

Abstract
The erythropoietin receptor (EpoR) is widely expressed but its renoprotective action is unexplored. To examine the role of EpoR in vivo in the kidney, we induced acute kidney injury (AKI) by ischemia-reperfusion in mice with different EpoR bioactivities in the kidney. EpoR bioactivity was reduced by knockin of wild-type human EpoR, which is hypofunctional relative to murine EpoR, and a renal tubule-specific EpoR knockout. These mice had lower EPO/EpoR activity and lower autophagy flux in renal tubules. Upon AKI induction, they exhibited worse renal function and structural damage, more apoptosis at the acute stage (<7 days), and slower recovery with more tubulointerstitial fibrosis at the subacute stage (14 days). In contrast, mice with hyperactive EpoR signaling from knockin of a constitutively active human EpoR had higher autophagic flux, milder kidney damage, and better renal function at the acute stage but, surprisingly, worse tubulointerstitial fibrosis and renal function at the subacute stage. Either excess or deficient EpoR activity in the kidney was associated with abnormal peritubular capillaries and tubular hypoxia, creating a “U-shaped” relationship. The direct effects of EpoR on tubular cells were confirmed in vitro by a hydrogen peroxide model using primary cultured proximal tubule cells with different EpoR activities. In summary, normal erythropoietin (EPO)/EpoR signaling in renal tubules provides defense against renal tubular injury maintains the autophagy-apoptosis balance and peritubular capillary integrity. High and low EPO/EpoR bioactivities both lead to vascular defect, and high EpoR activity overides the tubular protective effects in AKI recovery.
Keywords: AKI, autophagy, EpoR, peritubular capillary, tubulointerstitial fibrosis
INTRODUCTION
Erythropoietin (EPO) is a glycoprotein hormone that binds and signals through the erythropoietin receptor (EpoR) and is required for erythropoiesis (50, 60). EPO also has cytoprotective properties against ischemic injury in a variety of extra-hematopoietic tissues including brain, heart, and kidney (10, 23, 25, 47, 65, 79, 83, 88, 96). The cytoprotection by EPO has been proposed to involve other receptors such as the β-common receptor (βcR) (63). Whether the renoprotection conferred by EPO against ischemia-reperfusion injury (IRI) is dependent on EpoR has not been studied.
We previously showed that functional EpoR is present in rat kidney and in a renal tubular epithelial cell line (34). Overexpression of EpoR in renal cells protected cells against hydrogen peroxide (H2O2) cytotoxicity and apoptosis, while knockdown of endogenous EpoR rendered cells more vulnerable to H2O2-induced cytotoxicity and apoptosis, proving that EpoR mitigates kidney cell damage induced by oxidative stress (34). However, the protective role of EpoR in vivo has not been examined.
The pathophysiology of IRI-induced acute kidney injury (AKI) is complex and composed of renal tubular and endothelial cell injury, apoptosis and necrosis, infiltration of inflammatory cells, release of proinflammatory and profibrotic cytokines, and oxidative stress at the acute injury stage, followed by subsequent renal tubular regeneration, angiogenesis, and regression of inflammation and fibrosis at the recovery stage (3, 8, 22, 51, 54). EPO/EpoR signaling is potentially involved in the onset, progression, or recovery of AKI. We used mice where EpoR signaling activity was altered via genetic manipulation to examine whether EpoR signaling activity affects the severity of kidney damage and the outcome after IRI. We provide proof-of-concept data as well as some molecular mechanisms underlying how EpoR signaling modifies IRI-induced AKI and renal fibrosis post-AKI. In the acute phase, there is a direct positive relationship between EpoR level and activity in the renal tubule and renoprotection. The situation post-AKI differs. Post-AKI recovery is optimal at normal EpoR levels and worsened with both low and high EpoR activity creating a “U-shaped” relationship. The underlying molecular mechanisms are complex with higher EpoR levels promoting autophagy, which is beneficial, but high EpoR levels also cause vascular defects, which may cast a dominating effect postinjury and impair recovery.
MATERIALS AND METHODS
Animal lines.
All animal experiments were conducted strictly following the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. Animals were housed in a temperature-controlled room with a 12:12-h light-dark cycle and given ad libitum access to tap water and standard rodent chow before the experiments.
All mice were from a 129 S1/SVlmJ (129 SV) background with equal numbers of males and females at 10–12 wk old. Several lines with different levels of EpoR activity were used. Mice with conditional knockout of EpoR in renal tubules (Ksp-Cre;EpoRflox/flox, synonymous with RT-EpoR−/−) were generated by cross-mating floxed EpoR (EpoRflox/flox) mice (generously provided by Dr. H. Wu, University of California Los Angeles, Los Angeles, CA) (87) with Ksp-cadherin promoter driven Cre recombinase (Ksp–Cre) mice (82) (O’Brien Kidney Research Center at the University of Texas Southwestern).
Mice with global knockin of the mutant truncated human (MTH) EpoR after the first tyrosine residue of the intracellular domain with gain-of-function had hyperfunction of EpoR signal transduction (EpoRMTH/MTH, herein referred to as EpoRM/M). Mice with global knockin of the wild-type human (WTH) EpoR mice (hEpoRWTH/WTH, herein referred to as hEpoRW/W) had hypofunctional EpoR signal transduction compared with mice with wild-type of murine EpoR (19) (kindly provided by Dr. J. Prchal, University of Utah, Salt Lake City, UT). Genotyping of hEpoRM/M and hEpoRW/W mice was performed based on published literature (19). IRI was deployed in hEpoRW/W and hEpoRM/− but not in hEpoRM/M mice, because mice homozygous for knockin of the truncated gain-of-function human EpoR were severely polycythemic and hypertensive, while mice heterozygous for this mutant allele (hEpoRM/−) had only mildly elevated hematocrit and are normotensive; thus, for the purpose of our studies, the heterozygous state of hyperactive gain-of-function suffices (19). To examine the association of EpoR levels with autophagy activity in mouse kidney, RT-EpoR−/−, hEpoRW/W, and hEpoRM/− mouse lines were mated with GFP-LC3 reporter mouse line kindly provided by Dr. N. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan). New three animal lines were subject to bilateral IRI.
AKI model.
Bilateral IRI was conducted as described previously (33, 37). Sham animals underwent laparotomy of the same surgical duration and manipulation of the kidneys without arterial clamping.
Blood, urine, and kidney procurement.
At defined times after reperfusion, 24-h urine was collected in individual metabolic cages. At termination, mice were anesthetized with isoflurane and blood was collected in heparinized tubes and centrifuged (3,000 g for 5 min at 4°C), and plasma was stored at −80°C until analysis. Mice were euthanized after kidney harvest while still under anesthesia. One slice of the kidney was fixed with 4% paraformaldehyde and embedded in paraffin block for histological and immunohistological studies; the remainder was snap frozen in liquid N2 and stored at −80°C until analysis.
Plasma and urine chemistry of animals were measured with a Vitros Chemistry Analyzer (Ortho-Clinical Diagnosis, Rochester, NY). Plasma and urine creatinine was measured using a P/ACE MDQ capillary electrophoresis system and photodiode detector (Beckman-Coulter, Fullerton, CA) at 214 nm (66). Mouse plasma EPO was determined by ELISA with mouse erythropoietin quantikine ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instruction.
New antibodies against EpoR.
The synthetic human anti-EpoR antibody (Fab6) was isolated from a phage-displayed library and synthesized as previously described (34). A set of new mouse anti-human EpoR monoclonal antibodies was raised. Animal husbandry and immunizations adhered to animal protocol were approved by Oregon Healthand Science University Institutional Animal Care and Use Committee. In brief, female Balb/c mice (10 wk old) were immunized with three doses of intraperitoneal injection (10 μg hEpoR protein) given 3 wk apart. Mice were euthanized 4 days after the last dose, and splenocytes were harvested and fused with SP2/0 Ag14 myeloma cells. Hybridoma clones were grown in methylcellulose-containing HAT medium (Stem Cell Technologies, Vancouver, BC). A total of 576 isolated hybridoma clones were generated, picked, and transferred to 96-well plates. Supernatants were collected and screened by ELISA. Clones of particular interest were cryopreserved and expanded in culture in DMEM (GIBCO, Grand Island, NY) supplemented with 10% (vol/vol) fetal bovine serum. Supernatants from selected ELISA-positive clones were shipped to University of Texas Southwestern for further characterization in cells and tissue. The 1-B9 antibody was used for this study. We discuss it below.
Other antibodies.
Mouse monoclonal antibody against β-actin (β-actin) was from Sigma-Aldrich (St. Louis, MO); mouse monoclonal antibody against Akt, rabbit antibody against phospho-Akt, and rabbit antibody against cleaved caspase-3 were from Cell Signaling Technology (Danvers, MA); mouse monoclonal antibody against CD31 and rabbit polyclonal antibody against CD31 (CD31) were from Abcam (Cambridge, MA); goat polyclonal antibody against CD34 (CD34) was from R&D Systems; rabbit polyclonal antibody against human Ki67 (Ki67) and, mouse monoclonal antibody against connective tissue growth factor (CTGF) were from Abcam; mouse monoclonal antibody against common βCR was from R&D Systems; rabbit antibody against EpoR (EpoR, M-20) was from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit antibody against phospho- and total extracellular signal-regulated kinase (Erk) was from Cell Signaling Technology; mouse monoclonal antibody against hypoxia-inducible factor-1α (HIF1α) and rabbit polyclonal antibody against HIF2α were from Novus Biologicals (Littleton, CO); mouse antibody against phospho- c-Jun NH2-terminal kinase (JNK) and rabbit antibody against JNK were from Cell Signaling Technology; rabbit antibody against microtubule-associated light chain 3 (LC3) was from Novus Biologicals (Littleton, CO); guinea pig antibody against p62 (p62) was from Progen Biotechnik (Heidelberg, Germany); rabbit antibody against pimondazole with hydroxyprobe-1 kit [pimonidazole (PIM)] was from Pharmacia International (Belmont, MA); mouse monoclonal antibody against α-smooth muscle actin (α-SMA) was from Sigma-Aldrich (St. Louis, MO); rabbit monoclonal antibody against phospho-Stat5 was from Abcam; rabbit polyclonal antibody against VEGF-A (VEGF-A) for immunoblotting and goat polyclonal antibody against VEGF-A for immunohistochemistry were from Santa Cruz Biotechnology; and rat monoclonal antibody against flk-1/VEGFR2 (VEGFR2) was from BD PharMingen (San Jose, CA). Secondary antibodies coupled to horseradish peroxidase for immunoblotting, or to FITC, Alexa Fluor, or Cy5, and Syto 61 infra red fluorescent nuclear acid stain for immunohistochemistry were purchased from Molecular Probes/Invitrogen (Molecular Probes, Eugene, OR). FITC-conjugated Lotus tetragonobus lectin [LTL-lectin (LTL)] was from Vector Laboratories (Burlingame, CA).
