

Abstract
Zn2+ is an essential element for cell survival/growth, and its deficiency is linked to many disorders. Extracellular Zn2+ concentration changes participate in modulating fundamental cellular processes such as proliferation, secretion, ion transport, and cell signal transduction in a mechanism that is not well understood. Here, we hypothesize that the Zn2+-sensing receptor ZnR/G protein-coupled receptor 39 (GPR39), found in tissues where dynamic Zn2+ homeostasis takes place, enables extracellular Zn2+ to trigger intracellular signaling pathways regulating key cell functions in vascular cells. Thus, we investigated how extracellular Zn2+ regulates cell viability, proliferation, motility, angiogenesis, vascular tone, and inflammation through ZnR/GPR39 in endothelial cells. Knockdown of GPR39 through siRNA largely abolished Zn2+-triggered cellular activity changes, Ca2+ responses, as well as the downstream activation of Gαq-PLC pathways. Extracellular Zn2+ promoted vascular cell survival/growth through activation of cAMP and Akt as well as overexpressing of platelet-derived growth factor-α receptor and vascular endothelial growth factor A. It also enhanced cell adhesion and mobility, endothelial tubule formation, and cytoskeletal reorganization. Such effects from extracellular Zn2+ were not observed in GPR39−/− endothelial cells. Zn2+ also regulated inflammation-related key molecules such as heme oxygenase-1, selectin L, IL-10, and platelet endothelial cell adhesion molecule 1, as well as vascular tone-related prostaglandin I2 synthase and nitric oxide synthase-3. In sum, extracellular Zn2+ regulates endothelial cell activity in a ZnR/GPR39-dependent manner and through the downstream Gαq-PLC pathways. Thus, ZnR/GPR39 may be a therapeutic target for regulating endothelial activity.
Keywords: angiogenesis, cell signaling, gene regulation, inflammation, vascular tone regulation
INTRODUCTION
Zn2+ is an essential supplementary nutrient for health and is involved in structural and regulatory cellular functions (11, 24). It interacts with Zn-finger domains and acts as a cofactor for hundreds of enzymes (5, 50). It also binds to numerous membrane receptors, transporters, and channels, thereby regulating their activity (20). Zn2+ deficiency is linked to many disorders involving different organs and tissues, such as digestive, immune, neuronal, and vascular systems (43, 50, 53). Severe lack of Zn2+ may lead to attenuation of growth and sexual development (50). However, an excess of extracellular Zn2+ is toxic; for example, a massive release of synaptic Zn2+ induces neuronal cell death in brain ischemia (3, 7, 22). Moreover, dramatic changes in intracellular and extracellular Zn2+ concentrations occur during various pathophysiological syndromes such as myocardial infarction, hepatic renal failure, and neoplastic processes (52). Despite the important role of Zn2+ in cellular function and the fluctuations in Zn2+ concentrations, little is known about the cellular signaling mechanisms regulated by Zn2+ and its effect on cell functions and fate in endothelial cells (26, 28).
In vertebrate animals and human beings, endothelial cells are the main vascular cells for blood vessels (33). These cells are involved in many aspects of vascular biology; among them, major functions of vascular cells include formation and repair of blood vessels (angiogenesis) (56), vasoconstriction and vasodilation (vascular tone) (32), and inflammation (35, 49), as well as blood clotting and barrier functions (54), etc. Although Zn2+ plays an essential role in endothelial cell health and functions, the link between extracellular Zn2+ and cellular signaling pathways involved in these vascular functions is not well understood.
Recently, Zn2+ has emerged as an important signaling molecule, activating intracellular pathways and regulating cell fate in various cell types like epithelium and colonocyte. A Zn2+-sensing receptor (ZnR), also known as the G protein-coupled receptor GPR39, was identified, which is a Gq-coupled receptor mediating changes in extracellular Zn2+ and major intracellular signaling pathways (2, 9, 10, 13, 42, 44–46). It triggers inositol 1,4,5-trisphosphate-dependent release of intracellular Ca2+, leading to activation of mitogen-activated protein (MAP) and phosphoinositide 3 (PI3) kinase pathways. Moreover, GPR39 knockout mice exhibited symptoms of Zn2+-deficiency, including depression, accelerated gastric emptying, and increased fecal excretion (30, 31). However, how Zn2+ or ZnR/GPR39 signaling regulates endothelial activity is not well understood.
In the present work, we investigated the role of ZnR/GPR39 in Zn2+-regulated endothelial activity related to angiogenesis, endothelial permeability, vascular tone, and inflammation. They are endothelial cell viability, proliferation, mobility, and tubule formation as related to angiogenesis and regulation of key molecules involved in vasoconstriction and vasodilation as related to vascular tone, as well as expression and secretion of key molecules involved in inflammation. The findings herein provide evidence for a specific Zn2+-sensing mechanism involving cellular Ca2+ and PI3 signaling and, hence, controlling key vascular cellular processes.
MATERIALS AND METHODS
Solution preparation.
ZnCl2 solutions (10 mM) were prepared by dissolving ZnCl2 (Sigma-Aldrich) into deionized water and adjusting to neutral pH by NaOH. Solutions were filtered by 0.22-μm filter (Thermo Fisher Scientific) and autoclaved (Harvey Sterile Max, Thermo Scientific). ZnCl2-supplemented medium (pH 7.2) were prepared by diluting ZnCl2 stock solution with cell culture medium (26–28, 59). A physiologically relevant concentration of 25 µM was used as Zn2+ treatment group, as the serum concentration of Zn is in the range of 10–30 µM (14). Typical cell culture medium DMEM (Sigma-Aldrich) has no or only a trace amount of Zn2+ (<0.7 pM) so was used as the negative control (Zn2+ deficiency control).
Cell culture.
Primary human coronary artery endothelial cells (HCAECs) were ordered from ATCC. Three batches of cells from different healthy donors were used. The components of cell culture medium were basal medium, 5% fetal bovine serum, 1% endothelial cell growth supplement, and 1% penicillin-streptomycin. HCAECs were cultured in a flask (Falcon, BD Bioscience) until ~90% confluence was reached. Then cells were detached by trypsin-EDTA (Life Technologies), centrifuged (Sorvall Biofuge Stratos, Thermo Electron), and resuspended. The cell number was counted with a cell counter (Bio-Rad). Cells at passages 3–5 were used (26–28, 59). For all cell treatment experiments, cells were starved overnight in serum-free medium and then treated with different concentrations of Zn2+ in basal DMEM medium without serum.
Mouse GPR39−/− endothelial cells.
Mouse GPR39−/− endothelial cells were a gift from Capital Medical University. The endothelial cells were >99% pure, as measured by staining for the von Willebrand factor (endothelial cells) and negative immunostaining for smooth muscle cell α-actin (25). Cells at passages 3–5 from six different batches were used.
Cell viability.
