

Abstract
Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are key regulators of cell proliferation and organ size; however, their physiological contribution after liver injury has not been fully understood. In this study, we sought to determine the role of YAP and TAZ during liver recovery after ischemia-reperfusion (I/R). A murine model of partial (70%) I/R was used to induce liver injury and study the reparative and regenerative response. After liver injury, there was marked activation and proliferation of hepatic stellate cells. The Hippo pathway components, large tumor suppressor 1 (LATS1) and its adapter protein, Mps one binder 1 (MOB1), were inactivated during liver repair, and YAP and TAZ were activated selectively in hepatic stellate cells. Concurrently, the expression of connective tissue growth factor and survivin, both of which are YAP and TAZ target genes, were upregulated. Hepatic stellate cell expansion and concomitant activation of YAP and TAZ occurred only in the injured liver and were not observed in the nonischemic liver. Treatment of mice with verteporfin, an inhibitor of YAP and TAZ, decreased hepatic stellate cell proliferation, survivin, and cardiac ankyrin repeat protein expression. These changes were associated with a significant decrease in hepatocyte proliferation. The data suggest that liver repair and regeneration after I/R injury are dependent on hepatic stellate cell proliferation, which is mediated by YAP and TAZ.
NEW & NOTEWORTHY This study is the first to assess the proliferation of hepatic stellate cells (HSCs) after ischemia-reperfusion (I/R) injury and their role in the reparative and regenerative process. Here we show that the Hippo pathway is inactivated after I/R and that Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) activation is detected in HSC. HSC proliferation and expansion are prominent during liver recovery after I/R injury. Inhibition of YAP/TAZ activation with verteporfin reduces HSC proliferation and target gene expression and attenuates hepatocyte proliferation.
Keywords: hepatic stellate cell proliferation, liver injury, liver regeneration, survivin
INTRODUCTION
Hepatic ischemia-reperfusion (I/R) injury is a major complication after liver resection, liver transplantation, and trauma (14, 21). Hepatic I/R leads to an acute inflammatory response, including neutrophil infiltration followed by hepatocellular damage (22). Severe hepatic injury and insufficient liver regeneration after I/R cause liver dysfunction, resulting in high morbidity and mortality rate. In experimental models of liver I/R injury, peak hepatocellular injury usually occurs within 24 h after reperfusion. Shortly thereafter, the process of liver repair and regeneration begins and involves a complex array of signaling events and cellular remodeling (5, 18, 33). However, the precise cellular and molecular mechanisms of liver regeneration after I/R injury have not been well studied.
The Hippo signaling pathway and its downstream effectors, Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), have been identified as key regulators of cell proliferation and organ size (40). The pathway responds to a wide variety of upstream signals that dictate its activation state (39). The core component of the mammalian Hippo pathway is a kinase cascade in which mammalian Ste20-like kinases 1/2 (MST1/2) phosphorylates and activates large tumor suppressor 1/2 (LATS1/2) and its adapter protein, Mps one binder 1 (MOB1) (19, 25, 34). LATS1/2 then phosphorylates the transcriptional coactivators, YAP and TAZ, downregulating their function by increasing cytoplasmic retention in a 14-3-3-mediated manner as well as increasing their degradation by the proteasome (6, 16, 27). Nonphosphorylated YAP and TAZ can localize in the nucleus when Hippo pathway activation is reduced, resulting in binding with transcription factors such as TEAD family proteins. YAP/TAZ-induced genes are related to cell proliferation, antiapoptosis, and amplification of progenitor/stem cells (40, 41). In liver pathology, inhibition of YAP prevented hepatic stellate cell (HSC) activation in vitro, and pharmacological inhibition of YAP reduced liver fibrosis after carbon tetrachloride-induced chronic liver injury (26).
Nonparenchymal cells, including HSC, play important roles in liver regeneration. HSCs reside in the space of Disse between sinusoids and hepatocytes. In response to liver injury, quiescent HSCs are stimulated by reactive oxygen species, proinflammatory cytokines secreted from Kupffer cells, and apoptotic hepatocytes and transdifferentiate into myofibroblasts (7, 17). In models of hepatectomy, myofibroblasts contribute to hepatocellular regeneration through the release of mitogens (43). In the early period after I/R, HSCs amplify acute liver injury by increasing proinflammatory mediators (35); however, the role of HSCs in liver recovery and regeneration after I/R is unknown. Hippo pathway signaling has been linked to the activation of HSCs (26). In the present study, we sought to determine whether YAP and TAZ regulate HSC biology after acute liver injury and how HSCs impact liver repair and regeneration following I/R injury.
MATERIALS AND METHODS
Animals.
Male Balb/c mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice used for these experiments were 8–12 wk of age. This project was approved by the University of Cincinnati Animal Care and Use Committee and was in compliance with the National Institutes of Health guidelines.
Hepatic I/R injury model.
Partial hepatic ischemia was performed as described previously (23). Briefly, mice were anesthetized with sodium pentobarbital (60 mg/kg ip). A midline laparotomy was performed, and an atraumatic clip was used to interrupt blood supply to the left and median lobes of the liver. After 90 min of partial hepatic ischemia, the clip was removed to initiate hepatic reperfusion. Sham mice did not undergo this procedure. Mice were killed after the indicated periods of reperfusion, and blood and liver samples were taken for analysis. The left lobe was used as ischemic liver, and the right robe was used as nonischemic liver for histological analysis. In some experiments, mice were injected intraperitoneally with 10% dimethylsulfoxide solution (control) or 100 mg/kg verteporfin after 24 h of reperfusion and every 24 h thereafter.
Tissue analyses.
