

Abstract
Cardiac ischemia-reperfusion (I/R) damages the electron transport chain (ETC), causing mitochondrial and cardiomyocyte injury. Reversible blockade of the ETC at complex I during ischemia protects the ETC and decreases cardiac injury. In the present study, we used an unbiased proteomic approach to analyze the extent of ETC-driven mitochondrial injury during I/R. Isolated-perfused mouse (C57BL/6) hearts underwent 25-min global ischemia (37°C) and 30-min reperfusion. In treated hearts, amobarbital (2 mM) was given for 1 min before ischemia to rapidly and reversibly block the ETC at complex I. Mitochondria were isolated at the end of reperfusion and subjected to unbiased proteomic analysis using tryptic digestion followed by liquid chromatography-mass spectrometry with isotope tags for relative and absolute quantification. Amobarbital treatment decreased cardiac injury and protected respiration. I/R decreased the content (P < 0.05) of multiple mitochondrial matrix enzymes involved in intermediary metabolism compared with the time control. The contents of several enzymes in fatty acid oxidation were decreased compared with the time control. Blockade of ETC during ischemia largely prevented the decreases. Thus, after I/R, not only the ETC but also multiple pathways of intermediary metabolism sustain damage initiated by the ETC. If these damaged mitochondria persist in the myocyte, they remain a potent stimulus for ongoing injury and the transition to cardiomyopathy during prolonged reperfusion. Modulation of ETC function during early reperfusion is a key strategy to preserve mitochondrial metabolism and to decrease persistent mitochondria-driven injury during longer periods of reperfusion that predispose to ventricular dysfunction and heart failure.
NEW & NOTEWORTHY Ischemia-reperfusion (I/R) damages mitochondria, which could be protected by reversible blockade of the electron transport chain (ETC). Unbiased proteomics with isotope tags for relative and absolute quantification analyzed mitochondrial damage during I/R and found that multiple enzymes in the tricarboxylic acid cycle, fatty acid oxidation, and ETC decreased, which could be prevented by ETC blockade. Strategic ETC modulation can reduce mitochondrial damage and cardiac injury.
Keywords: fatty acid oxidation, mitochondria, tricarboxylic acid cycle
INTRODUCTION
Mitochondria are essential in maintaining cardiac function and metabolic homeostasis under physiological conditions (16, 19, 23, 27). Mitochondrial dysfunction, however, is also a critical factor in increasing myocardial injury during ischemia-reperfusion (I/R) (16, 19, 23, 27). The electron transport chain (ETC) sustains damage during I/R (24, 25). The damaged ETC increases cardiac injury by reducing energy production, enhancing generation of reactive oxygen species (ROS), and sensitizing to the opening of the mitochondrial permeability transition pore (16, 17, 23, 27). Rapid and reversible blockade of electron transport during ischemia with amobarbital (1, 8, 9) protects the ETC and subsequently decreases myocardial injury during I/R. Hence, transient shutdown of the ETC during ischemia attenuates the ETC-driven damage, providing clear evidence that the ETC is a major source of mitochondrial injury during I/R (7, 16).
In addition to the ETC, a variety of metabolic enzymes within the mitochondrial matrix are also damaged during I/R (20, 35, 36). Pyruvate dehydrogenase (PDH) is degraded in heart mitochondria during I/R (20, 35). Enzymes involving the tricarboxylic acid (TCA) cycle, including malate dehydrogenase (MDH), and fatty acid metabolism, including enoyl CoA dehydrogenase (ECDH), are also decreased after I/R (20). PDH is a gatekeeper for metabolic substrates from glucose metabolism that flow into the TCA cycle that is the main source of NADH supply to the ETC (36). Fatty acids are also key sources for NADH generation (36). Therefore, degradation of PDH in concert with enzymes of the TCA cycle and fatty acid oxidation can impair the generation of NADH that is a key source of reducing equivalents for the ETC. Decreases in the content of multiple pathway enzymes in series are likely to impair overall metabolic flux through the pathway. Thus, the decreased NADH supply coupled with the damaged ETC during I/R will likely compromise bioenergy production. In addition, impairment of metabolic enzymes can increase cardiac injury through redox imbalance within mitochondria and ultimately augment oxidative stress during I/R.
Blockade of electron transport protects the ETC during I/R (8, 32). In the present study, we asked if blockade of electron transport before ischemia can protect metabolic enzymes during I/R. A proteomic approach was used in the present study to identify the target enzymes that are degraded during I/R. The degradation of selected enzymes was confirmed using immunoblot analysis. Our study shows that blockade of electron transport before ischemia preserves the content of multiple matrix metabolic enzymes including PDH, MDH, succinate dehydrogenase (SDH), and ECDH. Thus, the ETC is the source of damage that contributes to widespread metabolic impairment of cardiac mitochondria after I/R.
METHODS
Preparation of mouse hearts for perfusion.
