

Abstract
Cystic fibrosis (CF) is the most common life-shortening genetic disease affecting ~1 in 3,500 of the Caucasian population. CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. To date, more than 2,000 CFTR mutations have been identified, which produce a wide range of phenotypes. The CFTR protein, a chloride channel, is normally expressed on epithelial cells lining the lung, gut, and exocrine glands. Mutations in CFTR have led to pleiotropic effects in CF patients and have resulted in early morbidity and mortality. Research has focused on identifying small molecules, or modulators, that can restore CFTR function. In recent years, two modulators, ivacaftor (Kalydeco) and lumacaftor/ivacaftor (Orkambi), have been approved by the U.S. Food and Drug Administration to treat CF patients with certain CFTR mutations. The development of these modulators has served as proof-of-concept that targeting CFTR by modulators is a viable therapeutic option. Efforts to discover new modulators that could deliver a wider and greater clinical benefit are still ongoing. However, traditional randomized controlled trials (RCTs) require large numbers of patients and become impracticable to test the modulators’ efficacy in CF patients with CFTR mutations at frequencies much lower than 1%, suggesting the need for personalized medicine in these CF patients.
Keywords: cystic fibrosis, cystic fibrosis transmembrane conductance regulator, ivacaftor, lumacaftor, N-of-1 study, personalized medicine
CYSTIC FIBROSIS AND CFTR
Cystic fibrosis (CF) is an autosomal recessive disease caused by loss or dysfunction of CF transmembrane conductance regulator (CFTR) Cl− channel activity. Normally, Cl− efflux through the CFTR channel helps establish an osmotic gradient, allowing water to flow into the luminal space (2, 28). Defects in CFTR function result in an inability to establish this osmotic gradient, leading to dehydration and build-up of a thick, viscous mucus layer (Fig. 1). Given the wide expression of CFTR in various organs throughout the body, CF presents as a wide-ranging and multisystem disease that is often difficult to treat. Although respiratory dysfunction is the most common cause of morbidity and mortality, the pancreas, liver, intestine, reproductive tract, and sweat glands are also affected (51). Common symptoms include pancreatic insufficiency, elevated sweat chloride levels, obstruction of the gut, and male infertility.
Fig. 1.

Defective cystic fibrosis transmembrane conductance regulator (CFTR) leads to buildup of thick, viscous mucus in the affected organs. The normal CFTR channel allows movement of chloride ions to the outside of the cell. The mutant CFTR channel does not allow movement of chloride ions, leading to the buildup of thick, viscous mucus on the outside of the cell.
LUNG
Respiratory manifestations of CF include a chronic cough, recurrent respiratory infections due to impaired mucociliary clearance, chronic inflammation leading to bronchiectasis, and pulmonary function tests consistent with obstructive lung disease. The chronic cycles of infection and inflammation ultimately result in respiratory failure (20). Progression of obstructive lung disease results in a declining trend in pulmonary function, typically shown by a decline in forced expiratory volume in 1 s (FEV1). Loss of innate immune response commonly results in chronic colonization in the lungs by pathogenic bacteria, including Pseudomonas aeruginosa, Hemophilus influenza, Staphylococcus aureus, and Klebsiella spp. and by fungal species such as Aspergillus.
PANCREAS
Pancreatic insufficiency resulting in decreased production of pancreatic enzymes is a common manifestation of CF (23). According to the 2015 Cystic Fibrosis Patient Registry Annual Report, nearly 87% of CF patients require some form of pancreatic enzyme replacement therapy (11a). Decreased levels of pancreatic enzymes lead to fat malabsorption, resulting in malnutrition, fat-soluble vitamin deficiency, and increased fat content in feces, producing characteristic oily and foul-smelling stools. As pancreatic tissue damage progresses, the pancreas can also lose its endocrine function, which leads to the development of CF-related diabetes mellitus.
LIVER
CFTR dysfunction also leads to development of CF-related liver disease (CFLD). CFLD is the third most common cause of mortality in CF patients, after respiratory problems and complications following transplantation, accounting for ~3% of all CF-related mortality (11a). CFTR is normally expressed on the apical membrane of cholangiocytes, the epithelial cells lining the bile duct, where CFTR is responsible for moving water and chloride ions into the bile duct. Loss of CFTR function leads to production of abnormally acidic and dehydrated bile, which can clog the bile ducts. Obstruction of hepatic bile ducts can trigger a proinflammatory response, leading to fibrosis, and eventually cirrhosis and portal hypertension. Moreover, once the acidic bile reaches the small intestine, it can cause damage to the intestinal lining (27).
GASTROINTESTINAL TRACT
Gastrointestinal (GI) tract manifestations of CF are primarily a result of dehydration of the intestinal lumen and prolonged postprandial acidity due to decreased bicarbonate secretion from the epithelial cells lining both the small intestine and the pancreas. The acidic environment can damage the cells lining the small intestine, as well as hinder the function of digestive enzymes within this region. Intestinal dehydration leads to the build-up of viscous mucus that can contribute to intestinal obstruction. Such an obstruction in neonatal CF patients can result in meconium ileus, which, if left untreated, can cause rupture of the intestine and sepsis. In adults, intestinal obstruction leads to distal intestinal obstructive syndrome, whereas milder obstructions result in constipation, which has been reported in nearly 50% of CF patients (19). Additionally, impaired GI function can lead to a form of microbial dysbiosis known as small intestinal bacterial overgrowth, which can result in diarrhea, macrocytic anemia, weight loss, as well as intestinal inflammation (19).
REPRODUCTIVE SYSTEM
Cystic fibrosis affects both the male and female reproductive systems. Nearly all males (over 95%) with CF are infertile due to improper development of the vas deferens, a condition known as congenital bilateral absence of the vas deferens (37). Infertility in female CF patients is due primarily to thick mucus accumulation within the cervical lumen, producing a barrier for sperm transport into the fallopian tubes. Evidence also suggests that inadequate fluid within the cervical lumen results in decreased sperm numbers in the oviduct (24).
SWEAT GLANDS
CFTR is also found within sweat glands where it is responsible for the reabsorption of chloride into sweat duct cells. Defective CFTR results in the inability to reabsorb the secreted chloride ions, which accumulate in the sweat duct along with sodium ions, which remain in the duct to maintain electroneutrality (35). This results in the characteristic salty skin of CF patients, and sweat chloride concentration is also used clinically to monitor CFTR function.
CFTR PROTEIN
CFTR is normally expressed on the apical membrane of epithelial cells where, upon cyclic nucleotide (cAMP and cGMP) stimulation, it actively transports Cl− and bicarbonate ions transepithelially. The CFTR gene, which is ~189 kb long, is located on the long arm of chromosome 7 at position q31.2, and produces a mature protein of 1,480 amino acids (55). The CFTR protein is known as a chloride channel consisting of five domains, including two membrane spanning domains (MSD1 and MSD2), two intracellular nucleotide binding domains (NBD1 and NBD2), and one intracellular regulatory domain (R) (Fig. 2). Each MSD subunit is composed of six transmembrane helices, which together form a central pore for ion conductance. Each NBD subunit can bind to and hydrolyze ATP. The R domain, which links two repeating MSD/NBD motifs, is a highly charged sequence containing multiple phosphorylation sites. CFTR activation requires R domain phosphorylation by various protein kinases, e.g., PKA and the hydrolysis of ATP by the NBDs (50).
Fig. 2.
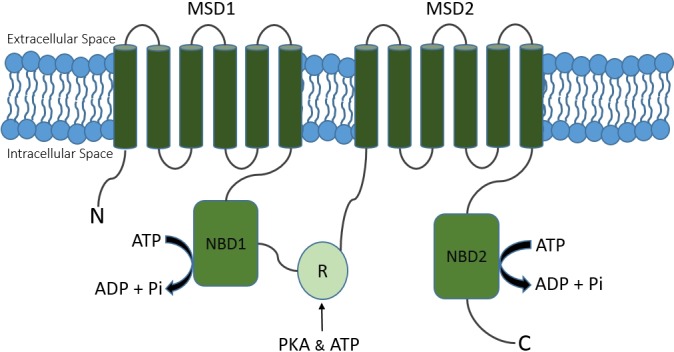
Schematic representation of CFTR. CFTR is a channel protein composed of two membrane spanning domains (MSD1 and MSD2), two nucleotide binding domains (NBD1 and NBD2), and one intracellular regulatory domain (R). N, NH2 terminal; C, COOH terminal.
CFTR MUTATIONS
More than 2,000 CF-causing mutations have been identified since the discovery of CFTR in 1989 [(46) and www.cftr2.org]. These mutations are grouped into six major classes based on their resulting defects (Fig. 3) (47).
Fig. 3.

CFTR mutation classification. Examples include common mutations in each class.
Class I (Synthesis)
CFTR mutations result in defective protein synthesis commonly caused by nonsense, frame-shift, or splice-site mutations leading to production of truncated CFTR protein. Class I mutations result in complete absence of CFTR at the cell surface and account for ~7% of all CF cases. Common examples include G542X, R553X, and W1282X.
Class II (Processing)
Mutations produce defects in posttranslational modification of CFTR, preventing CFTR trafficking to the cell surface and marking it for premature degradation. The most common Class II mutations include F508del, N1303K, and G85E. Class II mutations have an overall prevalence of ~85% within the CF population. The high prevalence of Class II mutations is due in large part to the F508del mutation, which is the most common CF-causing mutation present on at least one allele in ~70% and at both alleles in ~45% of all CF patients (www.cftr2.org).
Class III (Gating)
Mutations result in the inability of CFTR to pump out chloride ions in response to cAMP stimulation. This class accounts for ~3% of all CF cases and common mutations include G551D, V530F, and S549R.
Class IV (Conductance)
Mutations produce defects in the channel pore, resulting in decreased chloride conductance through the open pore. Class IV mutations account for less than 3% of all CF cases; the most common examples include R117H, R334W, and S1235R.