Kidney histology and histopathology.
Kidney tissues were fixed in 4% paraformaldehyde for 16 h at 4°C, and 4-μm sections of paraffin-embedded kidney tissues were stained with hematoxylin and eosin and trichrome. Histology was examined and photographed by a researcher blinded to the identity of the samples. A semiquantitative pathological scoring system was used as described previously (37, 66). The total score for each kidney was calculated by summation of all scores from 20 fields with a maximum numeric score of 30 at ×40 magnification. To evaluate renal fibrosis, 4-μm paraffin-embedded kidney sections were stained with Masson trichrome and then examined and photographed by a pathologist blinded to the experimental protocol. Fibrotic area and fibrosis intensity were quantified with ImageJ software with published methods (32, 33, 78).
Primary culture of renal proximal tubules.
Renal tubules were isolated from collagenase-digested cortical fragments of mouse kidneys using previously described protocols with modification (86). Briefly, renal cortexes were dissected visually on petri dishes on ice, and slices were digested with collagenase. After digestion, the supernatant was passed tandemly through two nylon sieves (pore size: 180 and 80 μm, Millipore). The tubules were seeded into plates and left undisturbed for 48 h at 37°C and 5% CO2. After 7 days, post-confluent cells were treated with H2O2 (200 μM; 24 h) in 10% fetal bovine serum. The culture media were collected for lactate dehydrogenase (LDH) determination (Cytotoxicity Detection Kit; TaKaRa Bio, Mountain View, CA).
Immunohistochemistry and immunoblotting in the kidney.
Four-micrometer sections of paraffin-embedded kidney were subjected to immunohistochemistry using standard protocols (35, 36). Total kidney lysate covering all kidney zones was prepared as described previously (35, 36, 66). Fifty micrograms of protein of kidney lysate was solubilized in Laemmli’s sample buffer and fractionated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with different primary antibodies and β-actin for loading control. The signal was visualized with the ECL kit (Perkin-Elmer LAS, Boston, MA). To quantify the number of proliferating cells, triple immunostaining for nuclei with Syto61, proliferating cells with Ki-67, and proximal tubules with FITC-labeled LTL-lectin was performed, and the numbers of Ki-67-positive cells in proximal tubules (LTL-positive) were counted.
Renal hypoxia.
Hypoxia in the kidney was examined with a hydroxyprobe-1 kit (PIM; Pharmacia International, Belmont, MA) with published methods (76). Briefly, PIM HCl was intraperitoneally injected (60 mg/kg body wt) 2 h before terminational study. Paraffin-embedded kidney sections were stained with PIM antibody (Pharmacia International) to detect PIM adducts in renal tubules, and hypoxia area was visualized with fluorescent microscopy.
Real time quantitative RT-qPCR.
Total RNA was extracted using RNAeasy kit (Qiagen, Germantown, MD) from mouse kidneys. Complimentary DNA (cDNA) was generated with Oligo-dT primers using SuperScript III First Strand Synthesis System (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. The primers used for qPCR are shown in Table 1 with the condition described in our previous publications (35, 36, 66). The reaction was performed in triplicate for each sample. Data are expressed at amplification number of 2−ΔΔCt by normalization of cyclophilin and comparison of controls.
Table 1.
Primers used for qPCR
| Forward Primer Sequence | Reverse Primer Sequence | |
|---|---|---|
| CD31 | 5′-TCC CCG AAG CAG CAC TCT T-3′ | 5′-ACC GCA ATG AGC CCT TTC T-3′ |
| EPO | 5′-CAT CTG CGA CAG TCG AGT TCT-3′ | 5′-CAC AAC CCA TCG TGA CAT TTT C-3′ |
| EpoR | 5′-GGA CCC TCT CAT CTT GAC GC-3′ | 5′-CTT GGG ATG CCA GGC CAG AT-3′ |
| NGAL | 5′-CCA GTT CGC CAT GGT ATT TT-3′ | 5′-CCT TGA GGC CCA GAG ACT-3′ |
| VEGF-A | 5′-CCA CGT CAG AGA GCA ACA TCA-3′ | 5′-TCA TTC TCT CTA TGT GCT GGC TTT-3′ |
| VEGFR2 | 5′-ACT GCA GTG ATT GCC ATG TTC T-3′ | 5′-TCA TTG GCC CGC TTA ACG-3′ |
| Cyclophilin | 5′-TGC TCT TTT CGC CGC TTG CT-3′ | 5′-TCT GCT GTC TTT GGA ACT TTG TCT G-3′ |
EPO, erythropoietin; EpoR, erythropoietin receptor; NGAL, neutrophil gelatinase-associated lipocalin.
Statistical analyses.
Data are expressed as means ± SD from six animals unless the number of samples was noted. Analysis was performed with Sigma Plot software (Systat Software, San Jose, CA). As appropriate, statistical analysis was performed using unpaired Student’s t-test or one-way ANOVA followed by the Student-Newman-Keuls post hoc test when applicable. P ≤ 0.05 was considered statistically significant.
RESULTS
IRI induced a transient increase in EpoR in the kidney.
The protective effect of EpoR/EPO signaling on ischemia injury has been described in several organs (10, 23, 25, 34, 47, 65, 74, 79, 83, 88, 91). We examined the biology of EpoR in AKI by first defining the time profile of alteration of native EpoR expression in the kidney after IRI (37). The changes in plasma creatinine (PCr) and blood urea nitrogen (BUN) are as expected (Fig. 1, A and B). Although we combined female and male mice and used mixed gender as one group, we did examine the gender effect on the severity of kidney injury induced by IRI. Male mice had more severe kidney injury and slower renal recovery than females (Table 2). The better renal function in female AKI mice might be due to renoprotection by estrogen and of testosterone on enhancement of susceptibility to ischemic kidney injury, respectively (38, 39, 43, 68, 75).
Fig. 1.

Transient increase in renal erythropoietin receptor (EpoR) expression in ischemic-reperfusion injury (IRI)-induced acute kidney injury (AKI). AKI was induced in wild-type (WT) mice by IRI via renal arterial clamping for 30 min, followed by reperfusion. At predetermined time points, 6 mice from each group were killed and blood were collected for the measurement of plasma creatinine (PCr; A) and blood urea nitrogen (BUN; B). Statistical significance was assessed by unpaired Student’s t-test and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups at same time point for A and B. C: mRNA of EpoR (left), EPO (middle), and neutrophil gelatinase-associated lipocalin (NGAL) (right) by quantitative polymerase chain reaction (qPCR). D: EpoR and β-common receptor (βCR) protein expression in the kidney. Left: representative immunoblots for EpoR determined by 3 EpoR antibodies (synthetic antibody Fab 6, commercial M-20, and monoclonal B9) and βCR. Right: summary of all immunoblots. Data are expressed as means ± SD (n = 6) from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups for C and D. E: characterization of new anti-EpoR monoclonal antibody. BaF3 cells, a murine interleukin 3 (IL-3)-dependent hematopoietic pro-B cell line, was purchased from American Type Culture Collection (Manassas, VA). BaF3-cell (EpoR-null) stably expressing Flag mouse EpoR cell line was generated by transfection of BaF3 cells with COOH-terminal Flag-tagged murine full-length EpoR retroviral vector. BaF3 cells transfected with empty vector only were used for negative control of EpoR expression. After confluence, cell lysate was prepared and subjected to immunoblot. Hybridoma supernatant from selected positive clones (by ELISA) was tested. Representative immunoblots for EpoR protein from 3 independent experiments using monoclonal anti-human EpoR antibodies (top), Flag-tagged EpoR protein with Flag antibody (middle), and β-actin protein (bottom) in native BaF3 cells and BaF3-expressing flag-tagged murine EpoR cells.
Table 2.
Renal function in WT male and female mice
| Both Genders | Female Mice | Male Mice | Female vs. Male Mice | |
|---|---|---|---|---|
| Plasma Cr, mg/dl | ||||
| Day 1 | 0.33 ± 0.04 | 0.31 ± 0.02 | 0.37 ± 0.03 | P = 0.021 |
| Day 4 | 0.12 ± 0.02 | 0.11 ± 0.01 | 0.14 ± 0.03 | P = 0.176 |
| Day 14 | 0.11 ± 0.02 | 0.09 ± 0.01 | 0.13 ± 0.02 | P = 0.036 |
| Plasma BUN, mg/dl | ||||
| Day 1 | 43.5 ± 5.6 | 39.4 ± 2.2 | 47.5 ± 3.9 | P = 0.035 |
| Day 4 | 21.5 ± 4.7 | 17.7 ± 3.5 | 25.8 ± 4.8 | P = 0.075 |
| Day 14 | 24.9 ± 4.2 | 20.8 ± 2.5 | 28.9 ± 3.9 | P = 0.038 |
Data are expressed as means ± SD from 3 mice from each gender, and statistical analysis was analyzed with unpaired Student’s t-test for the differences between female and male mice. Cr, creatine; BUN, blood urea nitrogen; WT, wild-type.
Both EpoR mRNA (Fig. 1C) and protein expression (Fig. 1D) were transiently increased from days 1 to 4 and returned to baseline at ~7 days. Due to previous experience with multiple ambiguous reagents, we used three anti-EpoR antibodies including a newly generated anti-EpoR monoclonal antibody to measure EpoR protein and also corroborated the results with EpoR mRNA. The specificity of our new anti-EpoR monoclonal antibody hEpoR0-1-B9 (1-B9) was validated and is shown in Fig. 1E. In contrast to EpoR, the renal mRNA level of its ligand EPO was transiently reduced at 24 h and returned to baseline in ~4 days (Fig. 1C). Neutrophil gelatinase-associated lipocalin (NGAL) transcript, a kidney injury marker (26), was dramatically increased in the kidney at 24 h and returned to near baseline in 1 wk (Fig. 1D). No change was observed in the expression of βCR protein (Fig. 1D), which has been postulated to mediate EPO cytoprotective effects in the brain and the heart (7, 14).
Mouse lines of different EpoR signaling activities generated by genetic manipulation.
