3K9me3, through interaction with a lysine methyltransferase, suppressor of variegation 3–9 homolog 1 (SUV39H1) (Yang et al. 2013). In INS-1 rat insulinoma cells (Merglen et al. 2004), enhanced H3K9me3 was detected at the promoter region of the membrane metallo-proteinase (Mme) gene with combined presence of menin, SUV39H1 and the chromatin remodeling protein DAXX (Feng et al. 2017b). The DAXX gene is frequently mutated in sporadic non-functioning PNETs (Chou et al. 2016, Jiao et al. 2011, Scarpa et al. 2017). Therefore, this study helps to integrate the potential involvement of multiple epigenetic regulators to prevent tumorigenesis.
H4R3me2s
PRMTs methylate arginine residues in histone H3 at positions 2, 8, 17, and 26; and histones H4 and H2A at positions 3 and 11, respectively (Yang and Bedford 2013). PRMTs are classified into two families. The type I family comprises the members 1, 2, 3, 4, 6 and 8, which catalyze mono-methylation and asymmetric dimethylation of arginines (Yang and Bedford 2013). The type II family consisting of PRMTs 5, 7, and 9 mediate monomethylation and symmetric dimethylation of arginines. In mammalian cells, PRMT5 is the predominant type II arginine methyltransferase which acts as a transcriptional repressor specifying H3R2me2 and H4R3me2 (Stopa et al. 2015). Menin has been shown to interact with PRMT5, coinciding with a repressive chromatin mark, symmetric dimethylation of H4R3 (H4R3me2s), at the promoter of the growth arrest specific 1(Gas1) gene (Gurung et al. 2013). GAS1 has been shown to direct the binding of the sonic hedgehog (SHH) ligand to its receptor patched 1 (PTCH1) to activate the pro-proliferative and oncogenic Hedgehog signaling pathway (Martinelli and Fan 2007). Thus, menin loss would coincide with increased GAS1 protein and oncogenic Hedgehog signaling. Further characterization of menin/PRMT5 showed that pharmacologic inhibition of Hedgehog signaling with the drug GDC-0449 reduced cell proliferation in insulinoma tumors of Men1f/f;RIP-Cre mice (Gurung et al. 2013). These data show that inhibition of Hedgehog signaling in PNETs could target the downstream effects from loss of menin/PRMT5 interaction, revealing a potential target for treating MEN1 tumors.
Histone acetylation and deacetylation
Histone acetyl transferases (HATs) catalyze acetylation at lysine residues in histone tails. There are five well characterized HAT families that can acetylate specific lysine residues of mammalian histones - p300/CBP, Gcn5-related N-acetyltransferase (GNAT) (GCN5/PCAF), MYST (for ‘MOZ, Ybf2/Sas3, Sas2 and Tip60) related HATs, the general transcription factor HATs which include the TFIID subunit TAF250, and nuclear receptor coactivators (SRCs) (Marmorstein and Zhou 2014). The acetylation marks on histones are biologically read by the bromodomain (BRD) contained in the bromodomain and extraterminal (BET) proteins comprising members BRD2, BRD3, BRD4, and BRDT. Histone acetylation generally favors active transcription that is reversed by the activity of histone deacetylases (HDACs) (Marmorstein and Zhou 2014). There are 18 HDACs that belong to four different subtypes classified on sequence similarities, shared catalytic mechanisms, and active sites (Seto and Yoshida 2014). Small molecule inhibitors (CPI-203, JQ1 and PFI-1) of bromodomain interactions with acetylated histone tails have been developed as potential therapeutics (Perez-Salvia and Esteller 2017). JQ1 binds to the acetyl-lysine recognition pocket of bromodomains 1 and 2 on BRD4 which displaces BRD4 from chromatin, thus preventing the ‘reading’ of the acetyl mark and blocking active transcription. CPI-203 is a primary amide analog of JQ1, and PFI-1 inhibits BRD2 and BRD4 (Perez-Salvia and Esteller 2017).
There is no direct evidence in MEN1-associated endocrine cells for the interaction of menin in protein complexes that are involved in specific histone acetylation or deacetylation. However, there is indirect evidence for the involvement of histone acetylation/deacetylation in endocrine cells that comes from the demonstration of growth inhibitory effects of HDAC inhibitors (HDCAi) (trichostatin-A (TSA), thailandepsin-A (TDP-A), sodium butyrate (NaB), valproic acid (VPA), and MS-275), and BET protein bromodomain inhibitors (BETi) (CPI201, PFI-1 and JQ1) in neuroendocrine tumor cell lines BON1, NCI-H720, NCI-H727, and QGP1 (Adler et al. 2009, Arvidsson et al. 2016, Baradari et al. 2006, Lines et al. 2017, Wong et al. 2014). BON1 is a human cell line derived from a peripancreatic lymph node metastasis of a pancreatic neuroendocrine carcinoid tumor (Townsend et al. 1993). NCI-H720 and NCI-H727 are derived from human atypical and typical bronchial carcinoid tumors, respectively (ATCC). QGP-1 is a carcinoembryonic antigen-producing cell line from a human pancreatic islet carcinoma (Kaku et al. 1980). Anti-tumor activity of the HDACi VPA and BETi CPI203 was also shown in a BON1 xenograft model (Greenblatt et al. 2007, Wong et al. 2014). Also, in vivo anti-tumor activity of JQ1 has been demonstrated in primary tumors (insulinomas) of the Men1f/f;RIP-Cre mouse model (Lines et al. 2017). These studies highlight the potential of targeting the epigenetic histone acetyl marks in treating MEN1-associated neuroendocrine tumors.
