

Abstract
The mTOR signaling pathway regulates protein synthesis and diverse aspects of neuronal morphology that are important for brain development and function. To identify proteins controlled translationally by mTOR signaling, we performed ribosome profiling analyses in mouse cortical neurons and embryonic stem cells upon acute mTOR inhibition. Among proteins whose translation was significantly affected by mTOR inhibition selectively in neurons, we identified the cytoskeletal regulator protein palladin, which is localized within the cell body and axons in hippocampal neurons. Knockdown of palladin eliminated supernumerary axons induced by suppression of the tuberous sclerosis complex protein TSC1 in neurons, demonstrating that palladin regulates neuronal morphogenesis downstream of mTOR signaling. Our findings provide novel insights into an mTOR-dependent mechanism that controls neuronal morphogenesis through translational regulation.
SIGNIFICANCE STATEMENT This study reports the discovery of neuron-specific protein translational responses to alterations of mTOR activity. By using ribosome profiling analysis, which can reveal the location and quantity of translating ribosomes on mRNAs, multiple aspects of protein translation were quantitatively analyzed in mouse embryonic stem cells and cortical neurons upon acute mTOR inhibition. Neurons displayed distinct patterns of ribosome occupancy for each codon and ribosome stalling during translation at specific positions of mRNAs. Importantly, the cytoskeletal regulator palladin was identified as a translational target protein of mTOR signaling in neurons. Palladin operates downstream of mTOR to modulate axon morphogenesis. This study identifies a novel mechanism of neuronal morphogenesis regulated by mTOR signaling through control of translation of the key protein palladin.
Keywords: axon, palladin, ribosome, translation
Introduction
Neuronal morphogenesis during development is critical for normal brain circuitry and function, and disruption of neuronal morphogenesis may lead to diverse brain disorders (Hu et al., 2014). Accumulating evidence suggests that precise regulation of protein synthesis is essential in the control of neuronal morphogenesis (Brittis et al., 2002; Bramham and Wells, 2007; Deglincerti and Jaffrey, 2012; Jung et al., 2012). The mTOR signaling pathway is a fundamental regulator of protein synthesis, and controls diverse aspects of neuronal morphology (Tavazoie et al., 2005; Costa-Mattioli and Monteggia, 2013; Takei and Nawa, 2014). The mTOR kinase protein complex mTORC1 promotes protein synthesis by phosphorylation of translation regulators, including S6K and 4EBP. TSC1 and TSC2 are critical negative regulators of mTOR kinase activity, and mutations of these genes cause the genetic disorder tuberous sclerosis complex (TSC). TSC features benign tumors, hypopigmented skin patches, and neurological disorders including epilepsy, intellectual disability, and autism spectrum disorders (ASDs). Mutations of TSC1 and TSC2 lead to hyperactivation of mTOR kinase and consequent disruption of neuronal morphogenesis (Tavazoie et al., 2005).
The mTOR signaling pathway modulates protein synthesis activity globally (Costa-Mattioli and Monteggia, 2013; Faller et al., 2015). However, recent studies have suggested that mTOR signaling may also regulate production of specific proteins. Upon stimulation, mTOR signaling rapidly induces translation of mRNAs encoding components of the protein synthesis machinery, therefore promoting general protein translation activity in the cell. Many of the proteins whose translation is augmented in response to mTOR activation are produced from mRNAs containing an oligo-pyrimidine motif at their 5′ end (5′TOP), which modulates initiation of translation depending on mTOR activity (Jefferies et al., 1994). This mechanism contributes to cell-type-specific functions through translational control of specific mRNAs bearing 5′TOP-like motifs (Hsieh et al., 2012; Thoreen et al., 2012; Olshen et al., 2013). In addition to promoting initiation of translation, mTOR regulates elongation of translation through modulation of elongation factors (Browne and Proud, 2004; Kenney et al., 2014). Recent studies have revealed that translation elongation factors may modulate production of specific proteins, with specific consequences in neurons (Park et al., 2008; Verpelli et al., 2010; Shepherd and Bear, 2011; Kenney et al., 2014). We thus hypothesized that the mTOR signaling pathway might control neuronal morphology by regulating production of specific proteins in neurons.
