

Abstract
The arginase enzyme developed in early life forms and was maintained during evolution. As the last step in the urea cycle, arginase cleaves l-arginine to form urea and l-ornithine. The urea cycle provides protection against excess ammonia, while l-ornithine is needed for cell proliferation, collagen formation, and other physiological functions. In mammals, increases in arginase activity have been linked to dysfunction and pathologies of the cardiovascular system, kidney, and central nervous system and also to dysfunction of the immune system and cancer. Two important aspects of the excessive activity of arginase may be involved in diseases. First, overly active arginase can reduce the supply of l-arginine needed for the production of nitric oxide (NO) by NO synthase. Second, too much l-ornithine can lead to structural problems in the vasculature, neuronal toxicity, and abnormal growth of tumor cells. Seminal studies have demonstrated that increased formation of reactive oxygen species and key inflammatory mediators promote this pathological elevation of arginase activity. Here, we review the involvement of arginase in diseases affecting the cardiovascular, renal, and central nervous system and cancer and discuss the value of therapies targeting the elevated activity of arginase.
I. INTRODUCTION
This introductory section will outline the role played by the ureohydrolase enzyme arginase in health and disease, emphasizing the involvement of arginase in disease and injury conditions that affect the cardiovascular system, the kidneys, neoplastic malignancies, and the brain and retina. Increases in arginase expression and activity have been reported in many diseases and syndromes. The activity of arginase was initially associated with liver function and later was found to be associated with malignancies. More recently, it has been linked with other disease states including those of the kidney, cardiovascular, and central nervous systems. The next sections will summarize research in these areas. New drug treatments are being developed to modulate the activity or expression of arginase. These will be discussed in the last section.
A. Arginase
The ureohydrolase arginase is a manganese-containing enzyme that catalyzes the final step in the urea cycle to dispose of toxic ammonia by converting l-arginine to l-ornithine and urea (229). Its importance in this cycle has long been recognized. Arginase is thought to have appeared first in bacteria, but it has persisted through evolution and is found in yeasts, plants, invertebrates, and vertebrates (53). The transfer of arginase from bacteria to eukaryotic cells has been suggested to have occurred via the mitochondria. Most plants, bacteria, yeasts, and invertebrates have only one arginase isoform, arginase 2 (A2), and it is located in the mitochondria. The majority of animals that metabolize excess nitrogen as urea also express arginase 1 (A1), and it is localized in the cytosol. In some vertebrates, A1 is expressed in the liver, red blood cells, and specific immune cell populations, whereas A2 is highly expressed in the kidney and is also expressed in some other tissues, including the brain and retina. Both isoforms can be induced by a variety of conditions.
A1 in humans comprises 322 amino acids (50), and A2 has 354 (73). Each isoform is encoded by a separate gene, and the two genes are located on separate chromosomes. The two isoforms have similar mechanisms of action, and they produce the same metabolites. They have greater than 60% homology in amino acid residues, and the areas critical to enzyme function are 100% homologous (220). High-resolution crystallographic analysis has shown that A1 and A2 are almost identical in structure. Both consist of three identical subunits, and the active site is located at the bottom of a 15 Å cleft (FIGURE 1). Binding of manganese ions at the bottom of the cleft is essential for enzyme activity. The protein folding of each subunit belongs to the α/β family and consists of a parallel, eight-stranded β-sheet that is flanked by numerous α-helices (3).
FIGURE 1.

A: a ribbon plot of the arginase trimer (liver arginase 1). The binuclear manganese cluster is represented by a pair of spheres in each monomer. B: the binuclear manganese cluster. Metal coordination interactions are indicated by green dotted lines, and the hydrogen bond between the metal-bridging hydroxide ion (red sphere) and Asp-128 is indicated by a white dotted line. Mn2+ is coordinated with square pyramidal geometry, leaving a vacant coordination site that permits octahedral coordination geometry as a means of transition state stabilization in catalysis. Mn2+ is coordinated with octahedral geometry. [From Kanyo et al. (104), with permission from Nature Publishing Group.]
Studies of arginase expression have demonstrated that the intracellular signaling events resulting in increased arginase expression/activity include activation of Rho kinase, mitogen-activated protein kinase, and protein kinase A; production of multiple cytokines [such as interleukin (IL)-13, IL-4, IL-6, IL-8, and tumor necrosis factor (TNF)-α]; and formation of reactive oxygen and nitrogen species and hypoxia (27, 66, 156, 161, 195, 223). Transcription factors regulating the expression of the A1 include signal transducer and activator of transcription (STAT)-6/STAT-3, foxhead box transcription factor (Fox)O4, hypoxia-inducible factor (HIF)-1, CCAAT/enhancer binding protein (C/EBP)β, and activating transcription factor (ATF)-2 (194, 197, 253), whereas transcription factors for A2 include extracellular signal-regulated kinase (ERK)5/cAMP response element binding protein (CREB), HIF-2, and interferon regulatory factor (IRF)-3 (10, 74, 116). Epigenetic control of A2 expression through altered histone deacetylase 2 activity also has been reported (157).
B. Arginase-Ornithine Pathway
Activity of arginase has two primary physiological functions: 1) detoxification of ammonia in the urea cycle and 2) production of ornithine needed for the synthesis of proline and polyamines (FIGURE 2) (229). Proline is produced through activity of ornithine aminotransferase (OAT) (141, 160) and is needed for collagen formation. Polyamines are produced through activity of ornithine decarboxylase (ODC), followed by spermidine and spermine synthases. Polyamines also can be generated from agmatine by activity of arginine decarboxylase (ADC). They have an essential role in cell proliferation and growth, and they are also needed for inflammation, wound healing, tissue repair, and neuronal development (120, 187).
FIGURE 2.

Scheme for the catabolism of l-arginine to l -ornithine/urea or l-citrulline/NO, production of polyamines and anabolism and catabolism of proline. Also shown are the pathway for synthesis of l-arginine from l-glutamine, the reversible pathway between l-ornithine and l-glutamine, and the recycling of l-citrulline into l-arginine. ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; NOS, nitric oxide synthase; OAT; ornithine aminotransferase; ODC, ornithine decarboxylase; OTC, ornithine transcarbamylase. Arginase (top left) is the final enzyme in the urea cycle within the liver, which restarts the cycle through the synthesis of l-citrulline from carbamoyl-phosphate (1) and l-ornithine (2) by OTC (middle). It should be noted that not all of these reactions occur within a given cell. In particular, the urea cycle is independent of the other reactions, i.e., l-arginine produced in the urea cycle is not a substrate for NOS and l-ornithine produced in the urea cycle is not a substrate for OAT or ODC.
In most cell types, l-citrulline is converted to l-arginine through the activity of argininosuccinate synthase (ASS) and argininosuccinate lyase (ASL). ASS and ASL are part of the urea cycle (153). Defective function of urea cycle enzymes in the liver can result in a toxic buildup of ammonia. If left untreated, disorders of the urea cycle can cause mental disorders, seizures, and death (46). Treatment of patients with defects in A1 activity involves decreasing their protein intake and supplementing their diet with essential amino acids. In severe cases, liver transplantation may be needed (46). As such, A1-deficient mice show a high lethality soon after birth (96). The effects of defects in A2 activity are not yet known. However, although viable, A2 knockout mice develop hypertension (92). Also, the expression of a rare A2 allele has been linked to a potential decrease in A2 expression and an increased risk of Alzheimer’s disease (AD) in men and an earlier age at onset in both men and women (81). It is not yet clear how this increased risk for AD relates to urea cycle function, but the authors suggest that the urea cycle induction may be protective in the context of AD. Despite the described effects of A2, mice globally deficient in A2 do not display observable phenotypes under normal conditions and breed at normal rates (199).
Wound repair is characterized by an acute phase that involves a burst of oxidative stress due to activation of resident macrophages that express high levels of the inducible isoform of nitric oxide synthase (iNOS or NOS2) and the phagocytic isoform of NADPH oxidase NOX2. NOS2 and NOX2 produce damaging levels of nitric oxide (NO) and superoxide, respectively. Both are important for eradicating pathogens (187). This acute response to injury is followed by a repair phase that begins within 3–5 days. During this phase, myeloid cell infiltration and arginase expression are increased (103). As explained above, arginase converts l-arginine to l-ornithine which is metabolized by OAT to form proline and ODC to form polyamines. Proline is used for collagen synthesis, whereas polyamines enhance cell proliferation. The balance between the consumption of l-arginine by arginase (for collagen and polyamine production) and NOS2 (for NO production) determines the outcome of wound repair, with arginase controlling the healing process and NOS2 regulating cytotoxic events.
Production of polyamines by arginase is also important for neuronal cell growth, development, and axon regeneration (57, 59, 120). After transection of the spinal cord, treatment of nerve grafts with acidic fibroblast growth factor has been found to promote increases in A1 expression/activity along with elevation of spermine formation within motor neurons and macrophages resulting in improved locomotor function (118). In contrast, hyperactivity of the arginase-ornithine pathway can be damaging in other contexts. For instance, arginase-induced increases in polyamines and proline production can lead to thickening, fibrosis, and stiffening of blood vessels and airways, hypertrophy and fibrosis of the heart and kidneys, neurodegeneration, and growth of tumors (51, 127, 164, 238). These adverse effects of overactive arginase are pathologically significant in diabetes, hypertension, aging, and cancer.
The metabolism of polyamines has also been shown to be involved in the ischemic injury of neuronal cells (95, 151, 152, 209, 228). Amino aldehydes, acrolein, and hydrogen peroxide are byproducts generated during the oxidation of spermine and spermidine by polyamine oxidases (FIGURE 3). These products of polyamine oxidation are highly toxic. All have been implicated in neuronal injury (191).
FIGURE 3.
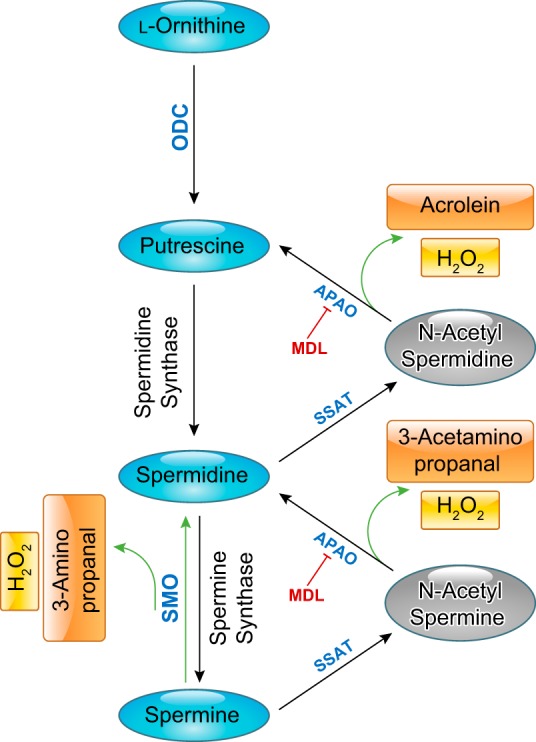
Flow chart of the l-ornithine pathway showing polyamine formation and polyamine oxidation. MDL, MDL-72,527 [N,N′-bis(2,3-butadienyl)-1,4-butanediamine]; ODC, ornithine decarboxylase; OAT, ornithine aminotransferase; SMO, spermine oxidase; APAO, N(1)-acetyl polyamine oxidase; SSAT, spermine spermidine acetyl transferase.
C. l-Arginine Metabolism: NO Synthase
In addition to the important roles of l-arginine in the urea cycle and formation of polyamines and proline, l-arginine is also the substrate for NO synthase (NOS). There are three isoforms of NOS: neuronal NOS (nNOS or NOS1), NOS2, and endothelial NOS (eNOS or NOS3). Two of these, NOS1 and NOS3, are expressed constitutively and require activation. In contrast, the inducible NOS2 is primarily regulated at the expression level by transcriptional and posttranscriptional mechanisms (221, 231, 232). Normal expression and function of NOS3 in vascular endothelial cells maintains proper blood flow by catalyzing the conversion of l-arginine into NO and l-citrulline. Therefore, NOS3 plays a critical role in vascular health.
Upon activation, NOS3 binds heme and tetrahydrobiopterin (BH4) at the NH2-terminal region. Active NOS3 binds calcium/calmodulin, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and nicotinamide adenine dinucleotide phosphate (NADP) at the COOH-terminal region (24, 62, 64) (FIGURE 4). Posttranslational modification of NOS3 is required for its localization to the plasma membrane. NOS3 myristoylation and subsequent palmitoylation direct it to membrane caveolae. However, neither modification is required for NO formation. In contrast, the cofactors BH4 and heme are both required for NO production. The subcellular localization of NOS3 in caveolae is believed to be important for its activity due to the proximity of the l-arginine transporter cationic amino acid transporter-1 (CAT-1) (36) and l-arginine recycling enzymes ornithine transcarbamylase (OTC), ASS, and ASL that support NO production (132). On the other hand, binding of caveolin-1 to NOS3 at caveolae inhibits its function. Activation of NOS3 is mediated by multiple protein-protein interactions and phosphorylation events as well as chemical, hormonal, and physical stimuli, including sheer stress.
FIGURE 4.

Scheme for the regulation of endothelial nitric oxide synthase (NOS3) and l-arginine catabolism to NO and l-citrulline, and for recycling of l-citrulline back to l-arginine. ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; BH4, tetrahydrobiopterin; CAT-1, cationic amino acid transporter-1; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; NADP, nicotinamide adenine dinucleotide phosphate.
Production of NO by NOS3 or NOS1 occurs at low to moderate rates. In contrast, NOS2 produces NO at high rates. This NOS2-derived NO is needed for alternative signaling mechanisms and microbial defense. NOS3-derived NO diffuses to vascular smooth muscle cells where it binds to and activates soluble guanylate cyclase (sGC). Activation of sGC triggers cGMP production, K+ efflux, and smooth muscle cell relaxation. Thus NOS3-derived NO regulates vascular tone. NOS3 also maintains proper blood flow by preventing leukocyte adhesion to the vascular endothelial cells and inhibiting platelet aggregation (44, 117, 170). Other roles of NO include host defense against microorganisms and malignancies, signaling by neuronal cells, remodeling of neuronal synapses, and posttranslational protein modification by protein nitrosylation (148).
l-Arginine needed for metabolic and physiological functions is provided through protein turnover and also from the diet. Thus l-arginine is considered to be a semi-essential amino acid. Due to its well-established role in enhancing NO production by NOS3, l-arginine is a popular supplement for athletes and physical fitness enthusiasts. Preclinical studies have shown that supplemental l-arginine therapy can improve erectile dysfunction and prevent or reverse endothelial dysfunction. However, some studies in animal models and humans have found no benefit or worsening of adverse outcomes during chronic l-arginine supplementation (127). These adverse effects are likely due to the counter-regulatory effects of l-arginine in increasing the expression/activity of arginase, which can reduce the supply of l-arginine for NOS. Under conditions of excessive arginase activity, arginase will compete with NOS for l-arginine, causing NOS uncoupling (FIGURE 5). In addition, it is known that chronic consumption of an excess of l-arginine and other amino acids induces aversion to otherwise nutritious foods (131).
FIGURE 5.

A: arginase and NOS compete for their common substrate l-arginine for the formation of their products. B: elevation of arginase activity by reducing the availability of l-arginine to NOS can reduce NO formation and uncouple NOS, causing it to produce more superoxide (O2.−), which rapidly reacts with NO to produce another potent oxidant, ONOO−. Both oxidants reduce levels of BH4, which is also required for normal NOS coupling. Elevated l-ornithine levels allow greater production of polyamines and proline.
D. The l-Arginine Paradox
The l-arginine paradox refers to the observation that treatment with supplemental l-arginine enhances NO-mediated biological effects in vascular endothelial cells despite the fact NOS3 is theoretically saturated with l-arginine. The concentration of l-arginine in cells is 100–800 μM, much greater than the levels needed to support the maximum activity of NOS3 (Km 3 μM) (229). Therefore, NOS3 should be saturated with l-arginine under physiological conditions. Further complicating the issue is the impact of arginase on NOS function. The l-arginine Km of arginase is 2 mM, roughly 1,000-fold that for NOS3. Therefore, arginase should not be able to compete with NOS3 for substrate (229). However, the Vmax of NOS3 is ~1,000 times less than that of arginase, which roughly equalizes their capability to metabolize l-arginine. This explains their ability to compete for the substrate (61, 229). However, given the high affinity of NOS3 for l-arginine, it is unlikely that the internal l-arginine concentration could drop to a level that would leave either enzyme in a situation of substrate deficiency. Nevertheless, the addition of extracellular l-arginine has been shown to improve endothelial-dependent relaxation in a variety of disease conditions characterized by vascular dysfunction (reviewed in Ref. 52). This paradox has been suggested to depend on the subcellular compartmentalization of l-arginine. However, studies manipulating the subcellular localization of arginase and NOS have failed to confirm this concept (54). Several studies indicate that plasma or extracellular levels of l-arginine and its transport into endothelial cells determine the concurrent NO production by NOS3 (200, 248). Impaired l-arginine uptake by the CAT-1 transporter also can limit the availability of l-arginine to NOS. This can occur either through reduced expression or function of CAT-1 (130) or via increased levels of competitors for CAT-1 transport such as l-ornithine, which has an affinity similar to that of l-arginine for the transporter (82).