Liver tissues were fixed in 10% neutral-buffered formalin, processed, and then embedded in paraffin for light microscopy. Sections were stained with hematoxylin and eosin (H and E) for histological examination and processed further for immunostaining. Immunohistochemical and immunofluorescent staining was performed as described previously (18). The antibodies were as follows: PCNA (sc-56; Santa Cruz Biotechnology, Dallas, TX), desmin (ab15200; Abcam, Cambridge, MA), F4/80 (ab6640, Abcam), α-smooth muscle actin (α-SMA) (ab5694, Abcam), YAP (no. 14074; Cell Signaling Technology, Danvers, MA), TAZ (HPA007415; Sigma-Aldrich, St. Louis, MO), CK19 (TROMA-III; DSHB, Iowa City, IO), and CK8/18 (ab 53280; Abcam). Evaluation of hepatocyte PCNA immunostaining was performed based on the percentage of positive nuclei of 400–600 hepatocytes from the five highest positive fields at high power (×400) and was expressed as hepatocyte PCNA labeling index. Evaluation of HSC PCNA immunostaining was performed by dual immunofluorescent staining based on the percentage of positive nuclei of desmin-staining cells from the eight highest positive fields at high power (×400) and was expressed as HSC PCNA labeling index. Quantitative morphometric analysis of immunostainings was performed with histologic sections at low power (×50) using ImageJ. Desmin and α-SMA staining areas were expressed as percentages of total area examined as described previously (8).
Liver content of platelet-derived growth factor-BB (PDGF-BB), transforming growth factor-β1 (TGF-β1), hepatocyte growth factor (HGF), and amphiregulin was assessed by ELISA (R&D Systems, Minneapolis, MN). Liver samples were weighed and immediately placed in 10 volumes (wt/vol) of homogenization buffer (protease inhibitor cocktail containing 10 nmol/l ethylenediaminetetraacetic acid, 2 mmol/l phenylmethylsulfonyl fluoride, 0.1 mg/ml soybean trypsin inhibitor, 1.0 mg/ml bovine serum albumin, and 0.002% sodium azide in isotonic phosphate-buffered saline, pH 7.0). Tissues were disrupted with a tissue homogenizer, and lysates were incubated at 4°C for 2 h. Samples were clarified by two rounds of centrifugation at 12,500 g for 10 min at 4°C.
Hepatocyte and hepatic stellate cell isolation.
Hepatocytes were isolated as previously described (32). Livers were perfused in situ with 45 ml liver perfusion media (Thermo Fisher Scientific, Florence, KY) followed by 45 ml of liver digestion media (Thermo Fisher Scientific). The liver was excised, minced, and strained through a steel mesh. The dispersed hepatocytes were collected by centrifugation at 50 g for 2 min at 4°C and washed twice with Williams media (Thermo Fisher Scientific). Hepatocytes were isolated via Percoll separation and washed with Williams media. HSCs were isolated as previously described (30). Livers were perfused and digested in situ with Hank’s balanced salt solution containing collagenase and protease. The liver was excised, minced, and strained through a steel mesh. The resulting cell suspension was centrifuged four times at 65 g for 2 min to remove hepatocytes and cellular debris. The supernatant was then centrifuged at 650 g for 10 min, and HSCs were clarified from the resulting pellet via Histodenz gradient centrifugation. Cytoplasm fractions and nuclear extracts were prepared from isolated hepatocytes and HSCs by using Nuclear Extraction Kit (Abcam). In in vitro study, primary HSCs were cultured in Dulbecco’s modified eagle medium containing 10% FBS, 10% horse serum, 100 mg/ml penicillin, and 100 U/ml streptomycin. HSCs were treated with or without verteporfin. Cell culture medium was changed every other day. HSC proliferation was determined by DNA incorporation of 5-bromo-2′deoxyuridine (BrdU) according to the manufacturer’s instructions (Roche, Mannheim, Germany). Cytotoxicity was determined by lactate dehydrogenase release according to the manufacturer’s instructions (Roche), and the percentage of cell death was calculated.
RT-PCR analysis.
Total RNA was extracted from liver tissue using Trizol (Invitrogen, Carlsbad, CA). cDNA was generated through the reverse transcription of 2 μg of RNA using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA). Samples were incubated at 25°C for 10 min, 37°C for 120 min, 85°C for 5 min to inactive the reverse transcriptase, and then cooled at 5°C for 5 min. Four microliters of diluted cDNA samples were used for quantitative two-step PCR using SYBR Green PCR Master Mix (Applied Biosystems). Each sample was analyzed in duplicate. 18S was used as a housekeeping control gene. Threshold cycles were automatically calculated by the iCycler iQ Real-Time Detection System. Threshold cycle values were normalized to the housekeeping control (18S) to give relative genomic equivalence. Primers utilized were as follows: αSMA, sense AAACAGGAATACGACGAAG, antisense CAGGAATGATTTGGAAAGGA; 18S sense AGTCCCTGCCCTTTGTACACA, antisense GATCCGAGGGCCTCAAAC.
Western blot analyses.
