

Abstract
We previously showed that testosterone (T) deficiency enhanced high-fat/low-carbohydrate diet (HFD)-induced hepatic steatosis in rats independent of insulin resistance and that T replacement reduced hepatic macrovesicular fat accumulation and inflammation. The present report explores the mechanism of Tʼs protective effects on HFD-induced steatohepatitis. Adult male rats were randomized into four treatment groups for 15 wk: intact rats on regular chow diet or HFD, and castrated rats on HFD with or without T replacement. Fatty acid β-oxidation and de novo synthesis were not changed by castration and T replacement, but expression of lipid export proteins ApoB100 and microsomal triglyceride transfer protein (MTP) was suppressed by HFD in both intact and castrated rats but restored by T replacement. Macrovesicular lipid droplet-related proteins perilipin 1 and fat-specific protein 27 were increased by HFD in castrated rats and suppressed by T replacement. Higher activation/expression of ER stress proteins (PERK, IRE-1α, JNK, NF-κB, and CHOP) was demonstrated in castrated rats fed HFD compared with intact animals, and T replacement suppressed these changes. We conclude that 1) HFD leads to ApoB100/MTP suppression reducing export of lipids; 2) castration promotes progression to steatohepatitis through activation of the ER stress pathway and enhancement of macrovesicular droplet protein expression; and 3) testosterone suppresses ER stress, inhibits the formation of macrovesicular lipid droplets, promotes lipid export, and ameliorates steatohepatitis induced by HFD and castration.
Keywords: ApoB100, ER stress, hepatic steatosis, lipid droplet, lipogenesis, testosterone deficiency
INTRODUCTION
In the Western world, nonalcoholic fatty liver disease (NAFLD) affects nearly one-third of the general population (11). NAFLD may progress from simple steatosis to steatosis with inflammation and liver cell injury (nonalcoholic steatohepatitis, NASH), or further progress to cirrhosis and hepatocellular carcinoma (2). NAFLD is strongly associated with Type 2 diabetes, obesity, and metabolic syndrome (3, 8, 33, 57). Higher body mass index in men is correlated with low total serum testosterone (T) levels (5, 6, 22). Approximately 20% to 50% of men with Type 2 diabetes and metabolic syndrome have hypogonadism. T treatment reverses the visceral fat accumulation that is associated with androgen deficiency (4, 37). Hypogonadal men are at higher risk to develop NASH, which may be attenuated by T treatment (1, 20, 21). Clinically, low-serum total testosterone level is independently associated with NAFLD, and the association remains unchanged even after adjustment for visceral adipose tissue and insulin resistance (26). Generally, the findings from the human studies are supported by a demonstration of hepatic steatosis in male chickens (12) and androgen receptor knockout (ARKO) male mice (15, 31, 64, 69).
The pathophysiology of T deficiency in inducing NAFLD is not known. Hepatic lipid accumulation results from an imbalance of lipid production and turnover. Sources of fat in the liver include fatty acids made by de novo synthesis, uptake of free fatty acids released by lipolysis, and fatty acids from exogenous (dietary) sources. Perturbations in gene expression related to fatty acid oxidation have been observed in both animal (51) and human (27) liver in NAFLD. De novo synthesis may contribute up to 30% of fat excreted in very low-density lipoprotein (VLDL) (66), and therefore, upregulation of endogenous lipid synthesis may play a role in lipid accumulation. Defective lipid export may also contribute, as NASH patients have been found to have low serum VLDL levels (17). The ER network is also an important site for synthesis and modulation of lipids in the liver (36, 45, 71). Lipid droplet formation from the ER mediated by specific lipid droplet proteins may also be implicated in the formation of macrovesicular droplet in NAFLD/NASH (16, 65). Hepatocyte damage, apoptosis, and inflammation also play key roles during NAFLD development and progression (59, 63). It is clear that T reduces hepatic fat, apoptosis, and inflammation, but the mechanism of T in this setting was unknown before this report.
Using a model of NAFLD in castrated rats fed a high-fat/low-carbohydrate diet (HFD), we have previously shown that hepatic steatosis occurred with elevated liver aminotransferases, increased macrovesicular fat accumulation, inflammation, and increased hepatocyte apoptosis (41). Of note, insulin resistance is not present in this castrated rat model of HFD-induced steatohepatitis, allowing us to study the effects of T independent of insulin action. Using this NAFLD model, we have shown that T replacement protects against the HFD-induced fat accumulation and the development of steatohepatitis. The objective of this study was to determine the mechanism of the T-protective effect against HFD-induced steatohepatitis in castrated rats. We hypothesized that the development of steatohepatitis in castrated male rats fed a HFD may result from a combination of increased lipid production and decreased turnover, increased macrovesicular lipid droplet formation, and overactivated endoplasmic reticulum stress; and that T replacement would ameliorate the severity of fat accumulation, cell apoptosis, and inflammation through reversal of these mechanisms.
MATERIALS AND METHODS
Animals and experimental design.
Animal handling and experimentation were in accordance with the recommendation of American Veterinary Medical Association, and protocols were approved by the Animal Care and Use Review Committee at the Los Angeles Biomedical Research Institute at Harbor-University of California, Los Angeles (Harbor-UCLA) Medical Center. As described previously (41), adult male Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA) and housed individually in temperature (22°C)- and humidity-controlled rooms and exposed to 12:12-h light-dark cycles. After a 2-wk adaptation period, the rats were randomized into four treatment groups: intact rats fed a regular chow diet (I+RCD) (n = 6), intact rats fed a HFD diet (I+HFD) (n = 8), castrated rats fed a HFD (C+HFD) (n = 7), and castrated rats with subcutaneous T implants fed HFD (C+HFD+T) (n = 7). The I+RCD group served as a baseline physiological control, while the I+HFD group served as a control for HFD effects. The HFD (30) was in liquid form with 1 kcal of energy per 1 ml of diet. The HFD provided 71% energy from fat, 18% energy from protein, and 11% from carbohydrates, while the RCD provided 16% energy from fat, 27% from protein, and 56% from carbohydrates. The fat composition was 60.9% corn oil, 35.7% olive oil, and 3.4% safflower oil (44.5% monounsaturated, 42.3% polyunsaturated, and 13.2% saturated fat). T replacement in castrated rats was accomplished using 3-cm T Silastic implants prepared from polydimethylsilozane tubing (OD, 3.18 mm; ID, 1.98 mm; Dow Corning, Midland, MI), packed with T (Sigma, St. Louis, MO), and sealed with Silastic medical adhesive A (Dow Corning) (35). The release rate of T from the Silastic implants was estimated to be ~30 μg·cm−1·day−1 and lasted for at least 6 mo (52). Every animal with an implant was examined to ensure that the implant remained intact for the duration of the experiment. Only animals whose implants remained intact and had circulating T levels indicative of functioning implants were included in this study.
After 15 wk, the rats were euthanized with an overdose of pentobarbital sodium after an overnight fast. Samples from liver lobes and plasma were snap-frozen in liquid nitrogen and stored at −80°C.
Quantitative real-time PCR.