In this study, we have discovered that mTOR signaling selectively regulates translation of distinct proteins in neurons including the cytoskeletal regulator palladin, which mediates mTOR-regulation of axon morphogenesis. Our findings provide insights into a translational mechanism that regulates neuronal morphology downstream of the mTOR signaling pathway.
Materials and Methods
Ribosome profiling.
Ribosome profiling was performed as described previously (Ingolia et al., 2012). Cortical neurons were prepared from E16 CD-1 (ICR, RRID:IMSR_CRL:22) mouse embryos (either sex), and cultured for 8 d in vitro and treated with the mTOR inhibitor Torin 1 (100 nm; Tocris Bioscience) or vehicle for 2.5 h, which allows to capture direct changes in mTOR-dependent protein synthesis before secondary feedback effects (Hsieh et al., 2012). EB3 mouse embryonic stem (ES) cells (RRID: CVCL_J647) were provided by RIKEN BRC (Niwa et al., 2002; Ogawa et al., 2004), and maintained in 2i-LIF medium. Subconfluent ES cells were treated with the mTOR inhibitor Torin 1 (100 nm) or vehicle for 2.5 h. After treatment with Torin 1 or vehicle (DMSO), cells were lysed in a buffer containing cycloheximide, and the polysome was processed for ribosome profiling analysis as described previously (Ingolia et al., 2012). Ribosome-mRNA complex was stabilized with cycloheximide upon lysis of cells, and followed by treatment with RNase I. The undigested ribosome-protected mRNA fragments were then subjected to sequencing library preparation. Next-generation sequencing analysis and subsequent computational analysis revealed codon-level ribosome distribution on mRNAs. For mRNA-seq, mRNAs were extracted from cells prepared in parallel and subjected to next generation sequencing library preparation. The libraries were sequenced with HiSeq2500 (Illumina) as single-end reads of 50 bp.
Data processing of ribosome profiling.
The short reads of the ribosome profiling and mRNA-seq were processed as follows. Adaptor sequence (CTGTAGGCACCATCAAT) was trimmed from the reads using BBDuk from the BBTools suite (U.S. Department of Energy Joint Genome Institute) with the following parameters: ktrim = r, k = 17, hdist = 1, mink = 10 minlength = 25, maxlength = 40. To remove reads derived from noncoding RNAs, the trimmed reads were aligned to mouse rRNA, tRNA, snRNA, snoRNA, and lncRNA sequences downloaded from RNAcentral database (The RNAcentral Consortium, 2017) using Bowtie 1.1.2 (Langmead et al., 2009; RRID:SCR_005476), and the aligned reads were discarded. Then, the remaining reads were aligned to the mouse genome (GRCm38.p5) using STAR (Dobin et al., 2013; RRID:SCR_015899). We allowed a maximum of two mismatches, and discarded unannotated and noncanonical splice junction reads with following parameters: outFilterMismatchNmax 2; outFilterIntronMotifs RemoveNoncanonicalUnannotated. The genomic annotation file containing splice junction information was obtained from GENCODE (release M12; Mudge and Harrow, 2015; RRID:SCR_014966). For transcript level ribosome occupancy analysis, we calculated changes of translation efficiency of each gene by comparing read counts of ribosome profiling and mRNA-seq. We performed three independent biological repeats and statistical analyses using Babel R package as reported (Olshen et al., 2013; RRID:SCR_004307). Previous study reported that acute mTOR inhibition moderately suppress translation of nearly all mRNAs (Thoreen et al., 2012). This effect is normalized by Babel package and only the mRNAs significantly affected by Torin 1 treatment were identified.
To assess precise positions of ribosome occupancy on mRNAs, uniquely aligning reads with length between 25 and 35 nt were used. The unique reads were mapped to CDS coordinates using mapToTranscripts function from GenomicFeatures R package (Lawrence et al., 2013). The reads were assigned to the positions corresponding to the first nucleotide of the P-site, by shifting the 5′ end of each read by +12 (for reads 25–30 nt long), +13 (31–34 nt) or +14 (35 nt) positions. The offsets for reads with various length were determined by Plastid (Dunn and Weissman, 2016). For this step, only the representative isoform for each gene registered in UCSC Table Browser knownCanonical track (Karolchik et al., 2004; RRID:SCR_005780) was used. Bioinformatic analysis of ribosome occupancy data was performed using custom R scripts that are available upon request.