Liver samples were homogenized in lysis buffer (10.0 mM HEPES, pH 7.9, 150.0 mM NaCl, 1.0 mM EDTA, 0.6% NP-40, 0.5 mM PMSF, 1.0 μg/ml leupeptin, 1.0 μg/ml aprotonin, 10.0 μg/ml soybean trypsin inhibitor, 1.0 μg/ml pepstatin, and 1.0% phosphatase inhibitor cocktail 2 from Sigma-Aldrich) on ice. Homogenates were centrifuged at 10,000 revolutions/min to remove cellular debris. Protein concentrations of each sample were determined using BCA protein assay kit (Pierce/ThermoFisher, Rockford, IL). Samples containing equal amounts of protein were resuspended in 5× SDS sample buffer, separated in a denaturing 7.5–12.5% polyacrylamide gel, and transferred to a 0.1 μm pore polyvinylidene difluoride membrane. Nonspecific binding sites were blocked with Tris-buffered saline (40.0 mM Tris, pH 7.6, 300.0 mM NaCl) with 0.1% Tween 20 containing 5% nonfat dry milk or 5% bovine serum albumin for 1 h at room temperature. Membranes were then incubated with antibodies to YAP (no. 14074; Cell Signaling Technology), phospho-YAP (Ser127, no. 13008; Cell Signaling Technology), TAZ (HPA007415; Sigma-Aldrich), phospho-TAZ (Ser89, sc-17610; Santa Cruz Biotechnology), phospho-LATS1 (Thr1079, bs-7913R; Bioss Antibodies, Woburn, MA), phospho-MOB1 (Thr35, no. 8699; Cell Signaling Technology), connective tissue growth factor (CTGF, sc-365970; Santa Cruz Biotechnology), survivin (sc-17779; Santa Cruz Biotechnology), Ankrd1 (bs-8074R; Bioss Antibodies), and β-actin (ab8227; Abcam) in Tris-buffered saline with 0.1% Tween 20. Membranes were washed and incubated with secondary antibodies conjugated to horseradish peroxidase. Immunoreactive proteins were detected by enhanced chemiluminescence.
Statistical Analysis.
All data are expressed as the means ± SE. Data were analyzed with a one-way ANOVA with subsequent Student’s t-test. Differences were considered significant when P < 0.05.
RESULTS
Liver repair and regeneration after I/R injury.
To determine the temporal and spatial patterns of nonparenchymal cells after I/R injury, histological analyses of the postischemic liver were performed (Fig. 1). As expected, mice subjected to I/R injury developed massive necrosis with infiltration of neutrophils. The liver had widespread damage, which destroyed zonal structure, and functional liver parenchyma was observed at the edge of the liver and in perivascular areas 24 h after I/R. Viable liver tissue progressively increased after that time point, with more functional liver mass observed at each of the 48-, 96-, and 168-h time points. To assess hepatocyte proliferation over this time course, sections were stained for PCNA (Fig. 1). As expected, increases in hepatocyte proliferation were observed 48 h after reperfusion and remained increased at 48-, 96-, and 168-h time points. To assess HSCs and Kupffer cells/macrophages, liver sections were stained for desmin and F4/80, respectively (Fig. 1). HSCs and macrophages shared a similar pattern of distribution with increased numbers of HSCs (desmin-positive cells) and macrophages (F4/80-positive cells) in periportal areas beginning 48 h after reperfusion. Subsequently, HSCs proliferated and were present in increasing numbers at the interface between viable liver parenchyma and necrotic areas. Interestingly, PCNA-positive hepatocytes were located prominently in the parenchyma, which was adjacent to HSCs and macrophages.
Fig. 1.

Temporal course of liver repair and regeneration after ischemia-reperfusion (I/R) injury. Hematoxylin and eosin (H and E) staining shows massive liver injury after 24 h of reperfusion with repair and restoration of nearly normal liver architecture 168 h after reperfusion. Hepatocyte proliferation was determined by immunohistochemical staining for proliferating cell nuclear antigen (PCNA) and shows robust proliferation of hepatocytes 48 and 96 h after reperfusion that tapers off at 168 h of reperfusion. Hepatic stellate cells (HSCs) and macrophages were detected by desmin and F4/80 staining, respectively. Proliferating hepatocytes were located prominently in the parenchyma adjacent to HSCs and macrophages. Original magnification is ×100 for H and E and ×200 for PCNA, desmin, and F4/80.
Because of the marked HSC expansion observed after I/R, we next measured the levels of PDGF-BB and TGF-β1 in the postischemic liver. PDGF-BB is a potent mitogen for HSCs (11), and TGF-β1 is a potent mediator of HSC activation (10). Both PDGF-BB and TGF-β1 increased 24 h after reperfusion (Fig. 2), before we observed increased numbers of HSC (Fig. 1).
Fig. 2.

Expression of liver platelet-derived growth factor-BB (PDGF-BB) and transforming growth factor-β (TGF-β) after ischemia-reperfusion (I/R) injury. Liver PDGF-BB and TGF-β protein levels were measured by ELISA. PDGF-BB and TGF-β levels increased during I/R liver injury and remained elevated during liver repair and regeneration. Data are means ± SE with n = 3–7 per group. *P < 0.05 compared with sham mice.
HSC proliferation and activation after I/R.
Macrophage recruitment after hepatic I/R has been previously described (4, 15); however, proliferation of HSCs after I/R injury and their role in the reparative and regenerative response have not been studied. Therefore, we investigated the proliferation and activation of HSCs after I/R injury during liver repair and regeneration. To analyze HSC proliferation, dual immunofluorescence staining for desmin and PCNA was performed (Fig. 3A). HSCs started to proliferate 24 h after reperfusion in periportal areas. HSC proliferation peaked at 48 h after reperfusion. After 96 h of reperfusion, proliferation decreased but remained significantly higher than sham controls (Fig. 3A). The same pattern of HSC proliferation after I/R was observed when desmin and PCNA were immunohistochemically stained in serial sections of liver (Fig. 3B). Dual immunofluorescence staining of desmin and E-cadherin showed no colocalization, suggesting no contribution of hepatoblasts (data not shown). In accordance with proliferation, morphometric analysis demonstrated that the number of desmin-positive cells significantly expanded within 96 h of reperfusion (Fig. 3B). Because α-SMA is a marker of activation of HSCs and myofibroblasts, we evaluated α-SMA expression in the ischemic liver. Expression of α-SMA mRNA was significantly (P < 0.05) upregulated after 96 h of reperfusion (fold α-SMA expression: sham, 1.00 ± 0.21; I/R 96 h, 5.60 ± 1.34). Immunostaining showed that α-SMA-positive cells appeared in periportal areas at 48 h, and they markedly increased at the interface between liver parenchyma and necrotic tissue at 96 h after reperfusion (Fig. 3C). The staining pattern for α-SMA was identical to that of desmin (Fig. 3B).