E. NOS Uncoupling
Under conditions where the amount of l-arginine needed for NO production is inadequate or if the cofactor BH4 is not available, NOS becomes uncoupled (178, 227). The uncoupled enzyme produces less NO and uses more molecular oxygen to generate superoxide (O2.−) (FIGURE 5B). When NO and O2.− are formed concurrently, they will react to produce the toxic oxidant peroxynitrite (ONOO−). The NO reaction with O2.− occurs much faster than NO can react with sGC or than O2.− can interact with superoxide dismutase (11). O2.− and ONOO− can oxidize BH4 to form BH2, which can also cause NOS3 uncoupling (107). Together, the decreases in NO and the increases in O2.− and ONOO− can lead to dysfunction of vascular endothelial cells (154).
The divergent roles of NO and ONOO− have been evident in the promotion or inhibition of anti-tumor immunity, respectively. Enhanced arginase activity may also contribute to altered immune function in conditions of trauma, infection, transplantation, autoimmunity, and cancer due to impairment of NOS2 function. For example, excessive arginase activity in myeloid cells in tumors can reduce the amount of l-arginine needed for the production of NO by NOS2, which may impair their T-cell responses and allow tumor cells to grow (164). Increased arginase expression/activity can also limit NOS2 expression in immune cells by decreasing the l-arginine needed for NOS2 translation (121).
II. ARGINASE IN CARDIOVASCULAR DISEASE
The progressive understanding of the far-reaching significance of NO in health and disease (113) and the recognition that overactive arginase could compete with NOS for l-arginine and reduce NO bioavailability have prompted the expansion of research on the role of arginase in conditions characterized by vascular endothelial dysfunction. Reduced vascular endothelial cell production of NO leads to impaired vasorelaxation and increased adhesion of leukocytes to the endothelium. Increases in the arginase product l-ornithine have been strongly implicated in hyperplasia of vascular smooth muscle cells along with vascular fibrosis and stiffening. In the sections that follow, we will review recent evidence for the role of these pathways in cardiovascular disease.
A. Hypertension
Hypertension is a major risk factor for cardiovascular disease. Elevated vascular resistance involves increased expression/activity of arginase along with reduced levels of NO, increased production of superoxide, and decreases in l-arginine and BH4. Studies in numerous experimental models of systemic hypertension have linked increases in vascular A1 expression and arginase activity to increases in blood pressure (213). Spontaneously hypertensive rats exhibit elevated vascular arginase activity and impaired endothelium-dependent vasorelaxation that can be reversed along with lowering of blood pressure by an arginase inhibitor (7). Angiotensin II-induced hypertension and vascular fibrosis and stiffening can be prevented or reduced by partial deletion of A1 or treatment with inhibitors of arginase or Rho kinase which is known to prevent the upregulation of A1 expression (16, 195). Additionally, in a DOCA-salt hypertensive animal model, partial A1 deletion or treatment with an arginase inhibitor blocked the rise in blood pressure (214).
B. Pulmonary Arterial Hypertension
Elevated arginase expression and activity have also been implicated in pulmonary arterial hypertension (PAH). Unlike systemic hypertension which primarily involves A1 upregulation, PAH has been particularly associated with increased expression of the A2 isoform (28, 34, 99). Again, a decrease in NO production is linked to an increase in arginase expression/activity (234). Additionally, the increase in A2 limits endothelium-dependent vasodilation of pulmonary segments in an experimental model of pulmonary embolism. In this model, arginase inhibitor treatment was shown to preserve the l-arginine supply, reduce pulmonary resistance (225), limit collagen deposition (75), and attenuate hypoxia-induced PAH (35, 98). Hypoxia has been recently reported to increase proliferation and A2 content of human pulmonary smooth muscle cells by a mechanism involving A2 inducement by AMPKα1 (235). HIF-2α, reported to be essential in hypoxia-induced PAH, appears to mediate PAH in mice, at least partially, via increases in expression and activity of pulmonary endothelial A1, not A2 (38).
Thus increases in arginase activity/expression seem to play a detrimental role in increasing blood pressure and causing endothelial dysfunction during pulmonary hypertension as well as systemic hypertension. Mechanisms underlying the differential involvement of the A1 and A2 isoforms in these conditions are unclear, but probably involve differences in the cellular and subcellular distribution. Further study is needed to clarify these mechanisms. Pulmonary hypertension also can result secondarily to hemolytic episodes such as occur in sickle cell disease.
C. Sickle Cell Disease
Sickle cell disease (SCD) is caused by autosomal recessive mutation of hemoglobin (HgS), which leads to deformation (sickling) of red blood cells (RBCs) and increased likelihood for rupture (165). With intravascular hemolysis, RBCs release significant amounts of A1, which greatly increases plasma arginase activity, resulting in reduced levels of plasma l-arginine, decreased tissue NO, and elevated l-ornithine (8). The concurrent release of hemogloblin, which is a potent scavenger of NO, adds to the reduction in tissue NO levels. Reduced endothelial NO, activation of the endothelial cells, and the sludging and trapping of sickled RBCs in capillaries add to the pathology. The arginase-induced increases in l-ornithine can drive cell proliferation and fibrosis. Clinical problems with SCD include anemia, stroke, severe vaso-occlusive pain episodes, ischemic attacks, acute chest syndrome, leg ulcers, as well as pulmonary hypertension (105).
Restoration of l-arginine bioavailability has been shown to be an effective means of reducing the pathology of SCD (139). Reports have indicated that chronic supplemental l-arginine or l-citrulline are effective at reducing the effects of SCD (140). l-Citrulline may be more effective clinically as it does not induce endothelial arginase, as does l-arginine (177). Treatment with arginase inhibitors is also a promising therapeutic strategy. A recent study reports that oral treatment of transgenic sickle cell mice with an arginase inhibitor, ABH, for 4 wk improved NO availability, reversed systemic and pulmonary and systemic vascular endothelial dysfunction (205).
D. Diabetic Vascular Disease
Diabetes mellitus has been shown to be linked to a number of cardiovascular complications in diabetic patients and contribute to significant morbidity and mortality. Patients with type 1 or type 2 diabetes commonly suffer from a variety of vascular pathologies, including impaired endothelial-dependent vasorelaxation, pathological remodeling of smooth muscle cells and the vascular wall, increased fibrosis, and decreased vascular compliance. Decreased levels of l-arginine have been observed in plasma collected from patients with diabetes and in vascular tissues collected from diabetic rats (80, 114, 163). Increases in the activity of arginase appear to be involved. Studies of the aorta as well as coronary and retinal arteries from animal models of diabetes have shown that increases in A1 expression and arginase activity are involved in the vascular dysfunction (55, 176, 178, 210).
Impairment of blood flow to the heart due to coronary artery disease (CAD) is a major feature of diabetes-associated vascular dysfunction. Coronary arteries isolated from diabetic patients have been found to express increased levels of A1 (5) and to exhibit impaired endothelium-dependent vasorelaxation that was improved by treatment with the arginase inhibitor l-NOHA (13). Also, studies in a rat model of type 2 diabetes showed that the arginase inhibitor nor-NOHA improved coronary microvascular function by increasing the supply of l-arginine and improving the bioavailability of NO (77). Furthermore, analysis of forearm blood flow in patients with CAD showed that infusion of nor-NOHA locally improved endothelium-dependent vasorelaxation (198). The beneficial effects of inhibiting arginase were especially evident in patients with type 2 diabetes.
Obesity and associated metabolic disorders, which are common to type 2 diabetes, also have devastating effects on cardiovascular function and are critical determinants of increased risk. In obesity, visceral adipose tissue expansion and inflammation have key roles in the cardiovascular dysfunction which involves increased production of proinflammatory cytokines, adipokines, and reactive oxygen species (ROS) (89, 226). Arginase expression in vascular, liver, and immune cells is enhanced by these processes and has been examined in models of high-fat-induced obesity and diabetes. One study using wild-type and endothelial cell-specific A1 knockout mice fed a high-fat/high-sucrose diet for 6 mo observed that impaired vascular relaxation along with increased vascular fibrosis, stiffness, and oxidative stress was associated with elevation of arginase expression/activity. Each of these alterations was prevented by systemic inhibition of arginase activity (ABH) or lack of A1 in vascular endothelial cells (VE-cadherin-Cre / A1-LoxP mice) (17). Further study of this model has shown the high-fat/high-sucrose-fed WT mice had elevated A1 expression and activity in the bone and bone marrow along with reductions in bone density, bone volume, and trabecular thickness. Concurrent treatment with the arginase inhibitor ABH prevented these changes (15).
Other recent studies focused on obesity-induced inflammation of the visceral adipose tissue (VAT) have reported that inhibitors of arginase (nor-NOHA or ABH) or lack of endothelial A1 also prevents or reduces high-fat/high-sucrose diet-induced increases in circulating inflammatory monocytes and macrophage infiltration into the VAT. In addition, these treatments or endothelial cell-specific A1 knockout blocked increases in proinflammatory macrophage (M1-like) and prevented increases in levels of inflammatory cytokines, chemokines, and cellular adhesion molecules (239). In diet-induced obesity, perivascular adipose tissue (PVAT) also has been reported to exhibit high levels of A1 and A2, low levels of l-arginine and NO and NOS3 uncoupling which were associated with impaired vasorelaxation (230). This impairment could be reversed by l-arginine supplementation or arginase inhibition.
E. Atherosclerosis
A critical, early event in atherosclerosis is endothelial dysfunction, which causes inflammation, vasoconstriction, and thrombus formation. Impaired vascular endothelial function leads to plaque formation and pathological remodeling of the arterial wall. Studies show that elevation of oxidized low-density lipoprotein (OxLDL) is critically involved in atherosclerosis (134, 182, 184). Increased levels of arginase expression and activity are also observed in atherosclerosis. This upregulation of arginase is thought to be mediated by OxLDL-induced activation of its receptor ‟lectin-like oxidized low-density lipoprotein receptorˮ (LOX-1) and RhoA/Rho kinase (ROCK), leading to increases in A2 expression. This LOX-1-mediated activation of A2 causes a reduction in NO generation due to uncoupling of NOS3 (182). Pharmacological blockade of LOX-1 and ROCK was found to attenuate the increase in arginase activity. Additionally, deletion of A2 in apolipoprotein E-deficient mice was shown to limit the development of atheromatous plaques, reduce levels of oxidative stress, and increase NO bioavailability, further implying the role of A2 in the pathology (184). The OxLDL-induced activation of A2 within vascular endothelial cells has been linked to the translocation of A2 from the mitochondria into the cytosol through processes involving activity of the mitochondrial processing peptidases (MPP). A putative MPP cleavage site in the A2 NH2-terminal may be involved in this A2 translocation. In support of this concept, MPP knockdown was shown to prevent the OxLDL-induced cytosolic translocation of A2, block NOS3 uncoupling, and improve vascular function in this model (156). OxLDL has also been reported to decrease levels of histone deacetylase 2, a factor that suppresses transcription of A2 (157). A2 expression has also been strongly implicated in the development of atherosclerosis. In contrast to these suggested damaging effects of macrophage A2, increases in macrophage A1 expression have been found to have a beneficial role in atherosclerotic plaque regression (58, 109). Thus targeting the two arginase isoforms selectively could offer a novel therapeutic strategy for limiting vascular pathologies associated with atherosclerosis (161, 204). The reported selective regulation of A2 versus NOS2 levels in macrophages via MAPK pathways (ERK vs. p38) may provide another strategy (100).
With regard to the differential involvement of A1 versus A2 in atherosclerosis and other types of vascular inflammatory conditions, it is worth noting that the literature about the expression of A1 in human macrophages is controversial. A similar controversy exists for NOS2. However, the sources of the macrophages used for different studies have been variable. Many studies have used blood-derived human monocytes, which has led some investigators to conclude major species differences exist in macrophages and that human macrophages are not able to produce NOS2/NO or arginase/l-ornithine (133). Also, analyses with human monocyte-macrophage cell lines or monocyte-derived macrophages have shown that these cells cannot be easily induced to produce NOS2 or NO (189). However, when human tissue macrophages have been studied, they have been found to be similar to macrophages from other vertebrate species (211). Recent studies of macrophages from patients with tuberculosis have confirmed that macrophages, like other human myeloid cells, express A1 and that A1 expression is involved in the progression or resolution of the pathologies (137).
F. Myocardial Ischemia-Reperfusion Injury
Ischemic injury of the myocardium, damage caused by coronary artery occlusion, is a major cause of morbidity and death, especially in patients with diabetes and atherosclerosis. Current strategies to revert myocardial infarction are focused on achieving rapid and effective reperfusion to minimize tissue damage (83). However, reperfusion may itself induce additional myocardial damage accompanied by induction of ROS, proinflammatory factors, and death of cardiomyocytes (217, 242). Impaired endothelial-dependent vasodilation due to a reduction in bioavailability of NO is a primary mechanism of ischemia-reperfusion (I/R) injury. Analyses in several in different animal models have demonstrated the critical role of increased arginase expression and activity in myocardial I/R injury (72, 78, 101, 217). Studies have shown that treatment with the arginase inhibitor nor-NOHA before ischemia, during late ischemia, and in early reperfusion restores NO bioavailability and markedly decreases the size of cardiac infarcts. However, it is important to note that the normal function of arginase in “healing” M2 macrophages is also important for tissue repair.
Like I/R injury, myocardial infarction (MI) sets off an inflammatory response involving endothelial cell activation and neutrophil adhesion, which can further result in myocardial cell death. A recent study reported that endothelial cell-specific knockout of the transcription factor FoxO4 in mice protected against MI-induced loss of cardiac function, neutrophil accumulation, and elevation of A1 expression. Furthermore, FoxO4 was shown to activate A1 transcription. Knockdown of FoxO4 in endothelial cell prevents hypoxia-induced reduction of NO levels and monocyte adhesion; these effects were reversed by ectopic expression of A1 (253).
G. Aging and Cellular Senescence
Excessive arginase activity has been linked to senescence of endothelial cell in both aging humans and experimental animals (14). Analyses in old rats have found increases in arginase activity in association with decreases in NO and increases in superoxide formation as compared with young rats (111). Moreover, acute treatment of old rats with inhibitors of both NOS and arginase was shown to prevent NOS3 uncoupling and reduce superoxide formation. Acceleration of endothelial cell senescence along with decreased NO production has also been shown to occur with chronic supplementation of l-arginine. This is likely to be due to upregulation of arginase (188). Interestingly, Xiong et al. (233) have reported that overexpression of A2 in vascular smooth muscle cells can induce their senescence by a mechanism involving activation of p66Shc and p53. This mechanism appears to be independent of ureohydrolase activity (233).
H. Erectile Dysfunction
Penile erection and the flaccidity process are mainly regulated by a neurophysiological event involving the relaxation and contraction of smooth muscle cells within the corpora cavernosa (CC). Studies have shown that NO is a key mediator of CC relaxation and penile erection (45). NO released from sinusoidal NOS3 in endothelial cells or NOS1 in nitrergic nerves causes relaxation of CC smooth muscle (26). While NO production regulates the intrinsic tone of the CC, arginase activity modulates CC tone by inhibiting the production of NO. This probably occurs due to competition between arginase and NOS for their common substrate l-arginine. While both A1 and A2 are expressed in the CC (19, 21, 213, 215), an increase in A2 expression is the primary mediator of erectile dysfunction (ED) (21, 39, 213). Elevated expression/activity of arginase, reduced NO synthesis, and decreased cavernosal relaxation is observed in CC of diabetic patients with ED (21). Conversely, inhibition of arginase maintains cellular l-arginine concentrations, which in turn enhances NOS activity and increases NO-dependent smooth muscle relaxation in human and rat penile CC (43, 112, 190). Furthermore, deletion of the A2 gene in mice abrogates diabetes-induced ED by improving endothelial cell- and nitrergic nerve-dependent relaxation of the CC (216).
Additionally, ED has been considered as an early marker of cardiovascular diseases (CVD) because it reflects endothelial dysfunction and impaired relaxation of smooth muscle cells in patients with ED and CVD. A recent study showed excessive plasma A1 and A2 levels in patients with ED compared with healthy control subjects (119). Also, the circulating levels of A2 were much higher than those of A1, suggesting that A2 plasma concentration may serve as a risk factor for ED (119).
In studies of the upstream mediators of arginase expression and activity, RhoA/ROCK and subsequent activation of p38 MAPK have been shown to be involved in elevation of arginase levels (27, 215, 216). Activated RhoA/ROCK pathway causes penile detumescence and ED in diabetes (20, 33, 215). However, partial deletion of the ROCK 2 gene in diabetic mice evoked less ROCK activity, lower levels of CC arginase activity, and reduced impairment of endothelial cell-dependent and nitrergic nerve-mediated relaxation as compared with diabetic wild-type mice (215). Taken together, these findings suggest that inhibiting arginase activity directly or by blocking the RhoA/ROCK pathway could offer important strategies for the treatment of ED.
III. ARGINASE IN RENAL DISEASE
A. Kidney Failure
The progression of kidney disease to end-stage renal failure is a major health problem. Several diseases including hypertension and diabetes are responsible for more than 40% of all kidney dysfunction or pathology in the United States (22). Current therapies include control of blood pressure and blood glucose levels, immunosuppression, and lifestyle changes. These strategies can delay but do not arrest the progression of renal failure. Immunosuppressive treatment is mainly recommended for renal transplant or dialysis patients in whom the illness has been caused by renal failure (2). Increased arginase activity seems to represent a key feature of kidney failure in diabetic nephropathy, hypertensive nephropathy, and glomerulonephritis (143). Thus it is important to identify the specific role of arginase in the progression of kidney failure.