In brief, total RNA was isolated using an RNAqueous-4PCR kit (Ambion, Thermo Fisher Scientific, Waltham, MA) and was DNase-treated. Using a Nanodrop spectrophotometer (Nanodrop Instruments, Wilmington, DE), we assessed the purity of the RNA by the visual appearance of the ethidium bromide-stained ribosomal bands and quantitated by light absorbance at 260 nm. Total RNA (1 µg) was reverse-transcribed into single-stranded cDNA using a TaqMan Gold RT-PCR kit (Thermo Fisher Scientific) at 50°C for 30 min in a total volume of 20 μl. Real-time PCR reactions were run in triplicate on 96-well plates using Applied Biosystems StepOne real-time PCR System (Applied Biosystems, Foster City, CA) following the manufacturer’s protocol. Reactions proceeded by activation of DNA polymerase at 95°C for 10 min, followed by 38 PCR denaturing cycles at 95°C for 15 s and annealing/extension at 60°C for 1 min. Relative levels of expression of each of the selected genes (relative fold-change vs. control group) were calculated using the ∆∆CT method. Results are expressed as means ± SE and are considered significant at P < 0.05. Sets of primers for the detection of different genes (SCD-1, CPT-1, DGAT-1, GPAT-1, and ApoB) were selected from published work (67) and obtained from RealtimePrimers (Elkins Park, PA). Supplemental Table S1 shows the primer sequences selected for the present study.
Mitochondria size analysis by transmission electron microscopy.
A midportion of middle lobe was fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at 4°C overnight, subsequently postfixed in 1% osmium tetroxide for 1 h. After dehydration, specimens were embedded in Epon 812 epoxy resin. The ultra-thin sections were stained with 3% uranyl acetate and lead nitrate and were examined with a HITACHI H-600 electron microscope at 75 kV. Livers of six to eight animals per group were studied. Mitochondria diameters were measured by a researcher blinded to the type of treatment that the rats received. Ten mitochondria, in as many hepatocytes, were measured for each rat at a magnification of ×30,000 (23).
De novo fatty acid synthesis and the desaturation index.
Animals received deuterium as a stable isotope tracer (28). Fourteen days before death, animals received a 2% (of lean mass) intraperitoneal injection of 99.9% deuterium with 0.9% sodium chloride (normal saline). Then, animals were provided 6% deuterium-enriched drinking water. The concentration of deuterium used was designed to maintain ~3% deuterium enrichment in body water. Blood was collected by tail bleeds to monitor enrichment. On day 14 of deuterium enrichment, rats were euthanized, and blood and tissues (subcutaneous fat, liver, and muscle) were collected for analysis of fatty acid synthesis.
Total lipids from plasma or tissue were saponified overnight in 200 proof ethanol and 30% KOH (1:1, wt/vol) to release all fatty acids, including those from phospholipids, triglycerides, and cholesteryl esters, and then they were extracted with petroleum ether using a previously published method (34). Fatty acids were derivatized as methyl esters using 0.5 N methanolic HCl (Supelco). Derivatized fatty acids were analyzed by gas chromatography/mass spectrometry (GC/MS) using a Hewlett-Packard model 5973 selective mass detector connected to a model 6890 gas chromatograph, with a Bpx70 column (30-m length, 250-μm diameter, 0.25-μm film thickness) from SGE (Austin, TX) (28). The GC conditions were as follows: helium flow rate, 1 ml/min and initial oven temperature, 150°C, which was programmed to increase at 3°C/min to a final temperature of 221°C. The retention times for palmitate, stearate, and oleate were 5.9, 8.6, and 9.2, respectively. Mass spectra were acquired using electron impact ionization and selective ion monitoring for the ion cluster corresponding to the mass-to-charge ratios (m/z) of 270 for palmitate. The mass isotopomer distribution was determined using a method described by Lee et al. (28), correcting for the contribution of the derivatizing agent and 13C natural abundance in the target compound. The mass isotopomer distribution was expressed in molar fractions (m0, m1, m2, m3) corresponding to the fraction of molecules that contain 0, 1, 2, 3 deuterium substitutions.
The calculation of the fraction of new synthesis (FNS) (de novo synthesis) has been described elsewhere (28, 67). In summary, FNS = ME (fatty acid)/(p × N). ME (molar enrichment) is calculated from the mass isotopomer distribution and represents the average number of deuterium atoms incorporated per molecule, p is the specific activity (enrichment) of deuterium in water, determined from the m2/m1 consecutive mass isotopomer ratio, and N represents the number of deuterium per molecules of fatty acid from body water (n = 21 for palmitate).
The desaturation index was calculated as the precursor-to-product ratio of newly synthesized (FNS) oleic and stearic acids as a measure of stearoyl-CoA desaturase 1 (SCD-1) activity (67). Palmitoleic acid was undetectable in most samples, so the palmitoleic/palmitic ratio was not determined.
Tissue preparation and Western blot analysis.
Briefly, liver tissues (~50 mg per sample) were homogenized in lysis buffer (0.25 M sucrose, 50 mM HEPES, 10 mM NaCl, 10 mM EDTA, 2 mM DTT) supplemented with protease inhibitors (Complete Protease Inhibitors; Roche, Basel, Switzerland). RIPA buffer (Santa Cruz Biotechnology, Santa Cruz, CA) was added after homogenization. Western blot analysis was performed as described previously (41). Proteins were denatured and separated by SDS-PAGE (Invitrogen, Carlsbad, CA). After transferring and blocking were completed, the immunoblot PVDF membrane (Bio-Rad, Hercules, CA) was probed using anti-PKR-like ER kinase (PERK) and phosphorylated PERK (pPERK), anti-inositol-requiring (IRE)-1α, anti-C/EBP homologous protein (CHOP, also known as GADD153), anti-JNK and phosphorylated JNK (pJNK), and anti-P65 of NF-κB (P65NF-κB) and phosphorylated P65NF-κB (pP65NF-κB), anti-perilipin 1 (PLIN1) (Santa Cruz Biotechnology); anti-fat specific protein 27(Fsp27, AKA CIDE C) (Abcam, Boston, MA); anti-phosphorylated IRE-1α (pIRE-1α) (Thermo Fisher Scientific); anti-apolipoprotein B (ApoB), or antimicrosomal triglyceride transfer protein (MTP) antibody (Abcam). After being washed and incubated with anti-rabbit (Amersham Biosciences, Piscataway, NJ), anti-goat, or anti-mouse (Abcam) secondary antibody, membranes were exposed to Hyperfilm ECL (Denville Scientific, Metuchen, NJ) with enhanced chemiluminescence solutions, per the manufacturer’s specifications (Amersham Biosciences). Band intensities were determined using Quantity One software from Bio-Rad (Hercules, CA). The specificity of each antibody has been validated by previous published work or by testing with positive control (anti-ApoB and anti-MTP, see Fig. 4, B and E). The detailed information about the antibodies used is shown in Supplemental Table S2.
Fig. 4.

Apolipoprotein B (ApoB) gene/protein expression and microsomal triglyceride transfer protein (MTP) protein expression in rat liver tissue. A: quantitative gene expression analysis of ApoB showed that castration and HFD decreased gene expression (P < 0.05, compared with I+RCD), but testosterone (T) replacement restored ApoB gene expression. B–D: density analysis of Western blots showed that HFD in intact and castrated animals had decreased apolipoprotein B-100 (ApoB100) protein expression (P < 0.05, compared with I+RCD), but T replacement attenuated the suppression of ApoB100 protein level in rat liver tissues. The changes of ApoB48 protein did not reach significance. Plasma was used as a positive control for anti-ApoB antibody. GAPDH was used as the loading control. E and F: density analysis of Western blot results showed that HFD and castration decreased MTP protein expression (P < 0.05, compared with I+RCD), but T replacement attenuated the suppression of MTP protein level in rat liver tissues. The mouse liver lysate was used as a positive control for anti-MTP antibody. GAPDH was used as the loading control. Values are expressed as means ± SE.