Cells were seeded in a 96-well cell culture plate (BD Biosciences) with 5,000 cells per well for 24 h to allow cell attachment. Medium was replaced by medium supplemented with different solutions and incubated for 24 h. Medium with 10% DMSO (Life Technologies) and medium alone were positive and negative controls, respectively. Another blank reference containing the same concentration of Zn ion solution without cells was used to exclude the interference of the ions. The 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrezolium bromide (MTT, Invitrogen) test was performed according to the manufacturer's protocol. Absorbance was measured at 570 nm, using a microplate reader (SpectraMax, Molecular Devices) (26–28, 59).
Cell proliferation.
A BrdU cell proliferation kit (Cell Signaling) was used for the cell proliferation test. Cells were seeded in a 96-well cell culture plate at 5,000 cells per well. After 24 h, medium was replaced by different solutions and incubated for 24 h to allow cell growth and doubling. The proliferation test was performed according to the manufacturer's protocol. Absorbance was measured at 450 nm. Positive control and negative control were medium without Zn ion supplement and medium without cells, respectively (26–28, 59).
Cell adhesion.
Cells were seeded onto a 24-well plate (Falcon, Corning). The final cell density was 50,000 cells per well. Cells were incubated at 37°C, 5% CO2, and 95% relative humidity for 5 h with the treatment of Zn2+. The time of 5 h should be sufficient for complete attachment of cells on a culture surface without cell division/growth during attachment. Then, the cell medium was removed, and cells were washed three times with Dulbecco’s phosphate-buffered saline(DPBS). Images of adhered cells were taken with a microscope (EVOS FL Cell Imaging System, AMG). The plate was sealed with self-sticking tape (Fisherbrand, Fisher Scientific). Then, the plate was put into a rotor inversely and centrifuged at 500 rpm for 5 min. Cells were washed with DPBS and fixed by 4% paraformaldehyde (Boston BioProducts). The images of the adhered cells were taken with a microscope (EVOS FL Cell Imaging System, AMG) and analyzed with ImageJ (National Institutes of Health, Bethesda, MD). At least 10 different fields were used for calculating adhered cell density and cell retention ratio (26–28, 59).
Cell migration.
Cells were seeded on a 12-well cell culture plate (BD Biosciences). The cytostatic agent hydroxyurea (250 µM) was added into culture medium to inhibit cell division/growth during migration. A straight line in a cell monolayer was created by scratching the surface using a p200 pipette tip (Thermo Scientific). Debris was removed by gently washing three times with DPBS, and cells were incubated with 3 ml of medium supplemented with different ion solutions. At 0, 6, and 24 h, optical images were taken using a phase contrast microscope (Advanced Microscopy). The width of the line at top, middle, and bottom positions was measured in Image-Pro Plus 6.0 (Media Cybernetics). The average cell migration rate was calculated as described before (26–28, 59).
Tubulogenesis assays.
Endothelial tubular differentiation assays were carried out according to a previously published method with some modifications (1). Briefly, cells (3,000 per sample) in phenol red-free medium containing 2% FCS were plated in 24-well plates precoated with 250 μl of growth factor-reduced, phenol red-free Matrigel (BD Biosciences) for 1 h at 37°C. After treatment, cells were incubated in a humidified incubator at 37°C and 5% CO2. After 16 h, bright-field images were captured from five random fields of view per sample at ×10 magnification. Tubule length and number of branching points for each condition were quantified using Image Pro-Plus 6.0 software.
RNA extraction.
Cells were seeded in 100-mm culture dishes (BD Technologies) and allowed to attach for 24 h. Then the cells were treated with medium and medium supplemented with ZnCl2, respectively, for 24 h. Cells were harvested, and total RNA was extracted using an RNeasy Mini Kit (Qiagen) and subsequently quantified using a spectrophotometer (Nanodrop 2000) with OD260/OD280 ratios between 1.9 and 2.1 (26–28, 59).
cDNA synthesis.
A total of 600 ng of RNA was used for reverse transcription using an RT2 First Strand Kit (Qiagen). Reverse transcription was performed in a thermo cycler (T100, Bio-Rad). Then, 91 μl of RNase-free water was added to the 20 μl of cDNA mix and stored at −20°C in a freezer (Puffer Bubbard, Thermo Scientific) (26–28, 59).
Real-time-PCR.
Gene expression analysis was performed in a CFX96 Touch RT-PCR Detection System (Bio-Rad) using the RT2 Profiler PCR array (Qiagen). The array includes 84 functional genes, five housekeeping genes, three reverse-transcription controls, and three positive PCR controls. A total of 25 μl of PCR components mix including cDNA, SYBR Green Mastermix, and RNase-free water was dispensed to the RT2 Profiler PCR Array plate. After initial heat activation (95°C, 10 min), cDNA was amplified as the following parameters: 95°C for 15 s and 60°C for 1 min. After the amplification, melting-curve analysis was performed using the default melting curve program. Only the genes with a single melting peak were chosen for final analysis. Data were analyzed by Bio-Rad CFX Manager 3.1 (Bio-Rad). The 2−ΔΔCT method was used to calculate gene fold changes. Real-time PCR was also used to estimate the mRNA levels of ZnR/GPR39. The level of target mRNA was normalized to the endogenous control β-actin. Primers and probes were from Dharmacon RNAi Technologies (Thermo Scientific): for GPR39, forward primer CATCTTCCTGAGGCTGA, reverse primer ATGATCCTCCGTCTGGTTG, probe TATGCTGGATGCCCAAC; and for actin, forward primer TGGAGAAAATCTGGCACCAC, reverse primer GGTCTCAAACATGATCTGG, probe ACCGCCAGAAGATGACC (26–28, 59).
Gene silencing.
Cells were seeded in 60-mm cell-culture dishes (density of 7 × 105 cells/plate) or in 96-well plates (1.5 × 104 cells/well) 24 h before transfection and maintained in standard culture medium as described above but without penicillin or streptomycin. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol and used 48 h posttransfection. Small interfering RNAs (siRNA) targeting human GPR39 (5′-CCAUGG AGUUCUACAGCAU-3′) were purchased from Santa Cruz Biotechnology. The corresponding scrambled siRNAs were used as controls (61).
Calcium response through imaging.
Cells were incubated with 3 µM Fura 2-acetoxymethyl ester for 30 min in Ringer’s solution with 0.1 BSA. After dye loading, the cells were washed in Ringer’s solution [sodium chloride (8.6 g/l), potassium chloride (0.3 g/l), and calcium chloride dihydrate (0.33 g/l)], and coverslider chambers were mounted into the microscope for imaging. All treatments with Zn2+ were applied in the absence of serum to avoid chelation of this ion. Fura 2 was excited at 340 and 380 nm and imaged with a 510-nm filter. Fura 2 fluorescence change was determined by the rate of initial normalized fluorescence change (ΔR/s) as previously described (2).