The Animal Care and Use Committees of the McGuire Veterans Affairs Medical Center and Virginia Commonwealth University approved the study. Male C57BL/6 mice (2–3 mo of age) were purchased from Harlan laboratory (Indianapolis, IN). Mice were anesthetized using pentobarbital sodium (100 mg/kg ip) and anticoagulated with heparin (1,000 IU/kg ip). Hearts were excised and perfused retrograde via the aorta in the Langendorff mode with modified Krebs-Henseleit (K-H) buffer oxygenated with 95% O2-5% CO2. Cardiac function was monitored with a balloon inserted into the left ventricle. In untreated hearts, the heart was perfused for 15 min with K-H buffer followed by 25 min of global ischemia at 37°C and 30 min of reperfusion. In the amobarbital-treated group, hearts were initially perfused 15 min with K-H buffer followed by 1 min of perfusion of amobarbital (2 mM) immediately before ischemia. Hearts in the time control (TC) group were only buffer perfused without I/R. A group of nonischemic hearts was perfused for 1 min with amobarbital (2 mM) after 15 min of equilibration perfusion followed by 55 min of perfusion. This group evaluated the potential impact of perfusion with amobarbital without ischemia. Hearts were paced at 420 beats/min during the 15-min equilibration period and after 10 min of reperfusion. During amobarbital perfusion, pacing was stopped (9).
Mitochondrial isolation and functional determination.
At the end of perfusion, mouse hearts were harvested and placed into buffer A [containing (in mM) 100 KCl, 50 MOPS, 1 EGTA, 5 MgSO4·7 H2O, and 1 ATP; pH 7.4] at 4°C. Cardiac mitochondria were isolated using the procedure of Palmer et al. (29) except that trypsin was used as the protease (8, 19). Cardiac tissue was finely minced and placed in buffer B (buffer A + 0.2% BSA) and homogenized with a polytron tissue processor (Brinkman Instruments, Westbury, NY) for 2.5 s at 10,000 rpm. The polytron homogenate was centrifuged at 500 g, the supernatant was saved for the isolation of subsarcolemmal mitochondria (SSM), and the pellet was washed. The combined supernatants were centrifuged at 3,000 g to sediment SSM. Interfibrillar mitochondria (IFM) were isolated by incubation of skinned myofibers, obtained after polytron treatment, with 5 mg/g (wet wt) trypsin for 10 min at 4°C. SSM and IFM were washed with buffer B and then suspended in 80 mM KCl, 50 mM MOPS, and 0.5 mM EGTA (26).
O2 consumption in mitochondria was measured using a Clark-type oxygen electrode at 30°C as previously described (25). SSM or IFM were incubated in 80 mM KCl, 50 mM MOPS, 1 mM EGTA, 5 mM KH2PO4, and 1 mg defatted dialyzed BSA/ml at pH 7.4. Glutamate (20 mM) + malate (10 mM) (complex I substrate) were used as complex I substrates to measure oxidative phosphorylation.
Isotope tags for relative and absolute quantification labeling.
Pooled SSM and IFM subpopulations (n = 4) from TC, I/R, and amobarbital + I/R hearts as well as an internal standard consisting of pooled protein from each individual group were lysed and precipitated overnight in acetone at −20°C, and pellets were resuspended in 20 μl of 0.5 M triethylammonium bicarbonate (pH 8.5) as previously described (12) with slight modifications. Protein contents were determined using a two-dimensional Quant Kit (Amersham, Piscataway, NJ), and 100 μg of each pooled sample were than denatured with 0.1% SDS and reduced with 5 mM Tris-(2-carboxyethyl)phosphine. After incubation for 1 h at 60°C, cysteines were blocked with 10 mM methyl methane thiosulfonate in isopropanol, and samples were incubated at room temperature for 10 min. Sequencing grade trypsin (10 μl, Applied Biosystems, Foster City, CA) was added at a trypsin-to-protein ratio of 1:20, and samples were incubated at 37°C overnight. Digested samples were labeled with isotope tags for relative and absolute quantification (iTRAQ) reagents using a four-track kit following the protocol provided by the vendor (Applied Biosystems).
After digestion and iTRAQ labeling, the ultracomplex protein digests were combined to create a 400-µg pooled protein digest sample that contained equal fractions of each of the four labeled samples for subsequent multidimensional protein identification technology analysis as previously described (2, 3, 13). After lyophilization, the digest mixture was reconstituted in strong cation exchange (SCX) loading buffer (5 mM ammonium formate in 20% acetonitrile, pH 3.0) to be fractionated with SCX SpinTips (Protea Biosciences, Morgantown, WV) per the manufacturer’s protocol. Briefly, the sample solution was loaded centrifugally onto the SCX SpinTip. The nonadsorbing solution that passed through the SCX SpinTip was collected. Eight different elution solutions were used to fractionate the peptides (20, 60, 100, 150, 200, 250, 400, and 500 mM ammonium formate in 10% acetonitrile) in a step-wise manner, for a total of nine sample fractions. The collected fractions were cleaned by repeated lyophilization, reconstituted in a 0.1 M acetic acid solution, and then lyophilized to dryness. Samples were reconstituted in a 5% acetonitrile-0.1% formic acid solution before analysis.
Mass spectrometry analyses with iTRAQ labeling.
The liquid chromatography-electrospray ionization mass spectrometry (MS) system used was a Shimadzu LC-20AD-HPLC (Tokyo, Japan) and QTrap5500-ESI mass spectrometer (AB Sciex, Toronto, ON, Canada) in MS/MS mode. Data acquisition and processing were performed using Analyst 1.5 software. Lyophilized SCX sample fractions were reconstituted in HPLC aqueous run buffer (0.1% trifluoroacetic acid and 2% acetonitrile) and injected onto a Kinetex C18 chromatographic column (100 × 2.1 mm, Phenomenex, Torrance, CA). Peptides were eluted from the column using an acetonitrile/formic acid gradient (2–90% acetonitrile in 120 min) and analyzed using a QTrap5500 mass spectrometer operated with Analyst 1.5 software. The MS acquisition was in data-dependent mode. The three most intense multiply charged ions with ion intensities above a threshold of 50,000 in each regular MS scan were chosen for MS/MS analyses. Spectra achieved a signal-to-noise ratio of ≥70.