B. Diabetic Nephropathy
Strong evidence indicates that at an early stage of diabetes, kidneys develop glomerular infiltration and hypertrophy. This is followed by thickening of the glomerular basement membrane, endothelial dysfunction, accumulation of the mesangial matrix, and enhanced excretion of urinary albumin, which leads to end-stage renal failure (155). Impairment of endothelial cell function is a common element of the vascular dysfunction observed in diabetic patients and animal models (40, 71).
Excessive arginase activity in the kidney is associated with endothelial dysfunction during diabetic nephropathy (244). The A2 isoform is highly expressed in kidney cells, whereas A1 is not detected (122, 143). The role of A2 in the kidney has been historically associated with the catabolism of excess of l-arginine, especially that produced through the gastrointestinal-kidney axis; however, new roles of A2 expression in the kidney are emerging. Recent studies have observed that pharmacological blockade or genetic deficiency of A2 provides kidney protection in diabetes as observed by reduced levels of albuminuria and renal histopathological changes, decreased blood urea nitrogen, and reduction of macrophage recruitment (143, 243). While increased kidney arginase activity is associated with reduced renal medullary blood flow in diabetes, alterations have not been seen in mice lacking A2 gene, suggesting that renal A2 reduces NO bioavailability in the kidney by catabolizing l-arginine (143).
Since reduced l-arginine bioavailability has been associated with NOS3 dysfunction and reduced NO production, oral supplementation with l-arginine or l-citrulline (precursor for l-arginine biosynthesis) are potential modalities to restore plasma l-arginine concentrations, thus improving vascular endothelial dysfunction. A study using supplementation of l-arginine or l-citrulline failed to increase NO bioavailability and to prevent and/or reverse markers of renal injury in type 1 diabetic mice despite successfully increasing of l-arginine levels in the plasma and kidney (245). The authors did not rule out the possibility that l-arginine uptake is impaired in a subpopulation of cells that express NOS3. Another possibility is that diabetes may lead to alterations in the subcellular compartmentation of proteins or enzymes of the arginine/NO pathway. This could lead to a lack of response to increases in extracellular l-arginine. In contrast, oral l-citrulline supplementation was shown to protect against kidney injury, improve the anti-inflammatory profile, and preserve nephron function during diabetes (179). Whether opposite results obtained with supplemental l-citrulline versus l-arginine reflect differences in the rodent models or differences in the efficacy of l-arginine or l-citrulline supplementation in altering the progression of diabetes is unclear.
C. Hypertensive Nephropathy
Growing evidence indicates that arginase plays a role in the pathophysiology of renal hypertension. However, the mechanisms of the pathology are unclear. A recent study observed that uni-nephrectomized rats display increased systolic blood pressure and progressive albuminuria. These changes were associated with increased renal arginase activity and excessive A1 expression in the glomeruli (129). However, no changes in arginase activity/expression were observed in kidney tissues from spontaneously hypertensive rats compared with their control group (6). Further study is required to define the specific involvement of arginase in hypertensive nephropathy.
IV. ARGINASE AND IMMUNE DYSFUNCTION AND CANCER
Numerous investigations have shown that patients with chronic inflammatory diseases or syndromes such as cancer, autoimmunity, infections, and physical trauma, among others, have impaired T lymphocyte responses (203). Increases in l-arginine metabolism, especially by tumor cells, myeloid cells, and recently in innate lymphoid cells 2 (ILC-2) (138, 172), have emerged as a primary mediator in the modulation of T-cell responses during pathologies associated with chronic inflammation. Although this active field is still developing, we will summarize the current understanding of the effect of l-arginine metabolism in the function of immune cells in cancer.
As noted above, l-arginine is the substrate for NOS and arginase. Additionally, l-arginine is a substrate for arginine:arginine decarboxylase (ADC) and arginine:glycine amidinotransferase (AGAT) (142) (FIGURE 6). Furthermore, ADC converts l-arginine to agmatine, which is converted to putrescine and urea by agmatinase. Additionally, AGAT represents the first and rate-limiting step in the de novo endogenous biosynthesis of creatine from l-arginine, which then feeds the production of ATP, especially in the muscle. Mammalian ADC is highly expressed in the brain (97, 254), while AGAT is expressed in muscle, brain, and heart (41, 94). The role of A1 and NOS2 in the modulation of immune responses through catabolism of l-arginine has been clearly established. While the impact of the other l-arginine-metabolizing enzymes in the regulation of immune tolerance remains to be investigated, it is predicted that they play a primary role in the regulation of inflammatory responses.
FIGURE 6.
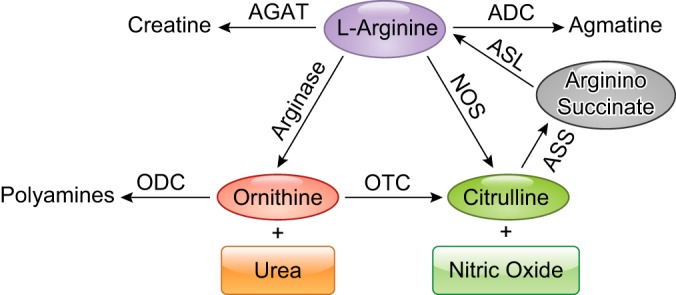
The enzymes that metabolize l-arginine and can affect l-arginine levels. Arginase 1 and 2 produce l-ornithine and urea. Nitric oxide synthase (NOS) produces nitric oxide (NO) and l-citrulline, which can be recycled back to l-arginine by arginosuccinate synthase (ASS) and arginosuccinate lyase (ASL). Arginine decarboxylase (ADC) produces agmatine. Arginine:glycine amidinotransferase (AGAT) produces creatinine. This diagram should not be interpreted to indicate that all of these enzymes are expressed simultaneously in a given cell type.
A. l-Arginine Metabolism and Myeloid Cell Function
A substantial literature has accumulated about the role of l-arginine in the function of macrophages and other myeloid populations, including monocytes, dendritic cells, polymorphonuclear cells, and myeloid-derived suppressor cells (MDSC). However, there are controversies about the relevance of l-arginine metabolism in human macrophages, as well as the dualistic and opposite effects of NO-producing “proinflammatory” (M1-like) macrophages versus l-ornithine-producing “prohealing” (M2-like) macrophages. Bone marrow-derived macrophages (BMDMs) or peritoneal macrophages are polarized into M1- or M2-like phenotypes upon activation with interferon-γ (IFN-γ) and Toll-like receptor (TLR) agonists or with IL-4 and IL-13, respectively (146, 181). Although this strategy has been used as a model to understand macrophage function in disease for years, new concepts have pointed out the difficulty of finding polarized M1 and M2 macrophages in physiological microenvironments (70, 150). Still, M1 responses are characterized by the production of NO after upregulation of NOS2, while M2 macrophages express A1 that produces l-ornithine needed for the synthesis of polyamines to promote cell proliferation or proline for collagen production. Thus, in murine macrophages, the expression of NOS2 and A1 is differentially regulated by Th1 and Th2 immune mediators, respectively (85, 147). The expression of the NOS2 transcript is induced through STAT-1, whereas A1 mRNA expression is upregulated through STAT-6/STAT-3, C/EBPβ, and PPARα. The mitochondrial isoform A2 is not significantly modulated by Th1 or Th2 cytokines (174). Although A2 is not significantly modulated through immune mediators, its role should not be ignored, as the reactions performed by A1 and A2 are highly similar (149). The inhibition of A1 leads to an increase in NOS2 expression and function, which consequently promotes NO production (32). Conversely, upregulation of A1 inhibits NOS activity and contributes to the pathophysiology of several disease processes, including vascular dysfunction and asthma (251). The inhibition of NOS2 expression by A1 appears to be mediated by l-arginine deprivation, which blocks NOS2 translation via increased GCN2 activity (121). Also, low levels of NO induce nitrosylation of cysteine residues in A1. This increases the biological activity of A1, further reducing l-arginine (186). Despite the fact that A1 and NOS can induce counterregulatory effects against each other, it has been reported that MDSC coexpress A1 and NOS2 as a mechanism to produce peroxynitrite that suppresses T-cell anti-tumor immune function (65), which is in agreement with the new concept that M1 and M2 processes can coexist as complementary mechanisms in immune cells.
Activation of murine macrophages with Th1 or Th2 cytokines also has different effects on the extracellular levels of l-arginine. Stimulation of peritoneal macrophages with IL-4 plus IL-13 induces an increase in their expression of A1 and CAT-2B. This results in a rapid increase in the uptake of extracellular l-arginine with the consequent reduction of l-arginine in the microenvironment. In contrast, macrophages stimulated with IFN-γ express NOS2 preferentially, do not increase CAT-2B, and do not deplete l-arginine from the microenvironment (174). Data from the A1 and A2 knockout mice have confirmed that A1 has a higher capacity to regulate the extracellular l-arginine (47, 96). Accordingly, in vitro experiments of cytokine-activated macrophages showed that only A1-producing macrophages, and not those expressing NOS2, caused l-arginine depletion from the medium. This also correlated with a decreased T-cell function and proliferation, caused by a prolonged loss of the T cell receptor ζ chain (CD3ζ) (69, 123, 135, 249). The addition of arginase inhibitors or exogenous l-arginine reversed the CD3ζ loss and reestablished T-cell proliferation (174).
B. Effects of l-Arginine Starvation in T Cells
The link between availability of l-arginine and T-cell responses was first established by experiments showing that both the thymic involution and decrease in T cells seen in mice undergoing extensive surgery were prevented after supplemental l-arginine injection (9). Later experiments in humans showed the induction of T-cell suppression after A1 release and deprivation of serum l-arginine following physical trauma, liver transplantation, or progression of cancer. Studies have also shown that culturing T cells in medium with reduced l-arginine levels led to a marked impairment in cell function (208). Conversely, the culture of T cells in high levels of l-arginine promoted their anti-tumor activity (68). Furthermore, T cells activated in an l-arginine-free environment developed all the alterations previously described in tumor-bearing mice and cancer patients, that is, decreased CD3ζ expression and impaired translocation of NFκB-p65 (250). Although initial reports suggested that the low T-cell function was due to a reduction in CD3ζ expression, T cells cultured in l-arginine-free media also exhibited increased IL-2 production and elevated expression of early activation markers CD25, CD69, CD122, and CD132. Also, when these T cells were activated with phorbol myristate acetate (PMA), which bypasses the T-cell receptor signaling, they failed to proliferate (250). This confirmed the suppressive effect triggered by the absence of l-arginine was not caused by a decrease in T-cell receptor signaling but rather by a general pathway that controlled the activity of T cells.
D-type cyclins (D1, D2, and D3) and cyclin-dependent kinases (cdk4 and cdk6) regulate T-cell progression through the early G1 phase and later S phase of the cell cycle (106). When T cells were cultured in l-arginine-free media, they were unable to upregulate cyclin D3 and cdk4. However, they increased cdk6 expression, which correlated with an arrest in the G0-G1 phase of the cell cycle (173). Interestingly, l-arginine starvation impaired the T-cell expression of cyclin D3 through decreases in mRNA stability and rate of translation (171, 173). Amino acid depletion blocks global translation of proteins by causing accumulation of empty aminoacyl-tRNA, activating the general control non-derepressible 2 (GCN2) kinase. GCN2 phosphorylates the translation initiation factor eIF2α. The phosphorylated eIF2α binds eIF2β with high affinity and blocks its ability to exchange GDP for GTP. This inhibits binding of the eIF2 complex to methionine aminoacyl-tRNA, leading to a global decrease in protein synthesis (86). T cells cultured in l-arginine-free media displayed a global decrease in translation, which was associated with increases in phospho-eIF2α (144). Also, the global decrease in translation impaired the expression of the RNA-binding protein HuR, which in turn reduced the stability of mRNA containing AUUA-rich elements (171). However, it is not known whether phospho-eIF2α and GCN2 are the only mediators of the arrest in translation induced by l-arginine starvation. In fact, recent studies suggested a potential role of mTOR signaling in these alterations (169).
In addition to the above effects on cell cycle progression, a recent study pointed to the role of l-arginine availability in the overall metabolic polarization of activated T cells (60). Stimulation of T cells under l-arginine starvation blocked their glycolytic function, without altering mitochondrial biogenesis or function (60). This result is in agreement with the inhibitory effect induced by A1-expressing cells, as glycolysis regulates expansion of activated T cells. Also, consistent with the effect of mitochondrial function in the activation of T cells, the culture of T cells under l-arginine starvation did not impact the expression of early activation mediators (84). Interestingly, l-arginine starvation appears to inhibit T-cell proliferation in vivo by inducing the accumulation of MDSC in a GCN2-dependent manner (60), adding a level of complexity in how amino acid starvation promotes tolerogenic responses in vivo.
C. Role of the De Novo Production of l-Arginine
Activated T cells and myeloid cells import substantial amounts of l-arginine. However, recent studies showed the importance of the synthesis of l-arginine as a partner mechanism to maintain the intracellular pool of l-arginine. The addition of l-citrulline, a metabolic precursor for l-arginine, rescued the antiproliferative effects of l-arginine starvation on T cells. Moreover, serum levels of l-citrulline increased after in vivo deprivation of l-arginine (60). l-Arginine synthesis from l-citrulline depends on the sequential interaction between the enzymes ASS and ASL. The rate-limiting enzyme ASS is constitutively expressed in T cells and upregulated in myeloid cells after activation with TLR agonists (60, 166). The function of ASS and ASL is to metabolize l-citrulline to resynthesize l-arginine. Indeed, NO-producing macrophages export l-citrulline, which is then reimported by myeloid cells and used in the de novo synthesis of l-arginine (166). Thus, when l-arginine availability is limited, l-citrulline is imported to support l-arginine production. Increased cell death has been observed in activated T cells cultured under l-arginine and l-citrulline starvation after silencing of ASS, suggesting the key role of this pathway in sustaining cellular viability during l-arginine deprivation (60). Furthermore, macrophages lacking ASS were found to lose their ability to block the growth of Mycobacterium tuberculosis due to a decrease in their ability to synthesize l-arginine needed for NO production (166). Interestingly, when M1 macrophages produce NO from l-citrulline recycling, A1 is no longer able to block NO production.
D. Deprivation of l-Arginine as a Therapy for Tumors
Seminal studies showed the efficacy of the depletion of the amino acid l-asparagine in the treatment of T- and B-cell leukemias. Similarly, recent preclinical and clinical studies have proposed the depletion of l-arginine as a therapy for several malignancies auxotrophic for this amino acid, including acute lymphoblastic leukemia, acute myeloid leukemia, melanoma, as well as liver and pancreatic carcinoma (63). The deprivation of l-arginine has been accomplished with pegylated forms of the mycoplasma-derived arginine deiminase (Peg-ADI) and A1 (Peg-A1). Peg-ADI has demonstrated antitumor activity, especially in tumors negative for ASS such as melanoma and hepatocellular carcinoma. However, ADI is immunogenic due to its bacterial origin, leading to self-reacting or blocking antibodies. Furthermore, Peg-ADI catabolizes l-arginine into l-citrulline and ammonia, a toxic product which causes neutropenia and neurological impairment. Also, it has been reported that tumors may gain the expression of ASS and become resistant to the Peg-ADI. Alternatively, one dose of Peg-A1 can reduce the levels of l-arginine in vivo for up to 7 days, without inducing noticeable toxicity, suggesting an increased half-life and enhanced capacity for depleting l-arginine (84). Also, no evidence of immunogenicity has been detected, providing an improvement in efficacy and safety profile. Peg-A1 induced significant anti-tumor effects in multiple preclinical and clinical models. Also, modified versions of Peg-A1 replacing its binding to Mn2+ with Co2+ shifts the optimal pH dependence of A1 from 8.5 to 7.5 and increases the overall catalytic activity, without affecting its anti-tumor effect. One point of important consideration is the fact that treatment with peg-A1 blocked tumor growth, but also impaired anti-tumor T-cell responses, suggesting a potential limitation that needs to be addressed.
V. ARGINASE AND NEUROVASCULAR DISEASE
The effect of elevated arginase activity in causing an increase in polyamine formation is known to have a beneficial impact on neuroprotection and neural regeneration in central nervous system (CNS) disease and injury conditions (48, 128). However, numerous studies have linked the upregulation of arginase to a variety of CNS diseases, including Alzheimer’s disease, multiple sclerosis, stroke, traumatic brain injury, Parkinson’s disease, and several retinal diseases. Studies using in situ hybridization and immunolocalization techniques have demonstrated the presence of both arginase isoforms in the brain (246). The authors found that A1 and A2 are both expressed in neurons. They noted that levels of A1 are higher than A2, but that A2 is usually coexpressed with A1. Arginase expression was especially high in the neurons of the cerebral cortex, cerebellum, pons, medulla, and spinal cord. Retinal cells also express both isoforms. Immunolocalization analysis showed A1 immunoreactivity in retinal neurons, glia, and vascular cells (158, 252) and abundant expression of A2 in the inner segments of the photoreceptors and horizontal cells (151, 152).
Production and release of NO is a critical feature of signaling by both neuronal and vascular cells and is especially important for maintaining proper blood flow within the CNS. Disruption of NO signaling is a major component of brain injury. NO produced by the different NOS isoforms has been shown to influence the progression of neural cell injury in different ways (212). High levels of NO produced through NOS2 activity during inflammatory conditions or NO formed by NOS1 can promote nitrative stress. This can lead to neuronal cell death. However, NO from NOS3 is required to maintain proper blood flow and inhibit aggregation of platelets and attachment of leukocytes to the vessel wall, thereby limiting oxidative stress and inflammation. On the other hand, arginase has been shown to be both a source and a target of oxidative stress and inflammation. As has been noted above, excessive arginase activity can increase oxidative stress and inflammation by reducing l-arginine supply leading to NOS uncoupling. On the other hand, inflammatory mediators and oxidative stress are known to cause increases in arginase expression (27, 141, 145). The involvement of arginase activity in CNS injury has been considered only recently. In the sections that follow, we explore the evidence that supports the involvement of the arginase pathways in neurovascular injury in both brain and retina.