Class V (mRNA Stability)
Mutations produce splicing errors that decrease the stability of CFTR mRNA and lead to decreased expression of CFTR at the cell surface. Class V mutations account for less than 3% of CF cases; the most common examples include A455E, c.1680-886A>G, and c.2657+5G>A.
Class VI (CFTR Stability)
Mutations cause instability of otherwise functional CFTR that has localized to the apical surface but has increased turnover rate. The most common examples of Class VI mutations include rescued F508del, Q1412X, and COOH-terminal truncated CFTRs [(11a, 40, 64) and www.cftr2.org].
These mutation classes may correspond to disease severity. For example, patients with two copies of Class I, II, or III mutations are typically pancreatic insufficient within the first few months of life, whereas patients with at least one Class IV, V, or VI mutation are pancreatic sufficient (1, 60).
CFTR MODULATORS
CFTR modulators are defined as small-molecule compounds capable of increasing CFTR function. These compounds being developed to treat CF include activators, potentiators, and correctors. CFTR activators work by increasing the intracellular levels of cAMP/cGMP, which are required to stimulate CFTR activity. CFTR potentiators increase the channel opening of CFTR that has localized to the cell surface. CFTR correctors act as protein chaperones to increase the amount of CFTR at the cell surface. The discovery of CFTR modulators typically involves high-throughput screening (HTS) assays followed by lead optimization assays (39).
In vivo clinical trials of modulators use a host of outcome measures, commonly including FEV1, sweat chloride levels (SwCl), Cystic Fibrosis Questionnaire-Revised (CFQ-R), and body mass index (BMI).
CFTR POTENTIATORS
CFTR potentiators are pharmacological compounds that increase the open probability of CFTR that has correctly localized to the cell surface (57). Ivacaftor (Kalydeco) is currently the only CFTR potentiator approved for clinical use; however, a host of potentiators currently in various stages of development have shown promising safety and efficacy results. Potentiators currently in clinical trials include a deuterated version of ivacaftor (CTP-656), QBW-251, and GLPG1837.
Ivacaftor (Kalydeco; VX-770)
Ivacaftor (Kalydeco) is the first CFTR modulator approved by the U.S. Food and Drug Administration (FDA). Currently, Kalydeco is for CF patients 2 yr or older with certain Class III (gating) or IV (conductance) mutations. Class III mutations include G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, and S549R, while the only Class IV mutation for which Kalydeco is approved is R117H (https://www.kalydeco.com). Combined, these mutations account for ~8% of all CF patients (11a). The discovery and subsequent FDA approval of ivacaftor served as proof-of-concept that targeting the underlying cause of CF is a viable therapeutic option for CF patients.
Ivacaftor was identified by Vertex Pharmaceuticals through HTS and lead optimization assay. In both recombinant Fisher rat thyroid (FRT) cells and more physiologically relevant human bronchial epithelial (HBE) cells derived from CF patients, ivacaftor was shown to potentiate CFTR-mediated Cl− secretion following forskolin (FSK) stimulation (53). In recombinant FRT cells expressing G551D- or temperature-rescued F508del-CFTR, the addition of ivacaftor following FSK stimulation resulted in an approximate fourfold and sixfold increase in transepithelial current (Isc), respectively. This response was blocked by the subsequent addition of CFTRinh-172, a CFTR blocker. In primary cultures of HBE cells expressing both G551D- and F508del-CFTR, the addition of ivacaftor following FSK stimulation resulted in an approximate 10-fold increase in Isc, equivalent to 48 ± 4% of non-CF HBE cell lines (53). In three out of six tested HBE cell lines expressing only F508del-CFTR, adding ivacaftor significantly increased Isc with a maximal response of 16 ± 4% of non-CF HBE (P < 0.05) (53). These in vitro results indicated a potential therapeutic benefit of ivacaftor to CF patients with Class II (processing) or Class III (gating) mutations and supported the introduction of ivacaftor into clinical trials with patients harboring such mutations.
Ivacaftor Phase 3 Clinical Trials
STRIVE, a randomized, double-blind, placebo-controlled clinical trial, was the first phase 3 clinical trial of ivacaftor and tested its efficacy in CF patients 12 yr or older with at least one copy of G551D-CFTR. The G551D-CFTR mutation is a Class III gating mutation found in ~3% of CF patients (www.cftr2.org). In this study, 161 subjects were 1:1 randomized to receive either Ivacaftor (150 mg q12 h; n = 83) or matching placebo (n = 78). Following a 24-wk treatment period, a treatment effect of +10.6 percentage points in ppFEV1 was reported (+10.4 in ivacaftor group, −0.2 in placebo group; P < 0.001). Additionally, there was a 55% reduction in pulmonary exacerbations (47 exacerbations in 28 subjects on ivacaftor, 99 exacerbations in 44 subjects on placebo; P = 0.001), an improvement in CFQ-R score, which exceeded the 4-point threshold for clinical importance (+5.9 points in ivacaftor group, +2.7 points in placebo group; P < 0.001), an average gain of 2.7 kg in body weight in the ivacaftor group compared with placebo (+3.1 kg in ivacaftor group, +0.4 kg in placebo group; P < 0.001), and a treatment effect on sweat chloride of −47.9 mmol/l from baseline (−48.7 mmol/l in ivacaftor group, −0.8 mmol/l in placebo group; P < 0.001) (44).
The ENVISION clinical trial tested the efficacy of ivacaftor in CF patients 6–11 yr old, again with at least one copy of G551D-CFTR. This double-blind, placebo-controlled trial, randomized 52 patients 1:1 to receive either ivacaftor (150 mg q12 h; n = 26) or matching placebo (n = 26). After 24 wk of treatment, a treatment effect of +12.5 percentage points in ppFEV1 was reported (+12.6 in ivacaftor group; +0.1 in placebo group; P < 0.001). Additionally, treatment effects of +1.9 kg from baseline body weight (+3.7 kg in ivacaftor group, +1.8 kg in placebo group; P < 0.001) and −54.3 mmol/l baseline sweat chloride (−55.5 mmol/l in ivacaftor group, −1.2 mmol/l in placebo group; P < 0.001) were reported. Although there was a numeric-score increase in the child version of CFQ-R (+6.3 points in ivacaftor group, +0.3 points in placebo group), this increase was not statistically significant in this younger patient population. This lack of significant improvement can be attributed to 1) higher baseline CFQ-R in this younger patient population and 2) small sample size in this trial as compared with the larger STRIVE clinical trial. The rate of pulmonary exacerbations also did not differ between the groups, although the rate in both groups was low (four events in ivacaftor groups, three events in placebo group) (13).
The KONNECTION study tested the efficacy of ivacaftor in CF patients with a non-G551D gating mutation on at least one allele. This two-part, randomized, double-blind and placebo-controlled study enrolled patients with nine different gating mutations: G1244E (n = 5), G1349D (n = 2), G178R (n = 5), G511S (n = 1), G970R (n = 4), S1251N (n = 8), S1255P (n = 2), S549N (n = 6), and S549R (n = 4). Following an 8-wk treatment period in part 1 of the study, there was a treatment effect of +10.7 ppFEV1 from baseline (+7.5 in ivacaftor group, −3.2 in placebo group; P < 0.0001), in addition to treatment effects of +0.7 kg/m2 in BMI (+0.7 kg/m2 in ivacaftor, +0.02 kg/m2 in placebo group; P < 0.0001), −49.2 mmol/l from baseline sweat chloride (−52.3 mmol/l in ivacaftor group; −3.1 mmol/l in placebo group; P < 0.0001), and a +9.6 point change in CFQ-R score (+8.9 points in ivacaftor group, −0.7 points in placebo group; P = 0.0004). Part 2 of the study was an open-laboratory, 16-wk extension period to assess the continuation of the observed effects through 24 wk of treatment. All patients in the extension period received ivacaftor 150 mg q12 h. G970R (n = 4) did not respond well to this treatment (ppFEV1 +2.55; SwCl −6.25; BMI +0.48; CFQ-R +1.4), although in vitro models predicted that it should (15).
The KONDUCT clinical trial tested the efficacy and safety of ivacaftor in CF patients aged 6 yr or older with at least one copy of the R117H-CFTR mutation. R117H-CFTR, which is present in ~3% of all CF patients, produces defects in both gating (Class III) and conductance (Class IV). In this double-blind, placebo-controlled, parallel-group study design, 69 subjects were randomized 1:1 to receive either ivacaftor 150 mg q12 h (n = 32 completed) or matching placebo (n = 35 completed) for 24 wk. Through 24 wk, no significant difference was found in either absolute change from baseline ppFEV1 (+2.6 in ivacaftor group, +0.5 in placebo group; P = 0.2) or change from baseline BMI (+0.49 kg/m2 in ivacaftor group, +0.23 kg/m2 in placebo group; P = 0.78). Significant improvements were seen in change from baseline in CFQ-R score (+7.6 points in ivacaftor group, −0.8 in placebo group; P = 0.009) and change from baseline in sweat chloride levels (−26.3 mmol/l in ivacaftor group, −2.3 mmol/l in placebo group; P < 0.001). Prespecified subgroup analysis based on age showed significant improvement in ppFEV1 for subjects ≥18 yr old (treatment difference +5.0 in ivacaftor group vs. placebo, n = 50; P = 0.01) but showed a significant decline in ppFEV1 in subjects 6–11 yr old (treatment difference −6.3 in ivacaftor group vs. placebo, n = 17; P = 0.03). These findings are potentially a result of a high baseline ppFEV1 in subjects 6–11 yr old (ppFEV1 = 95.8% at baseline) and a more progressed disease state in the adult subjects (ppFEV1 = 64.5% at baseline) (34).
The KIWI clinical trial was a two-part, open-label, phase 3 study to evaluate the safety, pharmacodynamics (PD), and pharmacokinetics (PK) of ivacaftor in CF patients between 2 and 5 yr old with at least one CFTR gating mutation. Results from this study expanded the use of ivacaftor to patients as young as 2 yr old (Table 1) (12).