Fig. 3.

Proliferation and activation of hepatic stellate cells (HSCs) after ischemia-reperfusion (I/R). HSC proliferation was assessed by 2 independent techniques (A and B) over a time course of I/R. Both demonstrate the same temporal pattern of HSC proliferation after I/R. A: HSC proliferation was determined by dual immunofluorescence staining for desmin (red) and proliferating cell nuclear antigen (PCNA, green). DAPI was used for nuclear staining. HSCs positive for PCNA are marked by arrows. Quantitative analysis of PCNA labeling was calculated as a percentage of PCNA-positive HSCs. Original magnification is ×630. B: HSC proliferation was determined by immunohistochemistry for desmin and PCNA in serial sections. Desmin-labeled area was measured as a percentage of total area. Original magnification is ×200. Representative HSCs are marked by arrows (red: PCNA positive, black: PCNA negative). Quantitative analysis of PCNA labeling was calculated as a percentage of PCNA-positive HSCs. Original magnification is ×630. C: activated HSCs were assessed by immunohistochemical staining for α-smooth muscle actin (α-SMA). Stellate cell activation increased during after I/R. α-SMA-labeled area was measured as a percentage of total area. Original magnification is ×200. For all graphs, data are means ± SE with n = 3–4. *P < 0.05 compared with sham mice.
Activation of YAP and TAZ in HSCs in the postischemic liver.
To examine whether YAP and TAZ contribute to liver cell proliferation after I/R injury, we measured YAP and TAZ expression by immunohistochemistry. In quiescent liver, YAP and TAZ were expressed in biliary epithelial cells, whereas hepatocytes showed low levels of expression (Fig. 4A). At 96 h after reperfusion in the postischemic liver, there was almost no change in YAP or TAZ expression in the nuclei of hepatocytes; however, we did observe nuclear staining in nonparenchymal cells (Fig. 4A). We then stained serial sections for YAP or TAZ with desmin, CK19, and CK8/18 to determine cell specificity. We observed a high level of nuclear staining of YAP and TAZ in desmin-positive cells (Fig. 4B), suggesting that YAP and TAZ were activated in HSCs after I/R. We also found that some of the CK19-positive reactive ductular cells and biliary epithelial cells have YAP/TAZ nuclear staining, whereas CK8/18-positive hepatocytes have fewer YAP- or TAZ-positive nuclei (Fig. 4B). In addition, we found that YAP and TAZ localized in the same HSC nuclei (Fig. 4C). We also confirmed the cell-specific YAP and TAZ expression by Western blot. Isolated hepatocytes and HSCs from mice were separated into cytoplasmic and nuclear fractions. In hepatocytes, I/R resulted in no changes in cytoplasmic expression of YAP, TAZ, phospho-YAP, or phospho-TAZ (Fig. 4D). Furthermore, there was no evidence that nuclear YAP and TAZ expression increased after I/R. In fact, nuclear YAP expression actually decreased in hepatocytes after I/R (Fig. 4D). In contrast, in HSC nuclear expression of YAP and TAZ increased markedly after I/R (Fig. 4D).
Fig. 4.

Cell-specific Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) activation in postischemic liver after ischemia-reperfusion (I/R). A: YAP and TAZ nuclear expression in sham liver and postischemic liver 96 h after I/R. Original magnification is ×200. PV, portal vein; CV, central vein. B: nuclear YAP and TAZ expression in hepatic stellate cells (HSCs), biliary epithelial cells, and reactive ductular cells was determined 96 h after I/R by staining of serial sections for YAP or TAZ with desmin, CK19, or CK8/18, respectively. Representative HSCs, biliary epithelial cells, and reactive ductular cells positive for YAP or TAZ are marked by arrows. Original magnification is ×400. C: concomitant activation of YAP and TAZ occurs in HSCs. Serial liver sections obtained 96 h after I/R were stained for YAP and TAZ. Representative HSCs positive for YAP and TAZ are marked by arrows. Original magnification is ×400. D: nuclear YAP and TAZ expression increases in HSCs, but not hepatocytes, after I/R. Cytoplasmic and nuclear fractions from hepatocytes and HSCs isolated 96 h after I/R were analyzed by Western blot and stained with YAP, phosphorylated YAP (Ser127), TAZ, and phosphorylated TAZ (Ser89).
Because phosphorylation of LATS1 and MOB1 leads to the activation of LATS1, which leads to cytoplasmic retention of YAP and TAZ, we assessed the expression of phosphorylated LATS1 (pLATS1) and phosphorylated MOB1 (pMOB1). In quiescent liver, the components of Hippo pathway are constitutively active, with a high baseline level of phosphorylated YAP and TAZ and their cytoplasmic retention (24, 42). We found that phosphorylated forms of LATS1 and MOB1 decreased significantly after reperfusion in the postischemic liver (Fig. 5A). Because CTGF and survivin are two major target genes of YAP and TAZ, we measured expression of these two proteins. As expected, expression of both CTGF and survivin was low in livers from sham control (Fig. 5B). However, after reperfusion in postischemic livers, CTGF and survivin expression increased significantly (Fig. 5B). CTGF expression remained high throughout the 120-h time course studied, whereas survivin expression peaked at 48 h and slowly decreased thereafter (Fig. 5B).
Fig. 5.