The resulting MS/MS spectra were analyzed using ABI Protein ProteinPilot software 4.0 (Applied Biosystems). The spectral data were searched against the mouse protein database (Uniprot_mouse_27Feb12 database customized to select for all mouse proteins) for identification of the peptides and corresponding proteins. In ProteinPilot, the sample type was selected as iTRAQ 4Plex for retrieval of the isotopic tag information from the mass spectra. After database correlation analysis, the proteins were grouped, scored, and normalized against one of four isotope correction factors. The Pro Group algorithm of ProteinPilot generated a protein score (ProtScore) that is a cumulative score from each of the peptides used by the algorithm in the protein identification. ProtScores above 2.0, 1.0, and 0.47 expressed percent confidence levels of >99%, >90%, and >66%, respectively. Each peptide match showed the iTRAQ isotopic labels, methyl methane thiosulfonate-labeled cysteines, and other posttranslational modification present as mass spectral shifts identified during the database correlation analysis. Each protein identified also showed the differential protein expression compared against the other iTRAQ labeled samples for relative quantitation (2).
Western blot analysis.
Mitochondrial proteins (25 μg/lane) were separated using 12% or 4–15% Tris-glycine gels (Bio-Rad, Hercules, CA) and transferred to a PVDF membrane (Millipore) using semidry transfer (Bio-Rad). Blots were incubated for 1 h at room temperature in 5% (wt/vol) nonfat dry milk (Bio-Rad) in 10 mM Tris (pH 7.5), 150 mM NaCl, and 0.1% Tween 20 followed by overnight incubation at 4°C with primary antibody. Primary antibodies [PDH (1:1,000), MDH (1:500), and enoyl coenzyme A hydratase, short chain, 1 mitochondrial (ECSH1; 1:1,000)] were purchased from Cell Signaling (Danvers, MA). After 1 h of incubation at room temperature with a 1:10,000 dilution of horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG F(ab)2 (GE Healthcare Life Sciences, Piscataway, NJ), blots were developed using ECL Plus Western blotting detection reagents (GE Healthcare Life Sciences). Membranes were digitally analyzed (Fujifilm LAS-4000, Fuji Photo Film, Tokyo, Japan) using Science Laboratory 2005 Multi Gauge version 3.1. Membranes were scanned every 10 s for 2 min. The membrane was reprobed (without “stripping”) with antibody to subunit 4 of cytochrome oxidase (1:2,500, Cell Signaling) used as a protein loading control. Background intensity adjustment, if performed, was always adjusted for the entire membrane. For the preparation of figures, membranes were cut horizontally as shown by the white lines.
Statistical analysis.
Data are expressed as means ± SE (31). For all analyses, differences between groups were compared by one-way ANOVA. When a significant F value was obtained, means were compared using the Student-Newman-Keuls test of multiple comparisons. Statistical significance in proteomic studies was calculated using the software contained within ABI Protein ProteinPilot Software 4.0. Statistical significance was defined as P < 0.05.
RESULTS
Blockade of electron transport decreased cardiac injury and protected mitochondrial respiration during I/R.
Amobarbital treatment immediately before ischemia decreased cardiac injury shown by decreased release of lactate dehydrogenase into the coronary effluent during reperfusion compared with untreated hearts. Contractile recovery was improved with increased left ventricular developed pressure and a decrease in left ventricular diastolic pressure (Table 1). Thus, blockade of electron transport decreased cardiac injury in mouse hearts during I/R, in line with previous observations (1, 7, 9).
Table 1.
Amobarbital treatment decreased cardiac injury and improved oxidative phosphorylation in mouse hearts after ischemia-reperfusion
| TC | I/R | AMO + I/R | |
|---|---|---|---|
| Cardiac function and cell injury | |||
| Left ventricular developed pressure, mmHg (preischemia) | 71 ± 3 | 70 ± 4 | 64 ± 4 |
| Percent recovery of left ventricular developed pressure | 89 ± 3 | 14 ± 5* | 38 ± 16* |
| Lactate dehydrogenase, mU·min−1·g−1 | 105 ± 17 | 599 ± 33* | 346 ± 108† |
| Subsarcolemmal mitochondria cardiac mitochondrial function | |||
| Protein yield, mg/g heart tissue | 14.0 ± 1.6 | 12.3 ± 1.6 | 10.3 ± 0.8* |
| State 3 (ADP stimulated) | 245 ± 19 | 135 ± 16* | 229 ± 24† |
| State 4 (ADP limited) | 42 ± 5 | 26 ± 5* | 45 ± 4† |
| RCR | 6.0 ± 0.5 | 5.6 ± 0.7 | 5.1 ± 0.2 |
| 2 mM ADP | 257 ± 15 | 126 ± 12* | 252 ± 25† |
| DNP stimulated | 232 ± 28 | 128 ± 14* | 232 ± 24† |
| Interfibrillar mitochondria cardiac mitochondrial function | |||
| Protein yield, mg/g heart tissue | 22.2 ± 0.3 | 21.3 ± 0.8 | 18.6 ± 0.9* |
| State 3 (ADP stimulated) | 259 ± 6 | 207 ± 24* | 302 ± 32† |
| State 4 (ADP limited) | 53 ± 4 | 45 ± 9 | 61 ± 6 |
| RCR | 5.0 ± 0.3 | 5.7 ± 1.5 | 4.9 ± 0.2 |
| 2 mM ADP | 308 ± 9 | 211 ± 21* | 344 ± 33† |
| DNP stimulated | 272 ± 12 | 199 ± 23* | 326 ± 31† |
Values are means ± SE; n = 4 for all groups. TC, time control perfusion without ischemia; I/R, ischemia-reperfusion without treatment; AMO, amobarbital administered before ischemia; DNP, dinitrophenol; RCR, respiratory control ratio.