To study the effects of EpoR activity on kidney injury and kidney recovery post-AKI, we used two murine lines of low EPO/EpoR signaling activity. The global hypofunctional model from knockin of wild-type human EpoR, which has lower activity than native murine EpoR (hEpoRWTH/WTH, referred to as hEpoRW/W), was previously described (19). In addition, we generated a renal tubule-specific deletion of EpoR mice (Ksp-Cre+;EpoRflox/flox, referred to as RT-EpoR−/−) mice.
hEpoRW/W mice exhibited weaker EPO/EpoR signaling activity in all tissues including hematopoietic cells as evidenced by the mild anemia with compensatory elevation of renal and plasma EPO (Table 3) (19). In the kidney, hEpoRW/W mice had lower EPO/EpoR signaling activity as shown by lower levels of renal phospho-Akt and phospho-Stat5 at baseline compared with WT mice (Fig. 2, A and B).
Table 3.
Comparison of hematocrit, plasma and renal EPO transcripts in mouse lines with different EpoR signaling activities
| Hematocrit, % | Plasma EPO, pg/ml | Kidney EPO mRNA (−2ΔΔ vs. WT) | |
|---|---|---|---|
| Global low EpoR signaling activity: knockin of human WT EpoR | |||
| WT | 46.8 ± 3.1 | 130.0 ± 13.4 | 1.0 |
| hEpoRW/W | 42.7 ± 2.8* | 212.8 ± 17.9** | 30.5 ± 3.1** |
| Low EpoR signaling activity in renal tubules: conditional knockout of mouse EpoR in the renal tubules | |||
| RT-EpoR+/+(WT) | 46.8 ± 2.3 | 121.8 ± 12.9 | 1.0 |
| RT-EpoR+/− | 48.2 ± 2.9* | 184.9 ± 14.4** | 3.8 ± 0.3** |
| RT-EpoR−/− | 50.6 ± 4.0** | 202.6 ± 20.8**# | 25.5 ± 1.4*## |
| Global high EpoR signaling activity: knockin of human mutant EpoR | |||
| WT | 47.1 ± 3.5 | 127.3 ± 10.1 | 1.0 |
| hEpoRM/− | 51.8 ± 4.1* | 89.8 ± 6.2** | 0.5 ± 0.2** |
Data are expressed as means ± SD from each group. Statistical significance was accepted when
P < 0.05,
P < 0.01 vs. WT mice;
P < 0.05,
P < 0.01 vs. RT-EpoR+/− mice by one-way ANOVA.
Fig. 2.

Measurement of EpoR signaling activity in three genetically modified mouse lines. A and B: EpoR signaling activity in the kidney of WT mice and hEpoRW/W mice at baseline. A: representative immunoblots for phospho-Akt (P-Akt, left), and summary of all immunoblots (right). Data are expressed as means ± SD (n = 6) from each group, statistical significance was assessed by unpaired Student’s t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups. B: representative immunofluorescent images from 3 independent experiments for phospho-Stat5 in renal cortex of WT mice and hEpoRW/W mice. Scale bar = 50 μm. C: generation and genotyping of specific knockout of EpoR in renal tubules. Mice with renal tubule-specific deletion of EpoR in (homozygous: Ksp-Cre+;EpoRflox/flox = RT-EpoR−/−; heterozygous: Ksp-Cre+;EpoRflox/+ = RT-Epo+/− and Ksp-Cre−;WT- EpoRflox/flox = RT-EpoR+/+) were generated after mating Ksp-Cre+;EpoRflox/+ with Ksp-Cre+;EpoRflox/+ mice. Left: PCR of extra-renal tissue (tail) displaying floxed EpoR and WT EpoR bands. Right: PCR results in the kidney and truncated EpoR transcript. Floxed EpoR and WT EpoR bands and truncated EpoR band were present in DNA samples extracted from kidney. D: representative fluorescent immunohistochemical image from 3 independent experiments. Paraffin-embedded kidney sections were stained with anti-Cre (green) and the nuclei with Cyto-61 (blue). E: EpoR expression in the kidney of mice. Representative immunoblot from 4 independent experiments for EpoR with several antibodies and for βcR in the kidney (left) and a summary of all immunoblots (right). F: representative immunoblots from 4 independent experiments for phospho-Akt (P-Akt, left) and a summary of all immunoblots (right). Data are expressed as means ± SD (n = 4) from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups for E and F. G: representative immunofluorescent images for phospho-Stat5 in the kidney. H and I: EpoR signaling activity in the kidney of WT mice and hEpoRM/− mice at baseline. H: representative immunoblots from 4 independent experiments for phospho-Akt (P-Akt, left) and a summary of all immunoblots (right). Data are expressed as means ± SD (n = 4) from each group, statistical significance was assessed by unpaired Student’s t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups. I: representative immunofluorescent images from 3 independent experiments for phospho-Stat5 in the kidney of mice. Bar scale = 50 μm.
In addition, we created a renal tubule-specific deletion of EpoR (RT-EpoR−/−) (Fig. 2C). Cre recombinase protein expression in the kidney is shown in Fig. 2D. RT-EpoR−/− mice had slightly higher hematocrit in an EpoR gene dose-dependent manner, associated with elevation of EPO mRNA in the kidney and EPO protein in plasma in RT-EpoR−/− mice (Table 3). The mechanism of augmentation of EPO and hematocrit is not known but may be in response to renal hypoxia due to vascular rarefaction in the kidney (see below). As expected, RT-EpoR−/− mice had much lower EpoR mRNA and protein in the kidney (Fig. 2E) as well as lower EpoR signaling activity (Fig. 2, F and G), supporting the functional consequences of reduced renal tubule EpoR.
We used a mouse line with knockin of mutant constitutively hyperactive human EpoR (hEpoRMTH/−, referred to as hEpoRM/−). These mice express a truncated EpoR devoid of negative regulatory mechanisms (19). Homozygous mice with knockin of mutant human EpoR (hEpoRMTH/MTH, referred to as hEpoRM/M) had severe polycythemia (19), which resembles primary familial congenital polycythemia in humans, and hence are not suitable for our experiments. In contrast, hEpoRM/− mice had much lower hematocrit than hEpoRM/M, but still higher hematocrit, lower plasma, and renal EPO expression than WT littermates (Table 3), which is consistent with the published data (19). Furthermore, hEpoRM/− mice had increased EpoR signaling activity in the kidney compared with WT mice at baseline (Fig. 2, H and I).
Altered EpoR signaling activity is associated with disturbed peritubular capillary structure.
EPO/EpoR signaling is required for normal angiogenesis and vasculogenesis. Defective EPO/EpoR signaling leads to vascular loss (4, 11, 13, 15, 55), and elevated EPO/EpoR signaling can result in excessive proliferation of capillary beds, which can lead to disruption of structure and loss of function (44, 81). In addition, AKI is associated with vascular rarefaction, which contributes to renal fibrosis and AKI-to-CKD progression (4, 13, 55). Thus we explored whether EpoR levels affect peritubular capillaries. We found lower vascular density in hEpoRW/W mice with reduced EpoR signaling in whole kidney. Interestingly, we also found lower vascular density in RT-EpoR−/− mice whose EpoR signaling activity was reduced selectively in renal tubules (Fig. 3A). Moreover, there was lower expression of CD31 mRNA and protein, an endothelial marker, vascular endothelial growth factor A (VEGF-A), and VEGFR2, the key receptor of VEGF-A in the kidney of hEpoRW/W and RT-EpoR−/− mice, compared with WT mice (Fig. 3, B–E), indicating that peritubular capillary rarefaction is associated with deficient VEGF-A/VEGFR2 signaling. Notably, hEpoRM/− mice showed higher density of but disorganized peritubular capillaries with thicker basement membrane as well as higher CD31, VEGF-A, and VEGFR2 (Fig. 3, A–E) and higher collagen IV with nonquantitative immunohistochemistry (Fig. 4A). These results support the notion that VEGF-A/VEGFR2, a key signaling pathway for vasculogenesis and angiogenesis, may be downstream of EPO/EpoR signaling (62, 94).
Fig. 3.

Both hypofunctional and hyperfunctional EpoR activity induced abnormal architecture of peritubular capillary structure and hypoxia in the kidney. A: representative micrographs from 3 independent experiments of capillary structure outlined by CD31 stain (white) in the kidney of hEpoRW/W mice, EpoR−/− mice, WT mice, and hEpoRM/− mice at baseline. Bar scale = 50 μm. B: representative immunoblots from 4 independent experiments for CD31, VEGF-A, and VEGFR2 protein in the kidney of hEpoRW/W mice, EpoR−/− mice, WT mice, and hEpoRM/− mice at baseline (left) and a summary of all immunoblots (right). C: representative immunofluorescent images from 3 independent experiments of CD34 (red) and VEGF-A (green) in kidneys of hEpoRW/W mice, EpoR−/− mice, WT mice, and hEpoRM/− mice at baseline. Bar scale = 20 μm. D: representative immunofluorescent images from 3 independent experiments of VEGFR2 stained by red fluorescence in cortex and outer medulla (OM) of hEpoRW/W mice, EpoR−/− mice, WT mice, and hEpoRM/− mice at baseline. Bar scale = 50 μm. E: the transcripts of CD31, VEGF-A, and VEGFR2 in kidneys of hEpoRW/W mice, EpoR−/− mice, WT mice, and hEpoRM/− mice at baseline were quantitatively analyzed with qPCR. Results are expressed at amplification number of 2−ΔΔCt by normalization of cyclophilin and comparison of WT mice. Data are expressed as means ± SD (n = 4) from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, ** P < 0.01, between 2 groups for B and E.
Fig. 4.