Chromatin remodeling
Histones and nucleosomes are subjected to remodeling by protein-complexes that contain ATP-dependent chromatin remodeling activity. Also, eukaryotic cells express histone variants, in addition to the “canonical” core histones, providing another layer of epigenetic regulation. Chromatin remodeling activity facilitates transcriptional initiation and elongation for the passage of RNA polymerase through nucleosomal arrays (Clapier and Cairns 2009). This is achieved by displacing histone dimers H2A/H2B or H3/H4 leading to the substitution of the core histones in the dimers with histone variants such as histone H2A.Z or H3.3 (Clapier and Cairns 2009, Szenker et al. 2011). In addition to controlling gene transcription, histone variants and their PTMs also contribute to nucleosome and chromatin structure and function (Szenker et al. 2011).
The evidence for the involvement of histone remodeling in MEN1-associated endocrine cells comes from sporadic non-functioning PNETs that show somatic mutations in death-domain-associated protein (DAXX) and its interacting partner, alpha thalassemia/mental retardation syndrome X-linked (ATRX) (Chou et al. 2016, Jiao et al. 2011, Scarpa et al. 2017). ATRX with its intrinsic ATPase activity, in collaboration with its partner DAXX, a highly specific histone chaperone, deposit the histone variant H3.3 in chromatin at telomeric and pericentromeric regions (Amorim et al. 2016). Telomeres protect chromosome ends to prevent the loss of DNA, and in neoplastic cells the maintenance of telomeres is required for immortality. Lengthening of telomeres in tumor cells can occur from excessive production of the telomerase enzyme or through alternative lengthening of telomeres (ALT), a process similar to homologous recombination (Amorim et al. 2016). ATRX/DAXX mutations, or loss of their expression in sporadic PNETs correlate with increased telomere length that can potentially support tumor progression (Kim et al. 2017, Marinoni et al. 2017, Scarpa et al. 2017, Singhi et al. 2017, VandenBussche et al. 2017). Some level of ALT was also detected in MEN1 mutation-positive sporadic PNETs (Scarpa et al. 2017). Whether ALT plays a role in the endocrine tumors of patients with the MEN1 syndrome remains to be determined.
DNA methylation and demethylation
DNA methylation is an epigenetic modification that occurs predominantly at CpG sites where a cytosine nucleotide is followed by a guanine nucleotide in which the cytosine is methylated to 5-methylcytosine (5-mC) (Li and Zhang 2014). Methylation at CpG islands (GC-rich DNA regions), is often located in promoter regions of genes. Aberrant hypo- or hyper- methylation of these promoter region CpG islands can result in gene overexpression or gene silencing, respectively. A family of DNA methyl transferases (DNMTs), designated DNMT1, DNMT3A, and DNMT3B mediate CpG methylation. While DNMT3A and DNMT3B establish de novo DNA methylation, DNMT1 serves to propagate and stably maintain the DNA methylation mark (Li and Zhang 2014). DNA methylation can be reversed by the ten-eleven translocation (TET) family of methylcytosine hydroxylases that convert 5mC to 5-hydroxymethylcytosine (5-hmC) (Li and Zhang 2014). Also, drugs such as 5-azacitidine (5AC) and 5-aza-2’-deoxycytidine (DAC) can block DNA methylation by directly inhibiting DNMTs (Sato et al. 2017).
The histone modification H3K4me3 has been shown to protect CpG islands from DNA methylation resulting in an antagonism between H3K4me3 and DNA methylation to regulate gene transcription (Cedar and Bergman 2009). Also, DNA methylation in transcription factor recognition sequences can alter transcription factor binding (Barlow and Bartolomei 2014). A study conducted in MIN6 mouse insulinomas cells (Miyazaki et al. 1990) showed that overexpression of menin increased the expression of the lncRNA Meg3 (Modali et al. 2015). This effect coincided with H3K4me3 at the Meg3 promoter, and hypomethylation of the central CpG of a cAMP response element (CRE) that binds the transcription factor CREB; the DNA hypomethylation prevented CREB binding (Modali et al. 2015, Zhao et al. 2006). Whether similar instances of reciprocal H3K4me3 and DNA methylation together with altered transcription factor binding are present in MEN1-associated endocrine cells remains to be determined.