For pause identification, a “pausing score” for each codon was calculated by dividing the ribosome occupancy in each codon by the average occupancy of codons across coding sequences (CDSs) of the same transcript. Codons with a pausing score >10 were considered to be ribosomal pausing sites. Venn diagrams for genes containing at least one pause were generated using VennDiagram R package (Chen and Boutros, 2011; RRID:SCR_002414). The first 15 and last 5 nt of each CDS were excluded due to accumulation of ribosomes at initiation and termination sites. Long stretches of codons without aligned reads are presumably caused by non-uniqueness of the sequence, which reduces average occupancy on codons excessively. To avoid false-positive detections of ribosome pausing due to this confound, a continuous stretch of codons to which no reads were mapped was collapsed to one codon. For these analyses, only well expressed genes (CDS with a median of >2 reads per 5 codon window) were considered as described previously (Ingolia et al., 2012).
For codon-specific ribosome occupancy analysis, we first calculated the number of reads containing each of the 61 aa-coding codons at positions corresponding to A-, P-, and E-sites of ribosomes. Codon-specific ribosome occupancy was normalized by the average occupancy of three 3′ side codons adjacent to the A-site, as reported (Nedialkova and Leidel, 2015). Codons in those positions have not been translated yet but are already inside the mRNA channel; therefore, they are not expected to bias codon frequencies. Codon-specific ribosome occupancy was then converted to percentage of total coding sequence reads in each sample, and the relative occupancy change was calculated respect to untreated cortical neuron. For this analysis, only in-frame reads from transcripts with at least 64 ribosome footprints mapped to them were included. The first and last 50 codons from each representative CDS were excluded from the analysis to avoid the potential bias at the beginning and end of genes.
5′TOP analysis.
Genome-wide analysis of 5′TOP was performed using R scripts, available upon request. To determine ratio of mRNAs bearing 5′TOP-like motif in all mRNAs transcribed from each gene, we assessed the 5′-end sequence of mRNAs and their abundance using the FANTOM5 quantitative transcription start site dataset (Lizio et al., 2015; RRID:SCR_002678). The transcription start site [CAGE tag starting site (CTSS)] datasets for neuronal differentiation of mouse ES cells was downloaded from the FANTOM5 database. For this analysis, mm9 mouse reference assembly was used for consistency with FANTOM5 data. Positions with <2 reads were filtered out from CTSS datasets to reduce noise. CTSSs with at least four pyrimidines in the 5′end 5 nt were identified as producing 5′TOP-like motif-containing mRNAs. For each dataset, total CTSS and 5′TOP-preceding CTSS were mapped within 500 bp to both directions of transcription start sites of canonical transcripts using GenomicAlignments' summarizeOverlaps function (Lawrence et al., 2013). The 5′TOP ratio was calculated as the division of 5′TOP-preceding CTSS count and total CTSS count for each gene and each dataset.
Antibodies.
For immunostaining, the following antibodies were used: anti-β III Tubulin (Tuj1; Covance, MMS-435P; RRID:AB_2313773), anti-Tau1 (Millipore, MAB3420; RRID:AB_94855), anti-palladin (Santa Cruz Biotechnology, sc-68900; RRID:AB_2158884; and Proteintech, 10853-1-AP; RRID:AB_2158782), and anti-GFP (rabbit antiserum generated against full-length eGFP, and NeuroMab, 75-132; RRID:AB_10672529).