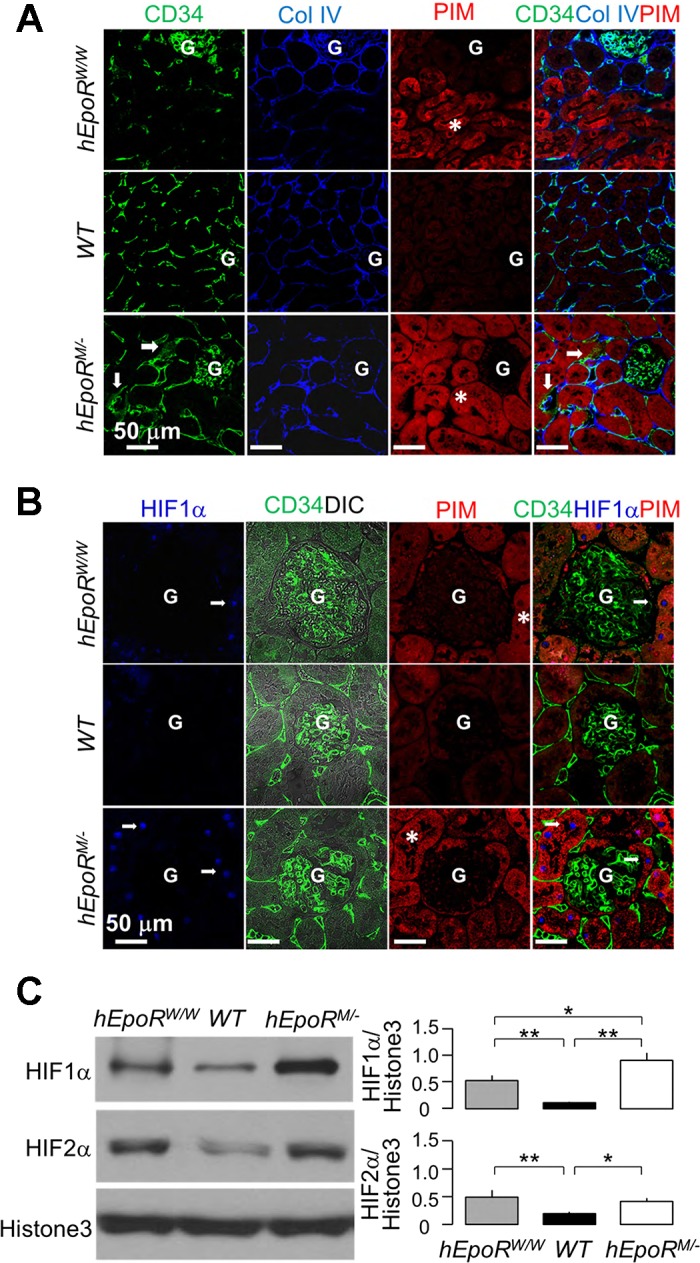
Abnormal EpoR signaling in the kidney leads to hypoxia in the kidney. hEpoRW/W mice, WT mice, hEpoRM/− mice, and hEpoRM/M mice at baseline were intraperitoneally injected with pimonidazole HCl (PIM). Two hours after injection, the kidneys were harvested for immunohistochemistry. Hypoxia was visualized with anti-PIM to detect PIM adducts in the renal tubules. A: representative immunofluorescent images from 3 independent experiments for CD34, Col IV, and PIM in the kidneys. CD34 (green), collagen IV (Col IV, blue), and PIM (red) were costained. Arrows indicate disorganized peritubular capillary with thicker basement membrane. Hypoxic areas are highlighted by massive PIM adducts (asterisks). G: glomerulus. Bar scale = 50 μm. B: representative immunofluorescent images from 3 independent experiments for hypoxia-inducible factor-1α (HIF1α), CD34, and PIM in the kidneys. CD34 (green), HIF1α (blue), and PIM (red) were costained. Arrows indicate nuclear stain pattern of HIF1α. The hypoxic areas are demonstrated by massive PIM adducts (asterisks). G: glomerulus. Bar scale = 50 μm. C: representative immunoblots from 4 independent experiments for HIF1α, HIF2α and histone 3 protein (loading control) in nuclear extracts from the kidney of hEpoRW/W mice, WT mice, hEpoRM/− mice, and hEpoRM/M mice at baseline (top) and a summary of all immunoblots (bottom). Data are expressed as means ± SD (n = 4) from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups.
Aberrant EpoR signaling activity is associated with renal tubular hypoxia.
As there were severe abnormal peritubular vascular structures in mice with either deficient or excessive EpoR signaling activity, we used the hydroxyprobe (PIM) to map hypoxia in the renal tubules to examine the relationship between EPO/EpoR signaling and the severity of renal tubular hypoxia at baseline. There were more PIM adducts in the renal tubules of diminished (hEpoRW/W mice) or excessive EpoR signaling (hEpoRM/+ and hEpoRM/M mice) compared with WT mice (Fig. 4A). Hypoxia was present in areas with less vessel density in hEpoRW/W mice, and in areas with the thicker peritubular, capillary basement membrane stained for collagen IV and disorganized capillary structure in hEpoRM/M mice. Therefore, disturbed peritubular capillary formation such as thicker capillary basement membrane and disorganized peritubular capillary in hEpoRM/M mice can lead to less efficient oxygen delivery to renal tubules and consequently tubule hypoxia even at baseline (Fig. 4A).
To further confirm hypoxia in the kidney of mice at baseline, we measured HIF1α and HIF2α, which are known to respond to hypoxia and contribute to renal fibrosis (27, 29, 58). We found that HIF1α expression was upregulated in highly PIM-labeled tubules (Fig. 4B). A similar pattern of HIF2α expression was also found (data not shown). Immunoblots of HIF showed higher HIF1α and HIF2α proteins in the kidney of both hEpoRW/W and hEpoRM/M mice compared with WT mice (Fig. 4C). Interestingly, both the EpoR-deficient and EpoR-hyperactive mice had vascular rarefaction or abnormal peritubular structure respectively, leading to impairment of oxygen delivery to renal tubules, renal tubular hypoxia, and HIF upregulation (Fig. 4, B and C).
Reduced renal EPO/EpoR signaling activity rendered the kidney more prone to ischemic injury and at higher risk of renal fibrosis.
To examine whether low EpoR signaling in the kidney influences kidney damage and recovery post-AKI, we conducted IRI in hEpoRW/W mice. hEpoRW/W mice had moderately but statistically higher PCr at the acute stage (2–7 days) and slower decline of PCr at the subacute stage (14 days) compared with WT mice (Fig. 5A), indicating that lower EPO/EpoR axis signaling is associated with more severe kidney damage and slower recovery after IRI. hEpoRW/W mice had more histologic damage at the acute stage (higher injury score in the outer medulla, Fig. 5B) and more fibrosis at the subacute stage compared with WT mice (Fig. 5C). Immunoblotting of fibrotic markers were consistent with the trichrome stain with earlier and more robust increase in α-SMA and CTGF in the kidney of hEpoRW/W mice than those in WT mice after IRI (Fig. 5D).
Fig. 5.

hEpoRW/W mice with low EpoR signaling activity had more severe kidney damage induced by IRI. AKI was induced in WT and hEpoRW/W mice by IRI. At predetermined time points, mice were killed, blood was collected and kidneys were harvested. A: plasma creatinine (PCr). Data are expressed as means ± SD (n = 6) from each group, statistical significance was assessed by unpaired Student’s t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups at same time point. B: histologic kidney damage in WT mice and hEpoRW/W mice. Representative micrographs from 4 independent experiments of hematoxylin and eosin (H&E) stain (top) in kidney cortex and OM and kidney injury scores (bottom) obtained in a blinded fashion at days 2 and 7 after IRI. Scale bar = 100 μm. Data are expressed as means ± SD (n = 4) from each group and statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups. C: renal fibrosis in WT mice and hEpoRW/W mice. Representative micrographs from 4 independent experiments of Trichrome stains (left) in kidney sections and renal fibrosis scores (right) obtained in a blinded fashion at day 14 after IRI. Scale bar = 200 μm. Data are expressed as means ± SD (n = 4) from each group, statistical significance was assessed by unpaired Student’s t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups. D: representative immunoblots from 4 independent experiments for α-smooth muscle actin (α-SMA) and connective tissue growth factor (CTGF) in the kidney (top) of WT mice and hEpoRW/W mice at predetermined time points after IRI. Summary of arbitrary units from all immunoblots (bottom). Data are expressed as means ± SD (n = 4) from each group, statistical significance was evaluated by unpaired Student’s t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups at same time point.
The hEpoRW/W knockin mouse has global replacement with the relatively hypofunctional human EpoR, so, strictly speaking, the observed effects can potentially be due to loss of EpoR in tissues other than the renal epithelia. In addition, we need to exclude the potential effects of mild anemia on renal outcome after IR. For this purpose, we conducted AKI model in a renal tubule-specific reduction of EpoR (RT-EpoR−/−) without anemia but actually a slightly higher hematocrit in an EpoR gene dose-dependent manner (Table 3).
After IRI induction, RT-EpoR−/− mice had the highest, and RT-EpoR+/+ mice the lowest, PCr levels with intermediate values in RT-EpoR+/− at the acute stage (Fig. 6A). Compared with RT-EpoR+/− and RT-EpoR+/+ mice, RT-EpoR−/− mice had the slowest recovery of PCr at the subacute stage (Fig. 6A), indicating more severe injury and impaired recovery of renal function in RT-EpoR−/− mice post-IRI.
Fig. 6.

Mice with low EpoR expression in renal tubules had more severe kidney damage and more renal tubulointerstitial fibrosis after acute kidney injury (AKI). AKI was induced in RT-EpoR+/+ mice, RT-EpoR+/− mice, and RT-EpoR−/− mice by bilateral IRI. At predetermined time points, were killed, blood was collected and kidneys were harvested. A: plasma creatinine (PCr). B: Representative micrographs from 4 independent experiments of H&E stain (top) of kidney cortex and OM and kidney injury scores (bottom) obtained in a blinded fashion at days 2 and 7 after IRI. Scale bar = 100 μm. C: renal fibrosis in RT-EpoR+/+ mice, RT-EpoR+/− mice, and RT-EpoR−/− mice. Representative microscopic images from 4 independent experiments of Trichrome stain (left) in kidney sections and renal fibrosis scores (right) obtained in a blinded fashion at day 14 after IRI. Scale bar = 200 μm. Data are expressed as means ± SD (n = 4) from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups for B and C. D: representative immunoblots from 4 independent experiments for α-SMA and CTGF in the kidney (top) of RT-EpoR+/+ mice, RT-EpoR+/− mice, and RT-EpoR−/− mice at predetermined time points after IRI and a summary of all immunoblots (bottom). Data are expressed as means ± SD from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, **P < 0.01 vs. RT-EpoR+/+ mice; #P < 0.05, ##P < 0.01 vs. RT-EpoR+/− mice at same time point for A (n = 6) and D (n = 4).
There were more severe histological changes in the kidney including renal tubular necrosis and tubular casts at the early stage and more interstitial infiltration at the subacute stage with higher injury score in RT-EpoR−/− mice compared with RT-EpoR+/− and RT-EpoR+/+ mice (Fig. 6B). Trichrome staining also showed more renal fibrosis in RT-EpoR−/− mice (Fig. 6C). Fibrotic markers (α-SMA and CTGF protein) in the kidney were significantly higher in RT-EpoR−/− mice, compared with RT-EpoR+/+ mice with normal and RT-EpoR+/− mice with intermediate levels of EpoR (Fig. 6D). Therefore, selective EpoR deficiency in renal tubules was associated with more severe kidney damage, delayed renal recovery, and increase in renal fibrosis post-IRI.