Several studies have shown indirect evidence for the participation of DNA methylation in MEN1-assocated endocrine cell types. DNA hypermethylation has been shown to occur in a subset of inherited MEN1 tumors, sporadic PNETs, sporadic pituitary tumors or sporadic parathyroid tumors at the promoter region of different genes (APC, CTNNB1, CDKN2A/p16, CDKN2B/p15, HIC1, MEG3, MLH1, RASSF1A, and RIZ1), with some also correlating with respective reduction of gene expression (Carling et al. 2003, Juhlin et al. 2010, Lindberg et al. 2008, Modali et al. 2015, Svedlund et al. 2012, Zhao et al. 2005). A genome-wide analysis of quantitative DNA methylation in parathyroid tissue samples showed global DNA hypermethylation in menin-null parathyroid tumors from MEN1 patients, and increased transcript levels of DNMT1, predicting increased DNMT1 activity in menin-null parathyroid cells accounting for the hypermethylated DNA (Yuan et al. 2016). Another study showed altered DNA methylation profiles in normal and pathologic parathyroid tissue with gene expression changes, and restored expression of hypermethylated genes upon treatment of primary cell cultures of parathyroid tumors with the DNA methylation inhibitor DAC (Starker et al. 2011). A few studies in endocrine/endocrine-related cell lines (BON1, H727, MIN6) have reported growth inhibition upon treatment with DNA methylation inhibitors 5AC or DAC (Alexander et al. 2010, Modali et al. 2015). These studies suggest DNA hypermethylation as an oncogenic event in MEN1-associated endocrine cell types, and therefore the potential benefit of DNA methylation inhibitors in treating PNETs, pituitary tumors and parathyroid tumors.
Non-coding RNA interactions
Non-coding RNAs (ncRNAs), as the name indicates, lack open reading frames (ORFs) and do not encode proteins. Short ncRNAs such as microRNA (miRNA) are processed from longer primary transcripts into small single-stranded molecules (20–24 nucleotides). They perform RNAi-mediated (RNA interference-mediated) gene silencing by complementary base-pair interactions targeting DNA in chromatin for RNA-induced transcriptional silencing (RITS), or messenger RNAs (mRNAs) post-transcriptionally for degradation, or by blocking translation (Cech and Steitz 2014). Long non-coding RNAs (lncRNAs) are transcripts of 200 bp or more in length that can interact with DNA and proteins whereby they also participate in epigenetic regulation (Kopp and Mendell 2018). Some lncRNAs have been shown to reside in chromatin-modifying and chromatin-remodeling protein complexes that silence gene expression. The eukaryotic ncRNAs play a dual role in epigenetic regulation - some miRNAs can silence genes that encode epigenetic factors such as DNMTs, and some lncRNAs can directly participate in chromatin modifying epigenetic control mechanisms such as in the chromatin inhibitory complex polycomb group (PcG) repressive complex 2 (PRC2) (Chen et al. 2015, Fabbri et al. 2007, Kaneko et al. 2014, Mondal et al. 2015).
In MEN1-associated endocrine cells (MIN6 and βlox5 cells), microRNA-24 (miR24) has been shown to target menin, and increased miR24 level was also shown to correlate with decreased menin level in the islets of Men1f/f;RIP-Cre mice and human parathyroid tumors (Grolmusz et al. 2017, Luzi et al. 2017, Luzi et al. 2012, Vijayaraghavan et al. 2014). This silencing of menin by a microRNA predicts downstream effects on chromatin modifications because menin acts as an epigenetic co-factor in chromatin modifying protein complexes. Low levels of the lncRNA MEG3 have been observed in mouse and human MEN1-associated PNETs, human sporadic PNETs, and in human sporadic pituitary tumors (Modali et al. 2015, Zhao et al. 2005). In MIN6 mouse insulinoma cells, Meg3 was demonstrated to co-immunoprecipitate with Ezh2 which is part of PRC2 for H3K27me3 establishment (Iyer et al. 2017). How these ncRNA interactions affect endocrine tumor epigenetics should be further investigated.
Targeting the pathologic epigenome of MEN1-associated endocrine cell types
Epigenome profiling for global epigenetic changes, such as in DNA methylation or histone modifications between normal and pathological samples, has demonstrated the presence of epigenetic changes in tumors and other disease states that could be exploited as targets for therapy (Jones et al. 2016). Epigenetic therapies that are under investigation to revert specific disease epigenomes to normal are designed to block the enzymatic activity of epigenetic regulators, disrupt interactions in chromatin modifying protein-complexes, or target specific epigenetic factors for degradation (Ahuja et al. 2016). However, designing of such therapeutics has to overcome several issues and challenges such as cell-specific targeting due to heterogeneity of the affected tissues, side effects, drug resistance, and achieving constant, consistent and long-lasting inhibition of the target (Dawson 2017). Potential therapies in the form of various small molecule chemical inhibitors that target epigenetic enzymes (e.g., DNMTi, HDACi, KTMi, KDMi, and BETi) have been developed. DNA hypomethylating agents (decitabine and azacitidine) that target DNMT1 and DNMT3A, and a few HDACi (panobinostat, vorinostat, and romidepsin) were approved by the US Food and Drug Administration (FDA) for the treatment of specific hematologic malignancies (Ahuja et al. 2016).