For immunoblotting, we used following antibodies: anti-palladin (Proteintech, 10853-1-AP; RRID:AB_2158782), anti-TSC1 (Cell Signaling Technology, 4906; RRID:AB_2209790 and Life Technology, 37-0400; RRID:AB_2533292), anti-S6 (Cell Signaling Technology, 2217; RRID:AB_331355), anti-phospho S6 (Cell Signaling Technology, Ser 240/244, 5364; RRID:AB_10694233), anti-Zfp36 (Santa Cruz Biotechnology, SC-374305; RRID:AB_10988939), anti-nucleoporin p62 (BD Biosciences, 610497; RRID:AB_397863), anti-Flag (Sigma-Aldrich, M2; RRID:AB_262044), anti-puromycin (Kerafast, 3RH11; RRID:AB_2620162), and anti-ERK (Cell Signaling Technology, 9102; RRID:AB_330744).
Genome editing.
The TSC1 gene was genetically manipulated using the CRISPR/Cas9 system (Ran et al., 2013). According to the inventors' direction, pSpCas9(BB)-2A-Puro (pX459) V2.0 was modified to target the TSC1 gene by inserting the annealed oligonucleotides 5′-CACCGCAACATGCCCCAGCAGCGAG-3′ and 5′-AAACCTCGCTGCTGGGGCATGTTGC-3′. After transfecting Neuro2A cells (RRID:CVCL_0470) with the CRISPR plasmid or control plasmid with FuGENE HD (Promega). as per the manufacturer's protocol, cells were replated and treated with puromycin. Cells were sparsely plated in 150 mm dish, and colonies were manually picked. The mutated genomic region was amplified with PCR using the primers 5′-TTCATGGGGTGAGCATATCTCCAG-3′ and 5′-CCCACATGGTCGAAAGGAAGAACT-3′. The mutation was first detected by T7 endonuclease 1 (New England BioLabs), and then the PCR product of the edited region was cloned into a plasmid by TA cloning (pCR2.1-TOPO, Life Technologies) to reveal the mutations by sequencing.
RNAi.
For knockdown of palladin, we have generated plasmids expressing short RNA hairpin targeting two distinct regions of palladin mRNA. Pairs of oligonucleotides 5′-GGTTTAAAGATGGGAAGCAGTCAAGTTAACTCTGCTTCCCATCTTTAAACCCTTTTTG-3′ and 5′-AATTCAAAAAGGGTTTAAAGATGGGAAGCAGAGTTAACTTGACTGCTTCCCATCTTTAAACC-3′, 5′-GAAAGAGCCCAGAAGAGCTTGCAAGTTAACGAAGCTCTTCTGGGCTCTTTCCTTTTTG-3′ and 5′-AATTCAAAAAGGAAAGAGCCCAGAAGAGCTTCGTTAACTTGCAAGCTCTTCTGGGCTCTTTC-3′ were cloned into U6 RNAi plasmid to generate palladin RNAi 1 and 2, respectively, as described previously (Gaudilliere et al., 2002). The TSC1 RNAi plasmid has been described previously (Mejia et al., 2013).
Immunoblotting analysis of isolated neurites.
Neurites were isolated as described before with modifications (Vidaki et al., 2017). Hippocampal neurons from E16 mice were plated on the top component of a hanging insert with 1 μm membrane pores (Merck, Millipore). The membrane was coated on both sides with poly-l-lysine. The cells were cultured for 5 d. To analyze proteins in the neurites, cell bodies were scraped from the top component of the filter, leaving the neurites at the bottom. After scraping the cells, Torin 1 was added to the neurites and incubated for 12 h, which allows us to evaluate changes of palladin protein abundance before neurites degenerate. Lysates of the neurites or whole cells were analyzed by immunoblotting and intensity of the bands was quantified.
Morphological analysis.
Hippocampal neurons from E16 or E17 mouse embryos were plated on coverslips coated with poly-l-lysine (Sigma-Aldrich) and cultured in Neurobasal media (ThermoFisher) supplemented with B27, glutamine, and penicillin/streptomycin. For axonal morphology analysis, hippocampal neurons were transfected 2–6 h after plating using a calcium phosphate method modified from a method described previously (Konishi et al., 2004) with RNAi or expression plasmids together with GFP expression plasmid to visualize transfected neurons. To count number of axons per cell, we identified axons with Tau1 immunostaining.
Experimental design and statistical analysis.