The reduction of EpoR in renal tubule is clearly affecting the vasculature and may engender secondary effects. To define if EpoR deficiency in the renal tubules per se directly renders renal tubules more susceptible to oxidative injury, renal tubules isolated from EpoRW/W or RT-EpoR−/− mice were placed in primary culture, and subjected to an in vitro model of H2O2-induced oxidative damage. Cells from EpoRW/W or RT-EpoR−/− mice had higher LDH release and cells from EpoRM/− mice had lower LDH release compared with cells from WT mice (Fig. 7A).
Fig. 7.

Association of EpoR activity in renal tubules with susceptibility to oxidative stress ex vivo. A: renal proximal tubules were isolated from mice and placed on primary culture. LDH concentration was determined in culture media after 24-h incubation of H2O2 (200 μM). Data are expressed as means ± SD (n = 4) from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups. A and B: AKI was induced in RT-EpoR+/+ mice, RT-EpoR+/− mice, and RT-EpoR−/− mice by bilateral IRI and kidneys were harvested at day 7 post-IRI for immunohistochemistry for Ki67 in the kidney. B: representative immunohistochemistry from 4 independent experiments for Ki67 expression in the kidney (left) of RT-EpoR+/+ mice and RT-EpoR−/− mice at day 7 after IRI. Summary of Ki67 data in renal epithelial cells (right). C: representative immunohistochemistry from 4 independent experiments for Ki67 expression in the kidney (left) of WT mice and hEpoRM/− mice at day 7 after IRI and a summary of percentage of Ki67 in renal epithelial cells (right). Arrows depict proliferating cells. Scale bar = 50 μm. Data are expressed as means ± SD (n = 4) each group, statistical significance was evaluated by unpaired Student t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups.
Furthermore, we explored the effect of EpoR activity in on cell proliferation renal tubules post-AKI by measuring Ki67, a cell proliferation marker in the kidney. Lower cell proliferation was found in both RT-EpoR−/− mice (Fig. 7B) and EpoRM/− mice (Fig. 7C), compared with their WT littermates. These data suggest that lower EpoR expression in the renal tubules confers vulnerability of the renal tubules to oxidative injury and impairs renal epithelial cell recovery. The impaired renal tubular regeneration in EpoRM/− mice conceivably resulted from abnormal peritubular capillary (Fig. 3) and tubular hypoxia (Fig. 4).
Hyperfunctional EPO/EpoR signaling reduced acute kidney damage but “paradoxically” increased renal fibrosis post-AKI.
Given that lower EPO/EpoR signaling renders the kidney more susceptible to IRI, it is important to test whether higher EPO/EpoR signaling activity protects against ischemic injury. To examine this hypothesis, we performed IRI in heterozygous hEpoRM/− mice. Upon IRI, hEpoRM/− mice had less elevation of PCr than WT mice at day 1 (Fig. 8A), consistent with less kidney damage (Fig. 8B). Surprisingly, in contrast to the rapid decline of PCr observed in WT mice after IRI, hEpoRM/− mice had elevated PCr 4 days post-IRI and had sustained elevation up to 2 wk compared with WT mice (Fig. 8A).
Fig. 8.

Hyperactive EpoR signaling reduced acute kidney damage but promoted tubulointerstitial changes in hEpoRM/− after IRI. At predetermined time points, mice from each genotype were killed, blood was collected, and kidneys were harvested. A: plasma creatinine (PCr). Data are expressed as means ± SD (n = 6) from each group, statistical significance was evaluated by unpaired Student’s t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups for same time point. B: acute kidney damage in WT mice and hEpoRW/W mice; representative micrographs from 4 independent experiments of H&E stain (top) of kidney cortex and OM and kidney injury scores (bottom) obtained in a blinded fashion at days 2 and 7 after IRI. Scale bar = 100 μm. C: renal fibrosis in WT mice and hEpoRW/W mice; representative microscopic images from 4 independent experiments of trichrome stain (left) in the kidney section and renal fibrosis scores (right) obtained in a blinded fashion at day 14 after IRI. Scale bar = 200 μm. Data are expressed as means ± SD from each group and statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups for B and C. D: representative immunoblots from 4 independent experiments for α-SMA and CTGF in the kidney (left) of WT mice and hEpoRM/− mice at predetermined time points after IRI and a summary of all immunoblots (right). Data are expressed as means ± SD from each group, statistical significance was evaluated by unpaired Student’s t-test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups for same time point for A and D.
There was milder change of kidney histology in hEpoRM/− compared with WT mice at day 2 (Fig. 8B). However, the more favorable phenotype did not persist. At day 7, hEpoRM/− mice had more severe histologic changes including proteinaceous, granular, and cellular casts, tubular epithelial necrosis, and tubulointerstitial infiltration than those at day 2 (Fig. 8B), indicating that the damage was exacerbated and kidney recovery was impaired after IRI. Compared with WT mice, hEpoRM/− mice had more renal fibrosis at 14 days post-IR (Fig. 8C) and higher expression of fibrotic markers (Fig. 8D). Taken together, hyperfunctional EPO/EpoR signaling activity delayed kidney recovery and promoted renal fibrosis post-IRI.
Altered EpoR signaling activity was associated with changes in autophagy and apoptosis in the kidney.
The association between injury and different levels of EpoR activity was clear, so we proceeded to explore some potential mechanisms. Because increased autophagy in renal tubules is associated with cytoprotection and inhibition of apoptosis (28, 31, 45, 48, 73), we sought to explore the link between EpoR activity and autophagy. Notably, in the kidneys of mice with low EpoR signaling activity (hEpoRW/W and RT-EpoR−/−), there were lower LC3 II/I ratios and higher p62 than in kidneys of WT mice, whereas in the kidney of mice with hyperfunctional EpoR signaling (hEpoRM/−), there were exact opposite changes (Fig. 9A). After IRI, hEpoRW/W mice and RT-EpoR−/− mice had lower, and hEpoRM/− mice had higher, LC3 puncta in the kidney at 2 days after IRI compared with WT mice (Fig. 9B). Additionally, hEpoRW/W mice and RT-EpoR−/− mice had higher, and hEpoRM/− mice had lower active caspase-3, a marker of apoptosis, in the kidney at 2 days after IRI (Fig. 9C). Collectively, the results suggest that normal EPO/EpoR signaling activity is required for maintenance of basal autophagy activity, and higher EpoR signaling suppresses apoptosis induced by IRI, which might be one cellular mechanism mediating renoprotection by EpoR.
Fig. 9.

Association of EpoR signaling activity with levels of autophagy and apoptosis in the kidney. A: states of renal autophagy in mice with different EpoR (hEpoRW/W mice, RT-EpoR−/− mice, WT mice, and hEpoRM/− mice) signaling activity at baseline. Representative immunoblots for phospho-ERK, total ERK, phospho-JNK, total JNK, LC3, and p62 in the kidney (left) and a summary of all immunoblots (right panel). AKI was induced in hEpoRW/W mice, RT-EpoR−/− mice, WT mice, and hEpoRM/− mice by bilateral renal ischemia of 30 min followed by reperfusion for 48 h (IRI) for evaluation of autophagy (B) and of apoptosis (C). B: GFP-LC3 puncta per renal tubule. Representative micrographic images of LC3 puncta (top), scale bar = 50 μm; bottom: summary of the number of GFP-LC3 puncta per renal tubule from analysis of 50 renal tubules in the zone of cortex plus OM in blind manner. C: representative immunoblots for caspase 3 in the kidney of AKI mice (left) and summary of all immunoblots (right). Data are expressed as means ± SD from each group, statistical significance was evaluated by one-way ANOVA followed by Student-Newman-Keuls post hoc test, and significance was accepted when *P < 0.05, **P < 0.01, between 2 groups.
DISCUSSION
EPO is required for maintenance of erythropoiesis through binding to EpoR. In addition, EPO also confers cytoprotection against oxidative stress and apoptosis in extraerythropoietic tissue (89). Whether EPO’s tissue-protective properties are mediated by EpoR or in combination with the βcR as a putative a coreceptor is not well established yet (14, 46, 80). The EPO working model is further complicated by the fact that EPO may act on the ephrin receptor EphB4 (53). The distribution of EpoR in nonhematopoietic tissue was debated due to the suboptimal quality of the anti-EpoR antibodies, although EpoR transcripts are unequivocally and widely present (2, 20, 34, 70, 87). In vitro studies from our (34) and other (9) laboratories showed that EpoR is cytoprotective in several kidney cell lines. Nevertheless, the in vivo effect of EpoR on IRI-induced AKI was never characterized. The clear phenotype with renal tubule deletion unequivocally testifies to the role of EpoR in the renal epithelium.
The current paper provides several novel findings; first, the reduction of EpoR activity in the kidney rendered it more susceptible to IRI at the acute stage, retarded renal recovery, and accelerated renal tubulointerstitial fibrosis. Second, excessive EpoR activity in the kidney decreased acute kidney damage at the acute stage but delayed recovery of kidney function and promoted renal fibrosis at the subacute stage. Third, EpoR-mediated protection against acute ischemia was associated with induction of autophagy, low EpoR signaling reduced autophagy and heightened apoptosis, and high EpoR signaling increased autophagy and suppressed apoptosis in the kidney. Fourth, hyperfunctional EpoR activity led to the aberrant structure of peritubular capillaries, renal tubular hypoxia, impaired renal tubular regeneration, and increased fibrosis post-IRI.
The consequences of altered EpoR signaling can result from two major effects of EpoR on renal tubules and peritubular capillaries during AKI onset and recovery. The effect of EpoR on autophagy in the renal tubules determines the severity of tubular damage and apoptosis induced by ischemic injury, while the effect of EpoR on peritubular capillaries affects the renal outcome (recovery vs. progression) after AKI. Therefore, precise and timely regulation of EPO/EpoR signaling activity in the kidney is important for prevention of ischemic kidney injury, sustenance of renal tissue perfusion and suppression of renal fibrosis, and retardation of AKI progression to CKD.