A. Ischemic Stroke
Disruptions of NO signaling are strongly implicated in tissue injury during ischemic stroke (67, 110, 224). The involvement of arginase in this pathology is also very likely given that overactive arginase will reduce the supply of l-arginine available to NOS. Indeed, studies in a rodent model of ischemic stroke have found that increases in arginase activity and A1 expression accompany the cerebral injury (167). Expression of A2 was not altered. The increases in A1 protein expression were localized to the lesion area and were correlated with an early and enduring upregulation of A1 in astroglia and activated macrophages. The increases in A1 were associated with increases in glial fibrillary acidic protein, a marker of glial cell activation. Delayed decreases in A1 were noted in neurons located close to the lesion area. In addition, the A1 distribution pattern was similar to that of brain-derived neurotrophic factor (BDNF). This suggests a potential role for A1 in neuroplasticity. Interestingly, a study of blood samples from patients with acute ischemic stroke has shown a positive association between levels of A1 expression, neutrophil-to-lymphocyte ratio, and stroke severity (162). Further study is needed to elucidate the specific role of A1 expression and activity in the ischemia-induced injury and subsequent tissue repair. Given the suggested role of A1 in M2-like macrophage-mediated repair function, it is possible that the increase in A1 expression represents a mechanism of repair. Further research using genetic strategies is needed to explore this possibility. A recent study shows that increases in A1 gene expression in blood cells are positively correlated with ischemic stroke severity (162). Relatively little is known about the involvement of the A2 isoform in stroke injury. However, one group has reported on work suggesting a protective role of A2 in cerebral ischemia (1). The authors found that deletion of the A2 gene resulted in higher infarction volumes and neurologic deficits in a model of permanent occlusion of the distal middle cerebral artery. The study did not address the molecular mechanisms of A2-mediated neuroprotection.
B. Traumatic Brain Injury
Altered NOS function and reduced NO formation have also been implicated in traumatic brain injury (TBI) (29, 67). Within minutes after the injury, levels of NO in the damaged brain tissue increase. Shortly thereafter, the NO levels decrease and remain below baseline values for periods ranging from hours to days. The NO decrease is significantly correlated with decreases in activity of NOS2 and NOS3 activity and reduced cerebral blood flow (CBF) (29). Studies have shown that treatment with supplemental l-arginine increases CBF and reduces signs of injury. This suggests that l-arginine depletion due to excessive arginase activity could be involved (30). Interestingly, impaired NO production along with increased arginase activity was correlated with the endothelial dysfunction in the mesenteric arteries at 24 h after TBI in a rat model, suggesting that blood vessels have a molecular memory of the neurotrauma (219). A subset of macrophages positive for A1 was shown to be induced and localized near the lesion 1 day after the injury, suggesting a role for A1 in the pathological response to TBI (88). However, further investigation using gene expression profiling to compare the A1-positive macrophages with A1-negative macrophage subpopulations in the same area showed that neither population had expression profiles consistent with previously described subsets of A1-positive “M2-like” healing macrophages or A1-negative “M1-like” inflammatory macrophage. Thus the role of A1-positive macrophages in CNS tissue injury and repair is not yet clear.
An analysis of CBF in a mouse model of TBI using arterial spin-labeling magnetic resonance imaging showed improved CBF in A2 knockout mice, suggesting the involvement of A2 in TBI-induced vascular dysfunction (18). The effects of the A2 knockout on tissue damage and neurological function were not reported. However, studies using a rat model of TBI observed enhanced polyamine catabolism following the injury (247). Furthermore, pharmacological blockade of the ornithine/polyamine pathway was found to limit cognitive impairment following TBI (180). The above results suggest a potential role for activation of the arginase/polyamine pathway and polyamine catabolism in TBI.
C. Alzheimer’s Disease
Alterations in the metabolism of l-arginine may also play a role in the pathogenesis of AD. However, studies of the relationship between arginase expression and AD have had conflicting results. One group reported that levels of A1 mRNA were increased in the frontal cortex of AD patients, but that A2 was unchanged (37). Another group reported that levels of A2 mRNA were increased in the frontal cortex while A1 was unchanged (81). Additionally, the second group described the presence of a rare A2 allele that was linked to an increase in the risk of early-onset AD in male subjects. Another study has described a correlation between age and region-specific decreases in NOS activity and expression and increases in arginase activity during AD (124). Thus increased arginase activity appears to be linked to the pathogenesis of AD, but whether the damage involves altered expression and/or function of A1 versus A2 is not yet clear. A recent report noted that sustained expression of A1 was protective in a mouse model of AD (90). In that study, the authors found that AAV-mediated overexpression of A1 in the hippocampus significantly reduced AD pathology through activation of autophagy. Furthermore, conditional depletion of A1 in myeloid cells resulted in worsening of the pathology. This is consistent with the results of studies by Cherry et al. (31) who showed that A1-positive microglia appear to participate in Aβ plaque clearance during sustained IL-1β inflammation. Further investigations are needed to define the specific role of arginase in AD and to determine its potential value as a disease biomarker.
D. Multiple Sclerosis
Visual dysfunction is an early sign of multiple sclerosis (MS). MS patients often develop optic neuritis that is characterized by impaired retinal function along with thinning of the nerve fiber layer and loss of retinal ganglion cells (RGCs) (185, 222). Studies in the experimental autoimmune encephalomyelitis (EAE) rat model of MS have reported that A1 expression and activity are increased in both brain and spinal cord. Increased production of ROS and increased expression of NOS2 were also observed (125, 126). The latter studies also found increases in arginase within the cerebrospinal fluid of MS patients (125). These results support the involvement of arginase and NO in the pathogenesis of acute neuroinflammation and suggest that they could be useful as biomarkers for disease progression. There is a growing body of evidence to suggest that myeloid-derived cells play important modulatory and effector roles in MS patients and animal models. A decreased level of A1 was observed in MDSCs from MS patients compared with healthy controls (25). Studies in a mouse model of EAE by Yang et al. (237) demonstrated that spermidine treatment could suppress the autoimmune response and alleviate clinical signs of EAE by regulating the infiltration of CD4+ T cells and macrophages. In that study, A1 expression was shown to be upregulated in macrophages in response to spermidine treatment. Interestingly, macrophages from mice treated with spermidine could also reverse EAE progression, while pretreatment of those macrophages with an arginase inhibitor abrogated the therapeutic effect. A recent study using models of EAE and spinal cord injury found that A1 is expressed in infiltrating myeloid cells but not in microglia (76).
E. Retinal Neurovascular Injury
Numerous retinal disease conditions, such as retinopathy of prematurity (ROP), diabetic retinopathy, glaucoma, and optic neuritis are characterized by neurovascular degeneration. Studies in a model of ROP have demonstrated a link between neurovascular injury and activity of the NOS and arginase pathways. ROP is a neurovascular disease that occurs in response to relative retinal hyperoxia conditions in the immature retina of premature and low-birth-weight babies. The hyperoxia-induced damage is characterized by degeneration of both neuronal and vascular cells and is followed by pathological vitreoretinal neovascularization. Analyses using a mouse model of oxygen-induced retinopathy (OIR) as a model for ROP have demonstrated the involvement of A2 in the hyperoxia-induced neurovascular injury (151, 207). These studies in mice with OIR showed that deletion of the A2 gene inhibited the death of both neuronal and vascular cells and improved retinal function. Increased A2 expression was observed in the retinal horizontal cells and photoreceptor cells of the hyperoxia-treated mice (151). Earlier studies had suggested that arginase activity could have a protective role in reducing retinal injury in this model. The authors reported a decrease in retinal cell death and improved astrocyte survival in TNF-α-deficient mice (206). The retinas from TNF-α-deficient mice exposed to OIR had decreased NOS2 along with increases in arginase activity and A2 mRNA expression as compared with the retinas of wild-type mice exposed to OIR. In contrast to this suggested protective function of A2 expression, experiments using A2 knockout mice exposed to OIR showed significant reductions in both neuronal and vascular injury (151, 207). These protective effects of the A2 gene deletion were accompanied by preservation of retinal neuronal function as shown by electroretinography (151).
Further analyses in the same model showed that the OIR treatment in wild-type mice stimulated increased spermine oxidase expression along with decreases in spermine, increases in spermidine, and increases in hydrogen peroxide formation as compared with normoxic controls. These results suggested that the hyperoxia exposure causes an increase in backward catabolism of polyamine by spermine oxidase (FIGURE 4) (152). Each of these alterations was markedly decreased in mice lacking the A2 gene. Moreover, inhibition of polyamine oxidase by treatment with MDL-72527 [N,N'-bis(2,3-butadienyl)-1,4-butanediamine] significantly improved neuronal survival. This suggests that polyamine catabolism is involved in the hyperoxia-induced neurodegeneration. Further studies using MDL-72527 in the same model showed that inhibition of polyamine oxidase also abrogated the hyperoxia-induced vascular damage (159). The vascular damage was shown to involve activation of microglial cells and increased release of inflammatory cytokines and chemokines. The MDL-72527 treatment significantly reduced both events.
Recent investigations using a mouse model of retinal I/R injury have further established the specific role of A2 expression in neurovascular injury (201). Deletion of the A2 gene markedly inhibited I/R-induced degeneration of the retinal ganglion cells, prevented microvascular degeneration, and preserved retinal electroretinographic function through a mechanism involving inhibition of ROS formation and blockade of programmed necrosis (necroptosis).
Previous investigations using the same mouse model have shown that NOS3 and peroxynitrite are also critically involved in hyperoxia-induced microvascular degeneration (23, 79). After 2 days of hyperoxia treatment, retinas of the wild-type mice showed marked degeneration of the immature capillaries in the posterior retina. Deletion of NOS3 or treatment with Nω nitro-l-arginine (l-NNA), an inhibitor of NOS, significantly attenuated this damage. Furthermore, NOS3 deletion also limited the hyperoxia-induced increases in the peroxynitrite biomarker nitrotyrosine as compared with the retinas of the wild-type mice (23), indicating that NOS3 activity is critically involved in the vascular pathology. NOS2 has also been strongly implicated in vascular injury during OIR. Experiments using NOS2-deficient mice and wild-type mice treated with a specific inhibitor of NOS2 (1400W) showed that NOS2 expression was highly increased during OIR injury and that NOS2 blockade improved the repair of the damaged vessels (192). Thus NOS function clearly plays a role in retinal vascular injury during OIR. The role of arginase in the pathology has also been demonstrated. As explained above, expression of the A2 isoform is critically involved in the hyperoxia-mediated vascular injury (207). This vascular protection in the A2-deficient retinas was accompanied by improved NOS function in combination with decreases in hyperoxia-induced increases in superoxide and peroxynitrite production, suggesting that the A2-induced injury involves uncoupling of NOS. These results suggest that the vascular protection in the retinas of the A2-deficient mice is attributable to a reduction in nitrative stress. While these studies using mice deficient in NOS3 or NOS2 have suggested primary roles for these isoforms in the hyperoxia-induced vascular injury (23, 192), nNOS (NOS1) is highly expressed in retinal neurons and could also be involved. Additional work is required to address the impact of A2 expression/activity on the function of NOS.
Analyses using experimental models of ocular inflammatory disease and type 1 diabetes have also shown correlations between elevated arginase activity and retinal dysfunction/injury (55, 158, 175, 252). Retinas of mice with inflammation induced by low-dose endotoxin treatment had increases in activity of arginase and expression of A1. These changes were accompanied by elevation of oxidative stress and inflammatory gene expression along with increased attachment of leukocytes to the retinal vessels. Double-knockout mice lacking one copy of the A1 gene together with both copies of the A2 were protected from these alterations (252). Also, the endotoxin-induced increase in expression of A1 was blunted by NOX2 deletion or inhibition of NADPH oxidase activity, suggesting that NOX2-dependent increases in oxidative stress and arginase activation are involved in the endotoxin-induced inflammation. Studies in mice rendered diabetic with streptozotocin and retinal endothelial cells treated with high glucose showed similar links between hyperglycemia, oxidative stress, inflammation, and increases in the expression and activity of arginase (158, 175). Furthermore, the elevations of oxidative stress and retinal inflammation in the diabetic retinas were accompanied by decreases in NO bioavailability. Additional studies using fundus imaging of retinal arterioles in living mice demonstrated a marked impairment of endothelium-dependent vasorelaxation in the diabetic retinas. This vascular dysfunction was markedly attenuated in mice lacking one copy of the A1 gene, indicating the involvement of A1 in the poor vascular function (55). Treatment of the mice with a specific inhibitor of arginase activity also prevented the diabetes-induced impairment of endothelial function, confirming a role for arginase in the dysfunction. Ex vivo experiments using pressure myography to measure endothelial-dependent vasodilation of retinal arteries isolated from diabetic rats showed similar beneficial effects of arginase blockade in preventing diabetes-induced vascular endothelial dysfunction (55). In summary, these results show that increases in arginase expression/activity play a critical role in the elevation of oxidative stress, inflammation, and vascular dysfunctions associated with endotoxin- or diabetes-induced retinopathy.
VI. THERAPEUTIC APPROACHES TO LIMIT OR ALTER ARGINASE FUNCTION
The early arginase inhibitors had many side effects due to their low potency and/or nonspecific actions (141). For example, α-difluoromethylornithine (DFMO) is a weak inhibitor of arginase but is nonspecific in that it is also a potent inhibitor for ODC (141). Thus, while DFMO treatment can increase NO production in models of elevated arginase activity, this effect could be mediated by its action in promoting ornithine accumulation which is known to inhibit arginase (93). Inhibition of ODC would also limit polyamine formation, which could reduce oxidative stress by preventing the backward metabolism of spermine and spermidine by polyamine oxidases. Another compound, NOHA (Nω-hydroxy-l-arginine), is a potent arginase inhibitor, but it is also an intermediate product in the metabolism of l-arginine by NOS to produce NO. Like NOHA, its analog Nor-NOHA is also a potent arginase inhibitor, but its half-life is much longer than that of NOHA. Neither analog inhibits NOS. Norvaline is another effective inhibitor of arginase, but it is also a substrate for amidotransferases (42). In addition to its role in the production of urea, activation of arginase is also involved in promoting the synthesis of polyamines, l-ornithine, l-proline, and l-glutamine. In fact, several amino acids inhibit arginase, including l-ornithine, l-leucine, l-valine, l-lysine, l-isoleucine, and l-norvaline. l-Ornithine is the most potent (91). In addition, l-citrulline increases NO formation but is also an allosteric inhibitor of arginase (196).
Several competitive arginase inhibitors are now available that have high specificity and do not inhibit NOS at the concentrations that are highly effective in inhibiting arginase. These compounds were developed by Christianson and colleagues (49, 104) based on their determination of the crystal structure of human A1 and A2. These investigators identified the full structures of both isoforms and discovered that their catalytic activity depends on a binuclear manganese cluster at the active site (49, 104). On the basis of this information, two highly selective arginase inhibitors were developed, BEC [S-(2-boronoethyl)-l-cysteine] and ABH [2(S)-amino-6-boronohexanoic acid] (4, 39). Both are boronic acid analogs of l-arginine, and both contain a boronic acid or N-hydroxyguanidinium head. Binding of this boronic head to the manganese cluster at the active catalytic site in arginase results in strong competitive inhibition of its activity (14).
While several specific inhibitors of arginase are available, none of them is isoform selective. This is a major limitation for their therapeutic value in view of the large amount of experimental data showing that arginase activity can be damaging in some contexts and protective in others. For instance, the expression and activity of A2 in macrophages has been linked to the development and progression of atherosclerosis, whereas expression of A1 in macrophages is thought to facilitate plaque involution (58, 109, 156, 182, 183). Macrophage expression of A1 is also very important for tissue repair after injury. Specific inhibition of A1 in liver cells could also trigger toxic effects. In contrast, excessive activity of A2 appears to be damaging in conditions of CNS disease and injury. Due to the lack of isoform-selective arginase inhibitors, RNA knockdown strategies have been used to specifically downregulate expression of A1 or A2. In a study of a mouse model of experimental asthma, knockdown of A1 using short hairpin RNA (shRNA) decreased A1 mRNA and protein expression and greatly reduced airway hyperresponsiveness by a mechanism involving decreases in IL-13 levels (236). In a study of erectile dysfunction in aged mice, adenovirus vector delivery of antisense A1 in the corpus cavernosum was found to improve erectile function (19). Anti-sense-mediated knockdown of A1 was also found to restore normal levels of NO production in endothelial cells exposed to high glucose conditions as a model for diabetes-induced vascular dysfunction (178). The search for isoform-selective arginase inhibitors is continuing and involves plant extract testing and structure-based inhibitor design (204).