Statistical analysis.
Statistical analyses were performed using the StatPlus 2007 Program (AnalystSoft, Vancouver, Canada) and SAS 9.3 (SAS Institute, Cary NC). Normally distributed data were analyzed by one-way ANOVA, and post hoc tests by Tukey-Kramer, with correction for multiple comparisons. Results are presented as the means ± SE. Statistical significance was construed at P < 0.05.
RESULTS
Characteristics of treatment groups.
The detailed study of the four groups of animals has been previously reported (41). The most important findings were circulating T levels in castrated animals were undetectable, while T levels in C+HFD+T group were approximately twofold higher compared with intact animals (41). I+HFD rats gained the most weight with the highest percentage of body fat. Body weight of C+HFD+T rats was similar to that of I+RCD. C+HFD rats demonstrated the lowest body weight gain, but a higher percentage body fat than C+HFD+T rats. T replacement led to an increase in percent lean body mass (41). Despite a lack of insulin resistance in this model, HFD-induced hepatic steatosis in castrated rats (C+HFD) was characterized by macrovesicular steatosis and increased inflammation and hepatocyte apoptosis compared with the intact (I+HFD) and T-replaced (C+HFD+T) animals, suggesting progression of hepatic steatosis to steatohepatitis in the presence of T deficiency (Fig. 1A) (41). Transmission electron microscopy (TEM) further confirmed that castration and HFD (C+HFD) resulted in more lipid droplets in hepatocytes compared with the intact (I+HFD) and T-replaced (C+HFD+T) animals (Fig. 1B).
Fig. 1.

Liver histology, mitochondrial size, and CPT-1α gene expression. A: liver histology in ×400 magnification stained with hematoxylin and eosin. Compared with the normal hepatocytes in intact rats fed a regular chow diet (I+RCD; n = 6), the intact rats fed a high-fat diet (I+HFD; n = 8) samples showed microvesicular lipid droplets and minimal macrovesicular fatty accumulation. Castrated rats fed a HFD (C+HFD; n = 7) showed more macrovesicular lipid droplets (arrowheads) and a focus of inflammation (black arrow). With testosterone (T) replacement, the C+HFD+T samples had fewer microvesicular and minimal macrovesicular lipid droplets, as well as fewer foci of inflammation compared with C+HFD. B: transmission electron microscopy (TEM) showing ultrastructure of hepatocyte: nucleus (empty star), the mitochondria (black arrow), the lipid droplets (star), and the endoplasmic reticulum (arrowhead). Image taken at magnification ×10,500. C: mitochondrial sizes in hepatocytes were determined at ×30,000 by TEM. No significant differences were found between groups of rats. D: CPT-1α gene expression. GAPDH was used as the loading control. Values are expressed as means ± SE.
Mitochondria morphology and fatty acid β-oxidation.
Mitochondrial morphology in hepatocytes at ×30,000 by TEM revealed no abnormalities in mitochondrial shape, size, or cristae, with no significant differences between groups of rats (Fig. 1, B and C). CPT-1α gene expression in liver tissue was not changed by HFD, castration, or T replacement (Fig. 1D). These results suggest that T does not play a role in fatty acid β-oxidation.
De novo synthesis of fatty acids, and expression of SCD-1, GPAT-1, and DGAT-1.
Palmitate is the main product of de novo synthesis, which is known to be suppressible by a high-fat diet. The fraction of new synthesis (FNS) of palmitate is a measure of the newly made fatty acids over the experimental period. The fraction of new synthesis of palmitate circulating in plasma was determined at various time points during the 14-day enrichment period. At each time point, animals on the HFD exhibited lower plasma FNS rates than I+RCD. On HFD, there were no differences in plasma FNS rates between castrated, intact rats, or T-replaced rats (Fig. 2). In the liver, FNS rates at day 14 followed a pattern similar to that of the plasma, with no differences between high-fat diet-fed groups. Similar patterns of suppression were observed in muscle and subcutaneous adipose tissues (Table 1).
Fig. 2.
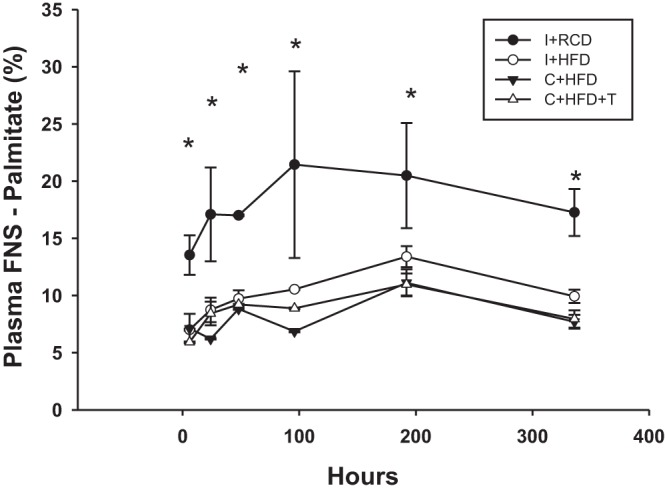
Fraction of new synthesis (FNS) of palmitate in plasma. This graph represents the FNS of palmitate circulating in the plasma at various time points during the 14-day deuterium enrichment period. At all of the time points, all three HFD-fed groups demonstrated lower FNS palmitate (i.e., lower circulating presence of de novo synthesized palmitate), in comparison to I+RCD group. *P < 0.05 compared with I+RCD.
Table 1.
Fatty acid fraction of new synthesis in plasma and body tissues
| Fraction of New Synthesis of Palmitate Over 14 Days [median %, (25–75% IQR)] | ||||
|---|---|---|---|---|
| Tissue | I+RCD | I+HFD | C+HFD | C+HFD+T |
| Plasma | 18 (16.7–19.6) | 8.1 (5.9–9.5)* | 7.3 (7.0–8.9)* | 10.1 (8.6–11.2)* |
| Liver | 25.9 (23.5–28.2) | 10.0 (6.8–10.4)* | 8.5 (7.4–10.6)* | 10.7 (9.8–12.8) |
| Muscle | 12.2 (9.2–13.2) | 5.3 (4.3–6.1) | 4.2 (3.7–5.2)* | 5.4 (4.9–5.7) |
| Subcutaneous adipose | 3.2 (2.6–3.4) | 1.7 (1.0–2.9) | 2.0 (1.3–2.7) | 1.4 (0.7–2.9) |
IQR, interquartile range. I+RCD, intact + regular chow diet; I+HFD, intact + high-fat diet; C+HFD, control + high-fat diet; C+HFD+T, control + high-fat diet + testosterone.
P < 0.05 compared with I+RCD.
SCD-1 is a lipogenic enzyme that converts the saturated fatty acids palmitate and stearate to monounsaturated fatty acids palmitoleate and oleate, respectively. Monounsaturated fatty acids are subsequently incorporated into triglycerides. SCD-1 gene expression levels in liver of intact and castrated rats fed HFD were suppressed ~60% and ~90%, respectively, compared with RCD animals (Fig. 3A, overall group comparison P < 0.01). The lower SCD-1 gene expression in castrated rats was not restored by T replacement (Fig. 3A). The isotopic desaturation index (oleic/stearic ratio, Fig. 3B) reflects SCD-1 conversion of newly synthesized (not dietary) stearic acid to oleic acid. HFD suppressed the desaturation index in I+HFD animals, did not significantly affect it in C+HFD, but suppressed it in C+HFD+T animals.