The correction of anemia was proposed to be one mechanism underlying EPO-mediated renoprotection (64). However, the beneficial effects of erythropoiesis-stimulating agents, EPO derivatives, or EpoR activators devoid of erythropoietic action on the outcome of AKI, are clearly dissociated from increased hematocrit (16), indicating that EPO-associated renoprotection is above and beyond stimulation of erythropoiesis (77). The present study provided further and direct evidence that renoprotection is not dependent on correction of anemia. Mice with renal tubule-specific EpoR deletion actually had slightly higher levels of renal EPO production and hematocrit but more severe ischemic kidney damage. Although increased viscosity can contribute to predisposition of the kidney to ischemia, this is highly unlikely from the very small increase in hematocrit. Our ex vivo data showing more severe cell injury in primary culture of renal tubules isolated from EpoR-deficient mice are congruent with in vitro finding of De Beuf et al. (17) that EPO-induced protection against oxidative stress was only present in cells with EpoR expression, supporting that EpoR is requisite for the protective effect of EPO.
Although there is evidence that EPO/EpoR signaling protects tissue against ischemia and hypoxia through suppression of apoptosis (34), there is limited, and in fact controversial, information about the effect of EPO/EpoR signaling on autophagy (5, 6, 41, 57, 95). In an experimental model of neonatal necrotizing enterocolitis, EPO reduced both excessive autophagy and apoptosis (95), suppressed hyperoxia-induced autophagy, and contained cell damage (5). In contrast, erythropoiesis-stimulating agents induce autophagy and confer renoprotection post-IRI (93). How EPO/EpoR signaling modulates autophagy at baseline is unknown. Because higher EpoR signaling increases phospho-Akt, which in turn elevates mammalian target of rapamycin (mTOR) activity and consequently downregulates autophagy flux, there should be an mTOR-independent signaling pathway through which EpoR modulates autophagy flux. In fact, we found higher ERK and JNK activation in hyperfunctional EpoR active mice. MAPK activation can dissociate Beclin1 and Bcl-2 complex, which is the key player to upregulate autophagy (71, 72, 90). The effect of ERK/JNK activation on autophagic flux may explain the renoprotection of ERK/JNK activation (21). Therefore, the final state of autophagy in the kidney is enhanced in mice with hyperfunctional EpoR because dissociation of Beclin1/Bcl-2 overrides the downregulation of autophagy from Akt/mTOR activation.
In addition to modulation of autophagic flux, MAPK and Akt signaling also affect kidney injury and recovery after AKI (67, 69). Akt activation is associated with suppression of cell apoptosis and promotion of renal fibrosis in cisplatin nephrotoxicity and IRI-induced AKI (52, 85). In vitro overexpression of Akt provokes cell senescence and elevates reactive oxygen species (40). Moreover, suppression of Akt phosphorylation accelerates tubular repair and inhibits renal fibrosis (92), and JNK signal activation by ischemia preconditioning contributes to renoprotection (67, 69). It has been proposed that Stat5 activation, one of the downstream effectors of EpoR, is required for EPO/EpoR cytoprotection (9, 34) and antiapoptosis (50). Within the EPO and EpoR circuits, HIFs are important new players in the defense against hypoxic injury. When activated, HIF also participates in renal fibrosis (27, 29). Thus, upon EPO binding to EpoR, diverse signaling pathways can be activated (59), and a finely tuned control of these signaling pathways is required to ensure renal recovery post-AKI, but how these pathways are timely and precisely controlled during different AKI stages is still largely unknown. These answers can only be secured after obtaining a mouse line with conditional inducible manipulation of EpoR.
AKI has profound deleterious effects on the renal endothelium, leading to peritubular capillary dysfunction, which is involved in sustaining ongoing ischemia, and in further renal tubular injury even after the initial hypoperfusion has ceased. Vascular rarefaction following IRI promotes progression to CKD post-AKI (42, 49). EPO or EpoR is crucial for angiogenesis and vasculogenesis from embryonic development through adulthood (1, 61). Either defective or excessive EPO/EpoR signaling activity can be associated with vascular pathology and diseases. Deficient EPO/EpoR signaling activity decreased vascular stability and caused vascular loss (12, 30) and impaired vasculogenesis and angiogenesis (62). All these effects are compatible with the vascular rarefaction observed in kidneys of both hEpoRW/W and RT-EpoR−/− mice. How defective EpoR signaling in the epithelium leads to peritubular capillary rarefaction is not known. Lower EpoR in the renal tubules decreased VEGF-A production in the renal tubules and reduction of VEGFR2 expression in the capillaries similar to that in vascular rarefaction due to VEGF deletion in renal tubules (18), supporting a model of cross talk between renal tubules and peritubular capillaries. Recent data in strong support of cross talk include deletion of VEGF-A in renal tubules, which led to VEGFR2 reduction in peritubular capillary and peritubular capillary rarefaction in the kidney (18).
Conversely, overexpression of EpoR or VEGF-A is associated with excess and disturbed capillary formation. Proliferative retinopathy from invasive and/or tissue-destructive neovascularization is partially due to higher EpoR expression in retinal vessels and by massive fibrosis partially due to excessive VEGF-A production (44, 81). We showed that hyperfunction of signaling in hEpoRM/− and hEpoRM/M mice induced disorganized neovascularization at baseline and accelerated renal fibrosis triggered by IRI, suggesting that hyperfunction of EpoR causes not only polycythemia (19) but also a microvasculopathy. The poor renal outcome is probably related, although not restricted, to two factors. First, disorganized peritubular papillary architecture with thicker capillary basement membrane resulting from overproduction of collagen in mice leads to renal hypoxia. Second, overproduction of HIFs and higher HIF signaling trigger renal fibrosis (27, 29). Interestingly, it has been shown that persistent excessive activation of autophagy may promote renal fibrosis in the unilateral ureter obstruction model (28, 56). Therefore, whether massive renal fibrosis post-AKI in mice with hyperfunctional EpoR activity results from higher autophagy flux remains to be determined. One study showed that a high dose of EPO significantly increased renal fibrosis and promoted AKI progression to CKD (24), which supports the idea that sustained high EPO/EpoR activity may be deleterious to the renal outcome after AKI. Therefore, a “blanket approach” of blind administration of erythropoiesis-stimulating agents post-AKI will unlikely lead to favorable outcomes.
Given that EpoR deficiency renders the kidney more susceptible to ischemic injury and promotes renal fibrosis, upregulation of EpoR may be a strategy to prevent against ischemic kidney injury. We previously showed that one of the molecular mechanisms underlying Klotho-mediated cytoprotection is upregulation of EpoR in a kidney cell line (34). We also documented that Klotho is renoprotective when it is given immediately after ischemia (37) and blocks AKI-to-CKD progression when given for the first 4 days after ischemia (84). Therefore, an in vivo AKI model is needed to examine whether EpoR is required for Klotho’s renoprotection.
In conclusion, EPO/EpoR signaling in the kidney plays a critical role in AKI development, recovery, and progression to CKD by acting on two main renal structures: renal tubules and peritubular capillaries. EpoR signaling participates in modulating autophagy flux in renal tubules to protect kidney against ischemic injury and in maintaining normal angiogenesis of peritubular capillaryies to promote renal recovery and to suppress fibrosis. Importantly, low EPO/EpoR signaling downregulates autophagy flux and increases renal susceptibility to ischemic injury whereas high EPO/EpoR signaling exerts the opposite effects. In contrast, the effects of EPO/EpoR signaling on peritubular capillary angiogenesis and secondary renal outcome post-AKI assume a U-shaped model, because both defective EpoR signaling and hyperfunctional EpoR signaling in renal tubules impair peritubular capillary architecture, induce renal tubular hypoxia, delay renal tubular regeneration, accelerate renal fibrosis, and promote AKI-to-CKD progression at the late stage. Theoretically, upregulation of EPO/EpoR signaling at the early stage would attenuate kidney damage, whereas downregulation of EPO/EpoR signaling after the acute stage would inhibit renal fibrosis, hence reducing progression from AKI to CKD. Therefore, it is conceivable that properly timed modulation of EPO/EpoR signaling activity independent of its erythropoietic actions can be considered as a novel strategy for treatment of AKI, promotion of renal recovery, and attenuation of renal fibrosis.
GRANTS
The studies were supported in part by the National Institutes of Health Grants R01-DK-091392, R01-DK-092461, R01-HL-089966, U01-HL-110964, and U01-HL-11146; George O’Brien Kidney Research Center at University of Texas Southwestern Medical Center and O’Brien Feasibility and Pilot Program Grant P30-DK-07938; Charles and Jane Pak Center Innovative Research Support; and Pak-Seldin Center for Metabolic Research.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.C.H., and O.W.M. conceived and designed the research; M.S., B.F., P.L., N.G., K.L.M., J.Y., Y-P.Z., M.T.G., and M.C.H. performed experiments; M.S., B.F., P.L., N.G., K.L.M., J.Y., L.J-S.H., S.S.S., Y-P.Z., M.T.G., P.R.S., O.W.M., and M.C.H. analyzed the data; M.S., B.F., N.G., K.L.M., J.Y., and M.C.H. prepared figures; O.W.M., and M.C.H. wrote the manuscript; L. J-S.H., S.S.S., P.R.S., O.W.M. and M.C.H., read and approved the final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. H. Wu (University of California Los Angeles. Los Angeles CA) for providing the floxed EpoR mouse line, Dr. J. Prchal (University of Utah, Salt Lake City, UT) for providing the EpoRMTH/MTH and hEpoRWTH/WTH mouse lines, and Dr. N. Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) for providing the transgenic GFP-LC3 reporter mouse. The authors are particularly grateful to Dr. R. Brekken and Dr. B. Levine (University of Texas Southwestern Medical Center) for extremely helpful advice throughout the study and to H. Cho and J. Paek for expert technical assistance.