All ribosome profiling was completed with samples of three independent experiments. All morphological analyses were completed from a minimum of three independent experiments. Statistical analyses were performed with R (RRID:SCR_001905). All histograms for morphological analyses are presented as mean + SEM unless otherwise noted. Student's t test was used for comparisons in experiments with two sample groups. In experiments with more than two sample groups, ANOVA was performed followed by Bonferroni's post hoc test for pairwise comparison among three and greater than three groups.
Results
Ribosome profiling analyses suggest palladin is a translational target of mTOR signaling
“Ribosome profiling” analysis can reveal quantitative distribution patterns of translating ribosomes on mRNAs in cells (Ingolia et al., 2012). We used this method to analyze neuron-specific effects of acute mTOR inhibition on the distribution of translating ribosomes on mRNAs (Fig. 1A). By comparing the effect of acute Torin 1 treatment of neurons and ES cells in ribosome profiling, we aimed to uncover cell-type-specific translational responses to alteration of mTOR activity (Fig. 1B,C). To identify neuron-specific translational targets of the mTOR signaling pathway, we searched for changes of ribosome distribution patterns upon acute treatment of mouse cortical neurons with Torin 1, and compared these changes with those in mouse ES cells. Because the mTOR signaling pathway controls initiation and elongation of translation, we analyzed both steps by interrogating three different aspects of translation. For translation elongation, we investigated codon-specific ribosome occupancy and ribosome stalling, which are two distinct aspects of translation associated with elongation. For translation initiation, we analyzed translation efficiency, which is determined by integrating ribosome occupancy throughout open reading frames (ORFs) and normalized by abundance of the corresponding mRNA.
Figure 1.

Ribosome-profiling analysis of neurons and ES cells upon mTOR inhibition. A, Schematics of ribosome profiling in this study. B, Torin 1 is an inhibitor of mTOR kinase, which inactivates translation machinery through downstream substrates. C, Lysates from neurons and ES cells treated with 100 nm Torin 1 or vehicle (control) for 2.5 h were immunoblotted with the phospho-S6 and S6 antibodies. Phosphorylation of S6 was significantly suppressed by the acute treatment with Torin 1. The phospho-S6 intensity was quantified and normalized by S6. The average relative abundance of three replicates was indicated.
First, we evaluated codon-specific ribosome occupancy on the 61 codons, through which we can examine relative translation elongation speed of each codon. The frequency of each codon appeared at the positions corresponding to the three tRNA accepting A-, P-, and E-sites of the ribosome was calculated for all footprints mapped to highly expressed transcripts, and normalized by overall expression and length of ORFs (Fig. 2A). When we compared the datasets in neurons, we observed only minor Torin 1-induced changes on ribosome occupancy for codons at A-, P-, and E-sites of the ribosome. An identical analysis in ES cells similarly showed little Torin 1-induced changes in codon-specific ribosome occupancy. These data suggest that mTOR does not globally alter the elongation rate for a particular codon during translation. Interestingly, ribosome occupancy for some codons appeared to be different between neurons and ES cells. For example, ribosome occupancy for histidine codons at A-site was increased in ES cells compared with neurons (Fig. 2A), suggesting that translation rate of histidine codons might be reduced in ES cells due to limited His-tRNA availability. Proline codons showed increased ribosome occupancy at P-site in ES cells compared with neurons (Fig. 2A), suggesting that peptidyl transfer on Pro-tRNAPro might be slower in ES cells than in neurons.
Figure 2.

Differences in ribosome occupancy distribution between ES cells and neurons. A, Mean change in codon-specific ribosome occupancy in untreated neurons, Torin 1-treated neurons, untreated ES cells, and Torin1-treated ES cells, for ribosomal A-site (top), P-site (middle), and E-site (bottom). Error bars represent SD (n = 3). B, C, Venn diagrams showing the number of genes that present a translational pause in the CDS body when comparing untreated and Torin 1-treated cortical neurons (B) or untreated cortical neurons and ES cells (C). The percentage of genes in each category respect to the combined number of pause-presenting genes is given in parentheses. D, E, Comparison of ribosome footprint profile over the CDS of Hsp90ab1 (D) and Gab1 (E) in ES cells and cortical neurons.