Fig. 3.

Quantitative gene expression analyses of stearoyl-CoA desaturase-1 (SCD-1), glycerol-3-phosphate acyltransferase-1 (GPAT-1), and diacylglycerol O-acyltransferase-1 (DGAT-1) in rat liver. A: HFD decreased SCD-1 (P < 0.05, compared with I+RCD), castration suppressed SCD-1 further (P < 0.05, compared with I+HFD), but testosterone (T) replacement did not change SCD-1 gene expression. B: isotopic desaturation index (oleic/stearic ratio) reflects SCD-1 conversion of newly synthesized stearic acid to oleic acid. The desaturation index was suppressed in I+HFD animals, not significantly affected in C+HFD, and suppressed C+HFD+T animals. C: C+HFD suppressed GPAT-1 gene expression (P < 0.05, compared with I+RCD), while T replacement induced GPAT-1 gene expression. D: DGAT-1 expression was unchanged. GAPDH was used as the housekeeping control. Values are expressed as means ± SE.
GPAT-1 is an enzyme that catalyzes the committed initial step in triglyceride synthesis, forming lysophosphatidic acid from glycerol-3-phosphate and newly synthesized saturated fatty acids (61). In animals on the HFD, GPAT-1 gene expression in liver was not suppressed in intact animals, but was suppressed in castrated animals (Fig. 3C, overall group comparison P < 0.05). T replacement restored the GPAT-1 gene expression compared with other groups (Fig. 3C, overall group comparison P < 0.05). DGAT-1 catalyzes triglyceride synthesis from diacylglycerol and long-chain fatty acids. DGAT-1 gene expression in liver tissue was not changed by HFD with or without castration or T replacement (Fig. 3D).
ApoB and MTP expression as markers of lipid export.
ApoB gene expression in liver tissue was suppressed by HFD in both intact and castrated animals (Fig. 4A, overall group comparison P < 0.05). T replacement restored the ApoB gene expression in liver compared with other groups fed HFD (Fig. 4A, P < 0.05). Correspondingly, protein expression of ApoB100 in liver was suppressed by HFD in both intact and castrated animals (Fig. 4, B and C, overall group comparison P < 0.05). T replacement restored the ApoB100 protein level in liver compared with other HFD groups (Fig. 4, B and C, P < 0.05). The protein level of ApoB48 in liver showed similar trends to ApoB100 in relationships between the different groups, but did not reach statistical significance (Fig. 4, B and D). Western blot analysis demonstrated that the protein level of MTP in liver was suppressed by HFD in both intact and castrated animals (Fig. 4, E and F, overall group comparison P < 0.05). T replacement restored the MTP protein level in liver compared with other HFD groups (Fig. 4, E and F, P < 0.05). The changes of the MTP protein levels between groups were consistent with the changes of ApoB, suggesting that the export of triglycerides was impaired in HFD-fed animals, but restored by T replacement in castrated rats.
Lipid droplet-related proteins PLIN1 and Fsp27.
Both PLIN1 and Fsp27 are lipid droplet-associated proteins that modulate lipid exchange and transfer to promote formation of larger lipid droplets (43, 53, 54, 65). HFD increased PLIN1 and Fsp27 protein expression in liver tissue in castrated rats (C+HFD) but not in intact animals fed with RCD or HFD (Fig. 5, A and B). T replacement suppressed the increase of PLIN1 and Fsp27 in liver compared with C+HFD group (Fig. 5, A and B, P < 0.01).
Fig. 5.

Expression of lipid droplet-related proteins, perilipin 1 (PLIN1) and fat-specific protein 27 (Fsp27). A and B: density analysis of Western blots showed that HFD in intact rats did not increase the expression of PLIN1 and Fsp27 (P > 0.05, compared with I+RCD) while C+HFD significantly induced expression of PLIN1 and Fsp27 proteins (P < 0.05, compared with I+HFD). T replacement suppressed expression of both proteins in rat liver tissues (P < 0.05, compared with C+HFD); GAPDH was used as the loading control. Values are expressed as means ± SE.
ER stress: activation of PERK, IRE1α, JNK, NF-κB, and CHOP expression.
PERK and IRE1α are ER-membrane transducers which, upon phosphorylation, activate the downstream pathways of ER stress, including the downstream targets of CHOP (proapoptotic transcription factor), JNK (involved in apoptosis and inflammation), and NF-κB (regulator of inflammation) (36, 45). PERK and IRE1α activation reflected by phosphorylation of both proteins was present in liver tissue in I+HFD (Fig. 6, A and B, overall group comparison P < 0.05). Castration appeared to increase activation of these proteins (C+HFD compared with I+HFD group, P < 0.05). T replacement attenuated the activation of PERK and IRE1α in liver compared with other groups (Fig. 6, A and B, overall group comparison P < 0.05). Correspondingly, protein expression of CHOP and phosphorylation of JNK and NF-κB in the liver were induced by HFD in intact animals, but even more so, in castrated animals (Fig. 6, C–E, overall group comparison P < 0.05). T replacement suppressed the expression or activation of these downstream targets of ER stress compared with other HFD groups (Fig. 6, C–E, P < 0.05). The changes of these ER stress transducers and their downstream targets between groups were consistent with each other, indicating the ER stress pathway is important in the development of hepatic steatosis in T deficiency. Although no apparent differences of the endoplasmic reticulum were identified between I+RCD, I+HFD, and C+HFD+T groups, the TEM images of the ultrastructure of hepatocyte (magnification ×30,000) showed contact between ER and lipid droplet and less organized ER in the C+HFD group (Fig. 6F).
Fig. 6.

Activation of endoplasmic reticulum (ER) stress proteins PKR-like ER kinase (PERK), inositol requiring-1α (IRE1α), c-jun NH2-terminal kinase (JNK), NF-κB, and expression of C/EBP homologous protein (CHOP). A–D: density analysis of Western blots showed that HFD in intact rats induced liver activation of PERK by phosphorylation (P < 0.05, compared with I+RCD), but not P65 of nuclear factor-κB (P65NF-κB), IRE1α, and JNK. C+HFD demonstrated further enhancement of all ER stress proteins, including PERK, IRE1α, JNK, and P65NF-κB (P < 0.05, compared with I+HFD). T replacement suppressed phosphorylation of all these proteins (P < 0.05, compared with C+HFD). E: expression of CHOP increased in I+HFD and was further increased in C+HFD, but suppressed by T replacement (compared with C+HFD, P < 0.05). GAPDH was used as the loading control. Values are expressed as means ± SE. F: TEM showing ultrastructure of hepatocyte: mitochondria (black arrow), lipid droplets (star), endoplasmic reticulum (arrowhead), and the contact between ER and lipid droplet (empty arrow). TEM magnification ×30,000.
DISCUSSION
The molecular mechanisms by which T deficiency is involved in the pathogenesis of simple steatosis and steatohepatitis independent of insulin resistance are poorly understood. Hepatic fat accumulation can result from increased fat supply to the liver, increased hepatic lipogenesis (including fatty acid de novo synthesis), reduced hepatic fatty acid β-oxidation, and/or suppressed lipid export from the liver (in the form of VLDL assembly and secretion) (18, 46). We have previously shown 1) food intake was not increased in castrated rats fed a HFD; 2) the enzymes regulating fatty acid synthesis were not increased; and 3) hepatic steatosis was not associated with insulin resistance in rats fed a high-fat, low-carbohydrate diet. The mechanism for the T-protective effect against steatohepatitis is, therefore, probably not attributable to excessive fat influx to the liver.