REFERENCES
- 1.Anagnostou A, Lee ES, Kessimian N, Levinson R, Steiner M. Erythropoietin has a mitogenic and positive chemotactic effect on endothelial cells. Proc Natl Acad Sci USA 87: 5978–5982, 1990. doi: 10.1073/pnas.87.15.5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anagnostou A, Liu Z, Steiner M, Chin K, Lee ES, Kessimian N, Noguchi CT. Erythropoietin receptor mRNA expression in human endothelial cells. Proc Natl Acad Sci USA 91: 3974–3978, 1994. doi: 10.1073/pnas.91.9.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 300: F721–F733, 2011. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bendix I, Schulze C, Haefen C, Gellhaus A, Endesfelder S, Heumann R, Felderhoff-Mueser U, Sifringer M. Erythropoietin modulates autophagy signaling in the developing rat brain in an in vivo model of oxygen-toxicity. Int J Mol Sci 13: 12939–12951, 2012. doi: 10.3390/ijms131012939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bi L, Hou R, Yang D, Li S, Zhao D. Erythropoietin protects lipopolysaccharide-induced renal mesangial cells from autophagy. Exp Ther Med 9: 559–562, 2015. doi: 10.3892/etm.2014.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohr S, Patel SJ, Shen K, Vitalo AG, Brines M, Cerami A, Berthiaume F, Yarmush ML. Alternative erythropoietin-mediated signaling prevents secondary microvascular thrombosis and inflammation within cutaneous burns. Proc Natl Acad Sci USA 110: 3513–3518, 2013. doi: 10.1073/pnas.1214099110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breggia AC, Wojchowski DM, Himmelfarb J. JAK2/Y343/STAT5 signaling axis is required for erythropoietin-mediated protection against ischemic injury in primary renal tubular epithelial cells. Am J Physiol Renal Physiol 295: F1689–F1695, 2008. doi: 10.1152/ajprenal.90333.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brines M, Cerami A. Discovering erythropoietin’s extra-hematopoietic functions: biology and clinical promise. Kidney Int 70: 246–250, 2006. doi: 10.1038/sj.ki.5001546. [DOI] [PubMed] [Google Scholar]
- 11.Chade AR. Renal vascular structure and rarefaction. Compr Physiol 3: 817–831, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Connor KM, Aderman CM, Smith LE. Erythropoietin deficiency decreases vascular stability in mice. J Clin Invest 118: 526–533, 2008. doi: 10.1172/JCI33813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clements ME, Chaber CJ, Ledbetter SR, Zuk A. Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS One 8: e70464, 2013. doi: 10.1371/journal.pone.0070464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coldewey SM, Khan AI, Kapoor A, Collino M, Rogazzo M, Brines M, Cerami A, Hall P, Sheaff M, Kieswich JE, Yaqoob MM, Patel NS, Thiemermann C. Erythropoietin attenuates acute kidney dysfunction in murine experimental sepsis by activation of the β-common receptor. Kidney Int 84: 482–490, 2013. doi: 10.1038/ki.2013.118. [DOI] [PubMed] [Google Scholar]
- 15.Dagher J, Du YP. Efficient and robust estimation of blood oxygenation levels in single cerebral veins. Med Biol Eng Comput 50: 473–482, 2012. doi: 10.1007/s11517-012-0886-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Beuf A, D’Haese PC, Verhulst A. Epoetin delta as an antifibrotic agent in the remnant kidney rat: a possible role for transforming growth factor beta and hepatocyte growth factor. Nephron, Exp Nephrol 115: e46–e59, 2010. doi: 10.1159/000313830. [DOI] [PubMed] [Google Scholar]
- 17.De Beuf A, Hou XH, D’Haese PC, Verhulst A. Epoetin delta reduces oxidative stress in primary human renal tubular cells. J Biomed Biotechnol 2010: 395785, 2010. doi: 10.1155/2010/395785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dimke H, Sparks MA, Thomson BR, Frische S, Coffman TM, Quaggin SE. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J Am Soc Nephrol 26: 1027–1038, 2015. doi: 10.1681/ASN.2014010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Divoky V, Liu Z, Ryan TM, Prchal JF, Townes TM, Prchal JT. Mouse model of congenital polycythemia: Homologous replacement of murine gene by mutant human erythropoietin receptor gene. Proc Natl Acad Sci USA 98: 986–991, 2001. doi: 10.1073/pnas.98.3.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott S, Busse L, Bass MB, Lu H, Sarosi I, Sinclair AM, Spahr C, Um M, Van G, Begley CG. Anti-Epo receptor antibodies do not predict Epo receptor expression. Blood 107: 1892–1895, 2006. doi: 10.1182/blood-2005-10-4066. [DOI] [PubMed] [Google Scholar]
- 21.Feliers D, Kasinath BS. Erk in kidney diseases. J Signal Transduct 2011: 768512, 2011. doi: 10.1155/2011/768512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghezzi P, Brines M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ 11, Suppl 1: S37–S44, 2004. doi: 10.1038/sj.cdd.4401450. [DOI] [PubMed] [Google Scholar]
- 24.Gobe GC, Bennett NC, West M, Colditz P, Brown L, Vesey DA, Johnson DW. Increased progression to kidney fibrosis after erythropoietin is used as a treatment for acute kidney injury. Am J Physiol Renal Physiol 306: F681–F692, 2014. doi: 10.1152/ajprenal.00241.2013. [DOI] [PubMed] [Google Scholar]
- 25.Gong H, Wang W, Kwon TH, Jonassen T, Li C, Ring T, FrøkiAEr J, Nielsen S. EPO and alpha-MSH prevent ischemia/reperfusion-induced down-regulation of AQPs and sodium transporters in rat kidney. Kidney Int 66: 683–695, 2004. doi: 10.1111/j.1523-1755.2004.00791.x. [DOI] [PubMed] [Google Scholar]
- 26.Haase-Fielitz A, Bellomo R, Devarajan P, Bennett M, Story D, Matalanis G, Frei U, Dragun D, Haase M. The predictive performance of plasma neutrophil gelatinase-associated lipocalin (NGAL) increases with grade of acute kidney injury. Nephrol Dial Transplant 24: 3349–3354, 2009. doi: 10.1093/ndt/gfp234. [DOI] [PubMed] [Google Scholar]
- 27.Haase VH. Hypoxia-inducible factor signaling in the development of kidney fibrosis. Fibrogenesis Tissue Repair 5, Suppl 1: S16, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He L, Livingston MJ, Dong Z. Autophagy in acute kidney injury and repair. Nephron Clin Pract 127: 56–60, 2014. doi: 10.1159/000363677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higgins DF, Kimura K, Iwano M, Haase VH. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle 7: 1128–1132, 2008. doi: 10.4161/cc.7.9.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holstein JH, Orth M, Scheuer C, Tami A, Becker SC, Garcia P, Histing T, Mörsdorf P, Klein M, Pohlemann T, Menger MD. Erythropoietin stimulates bone formation, cell proliferation, and angiogenesis in a femoral segmental defect model in mice. Bone 49: 1037–1045, 2011. doi: 10.1016/j.bone.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM, Hsu C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 37: 289–296, 2012. doi: 10.1097/SHK.0b013e318240b52a. [DOI] [PubMed] [Google Scholar]
- 32.Bian A, Shi M, Flores B, Gillings N, Li P, Yan SX, Levine B, Xing C, Hu MC. Downregulation of autophagy is associated with severe ischemia-reperfusion-induced acute kidney injury in overexpressing C-reactive protein mice. Plos One 12:e0181848 2017. doi: 10.1371/journal.pone.0181848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, Shelton J, Amaral AP, Faul C, Taniguchi M, Wolf M, Brand M, Takahashi M, Kuro-O M, Hill JA, Moe OW. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol 26: 1290–1302, 2015. doi: 10.1681/ASN.2014050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu MC, Shi M, Cho HJ, Zhang J, Pavlenco A, Liu S, Sidhu S, Huang LJ, Moe OW. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int 84: 468–481, 2013. doi: 10.1038/ki.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24: 3438–3450, 2010. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu MC, Shi M, Zhang J, Quiñones H, Griffith C, Kuro-o M, Moe OW. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 22: 124–136, 2011. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu MC, Shi M, Zhang J, Quiñones H, Kuro-o M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int 78: 1240–1251, 2010. doi: 10.1038/ki.2010.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutchens MP, Nakano T, Kosaka Y, Dunlap J, Zhang W, Herson PS, Murphy SJ, Anderson S, Hurn PD. Estrogen is renoprotective via a nonreceptor-dependent mechanism after cardiac arrest in vivo. Anesthesiology 112: 395–405, 2010. doi: 10.1097/ALN.0b013e3181c98da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikeda M, Swide T, Vayl A, Lahm T, Anderson S, Hutchens MP. Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex- and age-specific manner. Crit Care 19: 332, 2015. doi: 10.1186/s13054-015-1049-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imai Y, Takahashi A, Hanyu A, Hori S, Sato S, Naka K, Hirao A, Ohtani N, Hara E. Crosstalk between the Rb pathway and AKT signaling forms a quiescence-senescence switch. Cell Reports 7: 194–207, 2014. doi: 10.1016/j.celrep.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Jang W, Kim HJ, Li H, Jo KD, Lee MK, Yang HO. The neuroprotective effect of erythropoietin on rotenone-induced neurotoxicity in SH-SY5Y cells through the induction of autophagy. Mol Neurobiol 53: 3812–3821, 2016. doi: 10.1007/s12035-015-9316-x. [DOI] [PubMed] [Google Scholar]
- 42.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol 12: 1448–1457, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Kang KP, Lee JE, Lee AS, Jung YJ, Kim D, Lee S, Hwang HP, Kim W, Park SK. Effect of gender differences on the regulation of renal ischemia-reperfusion-induced inflammation in mice. Mol Med Rep 9: 2061–2068, 2014. doi: 10.3892/mmr.2014.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kase S, Saito W, Ohgami K, Yoshida K, Furudate N, Saito A, Yokoi M, Kase M, Ohno S. Expression of erythropoietin receptor in human epiretinal membrane of proliferative diabetic retinopathy. Br J Ophthalmol 91: 1376–1378, 2007. doi: 10.1136/bjo.2007.119404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaushal GP, Kaushal V, Herzog C, Yang C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 4: 710–712, 2008. doi: 10.4161/auto.6309. [DOI] [PubMed] [Google Scholar]