Because ribosome stalling may impact protein expression, localization and folding (Darnell and Klann, 2013; Wilson et al., 2016), we next investigated ribosome stalling events during translational elongation. We detected 784 and 480 genes with ribosome stalling events in neurons cultured without and with Torin 1, respectively (Fig. 2B). Ribosome stalling was observed in both conditions for a substantial fraction of genes (368 genes). Stalling in 416 and 112 genes was detected selectively in control neurons and Torin 1-treated neurons, respectively. However, upon manual review, stalling peaks exhibiting read numbers substantially altered by Torin 1-treatment were not evident. We also compared genes exhibiting ribosome stalling between neurons and ES cells (Fig. 2C). These analyses revealed fewer overlapping genes exhibiting ribosome stalling in both cell types than Torin 1-treated and untreated neurons. Interestingly, some mRNA loci showed a significant difference in pausing intensity despite similar levels of expression in the two cell types. For example, the Hsp90ab1 gene displayed higher pausing intensity at a specific site selectively in ES cells (Fig. 2D), whereas a stalling site in the Gab1 gene was neuron-specific (Fig. 2E). Together, these results suggest that mTOR regulation plays a minor role in the control of ribosome stalling on mRNAs, and developmental changes may have a larger impact on ribosome stalling.
Finally, we searched for Torin 1-induced changes on translational efficiency of transcripts, defined as the ratio of ribosome occupancy relative to transcript abundance. These analyses revealed modulation of protein translation upon acute mTOR inhibition specifically in neurons (Fig. 3). We found that ribosome occupancy of 561 and 54 mRNAs was significantly downregulated and upregulated, respectively, upon Torin 1-treatment in neurons (Fig. 3A). In ES cells, ribosome occupancy of 2338 and 31 mRNAs was significantly downregulated and upregulated, respectively, upon Torin 1 treatment (Fig. 3B). One hundred and thirty mRNAs were commonly downregulated in both cell types, whereas no overlap between upregulated mRNAs between the datasets were observed. Four hundred and thirty-one mRNAs were only downregulated in neurons, and 2208 mRNAs were only downregulated in ES cells. We compared our result with a previously performed ribosome profiling analysis with mouse embryonic fibroblasts (MEFs) upon inhibition of mTOR signaling (Thoreen et al., 2012; Fig. 3D). Among the reported 253 genes translationally downregulated by Torin 1 treatment in MEFs, 66% (167 genes) overlapped with the genes downregulated in ES cells (Fig. 3D). On the other hand, only 20% (50 genes) of those downregulated in MEFs upon Torin 1 treatment overlapped with the genes downregulated in neurons. Together, these data demonstrate that neurons respond differentially to the acute inhibition of mTOR signaling.