Several arginase inhibitors, including nor-NOHA, BEC, and ABH, are commercially available for preclinical use and have also been used in humans. However, so far clinical studies using arginase inhibitors in patients with cardiovascular disease have been limited to small-scale “proof-of-concept” studies involving local administration by cutaneous microdialysis or intra-arterial infusion. Promising results have been reported for treatment of patients with coronary artery disease and type 2 diabetes (115, 198), heart failure (168), hypertension (87), and after resuscitation following cardiac arrest (102). These observations suggest that inhibiting arginase activity could be beneficial in the treatment of cardiovascular disease. However, the effect of these inhibitors in larger clinical settings and the potential adverse effects of long-term treatment remains to be determined. Given the critical role for A1 in detoxifying ammonia in the urea cycle, it is very important to show that the treatment does not have an adverse effect on the hepatic urea cycle. Another possible limitation is that A1 inhibition will have an adverse effect on tissue repair functions that are compromised in many cardiovascular diseases. Thus, although increases in the expression/activation of arginase have been strongly linked to the development of multiple pathologies, the proper design of isotype-selective, cell-targeted inhibitors could facilitate the development of strategies to inhibit arginase as a therapy for disease.
VII. SUMMARY
The involvement of excessive activity of the arginase/ornithine pathway in cardiovascular and renal dysfunction and injury has been well documented by research in experimental models and human disease conditions. The involvement of these pathways in CNS disease and cancer has also been clearly demonstrated. Targeting specific elements in the arginase/ornithine pathways offers immense potential for the treatment of cardiovascular, renal, and CNS diseases as well as cancer. Studies in patients with cardiovascular disease have shown beneficial effects of local delivery of arginase inhibitors. As has been explained in the previous sections, the two arginase isoforms are 100% homologous in the areas critical for their activity. In addition, both isoforms are clearly involved in dysregulation of NOS function. However, A1 and A2 are encoded by different genes and localized to different intracellular compartments. Both isoforms are widely expressed in many cell types, and both are involved in multiple cellular functions. Analyses using mutant mice and specific gene knockdown have clearly demonstrated the differential involvement of the two isoforms in disease conditions. However, extensive evidence substantiates a positive role for A1 activity in maintaining normal cell growth, collagen synthesis, and neuronal development as well as tissue repair from injury. Therefore, the global inhibition of arginase activity for extended periods could be detrimental. Studies examining the subcellular and molecular regulation of arginase activity are underway. Knowledge gained from this work will facilitate the design of new strategies to limit the pathophysiological actions of overactive arginase. Further study is needed to provide a better understanding of the specific role of the two arginase isoforms and their downstream targets in health and disease and to identify better therapeutic strategies.
In closing, we provide a tabular overview of the current and potential therapeutic approaches to disease states related to arginase function. The pathological conditions associated with elevated arginase 1 and 2 expression and activity and the advantageous effects of suppressing activity or expression are shown in TABLE 1. Additionally, the emerging strategies for the differential treatment of cancers by targeting arginase activity or expression, or the metabolism of l-arginine, are given in TABLE 2.
Table 1.
Pathological conditions associated with detrimental effects of increases in arginase 1 and 2 expression and/or beneficial effects of arginase inhibitor or suppressor treatments
| Pathological Condition | Reference Nos. |
|---|---|
| Hypertension | 7, 16, 195, 213, 214 |
| Pulmonary artery hypertension | 28, 34, 35, 38, 75, 98, 99, 225, 234, 235 |
| Sickle cell disease | 8, 205 |
| Diabetic vascular disease | 5, 13, 15, 17, 55, 77, 176, 178, 198, 210 |
| Atherosclerosis | 156, 157, 182, 184 |
| Myocardial I/R injury | 72, 78, 101, 217, 253 |
| Aging/cellular senescence | 14, 111, 188, 233 |
| Erectile dysfunction | 21, 39, 43, 112, 119, 190, 213, 216 |
| Diabetic nephropathy | 143, 243, 244 |
| Hypertensive nephropathy | 129 |
| Cancer | 65, 93, 136, 141, 174, 241 |
| Ischemic stroke | 162, 167 |
| Traumatic brain injury | 18 |
| Alzheimer’s disease | 31, 37, 81, 90, 124 |
| Multiple sclerosis | 25, 76, 125, 126 |
| Retinal disease | 55, 151, 158, 201, 207, 252 |
Table 2.
Strategies for reducing cancer progression by targeting l-arginine metabolism
| Strategies |
|---|
| A. Increasing T cell anti-tumor immune function by decreasing myeloid derived suppressor cells (MDSC) number or activity |
| 1) DFMO: reduces the number of MDSC, inhibits ODC and arginase activity, and decreases polyamine formation (93, 136, 141, 241) |
| 2) Arginase inhibitors: increase l-arginine and reduce l-ornithine and polyamine levels (65, 174) |
| 3) NOS2 inhibitors: decrease NO formation (65, 202) |
| 4) Phosphodiesterase-5 : reduce A1 and NOS2 expression (193) |
| 5) Cyclooxygenase 2 inhibitors: reduce A1 expression and MDSC number (172, 218) |
| B. Reducing l-arginine availability to tumors auxotrophic for l-arginine (e.g., melanoma, hepatic carcinoma, acute lymphoblastic leukemias) by treatment with enzymes that metabolize l-arginine |
| 1) Pegylated arginine deiminase (Peg-ADI) (12, 56, 108) |
| 2) Pegylated arginase 1 (Peg-A1) (84, 144, 240) |
VIII. AREAS FOR FUTURE STUDY AND UNDERSTANDING
There are many questions that must be answered to better understand the functions of arginase and develop more effective strategies for targeting arginase activity as a therapy for diseases that affect the cardiovascular, renal, and nervous systems and cancers. These include the following:
-
•
How do the functions of arginase 1 and arginase 2 differ mechanistically? Both isoforms metabolize l-arginine to produce l-ornithine and urea, but they differ substantially in terms of their intracellular localization and tissue distribution. Does their production of the downstream products of ornithine metabolism also differ? These questions can be addressed using genetic strategies to manipulate the activity and distribution of each isoform, but development of isoform-specific inhibitors would greatly facilitate research in this area.
-
•
What is the physiological basis for conflicting reports on the arginase isoform (1 or 2) involved in vascular dysfunctions: impaired endothelial-dependent relaxation and stiffening? Species differences are known, but the isoform involved within a species has differed in some reports.
-
•
What are the specific roles of arginase 1 and arginase 2 in macrophages? Expression of arginase 1 is thought to have a healing role in the repair phase of wound healing, while expression of arginase 2 has been implicated in chronic inflammatory disease conditions. However, there are some reports that inflammatory macrophages express both isoforms. Does this mean that both are needed for different functions?
-
•
Is arginase 1 activity regulated at the posttranslational level as has been reported for arginase 2? In smooth muscle cells, arginase 2 activity has been shown to increase when the enzyme is translocated from the mitochondria into the cytosol. Is arginase 1 activity also modified by changes in its subcellular localization? Is activity of either isoform modified by phosphorylation events?
-
•
Are some arginase functions independent of the ureohydrolase activity? Studies using a catalytically inactive arginase 2 mutant in models of atherosclerosis found that arginase 2 expression induces senescence and apoptosis of smooth muscle cells independently of ureohydrolase activity. Does this phenomenon apply to arginase 1 or to other cell types and disease models?
-
•
How can the problems of differential effects arginase activity on cancers be managed therapeutically? Concurrent mixed neoplasms would complicate treatment.
-
•
Can circulating levels of arginase and/or its metabolic products serve as biomarkers for the development or progression of disease?
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant RO1HL070215 (to R. W. Caldwell), National Eye Institute Grant RO1EY011766 (to R. B. Caldwell and R. W. Caldwell), National Cancer Institute Grant RO1CA18485 (to P. C. Rodriguez), American Heart Association Grants 13SDG174100007 (to H. A. Toque) and 11SDG7440088 (to S. P. Narayanan), and United States Department of Veterans Affairs Grant I01BX003221 (to R. B. Caldwell). R. B. Caldwell is the recipient of a Research Career Scientist Award from the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
Present address for P. C. Rodriguez: Moffitt Cancer, 12902 Magnolia Dr., Tampa, FL 33612.
Address for reprint requests and other correspondence: R. W. Caldwell, Dept. of Pharmacology and Toxicology, Medical College of Georgia, Augusta University, 1459 Laney Walker Blvd., Augusta, GA 30912 (e-mail: wcaldwel@augusta.edu).
REFERENCES
- 1.Ahmad AS, Shah ZA, Doré S. Protective role of arginase II in cerebral ischemia and excitotoxicity. J Neurol Neurosci 7: 88, 2016. doi: 10.21767/2171-6625.100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altieri P, Sau G, Cao R, Barracca A, Menneas A, Micchittu B, Cabiddu G, Esposito P, Pani A. Immunosuppressive treatment in dialysis patients. Nephrol Dial Transplant 17, Suppl 8: 2–9, 2002. doi: 10.1093/ndt/17.suppl_8.2. [DOI] [PubMed] [Google Scholar]
- 3.Ash DE. Structure and function of arginases. J Nutr 134, Suppl: 2760S–2764S, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Baggio R, Emig FA, Christianson DW, Ash DE, Chakder S, Rattan S. Biochemical and functional profile of a newly developed potent and isozyme-selective arginase inhibitor. J Pharmacol Exp Ther 290: 1409–1416, 1999. [PubMed] [Google Scholar]
- 5.Bagi Z, Feher A, Dou H, Broskova Z. Selective up-regulation of arginase-1 in coronary arteries of diabetic patients. Front Immunol 4: 293, 2013. doi: 10.3389/fimmu.2013.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagnost T, Berthelot A, Alvergnas M, Miguet-Alfonsi C, André C, Guillaume Y, Demougeot C. Misregulation of the arginase pathway in tissues of spontaneously hypertensive rats. Hypertens Res 32: 1130–1135, 2009. doi: 10.1038/hr.2009.153. [DOI] [PubMed] [Google Scholar]
- 7.Bagnost T, Ma L, da Silva RF, Rezakhaniha R, Houdayer C, Stergiopulos N, André C, Guillaume Y, Berthelot A, Demougeot C. Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc Res 87: 569–577, 2010. doi: 10.1093/cvr/cvq081. [DOI] [PubMed] [Google Scholar]
- 8.Bakshi N, Morris CR. The role of the arginine metabolome in pain: implications for sickle cell disease. J Pain Res 9: 167–175, 2016. doi: 10.2147/JPR.S55571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbul A, Rettura G, Levenson SM, Seifter E. Arginine: a thymotropic and wound-healing promoting agent. Surg Forum 28: 101–103, 1977. [PubMed] [Google Scholar]
- 10.Barra V, Kuhn AM, von Knethen A, Weigert A, Brüne B. Apoptotic cell-derived factors induce arginase II expression in murine macrophages by activating ERK5/CREB. Cell Mol Life Sci 68: 1815–1827, 2011. doi: 10.1007/s00018-010-0537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87: 1620–1624, 1990. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beddowes E, Spicer J, Chan PY, Khadeir R, Corbacho JG, Repana D, Steele JP, Schmid P, Szyszko T, Cook G, Diaz M, Feng X, Johnston A, Thomson J, Sheaff M, Wu BW, Bomalaski J, Pacey S, Szlosarek PW. Phase 1 Dose-Escalation Study of Pegylated Arginine Deiminase, Cisplatin, and Pemetrexed in Patients With Argininosuccinate Synthetase 1-Deficient Thoracic Cancers. J Clin Oncol 35: 1778–1785, 2017. doi: 10.1200/JCO.2016.71.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol 300: H777–H783, 2011. doi: 10.1152/ajpheart.00831.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 108: 2000–2006, 2003. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- 15.Bhatta A, Sangani R, Kolhe R, Toque HA, Cain M, Wong A, Howie N, Shinde R, Elsalanty M, Yao L, Chutkan N, Hunter M, Caldwell RB, Isales C, Caldwell RW, Fulzele S. Deregulation of arginase induces bone complications in high-fat/high-sucrose diet diabetic mouse model. Mol Cell Endocrinol 422: 211–220, 2016. doi: 10.1016/j.mce.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatta A, Yao L, Toque HA, Shatanawi A, Xu Z, Caldwell RB, Caldwell RW. Angiotensin II-induced arterial thickening, fibrosis and stiffening involves elevated arginase function. PLoS One 10: e0121727, 2015. doi: 10.1371/journal.pone.0121727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhatta A, Yao L, Xu Z, Toque HA, Chen J, Atawia RT, Fouda AY, Bagi Z, Lucas R, Caldwell RB, Caldwell RW. Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc Res 113: 1664–1676, 2017. doi: 10.1093/cvr/cvx164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bitner BR, Brink DC, Mathew LC, Pautler RG, Robertson CS. Impact of arginase II on CBF in experimental cortical impact injury in mice using MRI. J Cereb Blood Flow Metab 30: 1105–1109, 2010. doi: 10.1038/jcbfm.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bivalacqua TJ, Burnett AL, Hellstrom WJ, Champion HC. Overexpression of arginase in the aged mouse penis impairs erectile function and decreases eNOS activity: influence of in vivo gene therapy of anti-arginase. Am J Physiol Heart Circ Physiol 292: H1340–H1351, 2007. doi: 10.1152/ajpheart.00121.2005. [DOI] [PubMed] [Google Scholar]
- 20.Bivalacqua TJ, Champion HC, Usta MF, Cellek S, Chitaley K, Webb RC, Lewis RL, Mills TM, Hellstrom WJG, Kadowitz PJ. RhoA/Rho-kinase suppresses endothelial nitric oxide synthase in the penis: a mechanism for diabetes-associated erectile dysfunction. Proc Natl Acad Sci USA 101: 9121–9126, 2004. doi: 10.1073/pnas.0400520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bivalacqua TJ, Hellstrom WJ, Kadowitz PJ, Champion HC. Increased expression of arginase II in human diabetic corpus cavernosum: in diabetic-associated erectile dysfunction. Biochem Biophys Res Commun 283: 923–927, 2001. doi: 10.1006/bbrc.2001.4874. [DOI] [PubMed] [Google Scholar]
- 22.Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 8: 29, 2010. doi: 10.1186/1478-7954-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks SE, Gu X, Samuel S, Marcus DM, Bartoli M, Huang PL, Caldwell RB. Reduced severity of oxygen-induced retinopathy in eNOS-deficient mice. Invest Ophthalmol Vis Sci 42: 222–228, 2001. [PubMed] [Google Scholar]
- 24.Bryan NS, Bian K, Murad F. Discovery of the nitric oxide signaling pathway and targets for drug development. Front Biosci (Landmark Ed) 14: 1–18, 2009. doi: 10.2741/3228. [DOI] [PubMed] [Google Scholar]
- 25.Cantoni C, Cignarella F, Ghezzi L, Mikesell B, Bollman B, Berrien-Elliott MM, Ireland AR, Fehniger TA, Wu GF, Piccio L. Mir-223 regulates the number and function of myeloid-derived suppressor cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol 133: 61–77, 2017. doi: 10.1007/s00401-016-1621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castela Â, Costa C. Molecular mechanisms associated with diabetic endothelial-erectile dysfunction. Nat Rev Urol 13: 266–274, 2016. doi: 10.1038/nrurol.2016.23. [DOI] [PubMed] [Google Scholar]
- 27.Chandra S, Romero MJ, Shatanawi A, Alkilany AM, Caldwell RB, Caldwell RW. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol 165: 506–519, 2012. doi: 10.1111/j.1476-5381.2011.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol 297: L1151–L1159, 2009. doi: 10.1152/ajplung.00183.2009. [DOI] [PubMed] [Google Scholar]
- 29.Cherian L, Hlatky R, Robertson CS. Nitric oxide in traumatic brain injury. Brain Pathol 14: 195–201, 2004. doi: 10.1111/j.1750-3639.2004.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cherian L, Robertson CS. l-Arginine and free radical scavengers increase cerebral blood flow and brain tissue nitric oxide concentrations after controlled cortical impact injury in rats. J Neurotrauma 20: 77–85, 2003. doi: 10.1089/08977150360517209. [DOI] [PubMed] [Google Scholar]
- 31.Cherry JD, Olschowka JA, O’Banion MK. Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J Neuroinflammation 12: 203, 2015. doi: 10.1186/s12974-015-0411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chicoine LG, Paffett ML, Young TL, Nelin LD. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol 287: L60–L68, 2004. doi: 10.1152/ajplung.00194.2003. [DOI] [PubMed] [Google Scholar]
- 33.Chitaley K, Wingard CJ, Clinton Webb R, Branam H, Stopper VS, Lewis RW, Mills TM. Antagonism of Rho-kinase stimulates rat penile erection via a nitric oxide-independent pathway. Nat Med 7: 119–122, 2001. doi: 10.1038/83258. [DOI] [PubMed] [Google Scholar]