In this study, we first studied mitochondrial morphology and β-oxidation of fatty acids (9) as potential indicators of mitochondrial dysfunction that may contribute to fat accumulation. Although the presence of crystalline inclusions and the decrease in cristae were reported in patients with NAFLD (49), we found no abnormalities in mitochondrial morphology. Lack of differences in CPT-1 gene expression levels in this study and our previously published data on lack of changes in PPARα protein expression suggest that the mechanism of development of castration and HFD-induced hepatic steatosis and protective effects of T are unrelated to mitochondrial fat oxidation. Our results contrast those from other studies demonstrating evidence of reduced β-oxidation in hepatic steatosis (10, 32). It is possible that any reduction of β-oxidation in our study was masked by potential induction mediated by dietary PUFA (13), and therefore, there were no differences compared with the group fed regular chow. However, it is difficult to directly compare the studies due to differences in hypogonadal animal models and in the methods of induction of NAFLD.
Next, we showed definitively that fatty acid synthesis is not enhanced by castration and HFD. Using stable isotopes, we demonstrated that the fraction of newly synthesized fatty acids was suppressed in all groups on the high-fat diet and that there were no changes in intact or castrated animals with or without T replacement. This is consistent with findings reported in our previous publication (41), in which we showed no change in SREBP-1 (68) and FAS (48) expression among castrated rats fed HFD with or without T treatment. The lack of upregulation of de novo synthesis may also be consistent with a lack of suppression of CPT-1 gene expression or reduced oxidation, as malonyl CoA (the product of the first step of de novo synthesis) is known to regulate CPT-1 as an inhibitor. Therefore, the development of steatohepatitis and the protective effect of T treatment in our animal model is not dependent on de novo synthesis in liver.
This study also investigated fatty acid modification and incorporation into triglycerides. SCD-1 is a key enzyme that produces monounsaturated fatty acids to be used as substrates in triglyceride synthesis (42). Liver SCD-1 is positively regulated by saturated fatty acids (42) and negatively by unsaturated fatty acids (14). The suppression of liver SCD-1 gene expression in the intact and castrated rats fed a HFD is, therefore, possibly due to negative regulation from the high proportion of polyunsaturated and monounsaturated fatty acids in the HFD (The 71% energy derived from fat comprises 44.5% monounsaturated, 42.3% polyunsaturated, and 13.2% saturated fatty acid.). Of note, SCD-1 gene expression was also further suppressed by castration in our rat model, although T replacement did not lead to improvement. We also calculated the isotopic desaturation index, the product-to-precursor ratio of newly synthesized fatty acids, as a reflection of SCD-1 activity (67). Under conditions of the HFD, the lack of statistically significant suppression of the isotopic desaturation index in C+HFD in the setting of low SCD-1 gene expression suggests a subnormal response among hypogonadal animals, but at the level of protein synthesis or enzyme activity. Although other studies suggest induction of lipogenic gene expression in NAFLD (10, 24, 25, 32), our study results may differ due to variation in dietary composition of the models studied. More studies are needed to determine whether there are androgen effects on hepatic SCD-1, and the potential role of T in dietary modulation by exposure to a healthier fat composition (rich in monosaturated and polyunsaturated fatty acids and low in saturated ones) in the HFD.
Together with the lack of significant suppression of the isotopic desaturation index, our finding of reduced GPAT-1 expression in castrated rats suggests that lipogenesis contributing to triglyceride synthesis may still be affected by T deficiency (Fig. 7). GPAT-1 is a mitochondrial enzyme that uses de novo synthesized fatty acids as substrates for triglyceride synthesis (39, 61), and overexpression of GPAT-1 in the rat leads to hepatic steatosis and insulin resistance (40). T replacement significantly induced GPAT-1 expression, suggesting that T possibly enhances new triglyceride for VLDL assembly. Although the liver triglyceride content per unit weight was not different in HFD groups (41), this study was limited in that we did not quantify total liver weight or size, and, therefore, cannot exclude the possibility of gross hepatomegaly as a reflection of lipid accumulation.
Fig. 7.

Mechanisms of action of testosterone (T) in inducing hepatic steatosis/steatohepatitis. In our study of HFD-induced steatosis and steatohepatitis in castrated rats, T replacement suppressed the endoplasmic reticulum (ER) stress pathway (PERK, IRE1α, JNK, NF-κB, and CHOP); inhibited production of lipid droplet proteins associated with large lipid droplets (Perilipin 1 and Fsp27); stimulated glycerol-3-phosphate acyltransferase-1 (GPAT-1) expression; and promoted lipid export through enhanced expression of ApoB100, apolipoprotein B-100 (ApoB100), and microsomal triglyceride transfer protein (MTP). CM, chylomicron; NEFA, nonesterified fatty acids; FAS, fatty acid synthase; SREBP-1, sterol regulatory element-binding protein 1; PERK, PKR-like ER kinase; IRE-1α, inositol requiring-1α; CHOP, C/EBP homologous protein; JNK, c-jun NH2-terminal kinase; P65NF-κB, P65 of nuclear factor-κB; PLIN1, perilipin 1; Fsp27, fat-specific protein 27; SCD1, stearoyl-CoA desaturase-1; GPAT1, glycerol-3-phosphate acyltransferase-1; DGAT1, diacylglycerol O-acyltransferase-1; LPA, lysophosphatidic acid; PL, phospholipid; LCFA, long-chain fatty acids; DAG, diacylglycerol; TG, triglyceride; PPARα, peroxisome proliferator-activated receptor α; CPT1a, carnitine palmitoyltransferase 1a; HSL, hormone-sensitive lipase; ATGL, adipose triglyceride lipase.
Our studies on ApoB and MTP expression indicate that T replacement enhances lipid turnover and export. Triglycerides made in the liver are exported to the circulation in VLDL (Fig. 7). ApoB100 is a constitutively expressed glycoprotein in hepatocytes, and is associated with lipid efflux through secretion in VLDL (19). Microsomal triglyceride transfer protein (MTP) is also a critical regulator for VLDL assembly and secretion. MTP gene knockout animals (47, 62) demonstrate decreased plasma lipoprotein levels, while overexpression of MTP cDNA in mouse liver led to increased hepatic secretion of VLDL containing apoB100 (29, 56). Our data demonstrated that ApoB gene and protein expression along with MTP protein expression were suppressed by HFD in both intact and castrated animals, suggesting that decreased ApoB 100 and MTP expression may have contributed toward impairment of hepatic lipid export. T replacement in castrated animals restored ApoB 100 and MTP expression, enhancing VLDL assembly in the ER for lipid efflux, and, thus, attenuated steatosis; however, this does not explain why these proteins are at decreased levels in intact animals.
Because HFD induced hepatic macrovesicular steatosis in castrated animals, we investigated the regulation of formation of lipid droplets. Lipid droplets are dynamic organelles that function not only as lipid repository and storage structures, but also in compartmentalization of lipids to protect cells from adverse consequences of overaccumulation (16). Droplet size and function are believed to be mediated by lipid droplet proteins that bind to their surfaces (43). PLIN1 and Fsp27 have been reported to be associated with larger lipid droplet formation in cells (43, 65), with PLIN1 activating Fsp27 in adipocytes (54). High Fsp27 expression has been observed in ob/ob mice with hepatic steatosis (38). In the present study, expression of PLIN1 and Fsp27 was unaffected by HFD in intact animals, but significant increases in both proteins occurred in castrated animals, which were reversed by T replacement. These data, coupled with the previously published liver histology (41), suggest that the development of macrovesicular steatosis in castrated animals is mediated by T deficiency and is attenuated through T replacement. Such findings of involvement of regulators of droplet size in hepatic steatosis associated with T deficiency have not been previously reported.