Table 1.
Summary of clinical trials: ivacaftor monotherapy
| Measure, Test, and Assay |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study Name | Trial Phase No. | Mutations | Age Group, yr | Sample Size | ppFEV1 | BMI | CFQ-R | SwCl | Conclusion (Failed or Approved) | Reference |
| STRIVE | 3 | G551D | ≥12 | 161 | +10.6% (P < 0.001) [24] | +2.7 kg (P < 0.001) [24] | +3.2 points (P < 0.001) [24] | −47.9 mmol/l (P < 0.001) [24] | Approved | (23) |
| ENVISION | 3 | G551D | 6–11 | 52 | +12.5% (P < 0.001) [24] | +1.9 kg (P < 0.001) [24] | +6.0 points (P > 0.05 [24] | −54.3 mmol/l (P < 0.0010 [24] | (24) | |
| KONNECTION | 3 | G1244E; G1349; G178R; G511S; G970R; S1251N; S1255P; S549N; S549R | ≥6 | 39 | +10.7% (P < 0.0001) [8] | +0.7 kg/m2 (P < 0.0001) [8] | +9.6 points (P < 0.0004) [8] | −49.2 mmol/l (P < 0.0001) [8] | (25) | |
| KONDUCT | 3 | R117H | ≥6 yr | 69 | +2.1% (P = 0.2) [24] | +0.26 kg/m2 (P = 0.78) [24] | +8.4 points (P = 0.009) [24] | −24.0 mmol/l (P < 0.001) [24] | (27) | |
| KIWI | 3 | G551D; S549N | 2–5 | 34 | +0.4 kg/m2 (P < 0.0001) [24] | −46.9 mmol/l (P < 0.0001) [24] | (28) | |||
| DISCOVER | 2 | Homozygous F508de1 | ≥12 | 140 | +1.7% (P = 0.15) [16] | −0.04 kg/m2 (P > 0.05) [16] | +1.3 points (P > 0.05) [16] | −2.9 mmol/l (P = 0.04) [16] | Failed | (29) |
Numbers within brackets indicate n. SwCl, sweat chloride levels.
Despite in vitro results demonstrating an increased Isc in F508del-CFTR expressing FRT and HBE cells upon FSK stimulation (53), no therapeutic benefit was observed in CF patients homozygous for F508del-CFTR. The DISCOVER trial was a phase 2 study to test the safety and efficacy of ivacaftor monotherapy in patients homozygous for F508del-CFTR. In this two-part, placebo-controlled, double-blind trial, 140 subjects were randomized 4:1 to receive either ivacaftor 150 mg q12 h for 16 wk or matching placebo (Part A), followed by an open-label extension period of 96 wk (Part B). Following 16 wk of treatment, there were no significant changes from baseline in ppFEV1 (treatment difference +1.7%; P = 0.15), CFQ-R score (treatment difference +1.3; P > 0.05), BMI (treatment difference −0.04 kg/m2; P > 0.05), or rate of pulmonary exacerbations (treatment difference 15%; P > 0.05). A small but significant change in sweat chloride from baseline to 16 wk was reported (treatment difference −2.9 mmol/l; P = 0.4); however, this change was not sustained through the Part B extension period (17).
CTP-656 (Deuterated Ivacaftor; D9-Ivacaftor)
Vertex Pharmaceuticals has acquired the rights to a deuterated version of ivacaftor (CTP-656; D9-ivacaftor), which gained orphan drug status from the FDA in January 2017 (http://www.accessdata.fda.govindex.cfm). In a two-part phase 1 crossover study, D9-ivacaftor was shown to have an improved PD profile compared with that of Kalydeco (ivacaftor). In part 1, eight healthy subjects received either Kalydeco 150 mg/day or D9-ivacaftor 150 mg/day after a high-fat meal for 7 days followed by crossover to the alternative treatment for an additional 7 days (sequence 1; n = 4: 7 days D9-ivacaftor → 7 days Kalydeco; sequence 2; n = 4: 7 days Kalydeco → 7 days D9-ivacaftor). Part 2 assessed multiple ascending doses of D9-ivacaftor (75 mg, 150 mg, and 225 mg), each administered daily after a high-fat meal for 7 days, compared with placebo (https://clinicaltrials.gov/ct2/show/NCT02599792?term=ctp-656&rank=2). Results presented during the 39th European Cystic Fibrosis Conference demonstrated improved PK for D9-ivacaftor compared with Kalydeco, including a 40% increase in half-life (D9-ivacaftor t1/2 of ~15 h; Kalydeco t1/2 of ~11 h), reduced clearance rate (D9-ivacaftor CL/F of 3.8 l/h vs. Kalydeco CL/F of 13.3 l/h), an approximately threefold increase in exposure (D9-ivacaftor area under the curve (AUC) of 44,916 ng·h−1·ml−1 vs. Kalydeco AUC of 12,925 ng·h−1·ml−1), and an approximately threefold increase in plasma concentrations at 24 h (D9-ivacaftor C24hrs of 712 ng/ml vs. Kalydeco C24hrs of 169 ng/ml). Data shown here are for 150-mg doses of both D9-ivacaftor and Kalydeco [(22, 52), http://www.concertpharma.com/wp-content/uploads/2016/06/ECFS-2016_presentation_final_10JUN16.pdf, http://www.concertpharma.com/wp-content/uploads/2015/10/CTP656-Ph1SAD-NACFC-08OCT2015.pdf, http://files.shareholder.com/downloads/ABEA-5J2EBT/0x0x896057/B374D978-58BD-4CC4-B0FF-97C8E583B8DB/CNCE_News_2016_6_10_General_Releases.pdf]. The improved PK profile of D9-ivacaftor along with a safety profile comparable to that of Kalydeco has allowed progression of D9-ivacaftor into a phase 2 clinical trial to evaluate its safety and efficacy in CF patients with CFTR gating mutations (52).
QBW-251
QBW-251 is a CFTR potentiator developed by Novartis Pharmaceuticals. In vitro results showed QBW-251 to be an effective potentiator for CFTR gating mutations, including F508del-CFTR. Promising in vitro results led to the initiation of a four-part phase 2 clinical trial to assess the safety, tolerability, and PK and PD profiles of single and multiple-ascending doses of QBW-251 in both healthy subjects and CF patients (11, 26). Enrolled subjects included patients heterozygous for CFTR mutations with residual channel activity (Class III, IV, V, and VI) or patients homozygous for F508del-CFTR, which is considered to be a Class II (processing) mutation but also has Class III (gating) and IV (conductance) defects (17, 54). Patients were randomized to receive either QBW-251 (150 mg, twice a day or 450 mg, twice a day) or matching placebo for 2 wk. Interim analysis on the first 40 patients who completed the 2-wk treatment period showed that QBW-251 was safe and well tolerated. Interestingly, after treatment with QBW-251 (450 mg, twice a day), CF patients (n = 12) with residual CFTR function showed significant increase in ppFEV1 (+7.3%; P = 0.004) compared with placebo (n = 10). Additionally, treatment with QBW-251 (150 mg, twice a day) resulted in a decrease in mean sweat chloride compared with placebo (−16.9 mmol/l; P = 0.029); however, no significant increase in ppFEV1 was reported for this dose. Patients homozygous for F508del-CFTR showed no improvement in ppFEV1 after receiving QBW-251 (450 mg, twice a day; n = 12), which finding played a role in the early termination of this trial (11, 26).
GLPG1837
During the 2016 North American Cystic Fibrosis Conference, Galapagos NV presented in vitro data regarding the development of GLPG1837, a CFTR potentiator identified via a YFP-based HTS assay (9). To characterize GLPG1837, a patch-clamp assay using CHO cells expressing either WT-, G551D-, or F508del-CFTR was used. Following preactivation of these cell lines with PKA and ATP, the addition of 3 μM GLPG1837 resulted in a 27.5 ± 3.0-fold increase in G551D-CFTR-mediated current, compared with a 12.8 ± 1.9-fold increase with ivacaftor (9, 25, 33). GLPG1837 was also characterized in patient-derived organoids (36). Crypts collected from rectal biopsies of patients heterozygous for either G551D/F508del-CFTR or S1251N/F508del-CFTR were used to generate the organoids. Following FSK stimulation, the introduction of GLPG1837 resulted in organoid swelling from both genotypes. GLPG1837 was shown to have an EC50 of 7.8 nM and 262 nM in S1251N/F508del-CFTR and G551D/F508del-CFTR-expressing organoids, respectively. GLPG1837 is currently in phase 2 clinical trials. SAPHIRA1 is a phase 2a, open-label study of multiple doses of GLPG1837 in subjects with at least one copy of G551D-CFTR, whereas SAPHIRA2 is an open-label study of two doses of GLPG1837 in subjects with at least one copy of S1252N-CFTR. SAPHIRA1 enrolled 26 patients, 25 of whom were on Kalydeco treatment before enrollment in the study. Following a 7-day washout period, subjects began a 4-wk course GLPG1837 with an increasing dose starting at 125 mg (twice a day) for the first week, 250 mg (twice a day) the second week, and 500 mg (twice a day) during the last 2 wk. GLPG1837 was found to be safe and well tolerated, with only mild to moderate adverse events being reported. A statistically significant and dose-dependent decrease in sweat chloride was reported in all dose groups. Following treatment at the highest dose level, subjects whose GLPG1837 concentration exceeded the organoid predicted target concentration had a sweat chloride decrease from postwashout baseline of 94 mmol/l to 52 mmol/l. Additionally, after 29 days of GLPG1837 treatment, ppFEV1 returned to prewashout baseline levels in patients taking Kalydeco (10). SAPHIRA2 enrolled seven patients with at least one copy of S1251N-CFTR, four of whom were on Kalydeco treatment before enrollment in this study. After a 7-day washout period, subjects began a 4-wk course of GLPG1837 with an increasing dose starting at 62.5 mg (twice a day) for the first 2 wk and 125 mg (twice per day) for the second 2 wk. GLPG1837 was again well tolerated and generally safe at both doses. Sweat chloride results were reported for five subjects. Four of five subjects had a decrease in sweat chloride level >15 mmol/l following 2 wk of 125-mg (twice per day) GLPG1837, and one subject had a decrease >50 mmol/l. Additionally, after the 4-wk GLPG1837-treatment period, ppFEV1 levels returned to prewashout levels seen in patients taking Kalydeco (14).