Western blot analysis.
Cells were washed twice with ice-cold PBS, followed by the addition of 200 μl of cell lysate medium. After collection of the cell lysate, protein concentrations were determined using the Bradford assay (Bio-Rad). Equal amounts of protein for each sample were applied to SDS-PAGE (10% gels). After electrophoresis, proteins were transferred onto nitrocellulose membranes (0.45 μm, Bio-Rad) and the membranes subsequently blocked with 5% (wt/vol) nonfat dried skimmed milk powder-TBST (TBS containing 0.05% Tween 20) for 1 h at room temperature. Primary antibodies against protein of interest and corresponding secondary antibodies conjugated to horseradish peroxidase (HRP) were applied. To confirm equal protein loading, membranes were stripped at 60°C for 30 min with a stripping buffer. The immunoblots were reblocked in 5% (wt/vol) nonfat dried skimmed milk powder-TBST followed by incubation with antibodies that react with β-actin, or total forms of proteins of interest. Bands were visualized using SuperSignal West Pico chemiluminescence detection reagents from Pierce Biotechnology (61).
Akt serine phosphorylation.
Cells were lysed using RIPA buffer after specified treatment. Proteins (20 µg) were analyzed by 4–15% Tris·HCl gel and transferred to nitrocellulose membranes (0.45 µm, Bio-Rad). The membranes were incubated overnight with primary rabbit polyclonal anti-mouse phospho-Akt (Ser473) antibody or rabbit polyclonal anti-mouse Akt antibody diluted in 5% nonfat milk or 5% BSA in TBS and then incubated with the corresponding secondary HRP-conjugated antibodies for 1 h. Immunoreactivity was detected using the ECL detection system (Amersham, Piscataway, NJ) (61).
cAMP direct immunoassay.
The concentration of cAMP was measured following the instructions of the cAMP Direct Immunoassay Kit (Abcam). Briefly, the cultured cells were scraped and dissociated completely and centrifuged at 14,000 g for 10 min to collect the supernatant as the testing sample. After being neutralized and acetylated with acetylating reagent and neutralizing buffer, respectively, 50 μl of standard cAMP (or testing samples were added to a G protein-coated 96-well plate and incubated with 10 μl of cAMP antibody at room temperature for 1 h with gentle agitation. Then 10 μl of cAMP-HRP was added, and the plates were incubated for another hour. The suspension was discarded, and the cells in the wells were washed with 1× cAMP assay buffer five times. The detecting reaction was conducted by incubating the cells with 100 μl of HRP for 1 h and stopped by adding 100 μl of 1 M HCl. Then, the reaction was checked by a microtiter plate reader at 450 nm. The absorbance of the substrate was also detected as background absorbance and subtracted from all standards and samples. The molar concentration of cAMP in cell pellets was determined from standard curves (60).
Immunofluorescence and cytoskeleton staining.
Cells were seeded in a 12-well cell culture plate and treated with medium supplemented with ZnCl2 for 24 h. An Image-iT Fix-Perm kit (Invitrogen) was used to fix cells. F-actin was stained with Actin Green 488 Ready Probes Reagent (Invitrogen). The cell nucleus was stained with the SlowFade Gold Anti-fade Reagent with DAPI (Invitrogen). The GPR39 was stained with rabbit anti-GPR39 antibody (Abcam) followed by Alexa Fluor 488 mouse anti-rabbit IgG (Invitrogen). Images were taken using an EVOS inverted fluorescent microscope (Advanced Microscopy). Fluorescence intensity of the cells was extracted by using ImageJ (version 1.49 software). Contrast of the representative images was autoadjusted using Image-Pro Plus 6.0 (60, 61).
ELISA analysis of proteins.
Cells were seeded with 4 ml of culture medium into a 60-mm petri dish (Primaria, Corning). After ~90% confluence was reached, cell culture medium was replaced by different treatment solutions. After 24 h, a 4-ml treatment solution was collected and centrifuged at 20,000 g. The supernatants were collected and aliquoted into 500-μl tubes. Then, 200 μl of cell lysis buffer with 1× protease, and phosphatase cocktail (Thermo Fisher Scientific) was added to the petri dish and the dish was put on ice. After a while, cell lysate was collected by cell scraper (Thermo Fisher Scientific) and centrifuged at 20,000 g. The supernatants were also collected and aliquoted. All samples were stored at −80°C until use. All ELISA reagents were bought from R&D Systems. All assays were performed according to their protocol. The absorbances were measured with a microplate reader (Molecular Devices) (26–28, 59).
Transendothelial electrical resistance.
Cells were cultured on inserts with collagen-coated polycarbonate in a transwell. The transwell had membrane filters with 0.4-µm pores (Corning Life Science, Lowell, MA). Transendothelial electrical resistance (TER) was measured using an Endohmeter (World Precision Instruments, Sarasota, FL). TER values are expressed in Ω·cm2. The resistance of collagen-coated inserts was subtracted from the resistance obtained in the presence of the endothelial cultures (61).
Permeability of the endothelial barrier.
We used fluorescein-isothiocyanate (FITC)-labeled dextran (40 kDa, Invitrogen) to measure permeability (61). Briefly, the medium in the upper chamber of an established monolayer was replaced with 500 µl of FITC-labeled dextran solution (2 mg/ml) in phenol-red-free DMEM. The lower chamber containing, 1,500 µl of phenol-red-free DMEM was sampled (20 µl) at 5-min intervals for 30 min. At each time point, 20-µl samples were taken from the upper chamber as well. The fluorescence intensity of each sample was determined using a fluorescence multiwell plate reader (PerkinElmer Victor 3) at excitation/emission wavelengths of 490/525 nm. The corresponding FITC-labeled dextran concentrations were determined from the standard curve. The endothelial monolayer permeability to dextran was expressed as a permeability coefficient in cm/s.
Statistical analysis.
All data are presented as means ± SD (n ≥ 5). One-way or two-way analysis of variance (ANOVA) followed by Tukey's post hoc test was used to determine statistically significant differences. An unpaired Student's t-test was also used to compare differences between two groups as appropriate. P < 0.05 was considered statistically significant.
RESULTS
Zn2+ stimulates intracellular Ca2+ release and activates downstream pathway mediated by ZnR/GPR39.