ER stress overactivation is an important contributor to both development of steatosis and progression to steatohepatitis (36, 45, 71). Insufficient adaptation to ER stress may result in adverse outcomes, such as fat accumulation, inflammation, and apoptosis (7), all of which have been found in HFD-induced steatohepatitis in our T-deficient rat model (41). ER stress led to increased downstream factors, such as JNK, NF-κB, and CHOP (also known as GADD153) (44), all known to be associated with apoptosis and inflammation, with CHOP implicated in the development of NASH (63). We showed that two main ER-transmembrane transducers, PERK and IRE1α, are activated by HFD in intact animals, and are further heightened in castrated animals, leading to subsequent activation of CHOP, JNK, and NF-κB. Therefore, overactivation of the ER stress pathway plays a crucial role in the progression of hepatic steatosis to steatohepatitis in castrated animals fed a HFD reversed by T replacement. The role of the type of dietary fats in hepatocyte ER stress has been previously demonstrated by Pagliassotti and colleagues (58, 60). They found that exposure of animals or cell lines to a high-saturated fat supply was associated with enhanced expression of endoplasmic reticulum stress markers, such as spliced X-box binding protein-1, glucose-regulated protein 78, and IRE1α, while a high-unsaturated fatty acid supply did not induce such changes. On the basis of those findings, we propose that ER stress in our model of NAFLD is primarily driven by T deficiency in the setting of a HFD, regardless of the type of fat. T replacement resulted in restoration of all of these factors to expression levels observed in intact animals. Moreover, these data add to the evidence that T replacement promotes overall ER health because VLDL assembly and lipid droplet formation also depend on the ER networks and are compromised by castration and restored by T replacement in the HFD environment.
We have shown that insulin resistance did not occur, and insulin signaling-related proteins did not change with castration or T replacement (41). While other T-deficient animal models demonstrated significant hepatic steatosis associated with insulin resistance (50, 70), our model of adult androgen deficiency-related steatohepatitis is different from other studies because a HFD with low carbohydrate did not induce insulin resistance, allowing us to examine the direct effects of T on hepatic steatosis. Sun and Lazar (55) summarized that the cause-and-effect relationship between hepatic steatosis and diabetes does not exist in all situations. By using the present model, our findings suggest that insulin resistance is not necessary for development of NAFLD and that the protective effect of T against NASH is primarily based on the influence on ER stress, lipid droplet formation, and lipid efflux without an influence on insulin signaling.
Male ARKO mice exhibited increased adiponectin, normal insulin sensitivity, and decreased lipolysis (15, 64). However, other studies reported that aging male ARKO mice developed obesity, insulin resistance, and increased liver and muscle triglyceride accumulation (31, 32). Liver-specific eugonadal male ARKO mice fed a HFD also developed hepatic steatosis and insulin resistance (32). The differences between these studies indicate that the androgen receptor may play a complicated and significant role in fatty liver and metabolic syndrome.
In summary (Fig. 7), our study demonstrates that HFD with androgen deficiency induces hepatic steatosis and progression to steatohepatitis through activation of the ER stress pathway, promotion of large lipid droplet formation, and suppression of ApoB- and MTP-mediated VLDL export. T replacement reverses these actions, resulting in subsequent amelioration of inflammation, apoptosis, and lipid accumulation. These direct effects of T are independent of insulin signaling.
GRANTS
The study was supported by a grant from the General Clinical Research Center (MO1 RR00425) to L. Nikolaenko and the UCLA Clinical and Translational Science Institute (1UL1TR000124) at Los Angeles Biomedical Research Institute (LA BioMed) and Harbor-UCLA Medical Center, and the Endocrine, Metabolism and Nutrition Training Grant (T32 DK-007571) and the Summer Student Fellowship Program at LA BioMed.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.J., C.W., L.N., M.D.-A., and R.S.S. conceived and designed research; Y.J., J.K.Y., L.N., J.N.C., and S.W.F. performed experiments; Y.J., J.K.Y., S.W.F., P.Y.L., and W.-N.P.L. analyzed data; Y.J., J.K.Y., C.W., S.W.F., Y.L., and W.-N.P.L. interpreted results of experiments; Y.J. and J.K.Y. prepared figures; Y.J. and J.K.Y. drafted manuscript; Y.J., J.K.Y., C.W., and R.S.S. edited and revised manuscript; Y.J., J.K.Y., C.W., and R.S.S. approved final version of manuscript.
ACKNOWLEDGMENTS
This study was presented in part at the Annual Meeting of the Endocrine Society in Chicago, June 2014 and the Annual Meeting of the Endocrine Society in Orlando, April 2017.
REFERENCES
- 1.Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology 39: 909–914, 2004. doi: 10.1002/hep.20140. [DOI] [PubMed] [Google Scholar]
- 2.Adams LA, Lymp JF, St. Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129: 113–121, 2005. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 3.Adams LA, Waters OR, Knuiman MW, Elliott RR, Olynyk JK. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. Am J Gastroenterol 104: 861–867, 2009. doi: 10.1038/ajg.2009.67. [DOI] [PubMed] [Google Scholar]
- 4.Allan CA, Strauss BJ, Burger HG, Forbes EA, McLachlan RI. Testosterone therapy prevents gain in visceral adipose tissue and loss of skeletal muscle in nonobese aging men. J Clin Endocrinol Metab 93: 139–146, 2008. doi: 10.1210/jc.2007-1291. [DOI] [PubMed] [Google Scholar]
- 5.Allan CA, Strauss BJ, Burger HG, Forbes EA, McLachlan RI. The association between obesity and the diagnosis of androgen deficiency in symptomatic ageing men. Med J Aust 185: 424–427, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Allen NE, Appleby PN, Davey GK, Key TJ. Lifestyle and nutritional determinants of bioavailable androgens and related hormones in British men. Cancer Causes Control 13: 353–363, 2002. doi: 10.1023/A:1015238102830. [DOI] [PubMed] [Google Scholar]
- 7.Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic Res 49: 1405–1418, 2015. doi: 10.3109/10715762.2015.1078461. [DOI] [PubMed] [Google Scholar]
- 8.Bellentani S, Marino M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann Hepatol 8, Suppl 1: S4–S8, 2009. [PubMed] [Google Scholar]
- 9.Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 25: 495–520, 2004. doi: 10.1016/j.mam.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Cai Z, Jiang X, Pan Y, Chen L, Zhang L, Zhu K, Cai Y, Ling Y, Chen F, Xu X, Chen M. Transcriptomic analysis of hepatic responses to testosterone deficiency in miniature pigs fed a high-cholesterol diet. BMC Genomics 16: 59, 2015. doi: 10.1186/s12864-015-1283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55: 2005–2023, 2012. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 12.Chen KL, Chi WT, Chu C, Chen RS, Chiou PW. Effect of caponization and testosterone implantation on hepatic lipids and lipogenic enzymes in male chickens. Poult Sci 86: 1754–1759, 2007. doi: 10.1093/ps/86.8.1754. [DOI] [PubMed] [Google Scholar]