P < 0.05 vs. the TC group;
P < 0.05 vs. the I/R group.
I/R decreased the rate of state 3 respiration in both SSM and IFM using glutamate + malate as complex I substrates compared with TC (Table 1). The decreased rate of uncoupled respiration (dinitrophenol-stimulated rate) in I/R-damaged mitochondria localized the defect at the ETC as previously described (22, 23, 25). I/R did not alter the protein yield of mitochondria (Table 1). Blockade of electron transport immediately before ischemia improved the rate of mitochondrial respiration in both SSM and IFM after I/R, supporting that blockade of electron transport decreases the damage of the ETC during I/R (Table 1), as previously observed (7, 9, 33). There was a slight decrease in the protein content of mitochondria. Amobarbital treatment for 1 min followed by nonischemic perfusion for 55 min did not alter oxidative phosphorylation (Table 2) nor the contents of PDH, ECSH1, and MDH2 in TC hearts (data not shown). Thus, the present study reproduced the phenotype of mitochondrial and cardiac protection previously observed after blockade of electron transport during ischemia (7, 9, 33).
Table 2.
Amobarbital treatment did not alter the oxidative phosphorylation in time control mouse hearts
| Subsarcolemmal Mitochondrial |
Interfibrillar Mitochondria |
|||
|---|---|---|---|---|
| TC | TC + AMO | TC | TC + AMO | |
| Glutamate + malate were used complex I substrates | ||||
| State 3 | 226 ± 8 | 223 ± 7 | 314 ± 17 | 330 ± 10 |
| State 4 | 52 ± 4 | 46 ± 4 | 63 ± 5 | 64 ± 1 |
| Respiratory control ratio | 4.4 ± 0.3 | 4.9 ± 0.4 | 5.1 ± 0.6 | 5.2 ± 0.3 |
| 2 mM ADP | 246 ± 23 | 241 ± 11 | 367 ± 27 | 387 ± 22 |
Values are means ± SE; n = 3 for each group. TC, time control perfusion without ischemia; AMO, amobarbital administered. P = not significant.
Proteomic analysis.
The primary parameter used to identify potential proteins in pathways responsible for mitochondrial respiration-mediated cell death was a minimum 10% decrease in protein expression measured in the I/R state versus the TC as well as a minimum 10% increase in amobarbital-treated I/R compared with untreated I/R. These percent changes were based on the comparison group in either analysis. For example, in the I/R group, we saw a 12% decrease in the expression of aconitase compared with the TC group, represented by a value of 0.88. Furthermore, our proteomic expression data were further stratified into groups of P values of significance. In the case of aconitase in the I/R versus TC analysis, the statistical significance of this result was a P value of 9.3 × 10−3, which falls under the P value group of 0.001 to <0.01. This aided us in demonstrating confidence in the protein expression patterns. A secondary parameter in our analysis was a restriction to just the IFM proteomic data. IFM showed the greatest percent changes, but overall similar results were observed in SSM for key results (data not shown).
ETC-mediated decreases in content of ETC subunits during I/R.
I/R leads to decreased activities of enzymes at the ETC in cardiac mitochondria (22, 25). Proteomic analysis showed that the contents of subunits of NADH dehydrogenase (flavoprotein 1 and iron-sulfur protein 3), complex III subunit (Rieske iron sulfur protein), and cytochrome oxidase subunits (5A and 6C) were decreased in I/R-damaged mitochondria compared with the TC (Table 3). Amobarbital treatment before ischemia preserved the content of these subunits in mitochondria after I/R compared with untreated hearts (Table 3). These results provide evidence that blockade of electron transport protects the ETC through mitigation of the subunit damage/degradation of respiratory complexes.
Table 3.
Electron transport subunits that were decreased by ischemia-reperfusion compared with the time control
| Electron Transport Chain | Protein Name | I/R-TC | AMO-I/R |
|---|---|---|---|
| Complex I | NADH dehydrogenase [ubiquinone] flavoprotein 1 | 0.73 | 1.26 |
| Complex I | NADH dehydrogenase [ubiquinone] iron-sulfur protein 3 | 0.77 | 1.30 |
| Complex III | Cytochrome b–c1 complex subunit Rieske | 0.79 | 1.22 |
| Complex IV | Cytochrome c oxidase subunit 5A | 0.73 | 1.34 |
| Complex IV | Cytochrome c oxidase subunit 6C | 0.77 | 1.36 |
TC, time control perfusion without ischemia; I/R, ischemia-reperfusion without treatment; AMO, amobarbital administered before ischemia.
ETC-mediated decreases in content of intermediary metabolism enzymes during I/R.