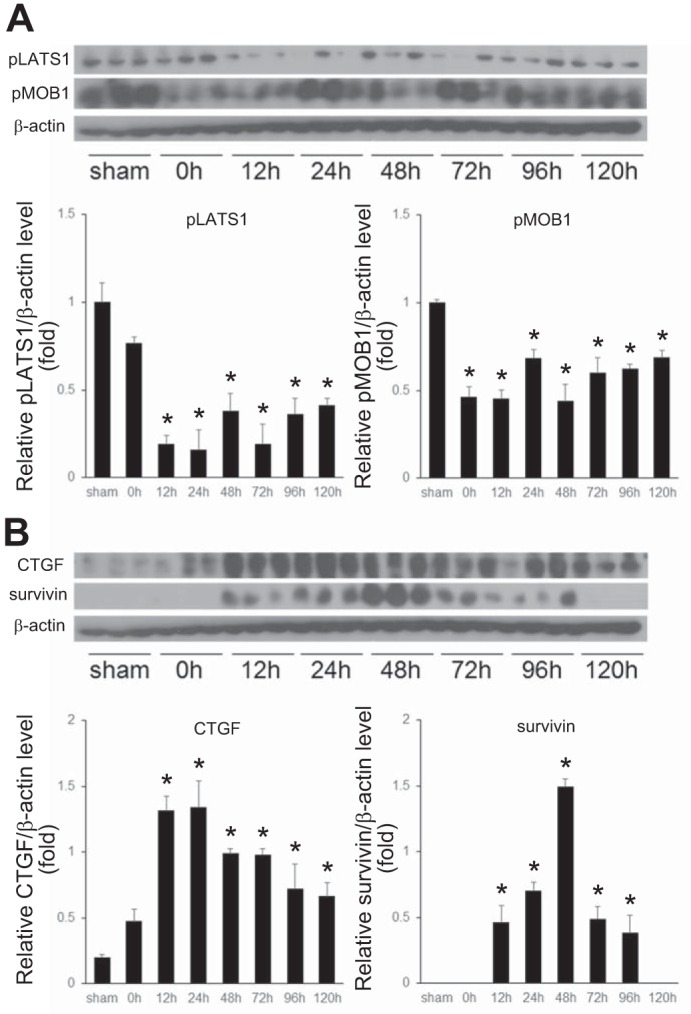
Hippo pathway and Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) target protein expression in postischemic liver after ischemia-reperfusion (I/R). A: I/R leads to reduced phosphorylation of the Hippo pathway proteins, large tumor suppressor 1 (LATS1) and Mps one binder 1 (MOB1). B: I/R leads to increased expression of the YAP/TAZ target genes, CTGF and survivin. Lysates of postischemic liver tissue were analyzed by Western blot. Results were quantitated by image analysis. Data are means ± SE with n = 3. *P < 0.05 compared with sham mice.
Previously, our laboratory reported that nonischemic liver lobes undergo regeneration with hepatocyte proliferation and increased liver mass in the absence of liver injury (23, 38). We sought to determine whether there was any HSC expansion that is related to YAP and TAZ activation in the nonischemic liver. Histological assessment of nonischemic liver showed no change in the numbers of nonparenchymal cells (Fig. 6A). Likewise, there was not a significant difference in YAP and TAZ activation between quiescent liver and nonischemic liver (Fig. 6B) and no notable changes in the phosphorylation of LATS1 or MOB1 (Fig. 6C). Finally, survivin expression was undetectable, and there was no increase in expression of CTGF until 120 h after reperfusion (Fig. 6D).
Fig. 6.

Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) and Hippo pathway regulation in nonischemic liver after ischemia-reperfusion (I/R). A: liver histopathology (H and E staining) in nonischemic liver after I/R showed no changes in liver architecture and no expansion of nonparenchymal cells. Original magnification is ×100. B: YAP and TAZ expression (immunohistochemistry) in nonischemic liver lobes after 96 h of reperfusion was minimal in hepatocytes. Original magnification is ×200. PV, portal vein; CV, central vein. C: no change in activation of Hippo pathway during I/R injury. Lysates of the nonischemic liver tissue were analyzed by Western blot and stained with phosphorylated large tumor suppressor 1 (LATS) and phosphorylated Mps one binder 1 (MOB1). D: expression of the YAP/TAZ target genes, CTGF and survivin, were assessed in lysates of nonischemic liver by Western blot. Results were quantitated by image analysis. Data are means ± SE with n = 3. *P < 0.05 compared with sham mice.
YAP and TAZ regulate HSC proliferation after I/R.
To examine the role of YAP and TAZ in liver repair and regeneration after I/R, mice were treated with verteporfin, which is a pharmacological inhibitor of YAP and TAZ (12, 37). Verteporfin was administered 24 h after reperfusion, after the peak of liver injury, so as to not interfere with the injury response. Treatment with verteporfin significantly reduced HSC proliferation in the postischemic liver (Fig. 7A), which was associated with a decrease in the number of desmin-positive cells (Fig. 7B). Subsequently, we also found that hepatocyte proliferation was significantly impaired by verteporfin in the ischemic liver (Fig. 7C). To investigate the cause of impaired hepatocyte proliferation, we measured liver HGF and amphiregulin levels, which are the main growth factors for hepatocyte proliferation. Verteporfin administration significantly reduced the expression of HGF but not amphiregulin in the postischemic liver (Fig. 7D). As expected, verteporfin had no effect on HSC or hepatocyte proliferation in the nonischemic liver (data not shown). Treatment with verteporfin had no effect on the expression of CTGF but significantly reduced survivin and cardiac ankyrin repeat protein (Ankrd1) expression in the postischemic liver (Fig. 7E).
Fig. 7.

Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) contribute to hepatic stellate cell (HSC) proliferation after ischemia-reperfusion (I/R). Mice were injected intraperitoneally with 10% dimethylsulfoxide solution (control) or 100 mg/kg verteporfin after 24 h of reperfusion and every 24 h thereafter. After 96 h of reperfusion, postischemic liver was analyzed. A: verteporfin treatment decreased HSC proliferation in ischemic liver after I/R. HSC proliferation was determined by dual immunofluorescence staining for desmin (red) and proliferating cell nuclear antigen (PCNA, green). DAPI was used for nuclear staining. Representative HSCs positive for PCNA are marked by arrows. Quantitative analysis of PCNA labeling was calculated as a percentage of PCNA-positive HSCs. Original magnification is ×630. B: verteporfin treatment decreased the number of HSCs in postischemic liver after I/R. Desmin-labeled cells were measured as a percentage of total area. Original magnification is ×200. C: verteporfin treatment decreased hepatocyte proliferation in postischemic liver after I/R. Hepatocyte proliferation was determined by immunohistochemical staining for PCNA. Original magnification is ×200. D: verteporfin treatment decreased growth factors in postischemic liver after I/R. Liver hepatocyte growth factor (HGF) and amphiregulin levels were analyzed by ELISA. E: verteporfin treatment decreased the expression of survivin and Ankrd1, but not connective tissue growth factor (CTGF). The lysates of ischemic liver were analyzed by Western blot. Results were quantitated by image analysis. Data are means ± SE with n = 3–4. *P < 0.05 compared with control group.
To confirm the effect of YAP and TAZ on HSC biology, we isolated HSCs from mouse liver and treated them with verteporfin to inhibit YAP and TAZ function in vitro. Verteporfin treatment dose dependently inhibited HSC proliferation (Fig. 8A) without cytotoxicity (Fig. 8B). Verteporfin treatment decreased the production of HGF by HSCs, whereas amphiregulin secretion was not observed in HSCs (Fig. 8C). Verteporfin treatment also decreased the expression of survivin and Ankrd1 but not CTGF (Fig. 8D).
Fig. 8.

Inhibition of Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) by verteporfin decreases hepatic stellate cell (HSC) proliferation and target gene expression in vitro. HSCs were isolated from mouse liver and treated with verteporfin or control culture medium for 5 days. A: verteporfin treatment decreased HSC proliferation as measured by DNA incorporation (n = 8). B: verteporfin treatment did not induce cytotoxicity (lactate dehydrogenase, LDH, release) of HSCs (n = 6). C: effect of verteporfin treatment (5 nM) on the release of hepatocyte growth factor (HGF) and amphiregulin by HSCs (n = 3). HGF and amphiregulin were measured by ELISA. Open bars, control; solid bars, verteporfin. D: effect of verteporfin treatment (5 nM) on expression of the YAP/TAZ target genes, CTGF, survivin, and Ankrd1, measured by Western blot. Results were quantitated by image analysis (n = 3). *P < 0.05 compared with control group.
DISCUSSION
To the best of our knowledge, this study is the first to assess the proliferation of HSCs after I/R injury and their role in the reparative and regenerative process. Our laboratory has previously shown that the process of liver regeneration after I/R injury is much different than that after partial hepatectomy, the latter being the current gold standard for the study of liver regeneration. However, hepatectomy is not a particularly clinically relevant model, and understanding the dynamics of tissue repair and how it is integrated with regeneration of functional liver mass is paramount.
After I/R, there is a large amount of dead tissue that must be remodeled. This process appears to be highly coordinated, with the clearance of necrotic cells coupled with the synthesis of new tissue matrix and the regeneration of hepatocytes. Our histological evidence supports this concept spatially and temporally, as recruited macrophages and proliferating HSCs occupied the same periportal areas at the interface between necrotic areas and areas of viable liver parenchyma with proliferating hepatocytes. Although HSCs have previously been shown to contribute to I/R injury (35), this is not altogether unexpected, as other cell types, including Kupffer cells and recruited macrophages, have also been implicated in the injury response. However, macrophages facilitate the process of liver repair and regeneration by clearing dead cells (29). Our data provide strong evidence that HSCs contribute to liver repair and regeneration after I/R injury.
Previous studies have assessed YAP in models of liver injury and regeneration. Liver-specific YAP deletion suppressed bile duct and hepatocyte proliferation and enhanced hepatocyte necrosis after bile duct ligation (3). Other studies showed YAP activation in association with HSC activation in a model of carbon tetrachloride-induced liver injury as well as in human liver fibrosis (26). YAP and TAZ share ~50% sequence homology, and both proteins are phosphorylated by the LATS kinase (20). During liver regeneration after partial hepatectomy, YAP and TAZ appear to have differential activation (13). In contrast, our study revealed concurrent YAP and TAZ activation in HSCs in the ischemic liver during liver repair and regeneration after I/R. These results were supported by our findings that phosphorylation of LATS1 and MOB1 was downregulated in the regenerative phase after I/R, suggesting that LATS kinase, one of the major components in Hippo pathway, was suppressed, leading to nuclear localization of YAP and TAZ in HSCs in the postischemic liver. On the contrary, we found no evidence of activation of the Hippo pathway in the nonischemic liver. Likewise, we found no evidence of HSC proliferation. The nonischemic liver is not injured yet does undergo regeneration (38), so our data suggest that HSCs may contribute preferentially to the remodeling and regeneration of the injured liver after I/R in a manner dependent on YAP and TAZ.