- 34.Cho WK, Lee CM, Kang MJ, Huang Y, Giordano FJ, Lee PJ, Trow TK, Homer RJ, Sessa WC, Elias JA, Lee CG. IL-13 receptor α2-arginase 2 pathway mediates IL-13-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 304: L112–L124, 2013. doi: 10.1152/ajplung.00101.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu Y, XiangLi X, Niu H, Wang H, Jia P, Gong W, Wu D, Qin W, Xing C. Arginase inhibitor attenuates pulmonary artery hypertension induced by hypoxia. Mol Cell Biochem 412: 91–99, 2016. doi: 10.1007/s11010-015-2611-z. [DOI] [PubMed] [Google Scholar]
- 36.Closs EI, Simon A, Vékony N, Rotmann A. Plasma membrane transporters for arginine. J Nutr 134, Suppl: 2752S–2759S, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation 3: 27, 2006. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cowburn AS, Crosby A, Macias D, Branco C, Colaço RD, Southwood M, Toshner M, Crotty Alexander LE, Morrell NW, Chilvers ER, Johnson RS. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci USA 113: 8801–8806, 2016. doi: 10.1073/pnas.1602978113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cox JD, Kim NN, Traish AM, Christianson DW. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat Struct Biol 6: 1043–1047, 1999. doi: 10.1038/14929. [DOI] [PubMed] [Google Scholar]
- 40.Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 108: 1527–1532, 2003. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 41.Cullen ME, Yuen AH, Felkin LE, Smolenski RT, Hall JL, Grindle S, Miller LW, Birks EJ, Yacoub MH, Barton PJ. Myocardial expression of the arginine:glycine amidinotransferase gene is elevated in heart failure and normalized after recovery: potential implications for local creatine synthesis. Circulation 114, Suppl: I16–I20, 2006. doi: 10.1161/CIRCULATIONAHA.105.000448. [DOI] [PubMed] [Google Scholar]
- 42.Davoodi J, Drown PM, Bledsoe RK, Wallin R, Reinhart GD, Hutson SM. Overexpression and characterization of the human mitochondrial and cytosolic branched-chain aminotransferases. J Biol Chem 273: 4982–4989, 1998. doi: 10.1074/jbc.273.9.4982. [DOI] [PubMed] [Google Scholar]
- 43.De A, Singh MF, Singh V, Ram V, Bisht S. Treatment effect of l-Norvaline on the sexual performance of male rats with streptozotocin induced diabetes. Eur J Pharmacol 771: 247–254, 2016. doi: 10.1016/j.ejphar.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 44.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest 96: 60–68, 1995. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dean RC, Lue TF. Physiology of penile erection and pathophysiology of erectile dysfunction. Urol Clin North Am 32: 379–395, 2005. doi: 10.1016/j.ucl.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deignan JL, Cederbaum SD, Grody WW. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol Genet Metab 93: 7–14, 2008. doi: 10.1016/j.ymgme.2007.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deignan JL, Livesay JC, Yoo PK, Goodman SI, O’Brien WE, Iyer RK, Cederbaum SD, Grody WW. Ornithine deficiency in the arginase double knockout mouse. Mol Genet Metab 89: 87–96, 2006. doi: 10.1016/j.ymgme.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 48.Deng K, He H, Qiu J, Lorber B, Bryson JB, Filbin MT. Increased synthesis of spermidine as a result of upregulation of arginase I promotes axonal regeneration in culture and in vivo. J Neurosci 29: 9545–9552, 2009. doi: 10.1523/JNEUROSCI.1175-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Costanzo L, Sabio G, Mora A, Rodriguez PC, Ochoa AC, Centeno F, Christianson DW. Crystal structure of human arginase I at 1.29-A resolution and exploration of inhibition in the immune response. Proc Natl Acad Sci USA 102: 13058–13063, 2005. doi: 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dizikes GJ, Grody WW, Kern RM, Cederbaum SD. Isolation of human liver arginase cDNA and demonstration of nonhomology between the two human arginase genes. Biochem Biophys Res Commun 141: 53–59, 1986. doi: 10.1016/S0006-291X(86)80333-3. [DOI] [PubMed] [Google Scholar]
- 51.Durante W. Role of arginase in vessel wall remodeling. Front Immunol 4: 111, 2013. doi: 10.3389/fimmu.2013.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol 34: 906–911, 2007. doi: 10.1111/j.1440-1681.2007.04638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dzik JM. Evolutionary roots of arginase expression and regulation. Front Immunol 5: 544, 2014. doi: 10.3389/fimmu.2014.00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elms S, Chen F, Wang Y, Qian J, Askari B, Yu Y, Pandey D, Iddings J, Caldwell RB, Fulton DJ. Insights into the arginine paradox: evidence against the importance of subcellular location of arginase and eNOS. Am J Physiol Heart Circ Physiol 305: H651–H666, 2013. doi: 10.1152/ajpheart.00755.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elms SC, Toque HA, Rojas M, Xu Z, Caldwell RW, Caldwell RB. The role of arginase I in diabetes-induced retinal vascular dysfunction in mouse and rat models of diabetes. Diabetologia 56: 654–662, 2013. doi: 10.1007/s00125-012-2789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res 62: 5443–5450, 2002. [PubMed] [Google Scholar]
- 57.Esch F, Lin KI, Hills A, Zaman K, Baraban JM, Chatterjee S, Rubin L, Ash DE, Ratan RR. Purification of a multipotent antideath activity from bovine liver and its identification as arginase: nitric oxide-independent inhibition of neuronal apoptosis. J Neurosci 18: 4083–4095, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feig JE, Vengrenyuk Y, Reiser V, Wu C, Statnikov A, Aliferis CF, Garabedian MJ, Fisher EA, Puig O. Regression of atherosclerosis is characterized by broad changes in the plaque macrophage transcriptome. PLoS One 7: e39790, 2012. doi: 10.1371/journal.pone.0039790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Filbin MT. The Muddle with MAG. Mol Cell Neurosci 8: 84–92, 1996. doi: 10.1006/mcne.1996.0047. [DOI] [PubMed] [Google Scholar]
- 60.Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, Sanchez-Pino D, Hernandez C, Wyczechowska DD, Ochoa AC, Rodriguez PC. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res 75: 275–283, 2015. doi: 10.1158/0008-5472.CAN-14-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Förstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, Kleinert H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 23: 1121–1131, 1994. doi: 10.1161/01.HYP.23.6.1121. [DOI] [PubMed] [Google Scholar]
- 62.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J 33: 829–837, 2012. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fultang L, Vardon A, De Santo C, Mussai F. Molecular basis and current strategies of therapeutic arginine depletion for cancer. Int J Cancer 139: 501–509, 2016. doi: 10.1002/ijc.30051. [DOI] [PubMed] [Google Scholar]
- 64.Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J 3: 2007–2018, 1989. [PubMed] [Google Scholar]
- 65.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12: 253–268, 2012. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, Chilian WM, Zhang C. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol 27: 1269–1275, 2007. doi: 10.1161/ATVBAHA.107.142521. [DOI] [PubMed] [Google Scholar]
- 67.Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KT. The role of the nitric oxide pathway in brain injury and its treatment–from bench to bedside. Exp Neurol 263: 235–243, 2015. doi: 10.1016/j.expneurol.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 68.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, Zamboni N, Sallusto F, Lanzavecchia A. l-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167: 829–842.e13, 2016. doi: 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghosh P, Sica A, Young HA, Ye J, Franco JL, Wiltrout RH, Longo DL, Rice NR, Komschlies KL. Alterations in NF kappa B/Rel family proteins in splenic T-cells from tumor-bearing mice and reversal following therapy. Cancer Res 54: 2969–2972, 1994. [PubMed] [Google Scholar]
- 70.Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 17: 34–40, 2016. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- 71.Goligorsky MS, Chen J, Brodsky S. Workshop: endothelial cell dysfunction leading to diabetic nephropathy : focus on nitric oxide. Hypertension 37: 744–748, 2001. doi: 10.1161/01.HYP.37.2.744. [DOI] [PubMed] [Google Scholar]
- 72.Gonon AT, Jung C, Katz A, Westerblad H, Shemyakin A, Sjöquist PO, Lundberg JO, Pernow J. Local arginase inhibition during early reperfusion mediates cardioprotection via increased nitric oxide production. PLoS One 7: e42038, 2012. doi: 10.1371/journal.pone.0042038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gotoh T, Sonoki T, Nagasaki A, Terada K, Takiguchi M, Mori M. Molecular cloning of cDNA for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line. FEBS Lett 395: 119–122, 1996. doi: 10.1016/0014-5793(96)01015-0. [DOI] [PubMed] [Google Scholar]
- 74.Grandvaux N, Gaboriau F, Harris J, tenOever BR, Lin R, Hiscott J. Regulation of arginase II by interferon regulatory factor 3 and the involvement of polyamines in the antiviral response. FEBS J 272: 3120–3131, 2005. doi: 10.1111/j.1742-4658.2005.04726.x. [DOI] [PubMed] [Google Scholar]
- 75.Grasemann H, Dhaliwal R, Ivanovska J, Kantores C, McNamara PJ, Scott JA, Belik J, Jankov RP. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am J Physiol Lung Cell Mol Physiol 308: L503–L510, 2015. doi: 10.1152/ajplung.00328.2014. [DOI] [PubMed] [Google Scholar]
- 76.Greenhalgh AD, Passos Dos Santos R, Zarruk JG, Salmon CK, Kroner A, David S. Arginase-1 is expressed exclusively by infiltrating myeloid cells in CNS injury and disease. Brain Behav Immun 56: 61–67, 2016. doi: 10.1016/j.bbi.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 77.Grönros J, Jung C, Lundberg JO, Cerrato R, Ostenson CG, Pernow J. Arginase inhibition restores in vivo coronary microvascular function in type 2 diabetic rats. Am J Physiol Heart Circ Physiol 300: H1174–H1181, 2011. doi: 10.1152/ajpheart.00560.2010. [DOI] [PubMed] [Google Scholar]
- 78.Grönros J, Kiss A, Palmér M, Jung C, Berkowitz D, Pernow J. Arginase inhibition improves coronary microvascular function and reduces infarct size following ischaemia-reperfusion in a rat model. Acta Physiol (Oxf) 208: 172–179, 2013. doi: 10.1111/apha.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gu X, Samuel S, El-Shabrawey M, Caldwell RB, Bartoli M, Marcus DM, Brooks SE. Effects of sustained hyperoxia on revascularization in experimental retinopathy of prematurity. Invest Ophthalmol Vis Sci 43: 496–502, 2002. [PubMed] [Google Scholar]
- 80.Hagenfeldt L, Dahlquist G, Persson B. Plasma amino acids in relation to metabolic control in insulin-dependent diabetic children. Acta Paediatr Scand 78: 278–282, 1989. doi: 10.1111/j.1651-2227.1989.tb11070.x. [DOI] [PubMed] [Google Scholar]
- 81.Hansmannel F, Sillaire A, Kamboh MI, Lendon C, Pasquier F, Hannequin D, Laumet G, Mounier A, Ayral AM, DeKosky ST, Hauw JJ, Berr C, Mann D, Amouyel P, Campion D, Lambert JC. Is the urea cycle involved in Alzheimer’s disease? J Alzheimers Dis 21: 1013–1021, 2010. doi: 10.3233/JAD-2010-100630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hatzoglou M, Fernandez J, Yaman I, Closs E. Regulation of cationic amino acid transport: the story of the CAT-1 transporter. Annu Rev Nutr 24: 377–399, 2004. doi: 10.1146/annurev.nutr.23.011702.073120. [DOI] [PubMed] [Google Scholar]
- 83.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123: 92–100, 2013. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hernandez CP, Morrow K, Lopez-Barcons LA, Zabaleta J, Sierra R, Velasco C, Cole J, Rodriguez PC. Pegylated arginase I: a potential therapeutic approach in T-ALL. Blood 115: 5214–5221, 2010. doi: 10.1182/blood-2009-12-258822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, Pearce EJ, Wynn TA. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol 167: 6533–6544, 2001. doi: 10.4049/jimmunol.167.11.6533. [DOI] [PubMed] [Google Scholar]
- 86.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6: 318–327, 2005. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 87.Holowatz LA, Kenney WL. Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilatation in hypertensive humans. J Physiol 581: 863–872, 2007. doi: 10.1113/jphysiol.2007.128959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hsieh CL, Kim CC, Ryba BE, Niemi EC, Bando JK, Locksley RM, Liu J, Nakamura MC, Seaman WE. Traumatic brain injury induces macrophage subsets in the brain. Eur J Immunol 43: 2010–2022, 2013. doi: 10.1002/eji.201243084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu H, Moon J, Chung JH, Kim OY, Yu R, Shin MJ. Arginase inhibition ameliorates adipose tissue inflammation in mice with diet-induced obesity. Biochem Biophys Res Commun 464: 840–847, 2015. doi: 10.1016/j.bbrc.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 90.Hunt JB Jr, Nash KR, Placides D, Moran P, Selenica ML, Abuqalbeen F, Ratnasamy K, Slouha N, Rodriguez-Ospina S, Savlia M, Ranaweera Y, Reid P, Dickey CA, Uricia R, Yang CG, Sandusky LA, Gordon MN, Morgan D, Lee DC. Sustained arginase 1 expression modulates pathological tau deposits in a mouse model of tauopathy. J Neurosci 35: 14842–14860, 2015. doi: 10.1523/JNEUROSCI.3959-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hunter A, Downs D. The inhibition of arginase by amino acids. J Biol Chem 157: 427–446, 1945. [Google Scholar]
- 92.Huynh NN, Andrews KL, Head GA, Khong SM, Mayorov DN, Murphy AJ, Lambert G, Kiriazis H, Xu Q, Du XJ, Chin-Dusting JP. Arginase II knockout mouse displays a hypertensive phenotype despite a decreased vasoconstrictory profile. Hypertension 54: 294–301, 2009. doi: 10.1161/HYPERTENSIONAHA.108.121731. [DOI] [PubMed] [Google Scholar]
- 93.Huynh NN, Harris EE, Chin-Dusting JF, Andrews KL. The vascular effects of different arginase inhibitors in rat isolated aorta and mesenteric arteries. Br J Pharmacol 156: 84–93, 2009. doi: 10.1111/j.1476-5381.2008.00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Item CB, Stöckler-Ipsiroglu S, Stromberger C, Mühl A, Alessandrì MG, Bianchi MC, Tosetti M, Fornai F, Cioni G. Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans. Am J Hum Genet 69: 1127–1133, 2001. doi: 10.1086/323765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ivanova S, Batliwalla F, Mocco J, Kiss S, Huang J, Mack W, Coon A, Eaton JW, Al-Abed Y, Gregersen PK, Shohami E, Connolly ES Jr, Tracey KJ. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc Natl Acad Sci USA 99: 5579–5584, 2002. doi: 10.1073/pnas.082609299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iyer RK, Yoo PK, Kern RM, Rozengurt N, Tsoa R, O’Brien WE, Yu H, Grody WW, Cederbaum SD. Mouse model for human arginase deficiency. Mol Cell Biol 22: 4491–4498, 2002. doi: 10.1128/MCB.22.13.4491-4498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iyo AH, Zhu MY, Ordway GA, Regunathan S. Expression of arginine decarboxylase in brain regions and neuronal cells. J Neurochem 96: 1042–1050, 2006. doi: 10.1111/j.1471-4159.2005.03544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang W, Sun B, Song X, Zheng Y, Wang L, Wang T, Liu S. Arginase inhibition protects against hypoxia–induced pulmonary arterial hypertension. Mol Med Rep 12: 4743–4749, 2015. doi: 10.3892/mmr.2015.3994. [DOI] [PubMed] [Google Scholar]
- 99.Jin Y, Calvert TJ, Chen B, Chicoine LG, Joshi M, Bauer JA, Liu Y, Nelin LD. Mice deficient in Mkp-1 develop more severe pulmonary hypertension and greater lung protein levels of arginase in response to chronic hypoxia. Am J Physiol Heart Circ Physiol 298: H1518–H1528, 2010. doi: 10.1152/ajpheart.00813.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jin Y, Liu Y, Nelin LD. Extracellular signal-regulated kinase mediates expression of arginase II but not inducible nitric-oxide synthase in lipopolysaccharide-stimulated macrophages. J Biol Chem 290: 2099–2111, 2015. doi: 10.1074/jbc.M114.599985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jung C, Gonon AT, Sjöquist PO, Lundberg JO, Pernow J. Arginase inhibition mediates cardioprotection during ischaemia-reperfusion. Cardiovasc Res 85: 147–154, 2010. doi: 10.1093/cvr/cvp303. [DOI] [PubMed] [Google Scholar]
- 102.Jung C, Quitter F, Lichtenauer M, Fritzenwanger M, Pfeil A, Shemyakin A, Franz M, Figulla HR, Pfeifer R, Pernow J. Increased arginase levels contribute to impaired perfusion after cardiopulmonary resuscitation. Eur J Clin Invest 44: 965–971, 2014. doi: 10.1111/eci.12330. [DOI] [PubMed] [Google Scholar]
- 103.Kämpfer H, Pfeilschifter J, Frank S. Expression and activity of arginase isoenzymes during normal and diabetes-impaired skin repair. J Invest Dermatol 121: 1544–1551, 2003. doi: 10.1046/j.1523-1747.2003.12610.x. [DOI] [PubMed] [Google Scholar]
- 104.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature 383: 554–557, 1996. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 105.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest 127: 750–760, 2017. doi: 10.1172/JCI89741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kato JY. Control of G1 progression by D-type cyclins: key event for cell proliferation. Leukemia 11, Suppl 3: 347–351, 1997. [PubMed] [Google Scholar]