CFTR CORRECTORS
CFTR correctors are pharmacological compounds that increase the delivery of CFTR to the cell surface (57). Lumacaftor is currently the only FDA-approved CFTR corrector; however, a number of other correctors are in various stages of development.
Lumacaftor (VX-809)
Lumacaftor, currently the only approved CFTR corrector, is available in a single pill with ivacaftor (marketed as Orkambi). Orkambi is indicated for CF patients 6 yr and older who are homozygous for F508del-CFTR (https://www.orkambi.com). Approximately 45% of CF patients are homozygous for F508del-CFTR (www.cftr2.org).
Lumacaftor was discovered by Vertex Pharmaceuticals via HTS assays for compounds that increased F508del-CFTR-mediated Cl− transport in a recombinant cell-based assay (54). In F508del-CFTR expressing FRT cells treated with lumacaftor for 48 h, an ~sevenfold increase in F508del-CFTR maturation was seen relative to vehicle-treated controls. F508del-CFTR maturation, characterized by an increase in molecular weight of CFTR via glycosylation, was expressed as a ratio of mature CFTR to total CFTR (mature + immature) (5). Additionally, the lumacaftor-treated FRT cells expressing F508del-CFTR demonstrated an approximately fivefold increase in transepithelial Cl− transport following FSK stimulation. Lumacaftor was also assessed in the more physiologically relevant HBE cell line model. HBE cells isolated from the lungs of seven CF patients homozygous for F508del-CFTR and incubated for 48 h with lumacaftor resulted in an approximately eightfold increase in CFTR maturation and an approximately fourfold increase in transepithelial Cl− transport. Compared with HBE cells derived from the lungs of non-CF patients, this corresponds to an increase of chloride transport from 3.4 ± 0.7 to 13.9 ± 2.3%. Moreover, lumacaftor was analyzed in combination with ivacaftor in F508del-CFTR HBE cells. The addition of ivacaftor to HBE cells expressing F508del-CFTR and incubated with lumacaftor for 48 h resulted in increased chloride transport, reaching levels equivalent to ~25% of non-CF HBE cells (54). These results served as evidence that lumacaftor can provide a therapeutic advantage to CF patients homozygous for F508del-CFTR. Furthermore, it supported the use of lumacaftor in combination with ivacaftor.
Lumacaftor Monotherapy
This phase 2a trial tested the safety and efficacy of lumacaftor monotherapy in subjects homozygous for F508del-CFTR. The randomized, double-blind, placebo-controlled study enrolled a total of 89 subjects into two groups. In group A, subjects were randomized 2:2:1 to receive either lumacaftor 25 mg/day (n = 18), 50 mg/day (n = 18), or a matching placebo for 28 days. In group B, subjects were randomized 2:2:1 to receive either lumacaftor 100 mg/day (n = 17), 200 mg/day (n = 19), or a matching placebo for 28 days. A total of 17 subjects received a placebo. Following 28 days of lumacaftor treatment, no significant improvement in ppFEV1 or CFQ-R score was reported in any of the dose groups. Mean percentage change from baseline in ppFEV1 was +0.07 in the placebo group and −2.46, −2.15, +0.32, and +0.47 in the 25-, 50-, 100-, and 200-mg dose groups, respectively (all P values >0.05). Change in CFQ-R score from baseline was +4.5 in the placebo group and −5.2, −6.3, −1.3, and +2.2 in the 25-, 50-, 100-, and 200-mg dose groups, respectively. A dose-dependent reduction in sweat chloride level was reported at 7 days of treatment; sweat chloride level was sustained throughout the 28-day treatment period but reversed following discontinuation of lumacaftor. Treatment difference in sweat chloride from baseline values through 28 days where +0.10, −4.61, −6.13, and −8.21 mmol/l in the 25-, 50-, 100-, and 200-mg groups, respectively. These changes were statistically significant compared with placebo in both the 100-mg (P < 0.05) and the 200-mg (P < 0.01) groups (Table 2) (7).
Table 2.
Summary of clinical trials: lumacaftor monotherapy
| Measure, Test, and Assay |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study Name | Trial Phase No. | Mutations | Age Group, yr | Sample Size | ppFEV1 | BMI | CFQ-R | Sweat Chloride | Conclusion (Failed or Approved) | References |
| Lumacaftor Monotherapy, 25 mg | 2 | Homozygous F508de1 | ≥18 | 18 | −2.53% (P > 0.05) [4] | −9.7 points (P > 0.05) [4] | +0.10 mmol/l (P > 0.05) [4] | Failed | (49) | |
| Lumacaftor Monotherapy, 50 mg | 2 | Homozygous F508de1 | ≥18 | 18 | −2.22% (P > 0.05) [4] | −10.8 points (P > 0.05) [4] | −4.61 mmol/l (P > 0.05) [4] | |||
| Lumacaftor Monotherapy, 100 mg | 2 | Homozygous F508de1 | ≥18 | 17 | +0.25% (P > 0.05) [4] | −5.8 points (P > 0.05) [4] | −6.13mmol/l (P < 0.05) [4] | |||
| Lumacaftor Monotherapy, 200 mg | 2 | Homozygous F508de1 | ≥18 | 19 | +0.40% (P > 0.05) [4] | −2.3 points (P > 0.05) [4] | −8.21 mmol/l (P < 0.01) [4] | |||
Numbers within brackets indicate n.
F508del-CFTR produces a processing defect, resulting in retention of CFTR in the endoplasmic reticulum (ER) and premature degradation, as well as defects in gating and stability for temperature-corrected CFTR that localizes to the cell surface (41). These characteristics of F508del-CFTR provide an explanation as to why lumacaftor monotherapy was not effective in patients homozygous for this mutation. Moreover, these characteristics indicate that, to better correct the F508del defect, a CFTR potentiator (e.g., lumacaftor) and corrector (e.g., ivacaftor) combination should be used.
Lumacaftor/Ivacaftor (Orkambi)
The TRAFFIC and TRANSPORT clinical trials were two phase 3, randomized, placebo-controlled, double-blind studies to evaluate the safety and efficacy of lumacaftor administered in combination with ivacaftor in CF patients aged 12 yr or older and homozygous for F508del-CFTR (Table 3). Combined, these studies randomized 1108 subjects in a 1:1:1 ratio to receive either 600 mg lumacaftor/day + 250 mg ivacaftor q12 h [LUM (600 mg/day)-IVA group], 400 mg lumacaftor q12 h + 250 mg ivacaftor q12 h [LUM (400 mg q12 h)–IVA group], or lumacaftor-matched placebo q12 h + ivacaftor-matched placebo q12 h for 24 wk. Significant improvement of ppFEV1 from baseline was reported as early as day 15 of treatment and maintained through the 24-wk study period in both treatment groups compared with placebo. Through 24 wk, the absolute change from baseline in ppFEV1 compared with placebo (n = 371) was +3.3 in the LUM (600 mg/day)–IVA group (n = 368; P < 0.001) and +2.8 in the LUM (400 mg q12 h)–IVA group (n = 369; P < 0.001) (59). Despite this significant improvement, it should be noted that the change in ppFEV1 in F508del-CFTR subjects treated with lumacaftor-ivacaftor was smaller than that observed in subjects with gating mutations, such as G551D-CFTR, treated with ivacaftor monotherapy (44). This difference can potentially be explained by the facts that 1) G551D-CFTR and other gating mutations correctly localize to the cell surface, whereas F508del-CFTR has multiple defects, including reduction in transport to the cell surface; 2) lumacaftor only partially restores F508del-CFTR processing, resulting in lower cell surface density of F508del-CFTR compared with G551D-CFTR; and 3) reduced stability of lumacaftor corrected F508del-CFTR when combined with potentiators such as ivacaftor (6, 56).
Table 3.
Summary of clinical trials: combination therapies
| Measure, Test, and Assay |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Study Name | Treatment | Trial Phase No. | Mutations | Age Group, yr | Sample Size | ppFEV1 | BMI | CFQ-R | SwCl | Conclusion (Failed or Approved) | References |
| TRAFFIC/TRANSPORT (pooled results) | LUM 600 mg qd + IVA 250 mg q12 h | 3 | Homozygous F508de1 | ≥12 | 368 | +3.3% (P < 0.001) [24] | +0.28 kg/m2 (P < 0.001) [24] | +3.1 points (P = 0.007) [24] | Approved | ** | |
| TRAFFIC/TRANSPORT (pooled results) | LUM 400 mg qd + IVA 250 mg q12 h | 3 | Homozygous F508de1 | ≥12 | 369 | +2.8% (P < 0.001) [24] | +0.24 kg/m2 (P < 0.001) [24] | +2.2 points (P = 0.05) [24] | |||
| EVOLVE | TEZ 100 mg qd + IVA 150 mg q12 h | 3 | Homozygous F508de1 | ≥12 | 248 | +4.0% (P < 0.0001) [24] | +0.06 kg/m2 (P = 0.4127) [24] | +5.1 points (P < 0.001) [24] | (57, 58) | ||
| EXPAND | TEZ 100 mg qd + IVA 150 mg q12 h | 3 | Homozygous F508de1 | ≥12 | 161 | +6.8% (P < 0.0001) [8] | +11.1 points (P < 0.0001) [8] | ||||
http://www.glpg.com/docs/view/586fdcb9edc8a-en. Numbers within brackets indicate n.