To determine the role of ZnR/GPR39, cells were transfected with siRNA against GPR39 to knock down its expression. We controlled GPR39 expression in endothelial cells as shown, with RT-PCR (Fig. 1A), Western blotting (Fig. 1B), and immunofluorescent staining (Fig. 1C) using siGPR39 but not scramble control. GPR39 appeared to be located largely on the cell membrane (Fig. 1C). The normal serum Zn2+ concentration is ~10–30 µM (14); thus, we used a Zn2+ concentration of 25 µM, which is in the vicinity of physiological condition, to stimulate the cells. Extracellular Zn2+ induced a significant Ca2+ response (Fig. 1D). Silencing of GPR39 was followed by a diminished Ca2+ response triggered by Zn2+. To further verify that the response was triggered via the Gαq-PLC pathway, as suggested before (2, 57), we used pharmacological inhibitors and monitored the Zn2+-dependent Ca2+ response. Inhibition of the Gαq using YM254890 (1 µM) blocked the Zn2+-dependent Ca2+ response almost completely (Fig. 1E). The PLCβ inhibitor u73122 (10 µM) largely abolished the signaling triggered by Zn2+, yet to a lower extent than the Gαq inhibitor or due to the fact that the PLC is downstream from the Gαq. Thus, inhibition of the latter suppressed the Ca2+ response more efficiently.
Fig. 1.

Zn2+ triggers Ca2+ response and downstream signaling through ZnR/GPR39 in endothelial cells. A: RT-PCR analysis of GPR39 expression in endothelial cells treated with different siRNA. B: Western blots showing the expression level of GPR39 in endothelial cells with different siRNA treatment. C: representative immunofluorescent images showing GPR39 expression in endothelial cells treated with different siRNA. (Scale bar = 20 µm.) D: rate of Ca2+ response from measurement of fluorescence change with Zn2+ (25 µM) treatment. E: rate of Ca2+ response from measurement of fluorescence change with treatment of Zn2+ (25 µM) and pharmacological inhibitors of Gαq and PLC. *P < 0.01 vs. controls at normal conditions in endothelial cells (n = 6).
Zn2+ promotes vascular cell viability and proliferation via ZnR/GPR39.
The effect of Zn2+ on endothelial survival and growth was tested by measuring cell viability and proliferation. Cell viability analysis using MTT revealed that Zn2+ significantly increased the survival rate of endothelial cells, and knockdown of GPR39 using siRNA dramatically counteracted such an effect induced by Zn2+ on cell viability (Fig. 2A). A similar result was observed for cell proliferation (Fig. 2B). Next, to further understand the mechanism of Zn2+ on cell survival/growth, we examined the intracellular cAMP level and the activation of the Akt pathway because they are the important modulators in cell viability. The level of cAMP was significantly enhanced after Zn2+ treatment (Fig. 2C), and Zn2+ also induced significant phosphorylation of Akt (Fig. 2D).
Fig. 2.
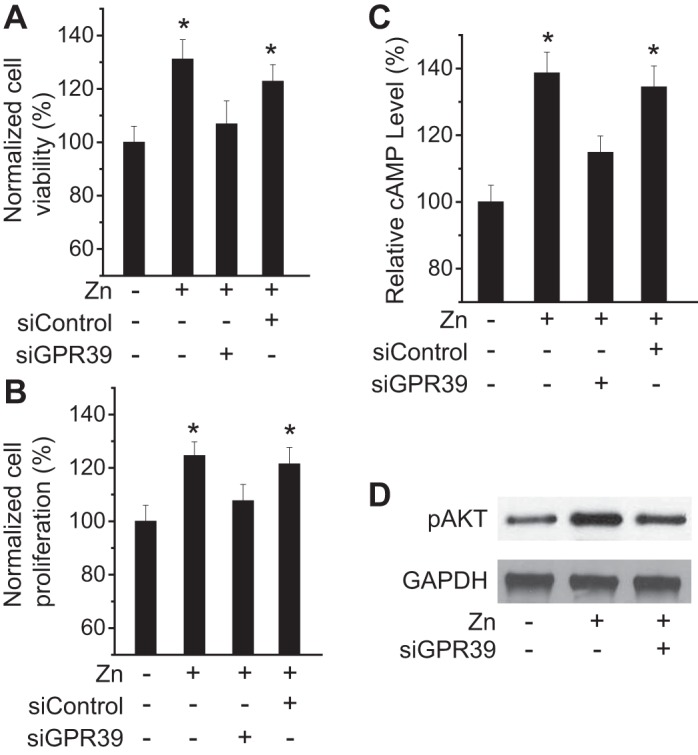
Zn2+ regulates endothelial cell viability and proliferation via ZnR/GPR39. A: cell viability of endothelial cells with Zn2+-deficient condition and Zn2+ (25 µM) treatment for 24 h. B: cell proliferation of endothelial cells with Zn2+-deficient condition and Zn2+ (25 µM) treatment for 24 h. C: intracellular cAMP level in endothelial cells with Zn2+-deficient condition and Zn2+ (25 µM) treatment for 24 h. D: Akt phosphorylation after Zn2+ (25 µM) treatment for 24 h in endothelial cells. *P < 0.05 vs. controls at Zn2+-deficient conditions in endothelial cells (n = 6).
Zn2+ promotes endothelial cell adhesion, mobility, and cytoskeletal reorganization and reduces transendothelial permeability via ZnR/GPR39.
Cell motility plays a pivotal role in various cellular functions and activity, such as division, migration, contraction, and angiogenesis. To elucidate the effect of Zn2+ on cell motility, we measured cell adhesion and mobility with and without extracellular Zn2+. The adhered cell density of endothelial cells increased ~30% after being treated with Zn2+ for 5 h compared with negative controls (Fig. 3A). Transfection with GPR39 siRNA significantly abolished such enhanced adhesion induced by Zn2+. Similar results were observed for cell migration rate (Fig. 3B). Zn2+ significantly increased cell migration rate, whereas knockdown of GPR39 by siRNA blocked such effects of Zn2+ on cell motility. Since cell movement is largely facilitated by cytoskeletal rearrangement, we next quantitatively measured the amount of F-actin stress fibers. Zn2+ treatment significantly promoted F-actin polymerization, leading to the emerging of F-actin stress fibers, as shown by representative images of endothelial cells (Fig. 3C). Quantitative analysis of F-actin fluorescence intensity revealed that significantly more F-actin appeared after Zn2+ treatment in cells, and siRNA knockdown of GPR39 abolished this phenomenon, as observed (Fig. 3D). Zn2+ treatment also significantly reduced transendothelial permeability but not for GRP39 knockdown cells, as shown by transendothelial electrical resistance (TER) (Fig. 3E) and permeability to dextran-40 (Fig. 3F).
Fig. 3.

Zn2+ promotes endothelial cell adhesion and mobility and cytoskeletal organization and reduces transendothelial permeability. A: normalized cell adhesion density after 5 h with or without Zn2+ (25 µM) and siRNA treatment. B: cell migration rate (µm/h) after 5 h with or without Zn2+ (25 µM) and siRNA treatment. C: representative image of F-actin fibers in endothelial cells with or without Zn2+. (Scale bar = 5 µm.) D: quantitative measurement of F-actin fluorescence density in endothelial cells. E: transendothelial electrical resistance (TER) with or without Zn2+ (25 µM) treatment. F: transendothelial permeability with or without Zn2+ (25 µM) treatment. *P < 0.05 vs. controls at Zn2+-deficient conditions in endothelial cells (n = 6).