- 107.Katusic ZS, d’Uscio LV, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci 30: 48–54, 2009. doi: 10.1016/j.tips.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kelly MP, Jungbluth AA, Wu BW, Bomalaski J, Old LJ, Ritter G. Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. Br J Cancer 106: 324–332, 2012. doi: 10.1038/bjc.2011.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A, Caligiuri G. Macrophage plasticity in experimental atherosclerosis. PLoS One 5: e8852, 2010. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Khan M, Dhammu TS, Matsuda F, Singh AK, Singh I. Blocking a vicious cycle nNOS/peroxynitrite/AMPK by S-nitrosoglutathione: implication for stroke therapy. BMC Neurosci 16: 42, 2015. doi: 10.1186/s12868-015-0179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol (1985) 107: 1249–1257, 2009. doi: 10.1152/japplphysiol.91393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim NN, Cox JD, Baggio RF, Emig FA, Mistry SK, Harper SL, Speicher DW, Morris SM Jr, Ash DE, Traish A, Christianson DW. Probing erectile function: S-(2-boronoethyl)-l-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry 40: 2678–2688, 2001. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]
- 113.Koshland DE., Jr The molecule of the year. Science 258: 1861, 1992. doi: 10.1126/science.1470903. [DOI] [PubMed] [Google Scholar]
- 114.Kövamees O, Shemyakin A, Pernow J. Amino acid metabolism reflecting arginase activity is increased in patients with type 2 diabetes and associated with endothelial dysfunction. Diab Vasc Dis Res 13: 354–360, 2016. doi: 10.1177/1479164116643916. [DOI] [PubMed] [Google Scholar]
- 115.Kövamees O, Shemyakin A, Pernow J. Effect of arginase inhibition on ischemia-reperfusion injury in patients with coronary artery disease with and without diabetes mellitus. PLoS One 9: e103260, 2014. doi: 10.1371/journal.pone.0103260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Krotova K, Patel JM, Block ER, Zharikov S. Hypoxic upregulation of arginase II in human lung endothelial cells. Am J Physiol Cell Physiol 299: C1541–C1548, 2010. doi: 10.1152/ajpcell.00068.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA 88: 4651–4655, 1991. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kuo HS, Tsai MJ, Huang MC, Huang WC, Lee MJ, Kuo WC, You LH, Szeto KC, Tsai IL, Chang WC, Chiu CW, Ma H, Chak KF, Cheng H. The combination of peripheral nerve grafts and acidic fibroblast growth factor enhances arginase I and polyamine spermine expression in transected rat spinal cords. Biochem Biophys Res Commun 357: 1–7, 2007. doi: 10.1016/j.bbrc.2007.02.167. [DOI] [PubMed] [Google Scholar]
- 119.Lacchini R, Muniz JJ, Nobre YTDA, Cologna AJ, Martins ACP, Tanus-Santos JE. Relationship between Arginase 1 and Arginase 2 levels and genetic polymorphisms with erectile dysfunction. Nitric Oxide 51: 36–42, 2015. doi: 10.1016/j.niox.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 120.Lange PS, Langley B, Lu P, Ratan RR. Novel roles for arginase in cell survival, regeneration, and translation in the central nervous system. J Nutr 134, Suppl: 2812S–2817S, 2004. [DOI] [PubMed] [Google Scholar]
- 121.Lee J, Ryu H, Ferrante RJ, Morris SM Jr, Ratan RR. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc Natl Acad Sci USA 100: 4843–4848, 2003. doi: 10.1073/pnas.0735876100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Levillain O, Balvay S, Peyrol S. Localization and differential expression of arginase II in the kidney of male and female mice. Pflugers Arch 449: 491–503, 2005. doi: 10.1007/s00424-004-1336-8. [DOI] [PubMed] [Google Scholar]
- 123.Li X, Liu J, Park JK, Hamilton TA, Rayman P, Klein E, Edinger M, Tubbs R, Bukowski R, Finke J. T cells from renal cell carcinoma patients exhibit an abnormal pattern of kappa B-specific DNA-binding activity: a preliminary report. Cancer Res 54: 5424–5429, 1994. [PubMed] [Google Scholar]
- 124.Liu P, Fleete MS, Jing Y, Collie ND, Curtis MA, Waldvogel HJ, Faull RL, Abraham WC, Zhang H. Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol Aging 35: 1992–2003, 2014. doi: 10.1016/j.neurobiolaging.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 125.Ljubisavljevic S, Stojanovic I, Pavlovic R, Pavlovic D. The importance of nitric oxide and arginase in the pathogenesis of acute neuroinflammation: are those contra players with the same direction? Neurotox Res 26: 392–399, 2014. doi: 10.1007/s12640-014-9470-3. [DOI] [PubMed] [Google Scholar]
- 126.Ljubisavljevic S, Stojanovic I, Pavlovic R, Sokolovic D, Pavlovic D, Cvetkovic T, Stevanovic I. Modulation of nitric oxide synthase by arginase and methylated arginines during the acute phase of experimental multiple sclerosis. J Neurol Sci 318: 106–111, 2012. doi: 10.1016/j.jns.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 127.Lucas R, Fulton D, Caldwell RW, Romero MJ. Arginase in the vascular endothelium: friend or foe? Front Immunol 5: 589, 2014. doi: 10.3389/fimmu.2014.00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ma TC, Campana A, Lange PS, Lee HH, Banerjee K, Bryson JB, Mahishi L, Alam S, Giger RJ, Barnes S, Morris SM Jr, Willis DE, Twiss JL, Filbin MT, Ratan RR. A large-scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone daidzeinas a clinically approved small molecule that can promote neuronal protection or regeneration via a cAMP-independent pathway. J Neurosci 30: 739–748, 2010. doi: 10.1523/JNEUROSCI.5266-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maquiaveli CC, da Silva ER, Rosa LC, Francescato HD, Lucon Júnior JF, Silva CG, Casarini DE, Ronchi FA, Coimbra TM. Cecropia pachystachya extract attenuated the renal lesion in 5/6 nephrectomized rats by reducing inflammation and renal arginase activity. J Ethnopharmacol 158, Pt A: 49–57, 2014. doi: 10.1016/j.jep.2014.09.042. [DOI] [PubMed] [Google Scholar]
- 130.Martens CR, Kuczmarski JM, Lennon-Edwards S, Edwards DG. Impaired l-arginine uptake but not arginase contributes to endothelial dysfunction in rats with chronic kidney disease. J Cardiovasc Pharmacol 63: 40–48, 2014. doi: 10.1097/FJC.0000000000000022. [DOI] [PubMed] [Google Scholar]
- 131.Maurin AC, Jousse C, Averous J, Parry L, Bruhat A, Cherasse Y, Zeng H, Zhang Y, Harding HP, Ron D, Fafournoux P. The GCN2 kinase biases feeding behavior to maintain amino acid homeostasis in omnivores. Cell Metab 1: 273–277, 2005. doi: 10.1016/j.cmet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 132.McDonald KK, Zharikov S, Block ER, Kilberg MS. A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the “arginine paradox”. J Biol Chem 272: 31213–31216, 1997. doi: 10.1074/jbc.272.50.31213. [DOI] [PubMed] [Google Scholar]
- 133.Mills CD, Thomas AC, Lenz LL, Munder M. Macrophage: SHIP of Immunity. Front Immunol 5: 620, 2014. doi: 10.3389/fimmu.2014.00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, Hayoz D, Ruffieux J, Rusconi S, Montani JP, Yang Z. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation 110: 3708–3714, 2004. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- 135.Mizoguchi H, O’Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science 258: 1795–1798, 1992. doi: 10.1126/science.1465616. [DOI] [PubMed] [Google Scholar]
- 136.Mohammed A, Janakiram NB, Madka V, Ritchie RL, Brewer M, Biddick L, Patlolla JM, Sadeghi M, Lightfoot S, Steele VE, Rao CV. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev Res (Phila) 7: 1198–1209, 2014. doi: 10.1158/1940-6207.CAPR-14-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Monin L, Griffiths KL, Lam WY, Gopal R, Kang DD, Ahmed M, Rajamanickam A, Cruz-Lagunas A, Zúñiga J, Babu S, Kolls JK, Mitreva M, Rosa BA, Ramos-Payan R, Morrison TE, Murray PJ, Rangel-Moreno J, Pearce EJ, Khader SA. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J Clin Invest 125: 4699–4713, 2015. doi: 10.1172/JCI77378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, Osborne LC, Hepworth MR, Tran SV, Rodewald HR, Shah H, Cross JR, Diamond JM, Cantu E, Christie JD, Pearce EL, Artis D. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol 17: 656–665, 2016. doi: 10.1038/ni.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morris CR. Alterations of the arginine metabolome in sickle cell disease: a growing rationale for arginine therapy. Hematol Oncol Clin North Am 28: 301–321, 2014. doi: 10.1016/j.hoc.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 140.Morris CR. New strategies for the treatment of pulmonary hypertension in sickle cell disease : the rationale for arginine therapy. Treat Respir Med 5: 31–45, 2006. doi: 10.2165/00151829-200605010-00003. [DOI] [PubMed] [Google Scholar]
- 141.Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol 157: 922–930, 2009. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Morris SM., Jr Regulation of enzymes of the urea cycle and arginine metabolism. Annu Rev Nutr 22: 87–105, 2002. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- 143.Morris SM Jr, Gao T, Cooper TK, Kepka-Lenhart D, Awad AS. Arginase-2 mediates diabetic renal injury. Diabetes 60: 3015–3022, 2011. doi: 10.2337/db11-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S, Wyczechowska D, Reiss K, Rodriguez PC. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia 27: 569–577, 2013. doi: 10.1038/leu.2012.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol 158: 638–651, 2009. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol 160: 5347–5354, 1998. [PubMed] [Google Scholar]
- 147.Munder M, Eichmann K, Morán JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol 163: 3771–3777, 1999. [PubMed] [Google Scholar]
- 148.Murad F. Discovery of some of the biological effects of nitric oxide and its role in cell signaling. Biosci Rep 19: 133–154, 1999. doi: 10.1023/A:1020265417394. [DOI] [PubMed] [Google Scholar]
- 149.Murray PJ. Amino acid auxotrophy as a system of immunological control nodes. Nat Immunol 17: 132–139, 2016. doi: 10.1038/ni.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20, 2014. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Narayanan SP, Suwanpradid J, Saul A, Xu Z, Still A, Caldwell RW, Caldwell RB. Arginase 2 deletion reduces neuro-glial injury and improves retinal function in a model of retinopathy of prematurity. PLoS One 6: e22460, 2011. doi: 10.1371/journal.pone.0022460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Narayanan SP, Xu Z, Putluri N, Sreekumar A, Lemtalsi T, Caldwell RW, Caldwell RB. Arginase 2 deficiency reduces hyperoxia-mediated retinal neurodegeneration through the regulation of polyamine metabolism. Cell Death Dis 5: e1075, 2014. doi: 10.1038/cddis.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Osowska S, Moinard C, Neveux N, Loï C, Cynober L. Citrulline increases arginine pools and restores nitrogen balance after massive intestinal resection. Gut 53: 1781–1786, 2004. doi: 10.1136/gut.2004.042317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315–424, 2007. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Palatini P. Glomerular hyperfiltration: a marker of early renal damage in pre-diabetes and pre-hypertension. Nephrol Dial Transplant 27: 1708–1714, 2012. doi: 10.1093/ndt/gfs037. [DOI] [PubMed] [Google Scholar]
- 156.Pandey D, Bhunia A, Oh YJ, Chang F, Bergman Y, Kim JH, Serbo J, Boronina TN, Cole RN, Van Eyk J, Remaley AT, Berkowitz DE, Romer LH. OxLDL triggers retrograde translocation of arginase2 in aortic endothelial cells via ROCK and mitochondrial processing peptidase. Circ Res 115: 450–459, 2014. doi: 10.1161/CIRCRESAHA.115.304262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pandey D, Sikka G, Bergman Y, Kim JH, Ryoo S, Romer L, Berkowitz D. Transcriptional regulation of endothelial arginase 2 by histone deacetylase 2. Arterioscler Thromb Vasc Biol 34: 1556–1566, 2014. doi: 10.1161/ATVBAHA.114.303685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Patel C, Rojas M, Narayanan SP, Zhang W, Xu Z, Lemtalsi T, Jittiporn K, Caldwell RW, Caldwell RB. Arginase as a mediator of diabetic retinopathy. Front Immunol 4: 173, 2013. doi: 10.3389/fimmu.2013.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Patel C, Xu Z, Shosha E, Xing J, Lucas R, Caldwell RW, Caldwell RB, Narayanan SP. Treatment with polyamine oxidase inhibitor reduces microglial activation and limits vascular injury in ischemic retinopathy. Biochim Biophys Acta 1862: 1628–1639, 2016. doi: 10.1016/j.bbadis.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pegg AE. The function of spermine. IUBMB Life 66: 8–18, 2014. doi: 10.1002/iub.1237. [DOI] [PubMed] [Google Scholar]
- 161.Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res 98: 334–343, 2013. doi: 10.1093/cvr/cvt036. [DOI] [PubMed] [Google Scholar]
- 162.Petrone AB, O’Connell GC, Regier MD, Chantler PD, Simpkins JW, Barr TL. The role of arginase 1 in post-stroke immunosuppression and ischemic stroke severity. Transl Stroke Res 7: 103–110, 2016. doi: 10.1007/s12975-015-0431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pieper GM, Dondlinger LA. Plasma and vascular tissue arginine are decreased in diabetes: acute arginine supplementation restores endothelium-dependent relaxation by augmenting cGMP production. J Pharmacol Exp Ther 283: 684–691, 1997. [PubMed] [Google Scholar]
- 164.Popovic PJ, Zeh HJ III, Ochoa JB. Arginine and immunity. J Nutr 137, Suppl 2: 1681S–1686S, 2007. [DOI] [PubMed] [Google Scholar]
- 165.Potoka KP, Gladwin MT. Vasculopathy and pulmonary hypertension in sickle cell disease. Am J Physiol Lung Cell Mol Physiol 308: L314–L324, 2015. doi: 10.1152/ajplung.00252.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, DeFreitas AA, Shirey KA, Reutterer B, Kernbauer E, Stockinger S, Decker T, Miyairi I, Vogel SN, Salgame P, Rock CO, Murray PJ. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe 12: 313–323, 2012. doi: 10.1016/j.chom.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Quirié A, Demougeot C, Bertrand N, Mossiat C, Garnier P, Marie C, Prigent-Tessier A. Effect of stroke on arginase expression and localization in the rat brain. Eur J Neurosci 37: 1193–1202, 2013. doi: 10.1111/ejn.12111. [DOI] [PubMed] [Google Scholar]
- 168.Quitter F, Figulla HR, Ferrari M, Pernow J, Jung C. Increased arginase levels in heart failure represent a therapeutic target to rescue microvascular perfusion. Clin Hemorheol Microcirc 54: 75–85, 2013. doi: 10.3233/CH-2012-1617. [DOI] [PubMed] [Google Scholar]
- 169.Raber PL, Sierra RA, Thevenot PT, Shuzhong Z, Wyczechowska DD, Kumai T, Celis E, Rodriguez PC. T cells conditioned with MDSC show an increased anti-tumor activity after adoptive T cell based immunotherapy. Oncotarget 7: 17565–17578, 2016. doi: 10.18632/oncotarget.8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 2: 1057–1058, 1987. doi: 10.1016/S0140-6736(87)91481-4. [DOI] [PubMed] [Google Scholar]
- 171.Rodriguez PC, Hernandez CP, Morrow K, Sierra R, Zabaleta J, Wyczechowska DD, Ochoa AC. l-Arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. J Immunol 185: 5198–5204, 2010. doi: 10.4049/jimmunol.1001224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med 202: 931–939, 2005. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rodriguez PC, Quiceno DG, Ochoa AC. l-Arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109: 1568–1573, 2007. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, Ochoa AC. l-Arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol 171: 1232–1239, 2003. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 175.Rojas M, Lemtalsi T, Toque HA, Xu Z, Fulton D, Caldwell RW, Caldwell RB. Nox2-induced activation of arginase and diabetes-induced retinal endothelial cell senescence. Antioxidants (Basel) 6: 43, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Romero MJ, Iddings JA, Platt DH, Ali MI, Cederbaum SD, Stepp DW, Caldwell RB, Caldwell RW. Diabetes-induced vascular dysfunction involves arginase I. Am J Physiol Heart Circ Physiol 302: H159–H166, 2012. doi: 10.1152/ajpheart.00774.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Romero MJ, Platt DH, Caldwell RB, Caldwell RW. Therapeutic use of citrulline in cardiovascular disease. Cardiovasc Drug Rev 24: 275–290, 2006. doi: 10.1111/j.1527-3466.2006.00275.x. [DOI] [PubMed] [Google Scholar]
- 178.Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, Caldwell RB, Caldwell RW. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res 102: 95–102, 2008. doi: 10.1161/CIRCRESAHA.107.155028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Romero MJ, Yao L, Sridhar S, Bhatta A, Dou H, Ramesh G, Brands MW, Pollock DM, Caldwell RB, Cederbaum SD, Head CA, Bagi Z, Lucas R, Caldwell RW. l-Citrulline protects from kidney damage in type 1 diabetic mice. Front Immunol 4: 480, 2013. doi: 10.3389/fimmu.2013.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rosi S, Ferguson R, Fishman K, Allen A, Raber J, Fike JR. The polyamine inhibitor alpha-difluoromethylornithine modulates hippocampus-dependent function after single and combined injuries. PLoS One 7: e31094, 2012. doi: 10.1371/journal.pone.0031094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol 166: 2173–2177, 2001. doi: 10.4049/jimmunol.166.4.2173. [DOI] [PubMed] [Google Scholar]
- 182.Ryoo S, Bhunia A, Chang F, Shoukas A, Berkowitz DE, Romer LH. OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis 214: 279–287, 2011. doi: 10.1016/j.atherosclerosis.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, Lim HK, Sohi J, Santhanam L, Soucy K, Tuday E, Baraban E, Ilies M, Gerstenblith G, Nyhan D, Shoukas A, Christianson DW, Alp NJ, Champion HC, Huso D, Berkowitz DE. Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res 102: 923–932, 2008. doi: 10.1161/CIRCRESAHA.107.169573. [DOI] [PubMed] [Google Scholar]