Additionally, pooled analysis of both studies showed significant improvements from baseline BMI [+0.28 kg/m2 in LUM (600 mg/day)–IVA group; +0.24 kg/m2 in LUM [400 mg q12 h]–IVA group; both P < 0.001] and rate (per 48 wk) of pulmonary exacerbations [reported as rate ratio relative to placebo: 0.70 in LUM (600 mg/day)–IVA group, P = 0.001; 0.61 in LUM (400 mg q12 h)–IVA group, P < 0.001] compared with placebo. Numerical improvements in CFQ-R scores were observed in both treatment groups of both studies; however, a significant improvement was seen only in the pooled analysis of the LUM (400 mg q12 h)–IVA group. Despite statistical significance, none of the treatment groups surpassed the four-point minimum clinical importance difference threshold [pooled analysis: +3.1 in LUM (600 mg/day)–IVA group, P = 0.007; +2.2 in LUM (400 mg q12 h)–IVA group, P = 0.05] (42).
Tezacaftor (VX-661)
In March 2017, Vertex Pharmaceutical announced results from two phase 3 clinical trials of tezacaftor (VX-661) administered in combination with ivacaftor in CF patients age 12 yr or older (http://investors.vrtx.com/releasedetail.cfm?releaseid=984388, http://investors.vrtx.com/releasedetail.cfm?releaseid=1019156). In the double-blind, placebo-controlled EVOLVE study, patients homozygous for F508del-CFTR were randomized in a 1:1 ratio to receive either 100 mg tezacaftor qAM + 150 mg ivacaftor q12 h (n = 248) or a matching placebo (n = 256) for 24 wk. Through 24 wk of treatment, a significant improvement in mean absolute change in ppFEV1 from baseline was observed (treatment difference +4.0%, P < 0.0001). Significant improvements were also reported in rate of pulmonary exacerbations (reported as rate ratio relative to placebo: 0.65, P = 0.0054), change from baseline CFQ-R scores (treatment difference +5.1 points, P < 0.0001), and change from baseline BMI (treatment difference +0.06 kg/m2, P = 0.4127). The EXPAND study was a double-blind, placebo-controlled, crossover study that enrolled patients with one copy of F508del-CFTR and a second CFTR mutation with residual function. In this study, subjects were randomized to receive either tezacaftor 100 mg/day + ivacaftor 150 mg q12 h (combination treatment group; n = 161), ivacaftor 150 mg q12 h (monotherapy group, n = 156), or matching placebo (n = 161) for 8 wk, followed by an 8-wk washout period, and another 8-wk treatment period in which patients were switched to the other one of the two treatments. Subjects receiving combination treatment showed a 6.8% increase in ppFEV1 compared with placebo following 8 wk of treatment (P < 0.0001), whereas subjects receiving ivacaftor monotherapy showed only a 4.7% increase compared with placebo (P < 0.0001). The improvement in ppFEV1 in the combination group compared with the ivacaftor monotherapy group was statistically significant (+2.1%, P < 0.0001). Additionally, a statistically and clinically significant improvement in mean absolute change CFQ-R score compared with placebo was reported (+11.1 points in combination treatment group, P < 0.0001; +9.7 points in ivacaftor monotherapy group, P < 0.0001). Additional phase 2 and 3 clinical trials to further evaluate the safety and efficacy of tezacaftor are currently under way.
VX-440, VX-152, and VX-659
Vertex Pharmaceutical is also developing next-generation correctors that include VX-440, VX-152, and VX-659. In Ussing chamber studies using HBE cells collected from five F508del homozygous patients, VX-440 and VX-152 restored chloride transport to ~75% and 65% of normal CFTR function, respectively, when used in triple combination with ivacaftor and tezacaftor. In comparison, treatment of these cells with Orkambi (ivacaftor/lumacaftor) resulted in chloride transport of ~25% of normal. VX-440 and VX-152 were also tested in HBE cell collected from three patients with one copy of F508del and a second mutation resulting in minimal CFTR function. Ussing chamber assays of these cells following introduction of either ivacaftor/tezacaftor/VX-152, ivacaftor/tezacaftor/VX-440, or Orkambi (ivacaftor/lumacaftor) resulted in correction of chloride transport to ~45%, 40%, and 15% of normal CFTR, respectively (presented in 29th NACFC by Vertex in investor meeting). These in vitro results along with phase 1 safety data of VX-440 and VX-152, each tested in 100 healthy subjects, supported the introduction of VX-440 and VX-152 into phase 2 clinical trials. VX-440 and VX-152 are currently in separate phase 2 trials designed to evaluate the safety and efficacy of these correctors in triple combination with ivacaftor and tezacaftor. Enrollment includes both patients homozygous for F508del-CFTR and patients who have one copy of F508del-CFTR and a second mutation resulting in a minimal CFTR function (F508del/Min-CFTR) (https://clinicaltrials.gov/ct2/show/NCT02951182?term=VX-440&rank=1, https://clinicaltrials.gov/ct2/show/NCT02951195?term=vx-152&rank=1, https://www.businesswire.com/news/home/20161025006561/en). In patients with F508del/Min-CFTR genotype, triple combination of VX-440 or VX-152 with ivacaftor and tezacaftor resulted in mean absolute change from baseline ppFEV1 of 12.0 and 9.7 percentage points, respectively. Additionally, patients homozygous for F508del-CFTR and already being treated with ivacaftor and tezacaftor experienced a 9.5 and 7.3 percentage point increase in ppFEV1 following addition of VX-440 or VX-152, respectively (https://www.businesswire.com/news/home/20170718006344/en/Vertex-Announces-Positive-Phase-1-Phase-2).
VX-659, another next-generation corrector being developed by Vertex Pharmaceuticals, has also shown promising preclinical results. In Ussing chamber assays of HBE cells isolated from four patients with one copy of F508del and a second mutation resulting in minimal CFTR function, VX-659 in triple combination with ivacaftor/tezacaftor resulted in chloride transport ~65% of normal CFTR, compared with ~50% with ivacaftor/tezacaftor/VX-440 treatment (54a). VX-659 has also shown higher maximal efficacy in HBE cells collected from patients homozygous for F508del (https://www.businesswire.com/news/home/20161025006561/en/). Phase 1 studies in patients with F508del/Min-CFTR genotype demonstrated a 9.6 percentage point increase in mean absolute ppFEV1 treatment with VX-659 in triple combination with ivacaftor/tezacaftor (https://www.businesswire.com/news/home/20170718006344/en/Vertex-Announces-Positive-Phase-1-Phase-2).
RARE CFTR MUTATIONS
Despite the recent advances in the development of CFTR modulators, currently only ~50% of CF patients are eligible for modulator therapy based on the mutations they carry [(15, 34, 44, 58) and www.cftr2.org]. This leaves a significant number of CF patients who are not indicated for modulator therapy based on current FDA guidelines. Additionally, many of the 2,000 CFTR mutations are so rare that conducting powerful RCTs to test the efficacy of modulators in these mutations is not a practical approach. This is best highlighted by the following examples in which cell-based model system has been a valuable tool to determine the class of CFTR mutants, to test the efficacy of the modulator, and to predict the clinical outcomes.
Cell Model
Similar to the modulator discovery, development, and preclinical evaluation, cell model plays an important role in determining the response of rare CFTR mutants to modulators for personalized medicine. There are two main cell-based model systems in use, nonpolarized cells cultured in dishes and polarized cells differentiated on membrane. FRT cells, human embryonic kidney (HEK) 293 cells, and other immortal cell lines have been used to transiently overexpress the CFTR mutants, followed by either biochemistry assays, such as Western blotting or physiological measurements such as patch clamping and iodide efflux. These results allow us to understand the class of CFTR mutants, which, in turn, help determine the type of modulators, potentiator or corrector, or both, that will be potentially therapeutically beneficial. However, these heterologous expression systems are not suitable in the context of personalized medicine due to their high likelihood of false-positive results and their inability to accurately represent patients’ genetic environment (38). These limitations can be overcome by using primary cell cultures obtained from CF patients along with the polarized cell model. Primary nasal epithelial cells, primary airway epithelial cells, primary bronchial epithelial cells, and intestinal stem cells (organoids) are the most common cells in use. Given that respiratory dysfunction is the most common cause of morbidity and mortality in CF patients, human bronchial epithelial cells are the gold standard of patient-derived cell cultures, because they are thought to most accurately represent the in vivo genetic environment, including the expression of CF modifier genes (43, 51). However, the use of human bronchial epithelial cells is hindered by their limited supply, invasive collection procedures, presence of non-CFTR Cl− channels (e.g., calcium-activated Cl− channels), and their relatively low CFTR expression compared with intestinal epithelial cells. Patient-derived intestinal epithelial cells collected via rectal biopsies are a minimally invasive alternative that have been shown to be effective in indicating a clinical response to CFTR modulators (21). More recently, the use of duodenum biopsy from CF patients has been described as well (3). Epithelial cells collected via rectal biopsies and the duodenum can be used to measure CFTR-mediated currents via either a Ussing chamber assay or, following the formation of organoids, a FSK-induced swelling assay (8, 16). More recently, advancements in using induced pluripotent stem cells have provided an opportunity to produce unlimited supplies of patient-specific cell cultures without the need for invasive procedures (62).
P67L
The P67L-CFTR is a rare CFTR mutation found in ~0.3% of CF patients (www.cftr2.org). P67L is generally associated with a mild CF phenotype, which typically presents with a later age of diagnosis, lower incidence of pancreatic insufficiency, better nutritional status, lower incidence of Pseudomonas aeruginosa infections, lower incidence of CF-related diabetes (CFRD), and equivocal sweat chloride levels that often produce negative results in newborn screening tests. However, P67L is not associated with an improved lung function as the rate of FEV1/forced vital capacity (FVC) decline in P67L patients is similar to that observed in patients homozygous for F508del-CFTR, a genotype associated with a severe CF phenotype [(30) and www.cftr2.org].