I/R altered multiple proteins involved in two mitochondrial metabolic pathways: TCA cycle (Fig. 1) and fatty acid oxidation (Fig. 2). PDH E1 α1-subunit, the magnesium-binding subunit of the tetramer responsible for catalyzing the conversion of pyruvate to acetyl CoA for the TCA cycle, was decreased in I/R compared with the TC (0.82) and preserved in amobarbital-treated ischemia compared with untreated I/R (1.2). Within the TCA cycle, protein segments for aconitase, isocitrate dehydrogenase, succinyl-CoA synthetase, succinyl-CoA dehydrogenase complex subunit B, fumarase, and MDH showed a decrease in I/R compared with TC perfusion. The contents of these enzymes were preserved in mitochondria from hearts treated with amobarbital immediately before ischemia. Within the fatty acid β-oxidation cycle, acyl CoA dehydrogenase, enoyl-CoA hydratase, and hyroxyacyl dehydrogenase were all decreased in I/R compared with the TC and preserved after I/R in mitochondria from hearts treated with amobarbital immediately before ischemia (Fig. 2).
Fig. 1.
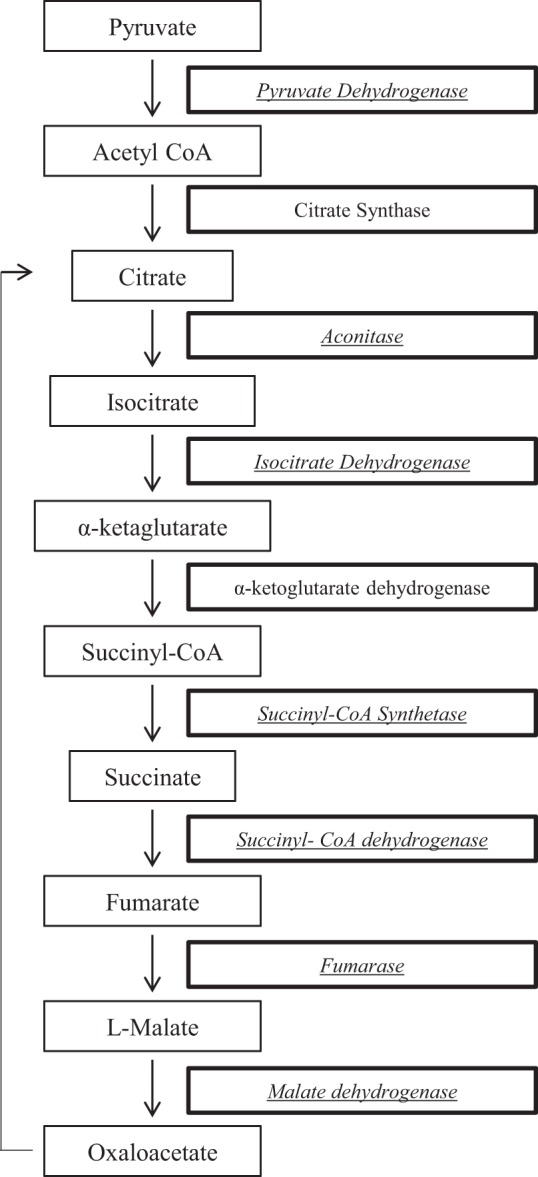
Summary of the enzymes in the tricarboxylic acid pathway that are altered during ischemia-reperfusion. The enzymes inside of the bold frame were decreased during ischemia-reperfusion. The content of the italic enzymes was protected by amobarbital treatment.
Fig. 2.

Summary of the enzymes involved in fatty acid oxidation that were altered during ischemia-reperfusion (I/R). The enzymes inside of the bold frame were decreased during I/R and their content protected by amobarbital (AMO) treatment.
Administration of amobarbital before ischemia protects PDH, MDH, and ECSH1 in both SSM and IFM after I/R.
Immunoblot analysis was used to confirm the changes in contents of selected enzymes. Compared with the TC, I/R markedly deceased the contents of PDH α-subunit, MDH, and ECSH1 in both SSM and IFM. I/R also increased the formation of cleaved PDH in both SSM and IFM (Figs. 3 and 4). Amobarbital treatment immediately before ischemia preserved the protein contents of PDH, MDH, and ECSH1 in both SSM and IFM compared with untreated hearts (Figs. 3 and 4). These results further support that electron transport is the upstream source leading to the degradation of these enzymes during I/R.
Fig. 3.

Immunoblot metabolic enzyme content in subsarcolemmal mitochondria (SSM). Compared with the time control, ischemia-reperfusion (I/R) markedly decreased the content of the full-length pyruvate dehydrogenase (PDH) α1-subunit and increased its cleaved products. A: amobarbital treatment maintained the PDH content in SSM after ischemia-reperfusion. I/R also decreased the contents of enoyl coenzyme A hydratase, short chain, 1 mitochondrial (ECSH1) and malate dehydrogenase 2 (MDH2) compared with the time control. B: amobarbital treatment partially restored the ECSH1 content compared with untreated hearts. Subunit 4 of cytochrome c oxidase (COX) was used as protein loading control. Background intensity was adjusted for the entire membrane in the membranes reprobed for subunit 4 of COX. Data are means ± SE; n = 3 in each group. *P < 0.05 vs. the time control or amobarbital-treated mitochondria; †P < 0.05 vs. the time control.
Fig. 4.

Immunoblot metabolic enzyme content in interfibrillar mitochondria (IFM). Compared with the time control, ischemia-reperfusion markedly decreased the content of the full-length pyruvate dehydrogenase (PDH) α1-subunit and increased its cleaved products. A: amobarbital treatment maintained the PDH content in IFM after ischemia-reperfusion. Ischemia-reperfusion also decreased the contents of enoyl coenzyme A hydratase, short chain, 1 mitochondrial (ECSH1) and malate dehydrogenase 2 (MDH2) compared with the time control. B: amobarbital treatment restored ECSH1 and MDH2 content compared with untreated hearts. Background intensity was adjusted for the entire membrane in the gels reprobed for subunit 4 of cytochrome oxidase. Data are means ± SE; n = 3 in each group. *P < 0.05 vs. time control or amobarbital treated mitochondria.