- 184.Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D, Shoukas A, Romer LH, Berkowitz DE. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res 99: 951–960, 2006. doi: 10.1161/01.RES.0000247034.24662.b4. [DOI] [PubMed] [Google Scholar]
- 185.Sakai RE, Feller DJ, Galetta KM, Galetta SL, Balcer LJ. Vision in multiple sclerosis: the story, structure-function correlations, and models for neuroprotection. J Neuroophthalmol 31: 362–373, 2011. doi: 10.1097/WNO.0b013e318238937f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, Balanson S, Ryoo S, Anderson M, Irani K, Khanday F, Di Costanzo L, Nyhan D, Hare JM, Christianson DW, Rivers R, Shoukas A, Berkowitz DE. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res 101: 692–702, 2007. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- 187.Satriano J. Arginine pathways and the inflammatory response: interregulation of nitric oxide and polyamines: review article. Amino Acids 26: 321–329, 2004. doi: 10.1007/s00726-004-0078-4. [DOI] [PubMed] [Google Scholar]
- 188.Scalera F, Closs EI, Flick E, Martens-Lobenhoffer J, Boissel JP, Lendeckel U, Heimburg A, Bode-Böger SM. Paradoxical effect of L-arginine: acceleration of endothelial cell senescence. Biochem Biophys Res Commun 386: 650–655, 2009. doi: 10.1016/j.bbrc.2009.06.091. [DOI] [PubMed] [Google Scholar]
- 189.Schneemann M, Schoeden G. Macrophage biology and immunology: man is not a mouse. J Leukoc Biol 81: 579, 2007. doi: 10.1189/jlb.1106702. [DOI] [PubMed] [Google Scholar]
- 190.Segal R, Hannan JL, Liu X, Kutlu O, Burnett AL, Champion HC, Kim JH, Steppan J, Berkowitz DE, Bivalacqua TJ. Chronic oral administration of the arginase inhibitor 2(S)-amino-6-boronohexanoic acid (ABH) improves erectile function in aged rats. J Androl 33: 1169–1175, 2012. doi: 10.2164/jandrol.111.015834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Seiler N. Oxidation of polyamines and brain injury. Neurochem Res 25: 471–490, 2000. doi: 10.1023/A:1007508008731. [DOI] [PubMed] [Google Scholar]
- 192.Sennlaub F, Courtois Y, Goureau O. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J Clin Invest 107: 717–725, 2001. doi: 10.1172/JCI10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 203: 2691–2702, 2006. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shatanawi A, Lemtalsi T, Yao L, Patel C, Caldwell RB, Caldwell RW. Angiotensin II limits NO production by upregulating arginase through a p38 MAPK-ATF-2 pathway. Eur J Pharmacol 746: 106–114, 2015. doi: 10.1016/j.ejphar.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD, Caldwell RB, Caldwell RW. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol 300: C1181–C1192, 2011. doi: 10.1152/ajpcell.00328.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shearer JD, Richards JR, Mills CD, Caldwell MD. Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am J Physiol Endocrinol Metab 272: E181–E190, 1997. [DOI] [PubMed] [Google Scholar]
- 197.Sheldon KE, Shandilya H, Kepka-Lenhart D, Poljakovic M, Ghosh A, Morris SM Jr. Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. J Immunol 191: 2290–2298, 2013. doi: 10.4049/jimmunol.1202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shemyakin A, Kövamees O, Rafnsson A, Böhm F, Svenarud P, Settergren M, Jung C, Pernow J. Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation 126: 2943–2950, 2012. doi: 10.1161/CIRCULATIONAHA.112.140335. [DOI] [PubMed] [Google Scholar]
- 199.Shi O, Morris SM Jr, Zoghbi H, Porter CW, O’Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol Cell Biol 21: 811–813, 2001. doi: 10.1128/MCB.21.3.811-813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shin S, Mohan S, Fung HL. Intracellular l-arginine concentration does not determine NO production in endothelial cells: implications on the “l-arginine paradox”. Biochem Biophys Res Commun 414: 660–663, 2011. doi: 10.1016/j.bbrc.2011.09.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shosha E, Xu Z, Yokota H, Saul A, Rojas M, Caldwell RW, Caldwell RB, Narayanan SP. Arginase 2 promotes neurovascular degeneration during ischemia/reperfusion injury. Cell Death Dis 7: e2483, 2016. doi: 10.1038/cddis.2016.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, Hailemichael Y, Jayaraman P, Myers JN, Grimm EA, Overwijk WW. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin Cancer Res 16: 1834–1844, 2010. doi: 10.1158/1078-0432.CCR-09-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol 16: 599–611, 2016. doi: 10.1038/nri.2016.80. [DOI] [PubMed] [Google Scholar]
- 204.Steppan J, Nyhan D, Berkowitz DE. Development of novel arginase inhibitors for therapy of endothelial dysfunction. Front Immunol 4: 278, 2013. doi: 10.3389/fimmu.2013.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Steppan J, Tran HT, Bead VR, Oh YJ, Sikka G, Bivalacqua TJ, Burnett AL, Berkowitz DE, Santhanam L. Arginase inhibition reverses endothelial dysfunction, pulmonary hypertension, and vascular stiffness in transgenic sickle cell mice. Anesth Analg 123: 652–658, 2016. doi: 10.1213/ANE.0000000000001378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Stevenson L, Matesanz N, Colhoun L, Edgar K, Devine A, Gardiner TA, McDonald DM. Reduced nitro-oxidative stress and neural cell death suggests a protective role for microglial cells in TNFalpha−/− mice in ischemic retinopathy. Invest Ophthalmol Vis Sci 51: 3291–3299, 2010. doi: 10.1167/iovs.09-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Suwanpradid J, Rojas M, Behzadian MA, Caldwell RW, Caldwell RB. Arginase 2 deficiency prevents oxidative stress and limits hyperoxia-induced retinal vascular degeneration. PLoS One 9: e110604, 2014. doi: 10.1371/journal.pone.0110604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Taheri F, Ochoa JB, Faghiri Z, Culotta K, Park HJ, Lan MS, Zea AH, Ochoa AC. l-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin Cancer Res 7, Suppl: 958s–965s, 2001. [PubMed] [Google Scholar]
- 209.Takano K, Ogura M, Nakamura Y, Yoneda Y. Neuronal and glial responses to polyamines in the ischemic brain. Curr Neurovasc Res 2: 213–223, 2005. doi: 10.2174/1567202054368335. [DOI] [PubMed] [Google Scholar]
- 210.Tawfik HE, El-Remessy AB, Matragoon S, Ma G, Caldwell RB, Caldwell RW. Simvastatin improves diabetes-induced coronary endothelial dysfunction. J Pharmacol Exp Ther 319: 386–395, 2006. doi: 10.1124/jpet.106.106823. [DOI] [PubMed] [Google Scholar]
- 211.Thomas AC, Mattila JT. “Of mice and men”: arginine metabolism in macrophages. Front Immunol 5: 479, 2014. doi: 10.3389/fimmu.2014.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev 61: 62–97, 2009. doi: 10.1124/pr.108.000547. [DOI] [PubMed] [Google Scholar]
- 213.Toque HA, Caldwell RW. New approaches to the design and discovery of therapies to prevent erectile dysfunction. Expert Opin Drug Discov 9: 1447–1469, 2014. doi: 10.1517/17460441.2014.949234. [DOI] [PubMed] [Google Scholar]
- 214.Toque HA, Nunes KP, Rojas M, Bhatta A, Yao L, Xu Z, Romero MJ, Webb RC, Caldwell RB, Caldwell RW. Arginase 1 mediates increased blood pressure and contributes to vascular endothelial dysfunction in deoxycorticosterone acetate-salt hypertension. Front Immunol 4: 219, 2013. doi: 10.3389/fimmu.2013.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Toque HA, Nunes KP, Yao L, Liao JK, Webb RC, Caldwell RB, Caldwell RW. Activated Rho kinase mediates diabetes-induced elevation of vascular arginase activation and contributes to impaired corpora cavernosa relaxation: possible involvement of p38 MAPK activation. J Sex Med 10: 1502–1515, 2013. doi: 10.1111/jsm.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Toque HA, Tostes RC, Yao L, Xu Z, Webb RC, Caldwell RB, Caldwell RW. Arginase II deletion increases corpora cavernosa relaxation in diabetic mice. J Sex Med 8: 722–733, 2011. doi: 10.1111/j.1743-6109.2010.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tratsiakovich Y, Yang J, Gonon AT, Sjöquist PO, Pernow J. Arginase as a target for treatment of myocardial ischemia-reperfusion injury. Eur J Pharmacol 720: 121–123, 2013. doi: 10.1016/j.ejphar.2013.10.040. [DOI] [PubMed] [Google Scholar]
- 218.Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 10: 464, 2010. doi: 10.1186/1471-2407-10-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Villalba N, Sackheim AM, Nunez IA, Hill-Eubanks DC, Nelson MT, Wellman GC, Freeman K. Traumatic brain injury causes endothelial dysfunction in the systemic microcirculation through arginase-1-dependent uncoupling of endothelial nitric oxide synthase. J Neurotrauma 34: 192–203, 2017. doi: 10.1089/neu.2015.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Vockley JG, Jenkinson CP, Shukla H, Kern RM, Grody WW, Cederbaum SD. Cloning and characterization of the human type II arginase gene. Genomics 38: 118–123, 1996. doi: 10.1006/geno.1996.0606. [DOI] [PubMed] [Google Scholar]
- 221.Vodovotz Y, Bogdan C, Paik J, Xie QW, Nathan C. Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor beta. J Exp Med 178: 605–613, 1993. doi: 10.1084/jem.178.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Walter SD, Ishikawa H, Galetta KM, Sakai RE, Feller DJ, Henderson SB, Wilson JA, Maguire MG, Galetta SL, Frohman E, Calabresi PA, Schuman JS, Balcer LJ. Ganglion cell loss in relation to visual disability in multiple sclerosis. Ophthalmology 119: 1250–1257, 2012. doi: 10.1016/j.ophtha.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wang L, Bhatta A, Toque HA, Rojas M, Yao L, Xu Z, Patel C, Caldwell RB, Caldwell RW. Arginase inhibition enhances angiogenesis in endothelial cells exposed to hypoxia. Microvasc Res 98: 1–8, 2015. doi: 10.1016/j.mvr.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Watanabe A, Sasaki T, Yukami T, Kanki H, Sakaguchi M, Takemori H, Kitagawa K, Mochizuki H. Serine racemase inhibition induces nitric oxide-mediated neurovascular protection during cerebral ischemia. Neuroscience 339: 139–149, 2016. doi: 10.1016/j.neuroscience.2016.09.036. [DOI] [PubMed] [Google Scholar]
- 225.Watts JA, Gellar MA, Fulkerson MB, Das SK, Kline JA. Arginase depletes plasma l-arginine and decreases pulmonary vascular reserve during experimental pulmonary embolism. Pulm Pharmacol Ther 25: 48–54, 2012. doi: 10.1016/j.pupt.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 226.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.White AR, Ryoo S, Li D, Champion HC, Steppan J, Wang D, Nyhan D, Shoukas AA, Hare JM, Berkowitz DE. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension 47: 245–251, 2006. doi: 10.1161/01.HYP.0000198543.34502.d7. [DOI] [PubMed] [Google Scholar]
- 228.Wood PL, Khan MA, Kulow SR, Mahmood SA, Moskal JR. Neurotoxicity of reactive aldehydes: the concept of “aldehyde load” as demonstrated by neuroprotection with hydroxylamines. Brain Res 1095: 190–199, 2006. doi: 10.1016/j.brainres.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 229.Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J 336: 1–17, 1998. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Xia N, Horke S, Habermeier A, Closs EI, Reifenberg G, Gericke A, Mikhed Y, Münzel T, Daiber A, Förstermann U, Li H. Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet-induced obese mice. Arterioscler Thromb Vasc Biol 36: 78–85, 2016. doi: 10.1161/ATVBAHA.115.306263. [DOI] [PubMed] [Google Scholar]
- 231.Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269: 4705–4708, 1994. [PubMed] [Google Scholar]
- 232.Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med 177: 1779–1784, 1993. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Xiong Y, Yu Y, Montani JP, Yang Z, Ming XF. Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l-arginine ureahydrolase activity: implications for atherosclerotic plaque vulnerability. J Am Heart Assoc 2: e000096, 2013. doi: 10.1161/JAHA.113.000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 18: 1746–1748, 2004. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 235.Xue J, Nelin LD, Chen B. Hypoxia induces arginase II expression and increases viable human pulmonary artery smooth muscle cell numbers via AMPKα1 signaling. Am J Physiol Lung Cell Mol Physiol 312: L568–L578, 2017. doi: 10.1152/ajplung.00117.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yang M, Rangasamy D, Matthaei KI, Frew AJ, Zimmmermann N, Mahalingam S, Webb DC, Tremethick DJ, Thompson PJ, Hogan SP, Rothenberg ME, Cowden WB, Foster PS. Inhibition of arginase I activity by RNA interference attenuates IL-13-induced airways hyperresponsiveness. J Immunol 177: 5595–5603, 2006. doi: 10.4049/jimmunol.177.8.5595. [DOI] [PubMed] [Google Scholar]
- 237.Yang Q, Zheng C, Cao J, Cao G, Shou P, Lin L, Velletri T, Jiang M, Chen Q, Han Y, Li F, Wang Y, Cao W, Shi Y. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ 23: 1850–1861, 2016. doi: 10.1038/cdd.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yang Z, Ming XF. Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front Immunol 5: 533, 2014. doi: 10.3389/fimmu.2014.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Yao L, Bhatta A, Xu Z, Chen J, Toque HA, Chen Y, Xu Y, Bagi Z, Lucas R, Huo Y, Caldwell RB, Caldwell RW. Obesity-induced vascular inflammation involves elevated arginase activity. Am J Physiol Regul Integr Comp Physiol 313: R560–R571, 2017. doi: 10.1152/ajpregu.00529.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, Pang R, Fan ST, Poon RT. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs 31: 99–107, 2013. doi: 10.1007/s10637-012-9807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ye C, Geng Z, Dominguez D, Chen S, Fan J, Qin L, Long A, Zhang Y, Kuzel TM, Zhang B. Targeting ornithine decarboxylase by alpha-difluoromethylornithine inhibits tumor growth by impairing myeloid-derived suppressor cells. J Immunol 196: 915–923, 2016. doi: 10.4049/jimmunol.1500729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 357: 1121–1135, 2007. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 243.You H, Gao T, Cooper TK, Morris SM Jr, Awad AS. Arginase inhibition mediates renal tissue protection in diabetic nephropathy by a nitric oxide synthase 3-dependent mechanism. Kidney Int 84: 1189–1197, 2013. doi: 10.1038/ki.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.You H, Gao T, Cooper TK, Morris SM Jr, Awad AS. Arginase inhibition: a new treatment for preventing progression of established diabetic nephropathy. Am J Physiol Renal Physiol 309: F447–F455, 2015. doi: 10.1152/ajprenal.00137.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.You H, Gao T, Cooper TK, Morris SM Jr, Awad AS. Diabetic nephropathy is resistant to oral l-arginine or l-citrulline supplementation. Am J Physiol Renal Physiol 307: F1292–F1301, 2014. doi: 10.1152/ajprenal.00176.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yu H, Iyer RK, Kern RM, Rodriguez WI, Grody WW, Cederbaum SD. Expression of arginase isozymes in mouse brain. J Neurosci Res 66: 406–422, 2001. doi: 10.1002/jnr.1233. [DOI] [PubMed] [Google Scholar]
- 247.Zahedi K, Huttinger F, Morrison R, Murray-Stewart T, Casero RA Jr, Strauss KI. Polyamine catabolism is enhanced after traumatic brain injury. J Neurotrauma 27: 515–525, 2010. doi: 10.1089/neu.2009.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zani BG, Bohlen HG. Transport of extracellular l-arginine via cationic amino acid transporter is required during in vivo endothelial nitric oxide production. Am J Physiol Heart Circ Physiol 289: H1381–H1390, 2005. doi: 10.1152/ajpheart.01231.2004. [DOI] [PubMed] [Google Scholar]
- 249.Zea AH, Ochoa MT, Ghosh P, Longo DL, Alvord WG, Valderrama L, Falabella R, Harvey LK, Saravia N, Moreno LH, Ochoa AC. Changes in expression of signal transduction proteins in T lymphocytes of patients with leprosy. Infect Immun 66: 499–504, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, Park HJ, Zabaleta J, Ochoa AC. l-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol 232: 21–31, 2004. doi: 10.1016/j.cellimm.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 251.Zhang C, Hein TW, Wang W, Miller MW, Fossum TW, McDonald MM, Humphrey JD, Kuo L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension 44: 935–943, 2004. doi: 10.1161/01.HYP.0000146907.82869.f2. [DOI] [PubMed] [Google Scholar]
- 252.Zhang W, Baban B, Rojas M, Tofigh S, Virmani SK, Patel C, Behzadian MA, Romero MJ, Caldwell RW, Caldwell RB. Arginase activity mediates retinal inflammation in endotoxin-induced uveitis. Am J Pathol 175: 891–902, 2009. doi: 10.2353/ajpath.2009.081115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhu M, Goetsch SC, Wang Z, Luo R, Hill JA, Schneider J, Morris SM Jr, Liu ZP. FoxO4 promotes early inflammatory response upon myocardial infarction via endothelial Arg1. Circ Res 117: 967–977, 2015. doi: 10.1161/CIRCRESAHA.115.306919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zhu MY, Iyo A, Piletz JE, Regunathan S. Expression of human arginine decarboxylase, the biosynthetic enzyme for agmatine. Biochim Biophys Acta 1670: 156–164, 2004. doi: 10.1016/j.bbagen.2003.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]