Our study also suggests that the proliferation and expansion of HSCs influence hepatocyte proliferation after I/R injury. Treatment of isolated HSCs with verteporfin, an inhibitor of YAP and TAZ, resulted in decreased HSC proliferation and decreased HSC expression of HGF in vitro. Likewise, verteporfin treatment decreased expression of HGF in the ischemic liver and was associated with reduced hepatocyte proliferation. In addition, our results suggest that YAP and TAZ regulate proliferation of HSCs in association with increased survivin and/or Ankrd1 expression, resulting in HSC expansion after I/R. Survivin is a member of the family of inhibitor of apoptosis proteins and also contributes to cell proliferation by regulating mitosis and cell division (1, 2, 28). Survivin expression is detected in HSCs during their activation (9). Ankrd1 has been shown to be expressed in fibrotic liver after carbon tetrachloride-induced liver injury (26). Survivin and Ankrd1 were expressed in isolated HSCs, and verteporfin treatment decreased their expression concomitant with decreased HSC proliferation. Interestingly, verteporfin did not affect CTGF expression in isolated HSCs or in the liver after I/R. CTGF is, not only regulated by YAP and TAZ, but also facilitated by the TGF-β signaling pathway and the epidermal growth factor receptor signaling system (31, 36). Although verteporfin treatment may affect cell types other than HSCs, we found little evidence of YAP and TAZ activation in hepatocytes but robust activation in HSCs in the injured liver. YAP and TAZ are also expressed in reactive ductular cells, and, although it is possible that these cells contribute to the reparative and regenerative response, they are seen in only limited areas around portal triads until 96 h after I/R. Therefore, it would seem that interference with YAP and TAZ by verteporfin in HSCs had a much greater influence on hepatocyte proliferation in the postischemic liver.
In summary, the present study demonstrates that, after I/R injury, HSC proliferation occurs preferentially in the injured liver. Furthermore, HSC proliferation in the postischemic liver is mediated by activation of YAP and TAZ. Finally, HSC expansion occurs in a spatially coordinated manner with Kupffer cells/macrophages at the interface of healthy and necrotic liver and contributes to hepatocyte proliferation.
GRANTS
This work was supported in part by National Institutes of Health Grant DK56029.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.K. and A.B.L. conceived and designed research; T.K. and R.M.S. performed experiments; T.K. and A.B.L. analyzed data; T.K. and A.B.L. interpreted results of experiments; T.K. prepared figures; T.K. and A.B.L. drafted manuscript; T.K., R.M.S., and A.B.L. edited and revised manuscript; T.K., R.M.S., and A.B.L. approved final version of manuscript.
REFERENCES
- 1.Altieri DC. Molecular circuits of apoptosis regulation and cell division control: the survivin paradigm. J Cell Biochem 92: 656–663, 2004. doi: 10.1002/jcb.20140. [DOI] [PubMed] [Google Scholar]
- 2.Baba HA, Wohlschlaeger J, Schmitz KJ, Nadalin S, Lang H, Benesch A, Gu Y, Biglarnia AR, Sotiropoulos GC, Takeda A, Takeda N, von Wnuck Lipinski K, Levkau B. Survivin is upregulated during liver regeneration in rats and humans and is associated with hepatocyte proliferation. Liver Int 29: 585–592, 2009. doi: 10.1111/j.1478-3231.2008.01911.x. [DOI] [PubMed] [Google Scholar]
- 3.Bai H, Zhang N, Xu Y, Chen Q, Khan M, Potter JJ, Nayar SK, Cornish T, Alpini G, Bronk S, Pan D, Anders RA. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology 56: 1097–1107, 2012. doi: 10.1002/hep.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baier PK, Baumgartner U, Hempel S, Wolff-Vorbeck G, von Dobschuetz E, Hopt UT. Kupffer cells infiltrate liver tissue early after ischemia-reperfusion and partial hepatectomy. Eur Surg Res 37: 290–297, 2005. doi: 10.1159/000089239. [DOI] [PubMed] [Google Scholar]
- 5.Barone S, Okaya T, Rudich S, Petrovic S, Tenrani K, Wang Z, Zahedi K, Casero RA, Lentsch AB, Soleimani M. Distinct and sequential upregulation of genes regulating cell growth and cell cycle progression during hepatic ischemia-reperfusion injury. Am J Physiol Cell Physiol 289: C826–C835, 2005. doi: 10.1152/ajpcell.00629.2004. [DOI] [PubMed] [Google Scholar]
- 6.Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell 11: 11–23, 2003. doi: 10.1016/S1097-2765(02)00776-1. [DOI] [PubMed] [Google Scholar]
- 7.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 115: 209–218, 2005. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beaussier M, Wendum D, Schiffer E, Dumont S, Rey C, Lienhart A, Housset C. Prominent contribution of portal mesenchymal cells to liver fibrosis in ischemic and obstructive cholestatic injuries. Lab Invest 87: 292–303, 2007. doi: 10.1038/labinvest.3700513. [DOI] [PubMed] [Google Scholar]
- 9.De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, Schwabe RF. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 132: 1937–1946, 2007. doi: 10.1053/j.gastro.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 10.Dooley S, Delvoux B, Lahme B, Mangasser-Stephan K, Gressner AM. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 31: 1094–1106, 2000. doi: 10.1053/he.2000.6126. [DOI] [PubMed] [Google Scholar]
- 11.Failli P, Ruocco C, De Franco R, Caligiuri A, Gentilini A, Giotti A, Gentilini P, Pinzani M. The mitogenic effect of platelet-derived growth factor in human hepatic stellate cells requires calcium influx. Am J Physiol 269: C1133–C1139, 1995. doi: 10.1152/ajpcell.1995.269.5.C1133. [DOI] [PubMed] [Google Scholar]
- 12.Gibault F, Corvaisier M, Bailly F, Huet G, Melnyk P, Cotelle P. Non-photoinduced biological properties of verteporfin. Curr Med Chem 23: 1171–1184, 2016. doi: 10.2174/0929867323666160316125048. [DOI] [PubMed] [Google Scholar]