As discussed above, various HDACi, BETi and DNMTi have been shown to inhibit the growth of endocrine/endocrine-related cells lines, and intraperitoneal injection of the BETi JQ1 has been shown to reduce cell proliferation and increase apoptosis in insulinomas of the Men1f/f;RIP-Cre mouse model (Adler et al. 2009, Alexander et al. 2010, Arvidsson et al. 2016, Baradari et al. 2006, Lines et al. 2017, Modali et al. 2015, Wong et al. 2014). Another study showed that lack of a histone demethylase Rbp2 in the Men1f/f;RIP-Cre mouse background could decrease insulinoma development (Lin et al. 2011). Rbp2 loss could potentially restore H3K4me3 that occurred in the insulinoma cells due to menin-deficiency. Furthermore, overexpression of RBP2 was detected in human PNETs or their metastases, implicating a target for histone demethylase inhibitor therapy (Maggi et al. 2016). These studies suggest the potential of epigenetic drug therapy of tumors arising in MEN1-associated endocrine cell types irrespective of whether the epigenetic status of the tumor cells is a direct consequence of menin loss or a consequence of the tumorigenesis process.
Perspectives
In this review, we aimed to survey the literature for evidence of epigenetic regulation in “MEN1-acssociated endocrine cell types” which mainly included information from the parathyroids, pituitary, pancreatic islets, and lung neuroendocrine cells with the intention of highlighting the direct effects of menin as an epigenetic co-factor or functionally related epigenetic effects. Whole genome/exome sequencing efforts have identified mutations in the MEN1 gene and/or genes associated with epigenetic control mechanisms in sporadic endocrine tumors - non-functioning and functioning PNETs, parathyroid adenoma, and lung neuroendocrine tumors. Future studies directed towards identifying the downstream effects from these mutations will help to understand the contribution of epigenetic changes in the pathogenesis of the specific endocrine tumors. Work conducted in cell lines derived from pancreatic or lung neuroendocrine tumors or their metastasis, and in vivo work in insulinomas of Men1f/f;RIP-Cre mice have established the role of menin as an epigenetic co-factor in the context of a few epigenetic modifications: H3K4me3, H3K9me3 and H4R3me2s, and DNA methylation. Given that DNA methylation and multiple histone modifications are modulated by menin raises the question whether epigenetic changes in MEN1-associated tumors would encompass multiple epigenetic marks. Future studies directed towards determining the changes in multiple histone modifications and/or DNA methylation in tumors (epigenome signatures) compared to corresponding normal cells could be valuable for understanding the tumorigenesis process and potential treatment opportunities. The challenges for conducting such studies for MEN1-associated tumors are the non-availability of ‘corresponding’ specific normal cell types, and the lack of relevant cell lines from human tumors. Perhaps generating mouse xenografts of human tumors could prove to be useful in establishing cell lines. A recent study has shown that in the context of human PNETs using a human hepatocyte growth factor (HGF) ligand facilitated the successful generation of xenografts in mice because PNETs express high levels of the HGF receptor c-MET (Krampitz et al. 2016). Similar methods could be used to establish cell lines from MEN1 and sporadic tumors. Such tumor models and cell lines will facilitate a deeper understanding of epigenetic changes during the tumorigenesis process to exploit epigenetic therapy as a treatment option for the tumors of MEN1-associated endocrine cell types.