Zn2+ promotes angiogenesis via ZnR/GPR39.
To explore the role of Zn2+ in angiogenesis, we examined endothelial tubule formation on growth factor-reduced Matrigel. Zn2+ treatment induced significantly more endothelial tubular differentiation compared with control groups without Zn2+ or with siGPR39 (Fig. 4A). Both endothelial cord length and branches with Zn2+ treatment were 30–40% more than negative controls and groups with siGPR39 (Fig. 4, B and C).
Fig. 4.

Zn2+ promotes vascular tubule formation. A: representative images showing tubule formation with or without Zn2+ (25 µM) and siRNA treatment. (Scale bar = 100 µm.) B: quantitative measurements of tubule length. C: quantitative measurements of tubule branching points. *P < 0.05 vs. controls at Zn2+-deficient conditions in endothelial cells (n = 6).
Zn2+ has no effect on endothelial activity in GPR39 knockout cells.
To further validate the role of GPR39 in extracellular Zn2+ sensing and binding, we investigated the effects of Zn2+ in mouse GPR39−/− endothelial cells. Immunofluorescent staining of GPR39 verified the absence of GPR39 in the knockout GPR39−/− cells (Fig. 5A). Addition of extracellular Zn2+ did not induce significant changes of Ca2+ signaling, cell survival/growth, cellular cAMP level, or cell adhesion and mobility in GPR39−/− cells (Fig. 5, B–D). Similarly, no significant changes of transendothelial permeability or angiogenesis were observed after Zn2+ treatment in GPR39−/− cells (Fig. 5, E and F).
Fig. 5.

Effects of Zn2+ on endothelial cell activity with knockout of GPR39. A: representative images of GPR39 staining and nuclei in wild-type (WT) and GPR39 knockout endothelial cells (GPR39−/−). (Scale bar = 20 µm.) B: rate of Ca2+ response from measurement of fluorescence change with Zn2+ (25 µM) treatment in WT and GPR39−/− cells. C: cell viability and cAMP activity of WT and GPR39−/− cells with Zn2+ (25 µM) treatment. D: cell adhesion density and mobility of WT and GPR39−/− cells with Zn2+ (25 µM) treatment. E: TER and permeability of WT and GPR39−/− monolayer with Zn2+ (25 µM) treatment. F: vascular tubule formation of WT and GPR39−/− cells with Zn2+ (25 µM) treatment. (Scale bar = 100 µm.) *P < 0.05 vs. GPR39−/− endothelial cells (n = 6).
Effects of Zn2+ on expression of key molecules related to typical endothelial activity.
To gain more information on molecular mechanisms behind the observed effects of Zn2+ on endothelial cells, we next studied the expression of key molecules related to typical endothelial activity at both gene and protein levels, using RT-PCR and ELISA, respectively.
To further understand the beneficial effect of Zn2+ on vascular cell survival and growth, we examined the related expression of platelet-derived growth factor-α receptor (PDGFRA) and vascular endothelial growth factor A (VEGFA). PDGFRA is a cell surface tyrosine kinase receptor for members of the platelet-derived growth factor family, and VEGFA is the main, dominant inducer to the growth of vascular endothelial cells. Extracellular Zn2+ significantly enhanced the expression of these genes and proteins involved in cell survival, apoptosis, and proliferation (Fig. 6A). Inhibition of ZnR/GPR39 expression abolished the enhanced expression of these molecules induced by Zn2+.
Fig. 6.

Effects of Zn2+ on genes and proteins related to vascular cell survival/growth and inflammation and vascular tone. A: mRNA and protein expression levels of PDGFRA and VEGFA in endothelial cells with treatment of Zn2+ (25 µM) and siGPR39. B: mRNA and protein expression levels of HMOX1, IL-10, SELL, and PECAM1 in endothelial cells with treatment of Zn2+ (25 µM) and siGPR39. C: mRNA and protein expression levels of PTGIS and NOS3 in endothelial cells with treatment of Zn2+ (25 µM) and siGPR39. *P < 0.01 vs. controls at Zn2+-deficient conditions for mRNA levels (n = 6). #P < 0.05 vs. controls at Zn2+-deficient conditions for protein levels (n = 6).
It is known that Zn2+ may participate in cell signaling regulation of vascular inflammation (28, 29). We thus studied the gene and protein levels of related genes after treatment of extracellular Zn2+. We found that both genes and proteins of heme oxygenase-1 (HMOX1) and IL-10 were significantly upregulated, whereas those of L-selectin (SELL) and platelet endothelial cell adhesion molecule (PECAM1) were significantly downregulated (Fig. 6B). Moreover, no significant regulations of intercellular adhesion molecule 1 (ICAM1) and C-C motif chemokine ligand 2 (CCL2) at the protein levels were observed despite the significant downregulation of the corresponding genes (data not shown). The levels of these inflammatory molecules were restored to normal if ZnR/GPR39 was silenced.
Another important function of vascular cells is vasoconstriction and vasodilation. To examine the role of Zn2+ in regulation of such vascular activity, we measured related gene and protein expression. Prostaglandin I2 synthase (PTGIS) and nitric oxide synthase-3 (NOS3) were significantly upregulated at both mRNA and protein levels (Fig. 6C). Blockage of Zn2+ signaling by knockdown of its receptor ZnR/GPR39 inhibited such effects induced by Zn2+.
DISCUSSION
Zn2+ is essential for cell survival and functions in many cells. Changes in Zn2+ concentration have a fundamental effect on numerous pathophysiological processes. The sensing and signaling of these changes may have an important homeostatic role. Moreover, many studies have underscored the importance of extracellular Zn2+ in cellular signaling associated with survival and proliferation (29). For instance, Zn2+ was linked to regulation of the mitogen-activated protein kinase (MAPK) and PI3 pathways. The specific receptor for extracellular Zn2+, ZnR/GPR39, is functional in various cell types, such as colonocytes, keratinocytes, neurons, pancreatic β cells, and salivary glands (2, 9, 10, 13, 42, 44–46). Here, we have shown that ZnR/GPR39 is essential for mediating Zn2+-dependent cellular signaling pathways leading to survival, proliferation, angiogenesis, inflammation, vascular tone, and other vascular activity. Silencing or knockout of ZnR/GPR39 diminished the Zn2+-dependent effect significantly but not completely, suggesting the possible existence of other putative Zn2+ receptor, e.g., transient receptor potential ion (melastatin) channel (TRPM)6/7 in vascular cells (21, 55, 58). TRPM6/7 is a ubiquitous membrane channel for cations including Ca2+, Mg2+, and Zn2+, etc. Although it is suggested that extracellular Zn2+ levels are low, Zn2+ released from the liver or in digested food may be a source for Zn2+ signaling (23). Moreover, Zn2+ is secreted by healthy epithelial cells with a robust release during epithelial injury (42). In addition, accumulation of Zn2+ happens in colonocytes triggered by oxidative stress (8). Interestingly, numerous Zn2+-containing vesicles stacked at the apical side of the cells, and this Zn2+ is required for cell survival (39).