P67L was historically believed to produce a Class IV (conductance) defect; however, functional analysis by Sabusap et al. (49) demonstrated that P67L mutation produced defects in CFTR protein folding, maturation, and gating but had near normal conductance. FRT cells expressing P67L-CFTR were shown to have decreased cell surface expression of CFTR compared with cells expressing WT-CFTR, indicating a Class II (processing) defect. Additionally, inside-out cell patch analysis showed decreased open probability of P67L-CFTR compared with WT-CFTR in FRT cells, indicating a Class III (gating) defect. These functional characteristics indicate that defects in P67L-CFTR may be corrected by ivacaftor and/or lumacaftor. Western blotting of cell lysates from WT-, F508del-, or P67L-CFTR expressing FRT cells treated with lumacaftor demonstrated a significant increase in P67L band C to levels comparable to WT-CFTR. Lumacaftor pretreated WT- and P67L-CFTR expressing FRT and primary nasal epithelial cells treated with ivacaftor demonstrated an increased open probability (Po) of cell surface localized CFTR (WT-CFTR: Po = 0.66 to 0.81 vs. P67L-CFTR: Po: 0.15 to 0.48; P = 0.03). Additionally, in polarized FRT cells and primary airway epithelial cells expressing P67L/F508del-CFTR and treated with ivacaftor and lumacaftor, Ussing chamber analysis demonstrated a significant increase in CFTR-mediated chloride transport to ~40–50% of WT-CFTR. In combination, these results further indicate that P67L-CFTR imparts defects in CFTR processing and gating and provide justification for the therapeutic use of ivacaftor and/or lumacaftor in patients with at least one copy of P67L-CFTR (49).
A 19-yr-old Scottish female with P67L/F508del-CFTR demonstrated improved clinical outcomes following initiation of ivacaftor. Prior to ivacaftor therapy, she was reported to have progressively worsening pulmonary function with FEV1 of 88% predicted at 18 yr old, forced expiratory flow midexpiratory phase of 75% predicted, increasing frequency of pulmonary exacerbations, with three to four incidents annually at onset of treatment, and worsening bronchiectasis. Sputum cultures were positive for chronic mucoid Pseudomonas aeruginosa infection that did not respond well to antibiotic treatment. A sweat chloride at 11 yr old showed elevated sweat chloride of 51 mmol/l. Additionally, pancreatic enzyme replacement therapy was initiated following development of pancreatic insufficiency. Following initiation of ivacaftor 150 mg, twice per day at 19 yr old, improvements in CF phenotypes were reported. Spirometry improvement was reported at a mean rate of 8.1% ppFEV1 per year compared with rate of change before ivacaftor therapy (P = 0.06), frequency of pulmonary exacerbations was reduced to one incident during the 1-yr follow-up period, sweat chloride was reduced to 25 mmol/l within 1 mo of ivacaftor therapy, and a 10% increase from pretreatment BMI was also reported (63). It should be noted that this patient started ivacaftor therapy before the approval of Orkambi; but given the in vitro results reported by Sabusap et al. (49), the combination of lumacaftor/ivacaftor compared with ivacaftor monotherapy may prove to be even more efficacious in patients with P67L-CFTR.
W1282X
W1282X-CFTR is a nonsense (Class I) mutation that introduces a premature termination codon (PTC), resulting in the deletion of part of NBD2 of CFTR (56). This mutation is found in ~1.7% of all CF patients but has a much higher prevalence in the Ashkenazi Jewish population with frequency over 45%, likely due to the founder effect [(61) and www.cftr2.org]. Class I CFTR mutations are generally associated with having a complete loss of CFTR activity, and, hence, a severe disease progression is commonly seen. However, nonsense mutations occurring in the COOH-terminal region of CFTR have been shown to have partial chloride channel function once adequate surface expression has been achieved (48). This is the case for W1282X, which results in the expression of ~85% of the 1,480 amino acid WT-CFTR.
These functional characteristics of W1282X-CFTR supported the idea that CFTR potentiators (e.g., ivacaftor) may have a therapeutic benefit in patients with this mutation. Furthermore, in vitro studies have shown increased CFTR activity following the addition of ivacaftor to W1282X-CFTR-expressing cells. In CF bronchial epithelial (CFBE41o-) cells expressing W1282X-CFTR, the addition of ivacaftor resulted in a significantly higher voltage clamp short circuit Isc following FSK stimulation compared with cells expressing R1162X-CFTR, a mutation that results in no CFTR function in the truncated state. Ivacaftor was further tested in primary nasal epithelial cells collected from a patient homozygous for W1282X-CFTR and again produced an increased Isc following FSK stimulation (31).
In consideration of these findings, a 31-yr-old female homozygous for W1282X-CFTR was treated with ivacaftor. Prior to initiation of ivacaftor therapy, the patient was reported to have progressively worsening CF symptoms approaching end-stage disease. Pre-ivacaftor spirometry showed FEV1 of 36% predicted, having declined from 47% predicted within the previous 5 yr. Additionally, the patient suffered from frequent pulmonary exacerbations (3.6 incidences per year), poor nutritional status with pre-ivacaftor BMI of 17 and declining despite nutritional support and insulin therapy for CFRD, Pseudomonas aeruginosa, and methicillin-resistant Staphylococcus aureus (MRSA) colonization and diffuse bronchiectasis despite aggressive therapy. In 2013, the patient started ivacaftor 150 mg (twice per day) therapy. Following 3 yr of ivacaftor therapy, FEV1 stabilized, but there was no significant improvement in FEV1, the rate of FEV1 decline, or sweat chloride levels (from 116 to 114 mEq/l) compared with preivacaftor baseline values. Additionally, the rate of pulmonary exacerbations declined from 3.6 incidences per year from preivacaftor baseline to 0.5 incidence per year with ivacaftor therapy (P < 0.05), and the patient also had a significant improvement in rate of BMI change (P < 0.001) and an improvement in nutritional status as BMI had increased to 20.3 (31).
G1208D
In 2016, Zhang et al. reported a clinical case of an African-American infant diagnosed with CF at birth following positive newborn screening. DNA sequencing revealed one copy of F508del-CFTR, while the second allele contained a missense mutation c.3623G→A, encoding G1208D-CFTR. G1208D-CFTR is a novel mutation with no prior documented cases or functional characteristics (www.cftr2.org). The patient was reported to have a mild CF phenotype with poor but steady weight gain, pancreatic sufficiency, intermediate sweat chloride levels, normal chest radiograph findings, and no hospitalizations for pulmonary exacerbations (65).
Western blot analysis of G1208D-CFTR expressing HEK293 cells showed comparable total CFTR levels to WT-CFTR expressing cells; however, G1208D-CFTR cells had an CFTR maturation efficiency (mature/total CFTR × 100%) of 57% compared with 90% in WT-CFTR-expressing cells. Additionally, PCR analysis of these cell lines showed no significant difference in mRNA levels, indicating the decreased cell surface expression of G1208D-CFTR was due to defects in protein maturation and processing (Class II defect). For G1208D-CFTR that does correctly localize to the cell surface, iodide efflux analysis demonstrated a further defect in channel function. Following FSK stimulation, G1208D-CFTR expressing HEK293 cells produced a significantly smaller iodide efflux compared with WT-CFTR, with a maximum efflux rate of 33% relative to WT-CFTR. These results demonstrated a further defect in channel function in G1208D-CFTR (65).
Given the Class II defects of G1208D-CFTR, Zhang et al. (65) tested the in vitro response of G1208D-CFTR expressing HEK293 cells to lumacaftor. The addition of lumacaftor resulted in a significant increase in total CFTR protein expression (1.5-fold increase compared with DMSO-treated control) and channel function (1.5-fold increase in maximal iodide efflux rate compared with DMSO-treated control) (65). The ability of lumacaftor to correct the G1208D-CFTR defect in vitro points to a potential therapeutic benefit in this patient with Orkambi (lumacaftor/ivacaftor) treatment.
S1045Y
S1045Y-CFTR mutation was identified in an African-American full-term male born in 2012 and diagnosed with CF following positive newborn screening (4). The patient had a mild CF phenotype with good weight gain and intermediate sweat chloride levels. Genotyping revealed that the patient had two CFTR mutations: S1045Y- and S926X-CFTR. The S926X-CFTR mutation introduces a premature stop codon, resulting in a truncated CFTR protein (Class I). The S1045Y-CFTR mutation had previously been identified in a Galician female patient homozygous for this mutation who had a severe course of CF (45).
S1045Y-CFTR traffics properly to the cell surface, as shown by similar Western blot band C and B expression in HEK293 cell lines expressing either WT- or S1045Y-CFTR. Despite its normal localization to the cell surface, CFBEo− cells expressing S1045Y-CFTR showed a 50% reduction in FSK-stimulated Isc currents in a Ussing chamber assay, indicating a 50% reduction in channel function. Given the heterozygous state of this patient, with the second mutation producing no functional CFTR, a 25% CFTR function was estimated for this patient. Further analysis of S1045Y-CFTR demonstrated that the introduction of tyrosine at position 1045 predisposes cell surface localized CFTR to rapid internalization by turning the sequence into a putative binding site for E3 ubiquitin ligase c-cbl following phosphorylation. This mutation has responded positively to in vitro addition of Orkambi, and plans are in place to test its efficacy in this patient (4).
The mutations discussed here are only a few examples of rare disease-causing CFTR mutations. There are numerous other rare mutations that are not discussed due to space limitations but that have shown positive results in either in vitro or in vivo experiments with CFTR modulators.
PERSONALIZED MEDICINE FOR CF PATIENTS
The examples above highlight the importance of personalized medicine in finding therapeutic options for CF patients with rare CFTR mutations. N-of-1 trials—blinded, randomized, crossover studies in which patients serve as their own controls—serve as a standardized clinical approach to test the efficacy of modulators in patients with either rare or de novo CFTR mutations. In N-of-1 studies, subjects receive treatment with both placebo and drug, usually for 14 days each, and clinical outcomes (e.g., SwCl, FEV1) are measured to determine the efficacy of the drug for the particular patient (Fig. 4). N-of-1 trials bypass many requirements in RCTs, which require homogenous study populations, the generalization of results to the larger population, considerations for the characteristics of an individual who may dictate response to an intervention, and the need to recruit and retain large sample sizes to obtain meaningful results (29, 32, 57).