ETC-mediated decreases in the contents of other proteins during I/R.
In addition to proteins involved in the two metabolic pathways, I/R also decreased contents of histone H2B type 1-A, H+ transporting F1 ATP synthase ε-subunit, myosin regulatory light chain 2 ventricular/cardiac muscle isoform, and ubiquinone biosynthesis protein COQ9 compared with TC. Amobarbital treatment preserved these protein contents compared with untreated hearts (Table 4).
Table 4.
Other peptides that were decreased by ischemia-reperfusion compared with the time control
| Protein Name | I/R-TC | AMO-I/R |
|---|---|---|
| Histone H2B type 1-A | 0.41 | 2.16 |
| H+ transporting F1 ATP synthase ε-subunit | 0.63 | 1.39 |
| Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 0.68 | 1.28 |
| Ubiquinone biosynthesis protein COQ9, mitochondrial OS = Mus musculus GN = Coq9 PE = 1 SV = 1 | 0.68 | 1.28 |
TC, time control perfusion without ischemia; I/R,ischemia-reperfusion without treatment; AMO amobarbital administered before ischemia.
DISCUSSION
We found a systematic decrease in the proteomic expression of multiple enzymes of two mitochondrial metabolic pathways of intermediary metabolism after I/R: the TCA cycle and fatty acid β-oxidation. These same pathways are protected when electron transport is blocked immediately before ischemia by the administration of amobarbital. Thus, blockade of electron transport before ischemia not only protects the ETC itself but also extends the protection during I/R to additional metabolic pathways. Hence, the present study advanced our understating of the mechanisms by which ETC-mediated processes augment cardiac injury during I/R. Thus, intervention to modulate mitochondrial respiration is a potentially important approach to reduce cardiac injury during I/R.
Proteomic approaches have been used to analyze mitochondrial protein changes during I/R (2, 14, 15, 20, 21, 30, 35). In buffer-perfused rabbit hearts, I/R led to alteration of 25 mitochondrial proteins involved in the TCA cycle, fatty acid metabolism, components of the ETC, and mitochondrial membrane proteins (20). This study detected changes using two-dimensional gel approaches. In the present study, the proteomic analysis allowed us to identify the TCA and fatty acid oxidation pathways that are affected during I/R. Most importantly, we identified that the mechanism of these decreases is due to ETC-dependent mechanisms that lead to the degradation of key enzymes in metabolic pathways upstream of the ETC itself. iTRAQ often underestimates alterations in relative quantification (3, 4). Thus, the use of a 10% decrease in content with restoration of content in the presence of blockade of electron transport was used in the iTRAQ analysis. Confirmation of changes in protein content with immunoblot analysis confirmed a >10% change (Figs. 3 and 4). Thus, proteomic study not only provides the approach to identify key protein targets of I/R injury but also provides insights into a strategy to protect these key pathways and alleviate the heart injury during I/R.
I/R leads to decreased complex I activity (9, 37, 39), but the mechanism by which I/R damages complex I remains unclear. In the present study, I/R resulted in a decrease in oxidative phosphorylation using complex I substrates, supporting that I/R damages complex I in mouse heart mitochondria. Our proteomic study shows that two subunits of complex I are damaged or degraded during I/R. Blockade of electron transport using amobarbital preserves respiration using complex I substrates (7). In a previous study (8), complex I activity was maintained in amobarbital-treated hearts after I/R. Indeed, in the present study, amobarbital treatment preserves the contents of complex I subunits in mouse heart mitochondria after I/R. These results further advance our understanding the mechanism whereby I/R leads to decreased complex I activity.
The ETC is a key source of ROS generation during I/R (9, 34). Complex III of the ETC is a dominant center to generate ROS (11, 18). In rat heart mitochondria, ischemia increases ROS generation accompanied by the decreased complex III activity (11, 18, 22). The decreased complex III activity is due to a decrease in the functional activity of the ISP protein (22), as demonstrated by a decrease in the electron paramagnetic resonance signal of the catalytic iron center. In the present study, the ISP subunit content was decreased in mouse heart mitochondria after I/R, consistent with our previous finding. Blockade of electron transport during ischemia protects the ISP in mouse heart mitochondria after I/R. Amobarbital decreases ROS generation in mitochondria after I/R, including from complex III (9, 33). These findings suggest that amobarbital treatment decreases ROS generation from mitochondria in part through protection of complex III.
Not only do the TCA cycle and fatty acid oxidation sustain ETC-dependent damage, but PDH does as well. Pyruvate is a main product of glycolysis under aerobic condition in cardiomyocytes. Within the mitochondrial matrix, pyruvate is converted to acetyl-CoA coupled to reduce NAD+ to NADH through decarboxylation catalyzed by PDH (35). NADH not only donates electrons to complex I at the ETC for oxidative phosphorylation but also affects substrate flexibility to switch between fatty acids and carbohydrates to meet the high energy demand of hearts (5, 35). The decreased PDH activity can increase cell injury by inhibiting respiration during reperfusion. Oxidation of carbohydrates has been shown to decrease cardiac injury during I/R. Thus, PDH impairment damages the substrate flexibility and likely augments cardiac injury after I/R. The TCA cycle oxidizes acetyl-CoA coupled to reduce NAD+ to NADH within the mitochondrial matrix (35). Decreased MDH and SDH will further decrease NADH generation and the subsequent substrate supply for the ETC. Amobarbital treatment protected PDH, MDH, and SDH in heart mitochondria. Thus, blockade of the electron transport also protects the enzymes upstream of the ETC to improve oxidative phosphorylation during reperfusion.