A growing body of evidence suggests that extracellular Zn2+ activates signaling transduction pathways that induce cell survival and proliferation. For example, Zn2+ upregulates the PI 3-kinase pathway, leading to activation of Akt in fibroblasts (18). It also induces transactivation of the epidermal growth factor receptor by Src in epithelial cells (40, 47). In addition, extracellular Zn2+ triggers the activation of extracellular signal-elated kinase (ERK)1/2, which, in turn, activates p21 and cyclin D1 in colonocytes (18). On the basis of the results from our and others' literature, we propose a mechanistic Zn2+ signaling pathway in endothelial cells (Fig. 7). As a natural ligand, extracellular Zn2+ activates the promiscuous ZnR/GPR39 and thereby triggers Gαq signaling pathways. Then the Gαq would subsequently activate the PLC, Akt, PI3, and MAPK signaling pathways. At the same time, it may induce an intracellular Ca2+ response. The elevated Ca2+ level will next trigger many other Ca2+-dependent signaling cascades, including ERK1/2 and Akt. Taken together, all these pathways would lead to the differential regulation of genes related to vascular functions, such as survival/growth, angiogenesis, inflammation, vascular tone, coagulation, etc.
Fig. 7.
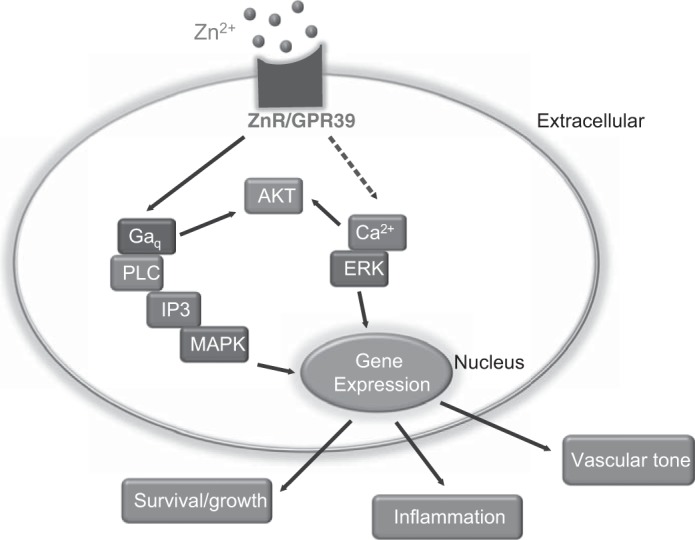
Proposed schematic pathway showing ZnR/GPR39-mediated Zn2+ signaling. The model suggests that ZnR/GPR39 is the major link between changes in extracellular Zn and physiological cell function in vascular cells, such as survival and proliferation, inflammation, and vascular tone.
To further explain the observed cellular function changes triggered by Zn2+ signaling, we evaluated the vascular functional gene profile as well as the corresponding protein expression. The enhanced cell viability and proliferation induced by Zn2+ could be explained by the increased levels of VEGFA and PDGFRA. VEGFA is a glycosylated mitogen that specifically acts on endothelial cells and has various effects, including mediating increased vascular permeability, inducing angiogenesis, vasculogenesis and endothelial cell growth, promoting cell migration, and inhibiting apoptosis (4). PDGFRA is a member of the platelet-derived growth factor family, and a mitogenic factor for cells of mesenchymal origin. Its deficiency leads to defects in oligodendrocytes, alveolar smooth muscle cells, and Leydig cells (19). Next, we examined the effect of Zn2+ on endothelial cell mobility including adhesion and migration. The actin cytoskeleton plays important roles in forming and maintaining the shape and structure of cells (48). The reorganization of cytoskeleton alters cell shape and motility (36). Several cellular processes, such as differentiation, growth, and apoptosis, are arbitrated by cell cytoskeletal organization and cell shape. The significant assembling of F-actin and cytoskeletal reorganization would provide support for the enhanced motility of vascular cells triggered by Zn2+.
Another important function of vascular cells is the mediation of inflammatory responses. Our results showed that various inflammatory molecules were upregulated or downregulated. Among them, some typical ones are HMOX1, IL-10, SELL, PECAM1, CCL2, IL-6, COX-2, and IL-1β. The ability of HMOX1 to catabolize free heme and produce carbon monoxide (CO) gives its anti-inflammatory properties by upregulation of IL-10 and IL-1 antagonist IL-1RA expression (34). IL-10 and IL-6 are major anti-inflammatory cytokines, whereas IL-1β is a proinflammatory cytokine (16, 24). The SELL gene encodes L-selectin, which is responsible for the recruitment of circulating leukocytes to the inflammation sites of endothelium (12). PECAM1 protein is a member of the immunoglobulin superfamily and is likely involved in leukocyte transmigration, angiogenesis, and integrin activation (6, 37, 51). CCL2 is a small cytokine that belongs to the C-C chemokine family. CCL2 recruits monocytes, memory T cells, and dendritic cells to the sites of inflammation produced by either tissue injury or infection (15, 38). Because various proinflammatory and anti-inflammatory molecules are differently regulated, it will need further studies to fully understand how Zn2+ would orchestrate the vascular inflammatory responses in different pathophysiological conditions.
Vascular tone refers to the degree of constriction experienced by a blood vessel relative to its maximally dilated state. Our results showed that Zn2+ also triggered the regulation of genes and proteins involved in vessel constriction and dilation including PTGIS and NOS3. PTGIS encodes a member of the cytochrome P-450 superfamily of enzymes, which catalyze the conversion of prostaglandin H2 to prostacyclin (prostaglandin I2), a potent vasodilator and inhibitor of platelet aggregation. NOS3 has a protective function in the cardiovascular system, which is attributed to NO production. Regulation of vascular tone is one of the best known roles of NO in the cardiovascular system. NO also has antithrombotic effects and antioxidant properties.
GRANTS
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (Grant 1R01 HL-140562). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.Z., Y.Z., B.M.F., L.T., and Y.-X.Q. conceived and designed research; D.Z. performed experiments; D.Z. analyzed data; D.Z., Y.S., and Y.-X.Q. interpreted results of experiments; D.Z. and Y.S. prepared figures; D.Z. drafted manuscript; D.Z., Y.S., and L.T. edited and revised manuscript; D.Z., Y.Z., B.M.F., L.T., and Y.-X.Q. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Y. Wang from Cornell University for advice on experimental design and manuscript preparations. We also thank Dr. Q. Wang for the generous gift of cells.