Fig. 4.

N-of-1 study design.
Recently, McGarry et al. (32) used a series of N-of-1 trials to test the efficacy of ivacaftor in seven CF patients with residual CFTR function mutations. Patients with Class III mutations, R117H-CFTR mutation, and those homozygous for F508del-CFTR were excluded. The seven patients had the following genotypes: F508del/1154insTC, G542X/3849+10kbC→T, F508del/Y563N, R334W/681delC, 1717-1G→A/G85E, 1717-1G→A/A455E, and F508del/A455E. Prior to initiating N-of-1 trials, human nasal epithelial (HNE) cells isolated from each subject were used to test the in vitro ability of ivacaftor to potentiate transepithelial Cl− current with the given mutations. The in vivo primary outcome was absolute change in sweat chloride from baseline.
From the seven subjects who completed the N-of-1 trials, changes in sweat chloride concentration (mmol/l) from baseline following ivacaftor use were significantly decreased for three subjects with F508del/1154insTC, G542X/3849+10kbC→T, and F508del/Y563N mutations (−40.8, −19.3, −14.8, respectively; all P < 0.001), were found to have no change in two subjects with R334W/681delC and 1717-1G→A/G85E mutations (−2.5 and +9.3, respectively; both P = 0.6), and significant increase in the remaining two subjects with 1717-1G→A/A455E and F508del/A455E mutations (+23.8 and +27.3, respectively; both P < 0.001). These results corresponded to the changes in Cl− current in ivacaftor-treated patient-derived HNE cells following FSK stimulation.
Although N-of-1 trials show a promising future in determining the modulator for CF patients with rare CFTR mutations, limitations/challenges exist due to the nature of N-of-1 trials. First, given the unique characteristics of each individual patient, e.g., genetic and disease stage, it could become difficult to determine the appropriate drug dose with acceptable efficacy and side effects. The second challenge is that the variations of the in vitro studies indicate that they may not always correspond to clinical outcomes. It is to be noted that developing cell lines for in vitro studies for these rare and complex patient disease conditions may also be a challenge, thereby increasing the difficulties to make a decision about pursuing the trials. Moreover, N-of-1 trials require the patient to be in relatively stable condition (i.e., lifestyle, clinical stability, and consistency of data collection) during the trials. Last but not least, another concern is that a carryover effect, which would impact the interpretation of results, may be present.
CONCLUSIONS
Recent advances in the development of CFTR modulators have served as proof-of-concept that targeting the underlying defect in CF is a viable therapeutic option for CF patients. Kalydeco (ivacaftor), a CFTR potentiator, and Orkambi (lumacaftor/ivacaftor), a CFTR corrector/potentiator combination, are the first two drugs to gain FDA approval for clinical use. These agents are known to be effective in only patients with certain CFTR mutations, leaving many others without similar treatment options. Additionally, given that many CF patients have CFTR mutations that occur at very low frequencies in the CF population, the use of RCTs to test the efficacy of modulators in these patients is not practical. N-of-1 trials provide a solution to this problem while challenges exist, and their use has resulted in patients being prescribed CFTR modulators who would otherwise not be treated with such compounds.
GRANTS
This research was funded by the National Institute of Diabetes and Digestive and Kidney Diseases Grants DK080834 and DK093045 (to A. P. Naren) and Cystic Foundation Grants HUANG17F0 (to Y. Huang), ARORA16F0 (to K. Arora), and NAREN14XX0, NAREN1610, and CLANCY15R0 (to A P. Naren).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.H., Y.H. and A.P.N. draft manuscript; M.H., Y.H. and K.M. prepared the figures and tables; M.H., Y.H., A.P.N., K.M., F.Y., and K.A. edited and revised manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. David Armbruster for proofreading and a critical review of the manuscript.
REFERENCES
- 1.Ahmed N, Corey M, Forstner G, Zielenski J, Tsui LC, Ellis L, Tullis E, Durie P. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut 52: 1159–1164, 2003. doi: 10.1136/gut.52.8.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson MP, Rich DP, Gregory RJ, Smith AE, Welsh MJ. Generation of cAMP-activated chloride currents by expression of CFTR. Science 251: 679–682, 1991. doi: 10.1126/science.1704151. [DOI] [PubMed] [Google Scholar]
- 3.Arora K, Huang Y, Mun K, Yarlagadda S, Sundaram N, Kessler MM, Hannig G, Kurtz CB, Silos-Santiago I, Helmrath M, Palermo JJ, Clancy JP, Steinbrecher KA, Naren AP. Guanylate cyclase 2C agonism corrects CFTR mutants. JCI Insight 2: 93686, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arora K, Yarlagadda S, Zhang W, Moon C, Bouquet E, Srinivasan S, Li C, Stokes DC, Naren AP. Personalized medicine in cystic fibrosis: genistein supplementation as a treatment option for patients with a rare S1045Y-CFTR mutation. Am J Physiol Lung Cell Mol Physiol 311: L364–L374, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834, 1990. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 6.Cholon DM, Quinney NL, Fulcher ML, Randell SH, Boucher RC, Gentzsch M. Potentiator ivacaftor impedes pharmacological correction of Delta F508 CFTR. Pediatr Pulmonol 48: 246ra96, 2014. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, De Boeck K, Donaldson SH, Dorkin HL, Dunitz JM, Durie PR, Jain M, Leonard A, McCoy KS, Moss RB, Pilewski JM, Rosenbluth DB, Rubenstein RC, Schechter MS, Botfield M, Ordoñez CL, Spencer-Green GT, Vernillet L, Wisseh S, Yen K, Konstan MW. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67: 12–18, 2012. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clancy JP, Szczesniak RD, Ashlock MA, Ernst SE, Fan L, Hornick DB, Karp PH, Khan U, Lymp J, Ostmann AJ, Rezayat A, Starner TD, Sugandha SP, Sun H, Quinney N, Donaldson SH, Rowe SM, Gabriel SE. Multicenter intestinal current measurements in rectal biopsies from CF and non-CF subjects to monitor CFTR function. PLoS One 8: e73905, 2013. doi: 10.1371/journal.pone.0073905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conrath K, Andrews M, van der Plas S, Sonck K, Gees M, Jans M, Nelles L, Gentzsch M, Wigerinck P. Novel potentiators for treating cystic fibrosis. Poster presented at 27th Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation, Salt Lake City, UT, October 17–19, 2013. [Google Scholar]
- 10.Conrath K, Gesson C, Allamassey L, Van de Steen O, Kanters D, De Kock H, De Boek C, Davies JC. Glpg1837 in subjects with cystic fibrosis and the S1251n or G551d mutation: results from phase 2a studies (Saphira 1 and 2). Poster presented at 31st Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation Indianapolis, IN, Nov. 2–4, 2017. [Google Scholar]
- 11.Coote K, Valdez-Misiolek R, Miraglia L, Fadul M, Welch G, Barnes W, Orth A, Dowling M, Strieter RM, Kazani S, Rowlands DJ. Qbw251 Is a Clinically Efficacious Cftr Potentiator across Residual Function and Gating-Defect Mutations: Translation of in Vitro Activity to Clinical Efficacy. Poster presented at 30th Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation Orlando, FL, Oct. 27–29, 2016 [Google Scholar]
- 11a.Cystic Fibrosis Foundation Cystic Fibrosis Foundation Patient Registry 2015 Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation, 2015. [Google Scholar]
- 12.Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, Southern KW, Robertson S, Green Y, Cooke J, Rosenfeld M. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 4: 107–115, 2016. doi: 10.1016/S2213-2600(15)00545-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, Mainz JG, Rodriguez S, Li H, Yen K, Ordoñez CL, Ahrens R. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 187: 1219–1225, 2013. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Boeck C, Van Braeckel E, Van der Ent CK, Verhulst S, Weersink EJ, Conrath K, Kanters D, Namour F, de Kock H, Van de Steen O. Glpg1837 in Subjects with Cystic Fibrosis (Cf) and the S1251n Mutation: Results from a Phase Iia Study (Saphira2). Poster presented at 30th Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation Orlando, FL, Oct 27–29, 2016. [Google Scholar]
- 15.De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, Higgins M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 13: 674–680, 2014. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de Jong NW, Bijvelds MJ, Scholte BJ, Nieuwenhuis EE, van den Brink S, Clevers H, van der Ent CK, Middendorp S, Beekman JM. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 19: 939–945, 2013. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- 17.Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordoñez CL, Geller DE. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 142: 718–724, 2012. doi: 10.1378/chest.11-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelfond D, Borowitz D. Gastrointestinal complications of cystic fibrosis. Clin Gastroenterol Hepatol 11: 333–342, 2013. doi: 10.1016/j.cgh.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168: 918–951, 2003. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 21.Graeber SY, Hug MJ, Sommerburg O, Hirtz S, Hentschel J, Heinzmann A, Dopfer C, Schulz A, Mainz JG, Tümmler B, Mall MA. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am J Respir Crit Care Med 192: 1252–1255, 2015. doi: 10.1164/rccm.201507-1271LE. [DOI] [PubMed] [Google Scholar]
- 22.Harbeson S, Nguyen S, Bridson G, Uttamsingh V, Wu L, Morgan AJ, Aslanian A, Braman V, Pilja L. Pharmacokinetic studies of deuterated analogs of Ivacaftor in preclinical models and healthy volunteers. Pediatr Pulmonol 50: 202, 2015. 25187271 [Google Scholar]