In the present study, we found that I/R led to degradation of metabolic enzymes within mitochondrial matrix. These results suggest that activated proteases within the matrix contribute to the degradation of these enzymes (37). Calpain 1 is a cysteine protease that has been localized within heart mitochondria in the intermembrane space and the matrix (10, 28, 37). Activation of calpain 1 within the intermembrane space cleaves apoptosis-inducing factor (AIF) to truncated AIF that is released into the cytosol to increase cell death during I/R (10, 28). The formation of the cleaved AIF is also a biomarker for mitochondrial calpain 1 activation (10, 28, 40). Interestingly, amobarbital treatment decreases the formation of the truncated AIF and its release from mitochondria (38), indicating that blockade of electron transport during ischemia prevents mitochondrial calpain 1 activation. I/R may degrade the metabolic enzymes with the matrix by activating mitochondrial calpain 1. Other proteases within mitochondrial matrix, including Lon, may also contribute to metabolic enzyme damage during I/R (30). In addition to enzyme subunit damage, the conformational change of enzyme including peptide misfolding can also lead to decreased enzyme activity.
Taken together, our proteomic approach framed the metabolic network affiliated with the ETC-mediated injury during I/R. Blockade of electron transport not only protects the ETC itself (9) but also protects metabolic pathways upstream of the ETC. Although amobarbital is a classic complex I inhibitor (6), there is a limitation to directly apply it to clinical use. Fortunately, alternative approaches to modulate complex I activity are emerging. Thus, understanding the mechanisms of cardioprotection by regulating mitochondrial respiration will facilitate the use of this therapeutic strategy to decrease cardiac injury in patients who suffer a ST segment elevation myocardial infarction followed by acute reperfusion therapy.
GRANTS
This work was supported by Office of Research and Development Medical Research Service Merit Review Awards 1IO1BX001355-01A1 and 2IO1BX001355-01A2 from the Department of Veterans Affairs (to E. J. Lesnefsky), Scientist Development Grant 11SDG5120011 and Grant-in-Aid 15GRNT24480123 from the American Heart Association (to Q. Chen), a Virginia Commonwealth University Clinical and Translational Science Award [National Institutes of Health (NIH) Grant UL1-TR-000058] and the Virginia Commonwealth University Center for Clinical and Translational Research Endowment Fund (to Q. Chen), NIH Grant R21-AG-054975-01 (to Q. Chen), NIH Grant R01-HL-128485 (to J. M. Hollander), and Virginia Commonwealth University’s Pauley Heart Center (to Q. Chen and E. J. Lesnefsky).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Q.C., J.M.H., and E.J.L. conceived and designed research; Q.C., J.T., and Y.H. performed experiments; Q.C., M.S.Y., J.T., J.M.H., and E.J.L. analyzed data; Q.C., M.S.Y., J.M.H., and E.J.L. interpreted results of experiments; Q.C. and M.S.Y. prepared figures; Q.C., M.S.Y., and E.J.L. drafted manuscript; Q.C., J.M.H., and E.J.L. edited and revised manuscript; Q.C., M.S.Y., J.M.H., and E.J.L. approved final version of manuscript.
REFERENCES
- 1.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, , et al. JT. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem 268: 18532–18541, 1993. [PubMed] [Google Scholar]
- 2.Baseler WA, Dabkowski ER, Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Croston TL, Powell M, Razunguzwa TT, Lewis SE, Schnell DM, Hollander JM. Reversal of mitochondrial proteomic loss in type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase. Am J Physiol Regul Integr Comp Physiol 304: R553–R565, 2013. doi: 10.1152/ajpregu.00249.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baseler WA, Dabkowski ER, Williamson CL, Croston TL, Thapa D, Powell MJ, Razunguzwa TT, Hollander JM. Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction. Am J Physiol Regul Integr Comp Physiol 300: R186–R200, 2011. doi: 10.1152/ajpregu.00423.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpi A, Menabò R, Kaludercic N, Pelicci P, Di Lisa F, Giorgio M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta 1787: 774–780, 2009. doi: 10.1016/j.bbabio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Chambers KT, Leone TC, Sambandam N, Kovacs A, Wagg CS, Lopaschuk GD, Finck BN, Kelly DP. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem 286: 11155–11162, 2011. doi: 10.1074/jbc.M110.217349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chance B, Williams GR, Hollunger G. Inhibition of electron and energy transfer in mitochondria. I. Effects of Amytal, thiopental, rotenone, progesterone, and methylene glycol. J Biol Chem 238: 418–431, 1963. [PubMed] [Google Scholar]
- 7.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol 292: C137–C147, 2007. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 8.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J Pharmacol Exp Ther 316: 200–207, 2006. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther 319: 1405–1412, 2006. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun 415: 533–538, 2011. doi: 10.1016/j.bbrc.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 278: 36027–36031, 2003. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 12.Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol 299: H529–H540, 2010. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dabkowski ER, Williamson CL, Bukowski VC, Chapman RS, Leonard SS, Peer CJ, Callery PS, Hollander JM. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am J Physiol Heart Circ Physiol 296: H359–H369, 2009. doi: 10.1152/ajpheart.