Acknowledgments
Funding:
Funding support for this review was from the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Declaration of interest:
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
References
- Adler JT, Hottinger DG, Kunnimalaiyaan M, Chen H. Combination therapy with histone deacetylase inhibitors and lithium chloride: a novel treatment for carcinoid tumors. Ann Surg Oncol. 2009;16:481–486. doi: 10.1245/s10434-008-0194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK. Exploring the tumors of multiple endocrine neoplasia type 1 in mouse models for basic and preclinical studies. Int J Endocr Oncol. 2014;1:153–161. doi: 10.2217/ije.14.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK. The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer. 2017a;24:T119–T134. doi: 10.1530/ERC-17-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK. Update on exploring the tumors of multiple endocrine neoplasia type 1 in mouse models for basic and preclinical studies. International Journal of Endocrine Oncology. 2017b;4:113–116. doi: 10.2217/ije.14.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK, Jothi R. Genome-wide characterization of menin-dependent H3K4me3 reveals a specific role for menin in the regulation of genes implicated in MEN1-like tumors. PLoS One. 2012;7:e37952. doi: 10.1371/journal.pone.0037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja N, Sharma AR, Baylin SB. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annual review of medicine. 2016;67:73–89. doi: 10.1146/annurev-med-111314-035900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander VM, Roy M, Steffens KA, Kunnimalaiyaan M, Chen H. Azacytidine induces cell cycle arrest and suppression of neuroendocrine markers in carcinoids. Int J Clin Exp Med. 2010;3:95–102. [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Caparros M-L, Jenuwein T, Reinberg D. Epigenetics. Cold Spring Harbor; New York: CSH Press, Cold Spring Harbor Laboratory Press; 2015. [Google Scholar]
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nature reviews. Genetics. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Amorim JP, Santos G, Vinagre J, Soares P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes (Basel) 2016;7 doi: 10.3390/genes7090066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson Y, Johanson V, Pfragner R, Wangberg B, Nilsson O. Cytotoxic Effects of Valproic Acid on Neuroendocrine Tumour Cells. Neuroendocrinology. 2016;103:578–591. doi: 10.1159/000441849. [DOI] [PubMed] [Google Scholar]
- Baradari V, Huether A, Hopfner M, Schuppan D, Scherubl H. Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocr Relat Cancer. 2006;13:1237–1250. doi: 10.1677/erc.1.01249. [DOI] [PubMed] [Google Scholar]
- Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harbor perspectives in biology. 2014;6 doi: 10.1101/cshperspect.a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ, Mei Y, Schumacher SE, Ben-David U, Chevalier A, et al. Landscape of Genomic Alterations in Pituitary Adenomas. Clin Cancer Res. 2017;23:1841–1851. doi: 10.1158/1078-0432.CCR-16-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busygina V, Suphapeetiporn K, Marek LR, Stowers RS, Xu T, Bale AE. Hypermutability in a Drosophila model for multiple endocrine neoplasia type 1. Hum Mol Genet. 2004;13:2399–2408. doi: 10.1093/hmg/ddh271. [DOI] [PubMed] [Google Scholar]
- Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai J, Peng Y, Li Y, Jiang X, Huang X, et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun. 2013;4:2810. doi: 10.1038/ncomms3810. [DOI] [PubMed] [Google Scholar]
- Carling T, Du Y, Fang W, Correa P, Huang S. Intragenic allelic loss and promoter hypermethylation of the RIZ1 tumor suppressor gene in parathyroid tumors and pheochromocytomas. Surgery. 2003;134:932–939. doi: 10.1016/s0039-6060(03)00422-7. discussion 939–940. [DOI] [PubMed] [Google Scholar]
- Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nature reviews. Genetics. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chen Z, Liu S, Tian L, Wu M, Ai F, Tang W, Zhao L, Ding J, Zhang L, Tang A. miR-124 and miR-506 inhibit colorectal cancer progression by targeting DNMT3B and DNMT1. Oncotarget. 2015;6:38139–38150. doi: 10.18632/oncotarget.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. Structural and functional coordination of DNA and histone methylation. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a018747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WC, Lin PH, Yeh YC, Shyr YM, Fang WL, Wang SE, Liu CY, Chang PM, Chen MH, Hung YP, et al. Genes involved in angiogenesis and mTOR pathways are frequently mutated in Asian patients with pancreatic neuroendocrine tumors. Int J Biol Sci. 2016;12:1523–1532. doi: 10.7150/ijbs.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, Carling T. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab. 2012;97:E1774–1781. doi: 10.1210/jc.2012-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science. 2017;355:1147–1152. doi: 10.1126/science.aam7304. [DOI] [PubMed] [Google Scholar]