- 13.Grijalva JL, Huizenga M, Mueller K, Rodriguez S, Brazzo J, Camargo F, Sadri-Vakili G, Vakili K. Dynamic alterations in Hippo signaling pathway and YAP activation during liver regeneration. Am J Physiol Gastrointest Liver Physiol 307: G196–G204, 2014. doi: 10.1152/ajpgi.00077.2014. [DOI] [PubMed] [Google Scholar]
- 14.Huguet C, Gavelli A, Bona S. Hepatic resection with ischemia of the liver exceeding one hour. J Am Coll Surg 178: 454–458, 1994. [PubMed] [Google Scholar]
- 15.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol 13: 316–327, 2016. doi: 10.1038/cmi.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LC, Yaffe MB. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J 19: 6778–6791, 2000. doi: 10.1093/emboj/19.24.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol 21, Suppl 3: S84–S87, 2006. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 18.Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, Edwards MJ, Lentsch AB. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 48: 1213–1223, 2008. doi: 10.1002/hep.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, Lim DS. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci USA 107: 8248–8253, 2010. doi: 10.1073/pnas.0912203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 28: 2426–2436, 2008. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemasters JJ, Thurman RG. Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol 37: 327–338, 1997. doi: 10.1146/annurev.pharmtox.37.1.327. [DOI] [PubMed] [Google Scholar]
- 22.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology 32: 169–173, 2000. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 23.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology 27: 1172–1177, 1998. doi: 10.1002/hep.510270226. [DOI] [PubMed] [Google Scholar]
- 24.Loforese G, Malinka T, Keogh A, Baier F, Simillion C, Montani M, Halazonetis TD, Candinas D, Stroka D. Impaired liver regeneration in aged mice can be rescued by silencing Hippo core kinases MST1 and MST2. EMBO Mol Med 9: 46–60, 2017. doi: 10.15252/emmm.201506089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, Johnson RL. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci USA 107: 1437–1442, 2010. doi: 10.1073/pnas.0911427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, Hoorens A, Reynaert H, Halder G, van Grunsven LA. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 63: 679–688, 2015. doi: 10.1016/j.jhep.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 27.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev 30: 1–17, 2016. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res 14: 5000–5005, 2008. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 29.Ohkubo H, Ito Y, Minamino T, Mishima T, Hirata M, Hosono K, Shibuya M, Yokomizo T, Shimizu T, Watanabe M, Majima M. Leukotriene B4 type-1 receptor signaling promotes liver repair after hepatic ischemia/reperfusion injury through the enhancement of macrophage recruitment. FASEB J 27: 3132–3143, 2013. doi: 10.1096/fj.13-227421. [DOI] [PubMed] [Google Scholar]
- 30.Quillin RC 3rd, Wilson GC, Nojima H, Freeman CM, Wang J, Schuster RM, Blanchard JA, Edwards MJ, Gandhi CR, Gulbins E, Lentsch AB. Inhibition of acidic sphingomyelinase reduces established hepatic fibrosis in mice. Hepatol Res 45: 305–314, 2015. doi: 10.1111/hepr.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rachfal AW, Brigstock DR. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol Res 26: 1–9, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Sakai N, Van Sweringen HL, Quillin RC, Schuster R, Blanchard J, Burns JM, Tevar AD, Edwards MJ, Lentsch AB. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology 56: 1468–1478, 2012. doi: 10.1002/hep.25768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin T, Kuboki S, Lentsch AB. Roles of nuclear factor-kappaB in postischemic liver. Hepatol Res 38: 429–440, 2008. doi: 10.1111/j.1872-034X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- 34.Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, Wang CY, Gao B, Jiang J, Yang Y. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci USA 107: 1431–1436, 2010. doi: 10.1073/pnas.0911409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart RK, Dangi A, Huang C, Murase N, Kimura S, Stolz DB, Wilson GC, Lentsch AB, Gandhi CR. A novel mouse model of depletion of stellate cells clarifies their role in ischemia/reperfusion- and endotoxin-induced acute liver injury. J Hepatol 60: 298–305, 2014. doi: 10.1016/j.jhep.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urtasun R, Latasa MU, Demartis MI, Balzani S, Goñi S, Garcia-Irigoyen O, Elizalde M, Azcona M, Pascale RM, Feo F, Bioulac-Sage P, Balabaud C, Muntané J, Prieto J, Berasain C, Avila MA. Connective tissue growth factor autocriny in human hepatocellular carcinoma: oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 54: 2149–2158, 2011. doi: 10.1002/hep.24587. [DOI] [PubMed] [Google Scholar]
- 37.Wang C, Zhu X, Feng W, Yu Y, Jeong K, Guo W, Lu Y, Mills GB. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am J Cancer Res 6: 27–37, 2015. [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson GC, Kuboki S, Freeman CM, Nojima H, Schuster RM, Edwards MJ, Lentsch AB. CXC chemokines function as a rheostat for hepatocyte proliferation and liver regeneration. PLoS One 10: e0120092, 2015. doi: 10.1371/journal.pone.0120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828, 2015. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol 13: 877–883, 2011. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22: 1962–1971, 2008. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16: 425–438, 2009. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu NL, Asahina K, Wang J, Ueno A, Lazaro R, Miyaoka Y, Miyajima A, Tsukamoto H. Hepatic stellate cell-derived delta-like homolog 1 (DLK1) protein in liver regeneration. J Biol Chem 287: 10355–10367, 2012. doi: 10.1074/jbc.M111.312751. [DOI] [PMC free article] [PubMed] [Google Scholar]