REFERENCES
- 1.Andrikopoulos P, Kieswich J, Harwood SM, Baba A, Matsuda T, Barbeau O, Jones K, Eccles SA, Yaqoob MM. Endothelial angiogenesis and barrier function in response to thrombin require Ca2+ influx through the Na+/Ca2+ exchanger. J Biol Chem 290: 18412–18428, 2015. doi: 10.1074/jbc.M114.628156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asraf H, Salomon S, Nevo A, Sekler I, Mayer D, Hershfinkel M. The ZnR/GPR39 interacts with the CaSR to enhance signaling in prostate and salivary epithelia. J Cell Physiol 229: 868–877, 2014. doi: 10.1002/jcp.24514. [DOI] [PubMed] [Google Scholar]
- 3.Assaf SY, Chung S-H. Release of endogenous Zn2+ from brain tissue during activity. Nature 308: 734–736, 1984. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 4.Barresi V, Di Gregorio C, Regiani-Bonetti L, Ponz-De Leon M, Barresi G, Vitarelli E. Stage I colorectal carcinoma: VEGF immunohistochemical expression, microvessel density, and their correlation with clinical outcome. Virchows Arch 457: 11–19, 2010. doi: 10.1007/s00428-010-0933-5. [DOI] [PubMed] [Google Scholar]
- 5.Berg JM, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science 271: 1081–1085, 1996. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 6.Cao G, O’Brien CD, Zhou Z, Sanders SM, Greenbaum JN, Makrigiannakis A, DeLisser HM. Involvement of human PECAM-1 in angiogenesis and in vitro endothelial cell migration. Am J Physiol Cell Physiol 282: C1181–C1190, 2002. doi: 10.1152/ajpcell.00524.2001. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW, Koh JY. Zinc and brain injury. Annu Rev Neurosci 21: 347–375, 1998. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- 8.Cima RR, Dubach JM, Wieland AM, Walsh BM, Soybel DI. Intracellular Ca2+ and Zn2+ signals during monochloramine-induced oxidative stress in isolated rat colon crypts. Am J Physiol Gastrointest Liver Physiol 290: G250–G261, 2006. doi: 10.1152/ajpgi.00501.2004. [DOI] [PubMed] [Google Scholar]
- 9.Cohen L, Asraf H, Sekler I, Hershfinkel M. Extracellular pH regulates zinc signaling via an Asp residue of the zinc-sensing receptor (ZnR/GPR39). J Biol Chem 287: 33339–33350, 2012. doi: 10.1074/jbc.M112.372441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen L, Sekler I, Hershfinkel M. The zinc sensing receptor, ZnR/GPR39, controls proliferation and differentiation of colonocytes and thereby tight junction formation in the colon. Cell Death Dis 5: e1307, 2014. doi: 10.1038/cddis.2014.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frederickson CJ, Koh J-Y, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci 6: 449–462, 2005. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 12.Fu JJ, Baines KJ, Wood LG, Gibson PG. Systemic inflammation is associated with differential gene expression and airway neutrophilia in asthma. OMICS 17: 187–199, 2013. doi: 10.1089/omi.2012.0104. [DOI] [PubMed] [Google Scholar]
- 13.Ganay T, Asraf H, Aizenman E, Bogdanovic M, Sekler I, Hershfinkel M. Regulation of neuronal pH by the metabotropic Zn2+-sensing Gq-coupled receptor, mZnR/GPR39. J Neurochem 135: 897–907, 2015. doi: 10.1111/jnc.13367. [DOI] [PubMed] [Google Scholar]
- 14.Ghasemi A, Zahediasl S, Hosseini-Esfahani F, Azizi F. Reference values for serum zinc concentration and prevalence of zinc deficiency in adult Iranian subjects. Biol Trace Elem Res 149: 307–314, 2012. doi: 10.1007/s12011-012-9445-2. [DOI] [PubMed] [Google Scholar]
- 15.Haukeland JW, Damås JK, Konopski Z, Løberg EM, Haaland T, Goverud I, Torjesen PA, Birkeland K, Bjøro K, Aukrust P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol 44: 1167–1174, 2006. doi: 10.1016/j.jhep.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav Immun 23: 309–317, 2009. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hershfinkel M, Silverman WF, Sekler I. The zinc sensing receptor, a link between zinc and cell signaling. Mol Med 13: 331–336, 2007. doi: 10.2119/2006-00038.Hershfinkel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoch RV, Soriano P. Roles of PDGF in animal development. Development 130: 4769–4784, 2003. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- 20.Huang EP. Metal ions and synaptic transmission: think zinc. Proc Natl Acad Sci USA 94: 13386–13387, 1997. doi: 10.1073/pnas.94.25.13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoue K, Branigan D, Xiong ZG. Zinc-induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J Biol Chem 285: 7430–7439, 2010. doi: 10.1074/jbc.M109.040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272: 1013–1016, 1996. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 23.Krebs NF. Overview of zinc absorption and excretion in the human gastrointestinal tract. J Nutr 130, Suppl: 1374S–1377S, 2000. doi: 10.1093/jn/130.5.1374S. [DOI] [PubMed] [Google Scholar]
- 24.Lee JH, Zhang Y, Zhao Z, Ye X, Zhang X, Wang H, Ye J. Intracellular ATP in balance of pro- and anti-inflammatory cytokines in adipose tissue with and without tissue expansion. Int J Obes 41: 645–651, 2017. doi: 10.1038/ijo.2017.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo S, Truong AH, Makino A. Isolation of mouse coronary endothelial cells. J Vis Exp 113: 2016. doi: 10.3791/53985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma J, Zhao N, Zhu D. Bioabsorbable zinc ion induced biphasic cellular responses in vascular smooth muscle cells. Sci Rep 6: 26661, 2016. doi: 10.1038/srep26661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma J, Zhao N, Zhu D. Biphasic responses of human vascular smooth muscle cells to magnesium ion. J Biomed Mater Res A 104: 347–356, 2016. doi: 10.1002/jbm.a.35570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J, Zhao N, Zhu D. Endothelial cellular responses to biodegradable metal zinc. ACS Biomater Sci Eng 1: 1174–1182, 2015. doi: 10.1021/acsbiomaterials.5b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr 130, Suppl: 1500S–1508S, 2000. doi: 10.1093/jn/130.5.1500S. [DOI] [PubMed] [Google Scholar]
- 30.Młyniec K, Budziszewska B, Holst B, Ostachowicz B, Nowak G. GPR39 (zinc receptor) knockout mice exhibit depression-like behavior and CREB/BDNF down-regulation in the hippocampus. Int J Neuropsychopharmacol 18: 18, 2014. doi: 10.1093/ijnp/pyu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moechars D, Depoortere I, Moreaux B, de Smet B, Goris I, Hoskens L, Daneels G, Kass S, Ver Donck L, Peeters T, Coulie B. Altered gastrointestinal and metabolic function in the GPR39-obestatin receptor-knockout mouse. Gastroenterology 131: 1131–1141, 2006. doi: 10.1053/j.gastro.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 32.Ospina JA, Duckles SP, Krause DN. 17β-Estradiol decreases vascular tone in cerebral arteries by shifting COX-dependent vasoconstriction to vasodilation. Am J Physiol Heart Circ Physiol 285: H241–H250, 2003. doi: 10.1152/ajpheart.00018.2003. [DOI] [PubMed] [Google Scholar]