- Debelenko LV, Brambilla E, Agarwal SK, Swalwell JI, Kester MB, Lubensky IA, Zhuang Z, Guru SC, Manickam P, Olufemi SE, et al. Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum Mol Genet. 1997;6:2285–2290. doi: 10.1093/hmg/6.13.2285. [DOI] [PubMed] [Google Scholar]
- Dreijerink KMA, Timmers HTM, Brown M. Twenty years of menin: emerging opportunities for restoration of transcriptional regulation in MEN1. Endocr Relat Cancer. 2017;24:T135–T145. doi: 10.1530/ERC-17-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Ma J, Hua X. Epigenetic regulation by the menin pathway. Endocr Relat Cancer. 2017a;24:T147–T159. doi: 10.1530/ERC-17-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Wang L, Sun Y, Jiang Z, Domsic J, An C, Xing B, Tian J, Liu X, Metz DC, et al. Menin and Daxx Interact to Suppress Neuroendocrine Tumors through Epigenetic Control of the Membrane Metallo-Endopeptidase. Cancer Res. 2017b;77:401–411. doi: 10.1158/0008-5472.CAN-16-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357 doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt DY, Vaccaro AM, Jaskula-Sztul R, Ning L, Haymart M, Kunnimalaiyaan M, Chen H. Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007;12:942–951. doi: 10.1634/theoncologist.12-8-942. [DOI] [PubMed] [Google Scholar]
- Grolmusz VK, Borka K, Kovesdi A, Nemeth K, Balogh K, Dekany C, Kiss A, Szentpeteri A, Sarman B, Somogyi A, et al. MEN1 mutations and potentially MEN1-targeting miRNAs are responsible for menin deficiency in sporadic and MEN1 syndrome-associated primary hyperparathyroidism. Virchows Arch. 2017 doi: 10.1007/s00428-017-2158-3. [DOI] [PubMed] [Google Scholar]
- Gurung B, Feng Z, Iwamoto DV, Thiel A, Jin G, Fan CM, Ng JM, Curran T, Hua X. Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res. 2013;73:2650–2658. doi: 10.1158/0008-5472.CAN-12-3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendy GN, Kaji H, Canaff L. Cellular functions of menin. Adv Exp Med Biol. 2009;668:37–50. doi: 10.1007/978-1-4419-1664-8_4. [DOI] [PubMed] [Google Scholar]
- Huang H, Sabari BR, Garcia BA, Allis CD, Zhao Y. SnapShot: histone modifications. Cell. 2014;159:458–458. e451. doi: 10.1016/j.cell.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482:542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Iyer S, Modali SD, Agarwal SK. The Long Non-Coding RNA MEG3 is an Epigenetic Determinant of Oncogenic Signaling in Functional Pancreatic Neuroendocrine Tumor Cells. Mol Cell Biol. 2017 doi: 10.1128/MCB.00278-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nature reviews. Genetics. 2016;17:630–641. doi: 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- Juhlin CC, Kiss NB, Villablanca A, Haglund F, Nordenstrom J, Hoog A, Larsson C. Frequent promoter hypermethylation of the APC and RASSF1A tumour suppressors in parathyroid tumours. PLoS One. 2010;5:e9472. doi: 10.1371/journal.pone.0009472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku M, Nishiyama T, Yagawa K, Abe M. Establishment of a carcinoembryonic antigen-producing cell line from human pancreatic carcinoma. Gan. 1980;71:596–601. [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell. 2014;53:290–300. doi: 10.1016/j.molcel.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Brosnan-Cashman JA, An S, Kim SJ, Song KB, Kim MS, Kim MJ, Hwang DW, Meeker AK, Yu E, et al. Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clin Cancer Res. 2017;23:1598–1606. doi: 10.1158/1078-0432.CCR-16-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F, Mendell JT. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell. 2018;172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmann C, Nunez FJ, Mendez F, Brosnan-Cashman JA, Meeker AK, Lowenstein PR, Castro MG. Mutated Chromatin Regulatory Factors as Tumor Drivers in Cancer. Cancer Res. 2017;77:227–233. doi: 10.1158/0008-5472.CAN-16-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampitz GW, George BM, Willingham SB, Volkmer JP, Weiskopf K, Jahchan N, Newman AM, Sahoo D, Zemek AJ, Yanovsky RL, et al. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc Natl Acad Sci U S A. 2016;113:4464–4469. doi: 10.1073/pnas.1600007113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, Buisson N, De Witte K, Salandre J, Lenoir G, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6:1177–1183. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harbor perspectives in biology. 2014;6:a019133. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenauer UD, Di Dalmazi G, Slater EP, Wieland T, Kuebart A, Schmittfull A, Schwarzmayr T, Diener S, Wiese D, Thasler WE, et al. Frequency and clinical correlates of somatic Ying Yang 1 mutations in sporadic insulinomas. J Clin Endocrinol Metab. 2015;100:E776–782. doi: 10.1210/jc.2015-1100. [DOI] [PubMed] [Google Scholar]
- Lin W, Cao J, Liu J, Beshiri ML, Fujiwara Y, Francis J, Cherniack AD, Geisen C, Blair LP, Zou MR, et al. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc Natl Acad Sci U S A. 2011;108:13379–13386. doi: 10.1073/pnas.1110104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Francis JM, Li H, Gao X, Pedamallu CS, Ernst P, Meyerson M. Kmt2a cooperates with menin to suppress tumorigenesis in mouse pancreatic islets. Cancer Biol Ther. 2016;17:1274–1281. doi: 10.1080/15384047.2016.1250986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Watanabe H, Peng S, Francis JM, Kaplan N, Pedamallu CS, Ramachandran A, Agoston A, Bass AJ, Meyerson M. Dynamic epigenetic regulation by menin during pancreatic islet tumor formation. Mol Cancer Res. 2015;13:689–698. doi: 10.1158/1541-7786.MCR-14-0457. [DOI] [PubMed] [Google Scholar]