- 13.Clarke SD. Polyunsaturated fatty acid regulation of gene transcription: a molecular mechanism to improve the metabolic syndrome. J Nutr 131: 1129–1132, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Enser M, Roberts JL. The regulation of hepatic stearoyl-coenzyme A desaturase in obese-hyperglycaemic (ob/ob) mice by food intake and the fatty acid composition of the diet. Biochem J 206: 561–570, 1982. doi: 10.1042/bj2060561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan W, Yanase T, Nomura M, Okabe T, Goto K, Sato T, Kawano H, Kato S, Nawata H. Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes 54: 1000–1008, 2005. doi: 10.2337/diabetes.54.4.1000. [DOI] [PubMed] [Google Scholar]
- 16.Farese RV Jr, Walther TC. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139: 855–860, 2009. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita K, Nozaki Y, Wada K, Yoneda M, Fujimoto Y, Fujitake M, Endo H, Takahashi H, Inamori M, Kobayashi N, Kirikoshi H, Kubota K, Saito S, Nakajima A. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 50: 772–780, 2009. doi: 10.1002/hep.23094. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Baró MR, Lewin TM, Coleman RA. Regulation of triglyceride metabolism. II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am J Physiol Gastrointest Liver Physiol 292: G1195–G1199, 2007. doi: 10.1152/ajpgi.00553.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greeve J. Inhibition of the synthesis of apolipoprotein B-containing lipoproteins. Handb Exp Pharmacol 170: 483–517, 2005. doi: 10.1007/3-540-27661-0_18. [DOI] [PubMed] [Google Scholar]
- 20.Haider A, Gooren LJ, Padungtod P, Saad F. Improvement of the metabolic syndrome and of non-alcoholic liver steatosis upon treatment of hypogonadal elderly men with parenteral testosterone undecanoate. Exp Clin Endocrinol Diabetes 118: 167–171, 2010. doi: 10.1055/s-0029-1202774. [DOI] [PubMed] [Google Scholar]
- 21.Hoyos CM, Yee BJ, Phillips CL, Machan EA, Grunstein RR, Liu PY. Body compositional and cardiometabolic effects of testosterone therapy in obese men with severe obstructive sleep apnoea: a randomised placebo-controlled trial. Eur J Endocrinol 167: 531–541, 2012. doi: 10.1530/EJE-12-0525. [DOI] [PubMed] [Google Scholar]
- 22.Jensen TK, Andersson AM, Jørgensen N, Andersen AG, Carlsen E, Petersen JH, Skakkebæk NE. Body mass index in relation to semen quality and reproductive hormones among 1,558 Danish men. Fertil Steril 82: 863–870, 2004. doi: 10.1016/j.fertnstert.2004.03.056. [DOI] [PubMed] [Google Scholar]
- 23.Kathirvel E, Morgan K, French SW, Morgan TR. Acetyl-l-carnitine and lipoic acid improve mitochondrial abnormalities and serum levels of liver enzymes in a mouse model of nonalcoholic fatty liver disease. Nutr Res 33: 932–941, 2013. doi: 10.1016/j.nutres.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Kelly DM, Nettleship JE, Akhtar S, Muraleedharan V, Sellers DJ, Brooke JC, McLaren DS, Channer KS, Jones TH. Testosterone suppresses the expression of regulatory enzymes of fatty acid synthesis and protects against hepatic steatosis in cholesterol-fed androgen-deficient mice. Life Sci 109: 95–103, 2014. doi: 10.1016/j.lfs.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Kelly DM, Akhtar S, Sellers DJ, Muraleedharan V, Channer KS, Jones TH. Testosterone differentially regulates targets of lipid and glucose metabolism in liver, muscle and adipose tissues of the testicular feminised mouse. Endocrine 54: 504–515, 2016. doi: 10.1007/s12020-016-1019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Kwon H, Park JH, Cho B, Kim D, Oh SW, Lee CM, Choi HC. A low level of serum total testosterone is independently associated with nonalcoholic fatty liver disease. BMC Gastroenterol 12: 69, 2012. doi: 10.1186/1471-230X-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Takayanagi R, Nakamuta M. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 20: 351–358, 2007. [PubMed] [Google Scholar]
- 28.Lee WN, Bassilian S, Lim S, Boros LG. Loss of regulation of lipogenesis in the Zucker diabetic (ZDF) rat. Am J Physiol Endocrinol Metab 279: E425–E432, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Liao W, Kobayashi K, Chan L. Adenovirus-mediated overexpression of microsomal triglyceride transfer protein (MTP): mechanistic studies on the role of MTP in apolipoprotein B-100 biogenesis. Biochemistry 38: 7532–7544, 1999. doi: 10.1021/bi9904196. [DOI] [PubMed] [Google Scholar]
- 30.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr 79: 502–509, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Lin HY, Xu Q, Yeh S, Wang RS, Sparks JD, Chang C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes 54: 1717–1725, 2005. doi: 10.2337/diabetes.54.6.1717. [DOI] [PubMed] [Google Scholar]
- 32.Lin HY, Yu IC, Wang RS, Chen YT, Liu NC, Altuwaijri S, Hsu CL, Ma WL, Jokinen J, Sparks JD, Yeh S, Chang C. Increased hepatic steatosis and insulin resistance in mice lacking hepatic androgen receptor. Hepatology 47: 1924–1935, 2008. doi: 10.1002/hep.22252. [DOI] [PubMed] [Google Scholar]
- 33.London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis 11: 55–74, 2007. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Lowenstein JM, Brunengraber H, Wadke M. Measurement of rates of lipogenesis with deuterated and tritiated water. Methods Enzymol 35: 279–287, 1975. doi: 10.1016/0076-6879(75)35165-3. [DOI] [PubMed] [Google Scholar]
- 35.Lue Y, Hikim AP, Wang C, Im M, Leung A, Swerdloff RS. Testicular heat exposure enhances the suppression of spermatogenesis by testosterone in rats: the “two-hit” approach to male contraceptive development. Endocrinology 141: 1414–1424, 2000. doi: 10.1210/endo.141.4.7416. [DOI] [PubMed] [Google Scholar]
- 36.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol 54: 795–809, 2011. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mårin P, Arver S. Androgens and abdominal obesity. Baillieres Clin Endocrinol Metab 12: 441–451, 1998. doi: 10.1016/S0950-351X(98)80191-2. [DOI] [PubMed] [Google Scholar]
- 38.Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, Gonzalez FJ. Hepatic steatosis in leptin-deficient mice is promoted by the PPARγ target gene Fsp27. Cell Metab 7: 302–311, 2008. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagle CA, An J, Shiota M, Torres TP, Cline GW, Liu ZX, Wang S, Catlin RL, Shulman GI, Newgard CB, Coleman RA. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J Biol Chem 282: 14807–14815, 2007. doi: 10.1074/jbc.M611550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, Romanelli AJ, Cline GW, Pongratz RL, Zhang XM, Choi CS, Coleman RA, Shulman GI. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab 2: 55–65, 2005. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Nikolaenko L, Jia Y, Wang C, Diaz-Arjonilla M, Yee JK, French SW, Liu PY, Laurel S, Chong C, Lee K, Lue Y, Lee WN, Swerdloff RS. Testosterone replacement ameliorates nonalcoholic fatty liver disease in castrated male rats. Endocrinology 155: 417–428, 2014. doi: 10.1210/en.2013-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ntambi JM. Dietary regulation of stearoyl-CoA desaturase 1 gene expression in mouse liver. J Biol Chem 267: 10925–10930, 1992. [PubMed] [Google Scholar]