- 23.Haupt ME, Kwasny MJ, Schechter MS, McColley SA. Pancreatic Enzyme Replacement Therapy Dosing and Nutritional Outcomes in Children with Cystic Fibrosis. J Pediatr 164: 1110–1115.e1, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Hodges CA, Palmert MR, Drumm ML. Infertility in females with cystic fibrosis is multifactorial: evidence from mouse models. Endocrinology 149: 2790–2797, 2008. doi: 10.1210/en.2007-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jih KY, Hwang TC. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci USA 110: 4404–4409, 2013. doi: 10.1073/pnas.1215982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazani S, Alcantara J, Debonnett L, Doucet J, Jones I, Kulmatycki K, Machineni S, Mostovy L, Nicholls I, Vegesna R, Verheijen J, Rowlands DJ. Qbw251 is a safe and efficacious Cftr potentiator for patients with cystic fibrosis. Am J Respir Crit Care 193: A7789, 2016. [Google Scholar]
- 27.Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Prz Gastroenterol 9: 136–141, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, Zaccolo M, Jalink K, Li M, Nelson DJ, Schuetz JD, Naren AP. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 131: 940–951, 2007. doi: 10.1016/j.cell.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Per Med 8: 161–173, 2011. doi: 10.2217/pme.11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacKenzie IER, Paquette V, Gosse F, George S, Chappe F, Chappe V. Modeling cystic fibrosis disease progression in patients with the rare CFTR mutation P67L. J Cyst Fibros 16: 335–341, 2017. doi: 10.1016/j.jcf.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Magne F, Durupt S, Nove-Josserand R, Bey-Omar F, Laoust L, Cottin V, Durieu I, Reynaud Q. Therapeutic benefit of ivacaftor in late cystic fibrosis caused by homozygous IVS8-5T CFTR polymorphism. J Cyst Fibros 16: 89–90, 2017. doi: 10.1016/j.jcf.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 32.McGarry ME, Illek B, Ly NP, Zlock L, Olshansky S, Moreno C, Finkbeiner WE, Nielson DW. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr Pulmonol 52: 472–479, 2017. doi: 10.1002/ppul.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mijnders M, Musch S, Peters F, Conrath K, Braakman I, Kleizen B. Mutations in the Second Cytoplasmic Loop of Cftr Suggest Distinct Mode of Action between Potentiators Vx-770 and Glpg1837. Poster presented at 31st Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation Indianapolis, IN, Nov 2–4, 2017. [Google Scholar]
- 34.Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, Rubenstein RC, Higgins M. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med 3: 524–533, 2015. doi: 10.1016/S2213-2600(15)00201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muhlebach MS, Clancy JP, Heltshe SL, Ziady A, Kelley T, Accurso F, Pilewski J, Mayer-Hamblett N, Joseloff E, Sagel SD. Biomarkers for cystic fibrosis drug development. J Cyst Fibros 15: 714–723, 2016. doi: 10.1016/j.jcf.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musch S, Ramalho A, Van der Plas S, Andrews M, Christophe T, Nelles L, De Boeck C, Conrath K. Measuring Potentiator Activity Using Organoids. Poster presented at 30th Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation Orlando, FL, Oct. 27–29, 2016. [Google Scholar]
- 37.Ong T, Marshall SG, Karczeski BA, Sternen DL, Cheng E, Cutting GR. Cystic Fibrosis and Congenital Absence of the Vas Deferens. In: GeneReviews, edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, and Ledbetter N. Seattle, WA: University of Washington, 1993. [Google Scholar]
- 38.Pedemonte N, Tomati V, Sondo E, Galietta LJ. Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol 298: C866–C874, 2010. doi: 10.1152/ajpcell.00404.2009. [DOI] [PubMed] [Google Scholar]
- 39.Pedemonte N, Zegarra-Moran O, Galietta LJ. High-throughput screening of libraries of compounds to identify CFTR modulators. Methods Mol Biol 741: 13–21, 2011. doi: 10.1007/978-1-61779-117-8_2. [DOI] [PubMed] [Google Scholar]
- 40.Penmatsa H, Frederick CA, Nekkalapu S, Conoley VG, Zhang W, Li C, Kappes J, Stokes DC, Naren AP. Clinical and molecular characterization of S1118F-CFTR. Pediatr Pulmonol 44: 1003–1009, 2009. doi: 10.1002/ppul.21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phuan PW, Yang B, Knapp JM, Wood AB, Lukacs GL, Kurth MJ, Verkman AS. Cyanoquinolines with independent corrector and potentiator activities restore ΔPhe508-cystic fibrosis transmembrane conductance regulator chloride channel function in cystic fibrosis. Mol Pharmacol 80: 683–693, 2011. doi: 10.1124/mol.111.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 135: 1610–1618, 2009. doi: 10.1378/chest.08-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramsey BW, Banks-Schlegel S, Accurso FJ, Boucher RC, Cutting GR, Engelhardt JF, Guggino WB, Karp CL, Knowles MR, Kolls JK, LiPuma JJ, Lynch S, McCray PB Jr, Rubenstein RC, Singh PK, Sorscher E, Welsh M. Future directions in early cystic fibrosis lung disease research: an NHLBI workshop report. Am J Respir Crit Care Med 185: 887–892, 2012. doi: 10.1164/rccm.201111-2068WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C, Elborn JS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365: 1663–1672, 2011. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rana-Díez P, Colón C, Alonso-Fernández JR, Solar A, Barros-Tizón JC, Barros-Casas D, Sirvent J, Carracedo A, Barros F. Three novel mutations in the CFTR gene identified in Galician patients. J Cyst Fibros 7: 520–522, 2008. doi: 10.1016/j.jcf.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 46.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073, 1989. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 47.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 352: 1992–2001, 2005. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 48.Rowe SM, Varga K, Rab A, Bebok Z, Byram K, Li Y, Sorscher EJ, Clancy JP. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol 37: 347–356, 2007. doi: 10.1165/rcmb.2006-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabusap CM, Wang W, McNicholas CM, Chung WJ, Fu L, Wen H, Mazur M, Kirk KL, Collawn JF, Hong JS, Sorscher EJ. Analysis of cystic fibrosis-associated P67L CFTR illustrates barriers to personalized therapeutics for orphan diseases. JCI Insight 1: e86581, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev 79, Suppl: S23–S45, 1999. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 51.Strausbaugh SD, Davis PB. Cystic fibrosis: A review of epidemiology and pathobiology. Clin Chest Med 28: 279–288, 2007. [DOI] [PubMed] [Google Scholar]
- 52.Uttamsingh V, Pilja L, Brummel CL, Grotbeck B, Cassella JV, Braman G. Ctp-656 Multiple Dose Pharmacokinetic Profile Continues to Support a Once-Daily Potentiator for Cystic Fibrosis Patients with Gating Mutations. Poster presented at 30th Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation Orlando, FL, Oct. 27–29, 2016. [Google Scholar]
- 53.Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 106: 18825–18830, 2009. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA 108: 18843–18848, 2011. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54a.Van Goor F. Precision Medicine in Cystic Fibrosis: Ivacaftor in Rare Mutations. Paper presented at 30th North American Cystic Fibrosis Conference Cystic Fibrosis Foundation Orlando, Florida, October 27–29, 2016 [Google Scholar]
- 55.Vankeerberghen A, Cuppens H, Cassiman JJ. The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions. J Cyst Fibros 1: 13–29, 2002. doi: 10.1016/S1569-1993(01)00003-0. [DOI] [PubMed] [Google Scholar]
- 56.Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, Hong JS, Pollard HB, Guggino WB, Balch WE, Skach WR, Cutting GR, Frizzell RA, Sheppard DN, Cyr DM, Sorscher EJ, Brodsky JL, Lukacs GL. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell 27: 424–433, 2016. doi: 10.1091/mbc.E14-04-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verkman AS, Galietta LJV. Chloride channels as drug targets. Nat Rev Drug Discov 8: 153–171, 2009. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vohra S, Shamseer L, Sampson M, Bukutu C, Schmid CH, Tate R, Nikles J, Zucker DR, Kravitz R, Guyatt G, Altman DG, Moher D; CENT Group . CONSORT extension for reporting N-of-1 trials (CENT) 2015 Statement. J Clin Epidemiol 76: 9–17, 2016. doi: 10.1016/j.jclinepi.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP; TRAFFIC Study Group; TRANSPORT Study Group . Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 373: 220–231, 2015. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walkowiak J, Sands D, Nowakowska A, Piotrowski R, Zybert K, Herzig KH, Milanowski A. Early decline of pancreatic function in cystic fibrosis patients with class 1 or 2 CFTR mutations. J Pediatr Gastroenterol Nutr 40: 199–201, 2005. doi: 10.1097/00005176-200502000-00022. [DOI] [PubMed] [Google Scholar]
- 61.Watson MS, Cutting GR, Desnick RJ, Driscoll DA, Klinger K, Mennuti M, Palomaki GE, Popovich BW, Pratt VM, Rohlfs EM, Strom CM, Richards CS, Witt DR, Grody WW. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med 6: 387–391, 2004. doi: 10.1097/01.GIM.0000139506.11694.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong AP, Bear CE, Chin S, Pasceri P, Thompson TO, Huan LJ, Ratjen F, Ellis J, Rossant J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol 30: 876–882, 2012. doi: 10.1038/nbt.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yousef S, Solomon GM, Brody A, Rowe SM, Colin AA. Improved clinical and radiographic outcomes after treatment with ivacaftor in a young adult with cystic fibrosis with the P67L CFTR mutation. Chest 147: e79–e82, 2015. doi: 10.1378/chest.14-1198. [DOI] [PubMed] [Google Scholar]
- 64.Zhang W, Fujii N, Naren AP. Recent advances and new perspectives in targeting CFTR for therapy of cystic fibrosis and enterotoxin-induced secretory diarrheas. Future Med Chem 4: 329–345, 2012. doi: 10.4155/fmc.12.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang X, Hothi JS, Zhang YH, Srinivasan S, Stokes DC, Zhang W. c.3623G > A mutation encodes a CFTR protein with impaired channel function. Respir Res 17: 8, 2016. doi: 10.1186/s12931-016-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]