- 46.Khan AI, Coldewey SM, Patel NS, Rogazzo M, Collino M, Yaqoob MM, Radermacher P, Kapoor A, Thiemermann C. Erythropoietin attenuates cardiac dysfunction in experimental sepsis in mice via activation of the β-common receptor. Dis Model Mech 6: 1021–1030, 2013. doi: 10.1242/dmm.011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim JH, Shim JK, Song JW, Song Y, Kim HB, Kwak YL. Effect of erythropoietin on the incidence of acute kidney injury following complex valvular heart surgery: a double blind, randomized clinical trial of efficacy and safety. Crit Care 17: R254, 2013. doi: 10.1186/cc13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22: 902–913, 2011. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kramann R, Tanaka M, Humphreys BD. Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. J Am Soc Nephrol 25: 1924–1931, 2014. doi: 10.1681/ASN.2013101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuhrt D, Wojchowski DM. Emerging EPO and EPO receptor regulators and signal transducers. Blood 125: 3536–3541, 2015. doi: 10.1182/blood-2014-11-575357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar S, Liu J, McMahon AP. Defining the acute kidney injury and repair transcriptome. Semin Nephrol 34: 404–417, 2014. doi: 10.1016/j.semnephrol.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuwana H, Terada Y, Kobayashi T, Okado T, Penninger JM, Irie-Sasaki J, Sasaki T, Sasaki S. The phosphoinositide-3 kinase gamma-Akt pathway mediates renal tubular injury in cisplatin nephrotoxicity. Kidney Int 73: 430–445, 2008. doi: 10.1038/sj.ki.5002702. [DOI] [PubMed] [Google Scholar]
- 53.Li C, Shi C, Kim J, Chen Y, Ni S, Jiang L, Zheng C, Li D, Hou J, Taichman RS, Sun H. Erythropoietin promotes bone formation through EphrinB2/EphB4 signaling. J Dent Res 94: 455–463, 2015. doi: 10.1177/0022034514566431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol 30: 268–277, 2010. doi: 10.1016/j.semnephrol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin SL, Chang FC, Schrimpf C, Chen YT, Wu CF, Wu VC, Chiang WC, Kuhnert F, Kuo CJ, Chen YM, Wu KD, Tsai TJ, Duffield JS. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol 178: 911–923, 2011. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12: 976–998, 2016. doi: 10.1080/15548627.2016.1166317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maiese K. Erythropoietin and mTOR: a “one-two punch” for aging-related disorders accompanied by enhanced life expectancy. Curr Neurovasc Res 13: 329–340, 2016. doi: 10.2174/1567202613666160729164900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayer G. Capillary rarefaction, hypoxia, VEGF and angiogenesis in chronic renal disease. Nephrol Dial Transplant 26: 1132–1137, 2011. doi: 10.1093/ndt/gfq832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore E, Bellomo R. Erythropoietin (EPO) in acute kidney injury. Ann Intensive Care 1: 3, 2011. doi: 10.1186/2110-5820-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moritz KM, Lim GB, Wintour EM. Developmental regulation of erythropoietin and erythropoiesis. Am J Physiol 273: R1829–R1844, 1997. [DOI] [PubMed] [Google Scholar]
- 61.Müller-Ehmsen J, Schmidt A, Krausgrill B, Schwinger RH, Bloch W. Role of erythropoietin for angiogenesis and vasculogenesis: from embryonic development through adulthood. Am J Physiol Heart Circ Physiol 290: H331–H340, 2006. doi: 10.1152/ajpheart.01269.2004. [DOI] [PubMed] [Google Scholar]
- 62.Nakano M, Satoh K, Fukumoto Y, Ito Y, Kagaya Y, Ishii N, Sugamura K, Shimokawa H. Important role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in mice. Circ Res 100: 662–669, 2007. doi: 10.1161/01.RES.0000260179.43672.fe. [DOI] [PubMed] [Google Scholar]
- 63.Nangaku M. Tissue protection by erythropoietin: new findings in a moving field. Kidney Int 84: 427–429, 2013. doi: 10.1038/ki.2013.140. [DOI] [PubMed] [Google Scholar]
- 64.Nemoto T, Yokota N, Keane WF, Rabb H. Recombinant erythropoietin rapidly treats anemia in ischemic acute renal failure. Kidney Int 59: 246–251, 2001. doi: 10.1046/j.1523-1755.2001.00485.x. [DOI] [PubMed] [Google Scholar]
- 65.Noguchi CT, Asavaritikrai P, Teng R, Jia Y. Role of erythropoietin in the brain. Crit Rev Oncol Hematol 64: 159–171, 2007. doi: 10.1016/j.critrevonc.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Panesso MC, Shi M, Cho HJ, Paek J, Ye J, Moe OW, Hu MC. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int 85: 855–870, 2014. doi: 10.1038/ki.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem 276: 11870–11876, 2001. doi: 10.1074/jbc.M007518200. [DOI] [PubMed] [Google Scholar]
- 68.Park KM, Kim JI, Ahn Y, Bonventre AJ, Bonventre JV. Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. J Biol Chem 279: 52282–52292, 2004. doi: 10.1074/jbc.M407629200. [DOI] [PubMed] [Google Scholar]
- 69.Park KM, Kramers C, Vayssier-Taussat M, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury, MAPK and MAPK kinase activation, and inflammation by remote transient ureteral obstruction. J Biol Chem 277: 2040–2049, 2002. doi: 10.1074/jbc.M107525200. [DOI] [PubMed] [Google Scholar]
- 70.Patterson SD, Rossi JM, Paweletz KL, Fitzpatrick VD, Begley CG, Busse L, Elliott S, McCaffery I. Functional EpoR pathway utilization is not detected in primary tumor cells isolated from human breast, non-small cell lung, colorectal, and ovarian tumor tissues. PLoS One 10: e0122149, 2015. doi: 10.1371/journal.pone.0122149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem 284: 2719–2728, 2009. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939, 2005. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 73.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 74: 631–640, 2008. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 74.Rodrigues CE, Sanches TR, Volpini RA, Shimizu MH, Kuriki PS, Camara NO, Seguro AC, Andrade L. Effects of continuous erythropoietin receptor activator in sepsis-induced acute kidney injury and multi-organ dysfunction. PLoS One 7: e29893, 2012. doi: 10.1371/journal.pone.0029893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodríguez F, Nieto-Cerón S, Fenoy FJ, López B, Hernández I, Martinez RR, Soriano MJ, Salom MG. Sex differences in nitrosative stress during renal ischemia. Am J Physiol Regul Integr Comp Physiol 299: R1387–R1395, 2010. doi: 10.1152/ajpregu.00503.2009. [DOI] [PubMed] [Google Scholar]
- 76.Rosenberger C, Rosen S, Paliege A, Heyman SN. Pimonidazole adduct immunohistochemistry in the rat kidney: detection of tissue hypoxia. Methods Mol Biol 466: 161–174, 2009. doi: 10.1007/978-1-59745-352-3_12. [DOI] [PubMed] [Google Scholar]
- 77.Rossert J, Eckardt KU. Erythropoietin receptors: their role beyond erythropoiesis. Nephrol Dial Transplant 20: 1025–1028, 2005. doi: 10.1093/ndt/gfh800. [DOI] [PubMed] [Google Scholar]
- 78.Rutkowski JM, Wang ZV, Park AS, Zhang J, Zhang D, Hu MC, Moe OW, Susztak K, Scherer PE. Adiponectin promotes functional recovery after podocyte ablation. J Am Soc Nephrol 24: 268–282, 2013. doi: 10.1681/ASN.2012040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 95: 4635–4640, 1998. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Santhanam AV, d’Uscio LV, Katusic ZS. Cardiovascular effects of erythropoietin an update. Adv Pharmacol 60: 257–285, 2010. doi: 10.1016/B978-0-12-385061-4.00009-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sapieha P, Joyal JS, Rivera JC, Kermorvant-Duchemin E, Sennlaub F, Hardy P, Lachapelle P, Chemtob S. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J Clin Invest 120: 3022–3032, 2010. doi: 10.1172/JCI42142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 13: 1837–1846, 2002. doi: 10.1097/01.ASN.0000016444.90348.50. [DOI] [PubMed] [Google Scholar]
- 83.Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, Kieswich J, Allen D, Harwood S, Raftery M, Thiemermann C, Yaqoob MM. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol 15: 2115–2124, 2004. doi: 10.1097/01.ASN.0000135059.67385.5D. [DOI] [PubMed] [Google Scholar]
- 84.Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, Liu Y, Levine B, Moe OW, Hu MC. αKlotho mitigates progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol 27: 2331–2345, 2016. doi: 10.1681/ASN.2015060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang J, Liu N, Tolbert E, Ponnusamy M, Ma L, Gong R, Bayliss G, Yan H, Zhuang S. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am J Pathol 183: 160–172, 2013. doi: 10.1016/j.ajpath.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Terryn S, Jouret F, Vandenabeele F, Smolders I, Moreels M, Devuyst O, Steels P, Van Kerkhove E. A primary culture of mouse proximal tubular cells, established on collagen-coated membranes. Am J Physiol Renal Physiol 293: F476–F485, 2007. doi: 10.1152/ajprenal.00363.2006. [DOI] [PubMed] [Google Scholar]
- 87.Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, Liu X, Wu H, Carmichael ST. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci 26: 1269–1274, 2006. doi: 10.1523/JNEUROSCI.4480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van der Meer P, Voors AA, Lipsic E, van Gilst WH, van Veldhuisen DJ. Erythropoietin in cardiovascular diseases. Eur Heart J 25: 285–291, 2004. doi: 10.1016/j.ehj.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 89.Vesey DA, Cheung C, Pat B, Endre Z, Gobé G, Johnson DW. Erythropoietin protects against ischaemic acute renal injury. Nephrol Dial Transplant 19: 348–355, 2004. doi: 10.1093/ndt/gfg547. [DOI] [PubMed] [Google Scholar]
- 90.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30: 678–688, 2008. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Westenbrink BD, Voors AA, Ruifrok WP, van Gilst WH, van Veldhuisen DJ. Therapeutic potential of erythropoietin in cardiovascular disease: erythropoiesis and beyond. Curr Heart Fail Rep 4: 127–133, 2007. doi: 10.1007/s11897-007-0030-5. [DOI] [PubMed] [Google Scholar]
- 92.Yang C, Cao Y, Zhang Y, Li L, Xu M, Long Y, Rong R, Zhu T. Cyclic helix B peptide inhibits ischemia reperfusion-induced renal fibrosis via the PI3K/Akt/FoxO3a pathway. J Transl Med 13: 355, 2015. doi: 10.1186/s12967-015-0699-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang C, Xu Z, Zhao Z, Li L, Zhao T, Peng D, Xu M, Rong R, Long YQ, Zhu T. A novel proteolysis-resistant cyclic helix B peptide ameliorates kidney ischemia reperfusion injury. Biochim Biophys Acta 1842: 2306–2317, 2014. doi: 10.1016/j.bbadis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 94.Yang Z, Wang H, Jiang Y, Hartnett ME. VEGFA activates erythropoietin receptor and enhances VEGFR2-mediated pathological angiogenesis. Am J Pathol 184: 1230–1239, 2014. doi: 10.1016/j.ajpath.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu Y, Shiou SR, Guo Y, Lu L, Westerhoff M, Sun J, Petrof EO, Claud EC. Erythropoietin protects epithelial cells from excessive autophagy and apoptosis in experimental neonatal necrotizing enterocolitis. PLoS One 8: e69620, 2013. doi: 10.1371/journal.pone.0069620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zou YR, Zhang J, Wang J, Peng L, Li GS, Wang L. Erythropoietin receptor activation protects the kidney from ischemia/reperfusion-induced apoptosis by activating ERK/p53 signal pathway. Transplant Proc 48: 217–221, 2016. doi: 10.1016/j.transproceed.2016.01.009. [DOI] [PubMed] [Google Scholar]