- Lindberg D, Akerstrom G, Westin G. Evaluation of CDKN2C/p18, CDKN1B/p27 and CDKN2B/p15 mRNA expression, and CpG methylation status in sporadic and MEN1-associated pancreatic endocrine tumours. Clin Endocrinol (Oxf) 2008;68:271–277. doi: 10.1111/j.1365-2265.2007.03034.x. [DOI] [PubMed] [Google Scholar]
- Lines KE, Stevenson M, Filippakopoulos P, Muller S, Lockstone HE, Wright B, Grozinsky-Glasberg S, Grossman AB, Knapp S, Buck D, et al. Epigenetic pathway inhibitors represent potential drugs for treating pancreatic and bronchial neuroendocrine tumors. Oncogenesis. 2017;6:e332. doi: 10.1038/oncsis.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzi E, Ciuffi S, Marini F, Mavilia C, Galli G, Brandi ML. Analysis of differentially expressed microRNAs in MEN1 parathyroid adenomas. Am J Transl Res. 2017;9:1743–1753. [PMC free article] [PubMed] [Google Scholar]
- Luzi E, Marini F, Giusti F, Galli G, Cavalli L, Brandi ML. The negative feedback-loop between the oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by mimicking the "Knudson's second hit". PLoS One. 2012;7:e39767. doi: 10.1371/journal.pone.0039767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi EC, Trillo-Tinoco J, Struckhoff AP, Vijayaraghavan J, Del Valle L, Crabtree JS. Retinoblastoma-binding protein 2 (RBP2) is frequently expressed in neuroendocrine tumors and promotes the neoplastic phenotype. Oncogenesis. 2016;5:e257. doi: 10.1038/oncsis.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini F, Giusti F, Tonelli F, Brandi ML. Management impact: effects on quality of life and prognosis in MEN1. Endocr Relat Cancer. 2017;24:T227–T242. doi: 10.1530/ERC-17-0203. [DOI] [PubMed] [Google Scholar]
- Marinoni I, Wiederkeher A, Wiedmer T, Pantasis S, Di Domenico A, Frank R, Vassella E, Schmitt A, Perren A. Hypo-methylation mediates chromosomal instability in pancreatic NET. Endocr Relat Cancer. 2017;24:137–146. doi: 10.1530/ERC-16-0554. [DOI] [PubMed] [Google Scholar]
- Marmorstein R, Zhou MM. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harbor perspectives in biology. 2014;6:a018762. doi: 10.1101/cshperspect.a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques P, Korbonits M. Genetic Aspects of Pituitary Adenomas. Endocrinol Metab Clin North Am. 2017;46:335–374. doi: 10.1016/j.ecl.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Martinelli DC, Fan CM. The role of Gas1 in embryonic development and its implications for human disease. Cell Cycle. 2007;6:2650–2655. doi: 10.4161/cc.6.21.4877. [DOI] [PubMed] [Google Scholar]
- Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–678. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- Modali SD, Parekh VI, Kebebew E, Agarwal SK. Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol Endocrinol. 2015;29:224–237. doi: 10.1210/me.2014-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal T, Subhash S, Vaid R, Enroth S, Uday S, Reinius B, Mitra S, Mohammed A, James AR, Hoberg E, et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat Commun. 2015;6:7743. doi: 10.1038/ncomms8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai MJ, Chruszcz M, Reddy G, Grembecka J, Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem. 2011;286:31742–31748. doi: 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey PJ, Nesbit MA, Rimmer AJ, Attar M, Head RT, Christie PT, Gorvin CM, Stechman M, Gregory L, Mihai R, et al. Whole-exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas. J Clin Endocrinol Metab. 2012;97:E1995–2005. doi: 10.1210/jc.2012-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey PJ, Nesbit MA, Rimmer AJ, Head RA, Gorvin CM, Attar M, Gregory L, Wass JA, Buck D, Karavitaki N, et al. Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 2013;98:E796–800. doi: 10.1210/jc.2012-4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olins DE, Olins AL. Chromatin history: our view from the bridge. Nature reviews. Molecular cell biology. 2003;4:809–814. doi: 10.1038/nrm1225. [DOI] [PubMed] [Google Scholar]
- Perez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics. 2017;12:323–339. doi: 10.1080/15592294.2016.1265710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncin J, Stevenaert A, Beckers A. Somatic MEN1 gene mutation does not contribute significantly to sporadic pituitary tumorigenesis. Eur J Endocrinol. 1999;140:573–576. doi: 10.1530/eje.0.1400573. [DOI] [PubMed] [Google Scholar]
- Romano R, Soong CP, Rose M, Costa-Guda J, Bellizzi J, Arnold A. EZH2 copy number and mutational analyses in sporadic parathyroid adenomas. Endocrine. 2017;55:985–988. doi: 10.1007/s12020-016-1142-z. [DOI] [PubMed] [Google Scholar]