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernández-Caggiano M, Prysyazhna O, Barallobre-Barreiro J, CalviñoSantos R, Aldama López G, Generosa Crespo-Leiro M, Eaton P, Doménech N. Analysis of mitochondrial proteins in the surviving myocardium after ischemia identifies mitochondrial pyruvate carrier expression as possible mediator of tissue viability. Mol Cell Proteomics 15: 246–255, 2016. doi: 10.1074/mcp.M115.051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gedik N, Krüger M, Thielmann M, Kottenberg E, Skyschally A, Frey UH, Cario E, Peters J, Jakob H, Heusch G, Kleinbongard P. Proteomics/phosphoproteomics of left ventricular biopsies from patients with surgical coronary revascularization and pigs with coronary occlusion/reperfusion: remote ischemic preconditioning. Sci Rep 7: 7629, 2017. doi: 10.1038/s41598-017-07883-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res 77: 334–343, 2008. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 17.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–831, 2009. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 18.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 278: 5557–5563, 2003. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 19.Hollander JM, Thapa D, Shepherd DL. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: influence of cardiac pathologies. Am J Physiol Heart Circ Physiol 307: H1–H14, 2014. doi: 10.1152/ajpheart.00747.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim N, Lee Y, Kim H, Joo H, Youm JB, Park WS, Warda M, Cuong DV, Han J. Potential biomarkers for ischemic heart damage identified in mitochondrial proteins by comparative proteomics. Proteomics 6: 1237–1249, 2006. doi: 10.1002/pmic.200500291. [DOI] [PubMed] [Google Scholar]
- 21.Kling A, Jantos K, Mack H, Hornberger W, Drescher K, Nimmrich V, Relo A, Wicke K, Hutchins CW, Lao Y, Marsh K, Moeller A. Discovery of novel and highly selective inhibitors of calpain for the treatment of Alzheimer’s disease: 2-(3-phenyl-1H-pyrazol-1-yl)-nicotinamides. J Med Chem 60: 7123–7138, 2017. doi: 10.1021/acs.jmedchem.7b00731. [DOI] [PubMed] [Google Scholar]
- 22.Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys 385: 117–128, 2001. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- 23.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol 33: 1065–1089, 2001. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 24.Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 280: H2770–H2778, 2001. doi: 10.1152/ajpheart.2001.280.6.H2770. [DOI] [PubMed] [Google Scholar]
- 25.Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 273: H1544–H1554, 1997. [DOI] [PubMed] [Google Scholar]
- 26.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275, 1951. [PubMed] [Google Scholar]
- 27.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88: 581–609, 2008. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozaki T, Tomita H, Tamai M, Ishiguro S. Characteristics of mitochondrial calpains. J Biochem 142: 365–376, 2007. doi: 10.1093/jb/mvm143. [DOI] [PubMed] [Google Scholar]
- 29.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252: 8731–8739, 1977. [PubMed] [Google Scholar]
- 30.Sepuri NBV, Angireddy R, Srinivasan S, Guha M, Spear J, Lu B, Anandatheerthavarada HK, Suzuki CK, Avadhani NG. Mitochondrial LON protease-dependent degradation of cytochrome c oxidase subunits under hypoxia and myocardial ischemia. Biochim Biophys Acta 1858: 519–528, 2017. doi: 10.1016/j.bbabio.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steel R, Torrie J. Principles and Procedures of Statistics. New York: McGraw-Hill, 1960. [Google Scholar]
- 32.Stewart S, Lesnefsky EJ, Chen Q. Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Transl Res 153: 224–231, 2009. doi: 10.1016/j.trsl.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka-Esposito C, Chen Q, Lesnefsky EJ. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Transl Res 160: 207–216, 2012. doi: 10.1016/j.trsl.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turrens JF, Beconi M, Barilla J, Chavez UB, McCord JM. Mitochondrial generation of oxygen radicals during reoxygenation of ischemic tissues. Free Radic Res Commun 13: 681–689, 1991. doi: 10.3109/10715769109145847. [DOI] [PubMed] [Google Scholar]
- 35.Ussher JR, Jaswal JS, Lopaschuk GD. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res 111: 628–641, 2012. doi: 10.1161/CIRCRESAHA.111.246371. [DOI] [PubMed] [Google Scholar]
- 36.Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, Wagg CS, Jaswal JS, Harris RA, Clanachan AS, Dyck JR, Lopaschuk GD. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res 94: 359–369, 2012. doi: 10.1093/cvr/cvs129. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Liu J, Ma A, Chen Y. Cardioprotective effect of berberine against myocardial ischemia/reperfusion injury via attenuating mitochondrial dysfunction and apoptosis. Int J Clin Exp Med 8: 14513–14519, 2015. [PMC free article] [PubMed] [Google Scholar]
- 38.Xu A, Szczepanek K, Hu Y, Lesnefsky EJ, Chen Q. Cardioprotection by modulation of mitochondrial respiration during ischemia-reperfusion: role of apoptosis-inducing factor. Biochem Biophys Res Commun 435: 627–633, 2013. doi: 10.1016/j.bbrc.2013.05.033. [DOI] [PubMed] [Google Scholar]
- 39.Yang M, Stowe DF, Udoh KB, Heisner JS, Camara AK. Reversible blockade of complex I or inhibition of PKCβ reduces activation and mitochondria translocation of p66Shc to preserve cardiac function after ischemia. PLoS One 9: e113534, 2014. doi: 10.1371/journal.pone.0113534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297: 259–263, 2002. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]