- 33.Patsch C, Challet-Meylan L, Thoma EC, Urich E, Heckel T, O’Sullivan JF, Grainger SJ, Kapp FG, Sun L, Christensen K, Xia Y, Florido MH, He W, Pan W, Prummer M, Warren CR, Jakob-Roetne R, Certa U, Jagasia R, Freskgård PO, Adatto I, Kling D, Huang P, Zon LI, Chaikof EL, Gerszten RE, Graf M, Iacone R, Cowan CA. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol 17: 994–1003, 2015. doi: 10.1038/ncb3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem 286: 16374–16385, 2011. doi: 10.1074/jbc.M110.207738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7: 803–815, 2007. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 36.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science 326: 1208–1212, 2009. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Privratsky JR, Newman PJ. PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res 355: 607–619, 2014. doi: 10.1007/s00441-013-1779-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475: 222–225, 2011. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ranaldi G, Ferruzza S, Canali R, Leoni G, Zalewski PD, Sambuy Y, Perozzi G, Murgia C. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J Nutr Biochem 24: 967–976, 2013. doi: 10.1016/j.jnutbio.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 40.Samet JM, Dewar BJ, Wu W, Graves LM. Mechanisms of Zn2+-induced signal initiation through the epidermal growth factor receptor. Toxicol Appl Pharmacol 191: 86–93, 2003. doi: 10.1016/S0041-008X(03)00219-9. [DOI] [PubMed] [Google Scholar]
- 42.Sharir H, Zinger A, Nevo A, Sekler I, Hershfinkel M. Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J Biol Chem 285: 26097–26106, 2010. doi: 10.1074/jbc.M110.107490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen H, Oesterling E, Stromberg A, Toborek M, MacDonald R, Hennig B. Zinc deficiency induces vascular pro-inflammatory parameters associated with NF-kappaB and PPAR signaling. J Am Coll Nutr 27: 577–587, 2008. doi: 10.1080/07315724.2008.10719741. [DOI] [PubMed] [Google Scholar]
- 44.Sunuwar L, Asraf H, Donowitz M, Sekler I, Hershfinkel M. The Zn2+-sensing receptor, ZnR/GPR39, upregulates colonocytic Cl− absorption, via basolateral KCC1, and reduces fluid loss. Biochim Biophys Acta 1863: 947–960, 2017. doi: 10.1016/j.bbadis.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sunuwar L, Gilad D, Hershfinkel M. The zinc sensing receptor, ZnR/GPR39, in health and disease. Front Biosci (Landmark Ed) 22: 1469–1492, 2017. doi: 10.2741/4554. [DOI] [PubMed] [Google Scholar]
- 46.Sunuwar L, Medini M, Cohen L, Sekler I, Hershfinkel M. The zinc sensing receptor, ZnR/GPR39, triggers metabotropic calcium signalling in colonocytes and regulates occludin recovery in experimental colitis. Philos Trans R Soc Lond B Biol Sci 371: 20150420, 2016. doi: 10.1098/rstb.2015.0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tal TL, Graves LM, Silbajoris R, Bromberg PA, Wu W, Samet JM. Inhibition of protein tyrosine phosphatase activity mediates epidermal growth factor receptor signaling in human airway epithelial cells exposed to Zn2+. Toxicol Appl Pharmacol 214: 16–23, 2006. doi: 10.1016/j.taap.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 48.Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 113: 37–47, 2003. doi: 10.1016/S0092-8674(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 49.Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res 79: 581–588, 2008. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 50.Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiol Rev 73: 79–118, 1993. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- 51.Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol 15: 692–704, 2015. doi: 10.1038/nri3908. [DOI] [PubMed] [Google Scholar]
- 52.Vikbladh I. Studies on zinc in blood. Scand J Clin Lab Invest 2: 143–148, 1950. doi: 10.3109/00365515009051850. [DOI] [PubMed] [Google Scholar]
- 53.Wapnir RA. Zinc deficiency, malnutrition and the gastrointestinal tract. J Nutr 130, Suppl: 1388S–1392S, 2000. doi: 10.1093/jn/130.5.1388S. [DOI] [PubMed] [Google Scholar]
- 54.Webb NJ, Bottomley MJ, Watson CJ, Brenchley PE. Vascular endothelial growth factor (VEGF) is released from platelets during blood clotting: implications for measurement of circulating VEGF levels in clinical disease. Clin Sci (Lond) 94: 395–404, 1998. doi: 10.1042/cs0940395. [DOI] [PubMed] [Google Scholar]
- 55.Wrighton KH. Epigenetics: the TRPM7 ion channel modifies histones. Nat Rev Mol Cell Biol 15: 427, 2014. doi: 10.1038/nrm3824. [DOI] [PubMed] [Google Scholar]
- 56.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature 407: 242–248, 2000. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 57.Yasuda S, Miyazaki T, Munechika K, Yamashita M, Ikeda Y, Kamizono A. Isolation of Zn2+ as an endogenous agonist of GPR39 from fetal bovine serum. J Recept Signal Transduct Res 27: 235–246, 2007. doi: 10.1080/10799890701506147. [DOI] [PubMed] [Google Scholar]
- 58.Yogi A, Callera GE, Antunes TT, Tostes RC, Touyz RM. Transient receptor potential melastatin 7 (TRPM7) cation channels, magnesium and the vascular system in hypertension. Circ J 75: 237–245, 2011. doi: 10.1253/circj.CJ-10-1021. [DOI] [PubMed] [Google Scholar]
- 59.Zhao N, Zhu D. Endothelial responses of magnesium and other alloying elements in magnesium-based stent materials. Metallomics 7: 118–128, 2015. doi: 10.1039/C4MT00244J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu D, Su Y, Young ML, Ma J, Zheng Y, Tang L. Biological responses and mechanisms of human bone marrow mesenchymal stem cells to Zn and Mg biomaterials. ACS Appl Mater Interfaces 9: 27453–27461, 2017. doi: 10.1021/acsami.7b06654. [DOI] [PubMed] [Google Scholar]
- 61.Zhu D, Wang Y, Singh I, Bell RD, Deane R, Zhong Z, Sagare A, Winkler EA, Zlokovic BV. Protein S controls hypoxic/ischemic blood-brain barrier disruption through the TAM receptor Tyro3 and sphingosine 1-phosphate receptor. Blood 115: 4963–4972, 2010. doi: 10.1182/blood-2010-01-262386. [DOI] [PMC free article] [PubMed] [Google Scholar]