- Rowley MJ, Corces VG. The three-dimensional genome: principles and roles of long-distance interactions. Curr Opin Cell Biol. 2016;40:8–14. doi: 10.1016/j.ceb.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Issa JJ, Kropf P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb Perspect Med. 2017;7 doi: 10.1101/cshperspect.a026948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacheri PC, Davis S, Odom DT, Crawford GE, Perkins S, Halawi MJ, Agarwal SK, Marx SJ, Spiegel AM, Meltzer PS, et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006;2:e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71. doi: 10.1038/nature21063. [DOI] [PubMed] [Google Scholar]
- Schussheim DH, Skarulis MC, Agarwal SK, Simonds WF, Burns AL, Spiegel AM, Marx SJ. Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends Endocrinol Metab. 2001;12:173–178. doi: 10.1016/s1043-2760(00)00372-6. [DOI] [PubMed] [Google Scholar]
- Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simbolo M, Mafficini A, Sikora KO, Fassan M, Barbi S, Corbo V, Mastracci L, Rusev B, Grillo F, Vicentini C, et al. Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J Pathol. 2017;241:488–500. doi: 10.1002/path.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhi AD, Liu TC, Roncaioli JL, Cao D, Zeh HJ, Zureikat AH, Tsung A, Marsh JW, Lee KK, Hogg ME, et al. Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin Cancer Res. 2017;23:600–609. doi: 10.1158/1078-0432.CCR-16-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starker LF, Svedlund J, Udelsman R, Dralle H, Akerstrom G, Westin G, Lifton RP, Bjorklund P, Carling T. The DNA methylome of benign and malignant parathyroid tumors. Genes Chromosomes Cancer. 2011;50:735–745. doi: 10.1002/gcc.20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72:2041–2059. doi: 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedlund J, Koskinen Edblom S, Marquez VE, Akerstrom G, Bjorklund P, Westin G. Hypermethylated in cancer 1 (HIC1), a tumor suppressor gene epigenetically deregulated in hyperparathyroid tumors by histone H3 lysine modification. J Clin Endocrinol Metab. 2012;97:E1307–1315. doi: 10.1210/jc.2011-3136. [DOI] [PubMed] [Google Scholar]
- Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97:2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- Townsend CM, Jr, Ishizuka J, Thompson JC. Studies of growth regulation in a neuroendocrine cell line. Acta Oncol. 1993;32:125–130. doi: 10.3109/02841869309083900. [DOI] [PubMed] [Google Scholar]
- van Nuland R, Smits AH, Pallaki P, Jansen PW, Vermeulen M, Timmers HT. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol. 2013;33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandenBussche CJ, Allison DB, Graham MK, Charu V, Lennon AM, Wolfgang CL, Hruban RH, Heaphy CM. Alternative lengthening of telomeres and ATRX/DAXX loss can be reliably detected in FNAs of pancreatic neuroendocrine tumors. Cancer Cytopathol. 2017;125:544–551. doi: 10.1002/cncy.21857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan J, Maggi EC, Crabtree JS. miR-24 regulates menin in the endocrine pancreas. Am J Physiol Endocrinol Metab. 2014;307:E84–92. doi: 10.1152/ajpendo.00542.2013. [DOI] [PubMed] [Google Scholar]
- Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bender A, Wang P, Karakose E, Inabnet WB, Libutti SK, Arnold A, Lambertini L, Stang M, Chen H, et al. Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat Commun. 2017;8:767. doi: 10.1038/s41467-017-00992-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C, Laddha SV, Tang L, Vosburgh E, Levine AJ, Normant E, Sandy P, Harris CR, Chan CS, Xu EY. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 2014;5:e1450. doi: 10.1038/cddis.2014.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Hua X. Menin represses tumorigenesis via repressing cell proliferation. Am J Cancer Res. 2011;1:726–739. [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Yang YJ, Song TY, Park J, Lee J, Lim J, Jang H, Kim YN, Yang JH, Song Y, Choi A, et al. Menin mediates epigenetic regulation via histone H3 lysine 9 methylation. Cell Death Dis. 2013;4:e583. doi: 10.1038/cddis.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Sanchez Claros C, Suzuki M, Maggi EC, Kaner JD, Kinstlinger N, Gorecka J, Quinn TJ, Geha R, Corn A, et al. Loss of MEN1 activates DNMT1 implicating DNA hypermethylation as a driver of MEN1 tumorigenesis. Oncotarget. 2016;7:12633–12650. doi: 10.18632/oncotarget.7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Dahle D, Zhou Y, Zhang X, Klibanski A. Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J Clin Endocrinol Metab. 2005;90:2179–2186. doi: 10.1210/jc.2004-1848. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhang X, Zhou Y, Ansell PJ, Klibanski A. Cyclic AMP stimulates MEG3 gene expression in cells through a cAMP-response element (CRE) in the MEG3 proximal promoter region. Int J Biochem Cell Biol. 2006;38:1808–1820. doi: 10.1016/j.biocel.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Zhuang Z, Vortmeyer AO, Pack S, Huang S, Pham TA, Wang C, Park WS, Agarwal SK, Debelenko LV, Kester M, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 1997;57:4682–4686. [PubMed] [Google Scholar]
- Zoghbi HY, Beaudet AL. Epigenetics and Human Disease. Cold Spring Harbor perspectives in biology. 2016;8 doi: 10.1101/cshperspect.a019497. [DOI] [PMC free article] [PubMed] [Google Scholar]