- 43.Okumura T. Role of lipid droplet proteins in liver steatosis. J Physiol Biochem 67: 629–636, 2011. doi: 10.1007/s13105-011-0110-6. [DOI] [PubMed] [Google Scholar]
- 44.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11: 381–389, 2004. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 45.Pagliassotti MJ. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu Rev Nutr 32: 17–33, 2012. doi: 10.1146/annurev-nutr-071811-150644. [DOI] [PubMed] [Google Scholar]
- 46.Postic C, Girard J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab 34: 643–648, 2008. doi: 10.1016/S1262-3636(08)74599-3. [DOI] [PubMed] [Google Scholar]
- 47.Raabe M, Flynn LM, Zlot CH, Wong JS, Véniant MM, Hamilton RL, Young SG. Knockout of the abetalipoproteinemia gene in mice: reduced lipoprotein secretion in heterozygotes and embryonic lethality in homozygotes. Proc Natl Acad Sci USA 95: 8686–8691, 1998. doi: 10.1073/pnas.95.15.8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Radenne A, Akpa M, Martel C, Sawadogo S, Mauvoisin D, Mounier C. Hepatic regulation of fatty acid synthase by insulin and T3: evidence for T3 genomic and nongenomic actions. Am J Physiol Endocrinol Metab 295: E884–E894, 2008. doi: 10.1152/ajpendo.90438.2008. [DOI] [PubMed] [Google Scholar]
- 49.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120: 1183–1192, 2001. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 50.Senmaru T, Fukui M, Okada H, Mineoka Y, Yamazaki M, Tsujikawa M, Hasegawa G, Kitawaki J, Obayashi H, Nakamura N. Testosterone deficiency induces markedly decreased serum triglycerides, increased small dense LDL, and hepatic steatosis mediated by dysregulation of lipid assembly and secretion in mice fed a high-fat diet. Metabolism 62: 851–860, 2013. doi: 10.1016/j.metabol.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 51.Serviddio G, Giudetti AM, Bellanti F, Priore P, Rollo T, Tamborra R, Siculella L, Vendemiale G, Altomare E, Gnoni GV. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid β-oxidation in rats fed a methionine-choline deficient diet. PLoS One 6: e24084, 2011. doi: 10.1371/journal.pone.0024084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stratton LG, Ewing LL, Desjardins C. Efficacy of testosterone-filled polydimethylsiloxane implants in maintaining plasma testosterone in rabbits. J Reprod Fertil 35: 235–244, 1973. doi: 10.1530/jrf.0.0350235. [DOI] [PubMed] [Google Scholar]
- 53.Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 47: 1936–1946, 2008. doi: 10.1002/hep.22268. [DOI] [PubMed] [Google Scholar]
- 54.Sun Z, Gong J, Wu H, Xu W, Wu L, Xu D, Gao J, Wu JW, Yang H, Yang M, Li P. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat Commun 4: 1594, 2013. doi: 10.1038/ncomms2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun Z, Lazar MA. Dissociating fatty liver and diabetes. Trends Endocrinol Metab 24: 4–12, 2013. doi: 10.1016/j.tem.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tietge UJ, Bakillah A, Maugeais C, Tsukamoto K, Hussain M, Rader DJ. Hepatic overexpression of microsomal triglyceride transfer protein (MTP) results in increased in vivo secretion of VLDL triglycerides and apolipoprotein B. J Lipid Res 40: 2134–2139, 1999. [PubMed] [Google Scholar]
- 57.van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, London R, Peduto T, Chisholm DJ, George J. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology 48: 449–457, 2008. doi: 10.1002/hep.22350. [DOI] [PubMed] [Google Scholar]
- 58.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 147: 943–951, 2006. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Ausman LM, Russell RM, Greenberg AS, Wang XD. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J Nutr 138: 1866–1871, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291: E275–E281, 2006. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 61.Wendel AA, Cooper DE, Ilkayeva OR, Muoio DM, Coleman RA. Glycerol-3-phosphate acyltransferase (GPAT)-1, but not GPAT4, incorporates newly synthesized fatty acids into triacylglycerol and diminishes fatty acid oxidation. J Biol Chem 288: 27299–27306, 2013. doi: 10.1074/jbc.M113.485219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wetterau JR, Gregg RE, Harrity TW, Arbeeny C, Cap M, Connolly F, Chu CH, George RJ, Gordon DA, Jamil H, Jolibois KG, Kunselman LK, Lan SJ, Maccagnan TJ, Ricci B, Yan M, Young D, Chen Y, Fryszman OM, Logan JV, Musial CL, Poss MA, Robl JA, Simpkins LM, Slusarchyk WA, Sulsky R, Taunk P, Magnin DR, Tino JA, Lawrence RM, Dickson JK Jr, Biller SA. An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science 282: 751–754, 1998. doi: 10.1126/science.282.5389.751. [DOI] [PubMed] [Google Scholar]
- 63.Willy JA, Young SK, Stevens JL, Masuoka HC, Wek RC. CHOP links endoplasmic reticulum stress to NF-κB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol Biol Cell 26: 2190–2204, 2015. doi: 10.1091/mbc.E15-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yanase T, Fan W, Kyoya K, Min L, Takayanagi R, Kato S, Nawata H. Androgens and metabolic syndrome: lessons from androgen receptor knock out (ARKO) mice. J Steroid Biochem Mol Biol 109: 254–257, 2008. doi: 10.1016/j.jsbmb.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 65.Yang H, Galea A, Sytnyk V, Crossley M. Controlling the size of lipid droplets: lipid and protein factors. Curr Opin Cell Biol 24: 509–516, 2012. doi: 10.1016/j.ceb.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 66.Yang LY, Kuksis A, Myher JJ, Steiner G. Contribution of de novo fatty acid synthesis to very low density lipoprotein triacylglycerols: evidence from mass isotopomer distribution analysis of fatty acids synthesized from [2H6]ethanol. J Lipid Res 37: 262–274, 1996. [PubMed] [Google Scholar]
- 67.Yee JK, Lee WN, Han G, Ross MG, Desai M. Organ-specific alterations in fatty acid de novo synthesis and desaturation in a rat model of programmed obesity. Lipids Health Dis 10: 72, 2011. doi: 10.1186/1476-511X-10-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yellaturu CR, Deng X, Cagen LM, Wilcox HG, Park EA, Raghow R, Elam MB. Posttranslational processing of SREBP-1 in rat hepatocytes is regulated by insulin and cAMP. Biochem Biophys Res Commun 332: 174–180, 2005. doi: 10.1016/j.bbrc.2005.04.112. [DOI] [PubMed] [Google Scholar]
- 69.Yu IC, Lin HY, Liu NC, Wang RS, Sparks JD, Yeh S, Chang C. Hyperleptinemia without obesity in male mice lacking androgen receptor in adipose tissue. Endocrinology 149: 2361–2368, 2008. doi: 10.1210/en.2007-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang H, Liu Y, Wang L, Li Z, Zhang H, Wu J, Rahman N, Guo Y, Li D, Li N, Huhtaniemi I, Tsang SY, Gao GF, Li X. Differential effects of estrogen/androgen on the prevention of nonalcoholic fatty liver disease in the male rat. J Lipid Res 54: 345–357, 2013. doi: 10.1194/jlr.M028969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang XQ, Xu CF, Yu CH, Chen WX, Li YM. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 20: 1768–1776, 2014. doi: 10.3748/wjg.v20.i7.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]