Figure 3.
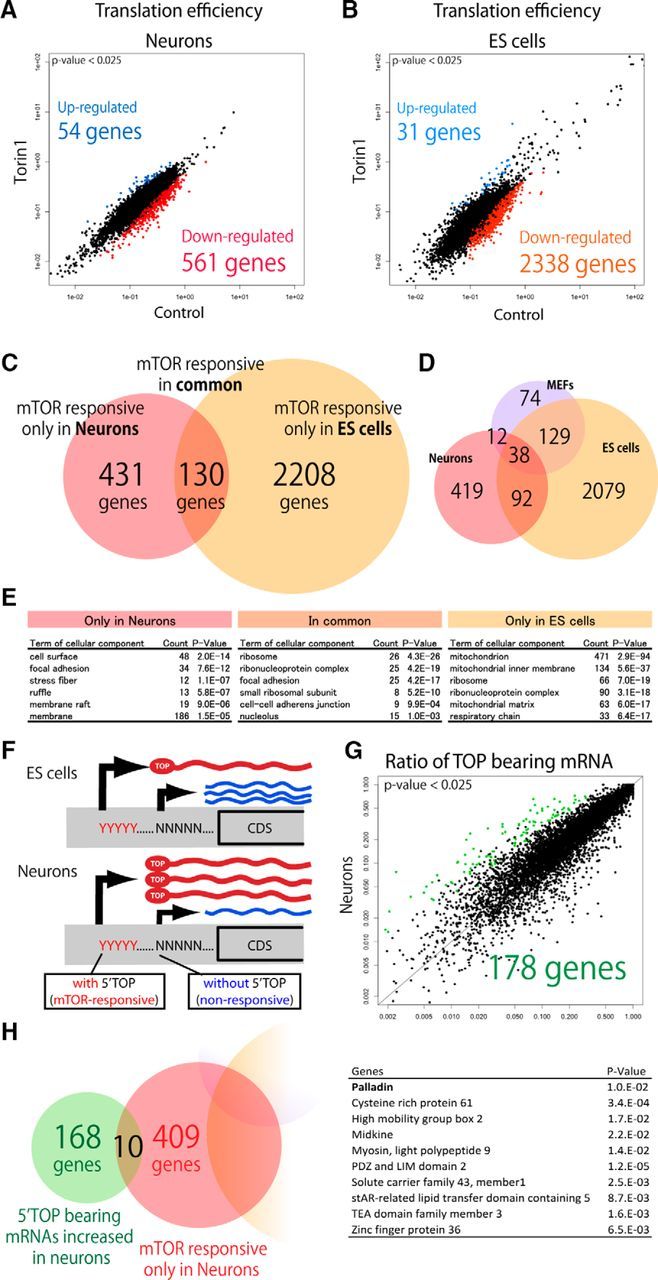
Differences in translation efficiency of ORFs between ES cells and neurons. A, B, Scatter plots of translation efficiency of highly expressed genes in neurons (A) and ES cells (B). Significantly downregulated or upregulated genes are highlighted in colors. C, Venn diagram illustrating overlap of genes whose translational efficiency was significantly downregulated by Torin 1-treatment in neurons and ES cells. D, Venn diagram illustrating overlap of genes whose translational efficiency was significantly downregulated by Torin 1-treatment in this report (neurons and ES cells) and a previous study (MEFs; Thoreen et al., 2012). E, Categories of gene ontology significantly enriched in the genes that are affected by Torin 1 specifically in neurons, in both neurons and ES cells, and specifically in ES cells. F, Schematic of alternative transcription start sites producing mRNAs with and without 5′TOP sequences at a different ratio in different cell types. G, A scatter plot of ratio of mRNAs containing 5′TOP in neurons and ES cells. H, Venn diagram of genes that are mTOR-regulated selectively in neurons and genes that generate mRNAs containing 5′TOP to a greater extent in neurons than ES cells. The overlapping 10 genes are listed with p values.
Gene ontology (GO) enrichment analysis was performed to detect enrichment of cellular functions of the translationally downregulated genes in our ribosome profiling analyses (Fig. 3E). Genes encoding translation machinery proteins were enriched among those affected by Torin 1 commonly in neurons and ES cells. Consistent with our results, previous studies have demonstrated components of the translational machinery are common targets of mTOR signaling (Hsieh et al., 2012; Gentilella et al., 2015). Among the genes affected by Torin 1 specifically in ES cells, genes related to energy metabolism and mitochondrial function were enriched (Fig. 3E). Interestingly, genes encoding cytoskeletal regulators related to “focal adhesion” and “stress fiber” were enriched among those genes affected by Torin 1-treatment only in neurons (Fig. 3E), suggesting that translational targets of the mTOR signaling pathway might directly regulate neuronal morphology through control of cytoskeletal structures.
Alternative transcriptional start sites (TSS) often produce mRNAs bearing different 5′-terminal sequences, and thus selection of TSS may determine whether mRNAs contain 5′ terminal oligo-pyrimidine tract (5′TOP) sequences (Thoreen et al., 2012). To identify candidate proteins translationally regulated by mTOR specifically in neurons with a method distinct from ribosome profiling analysis, we performed a global in silico search for genes switching TSSs to produce mRNAs with 5′TOP sequences more during neural development. We used the FANTOM5 CAGE peaks time course dataset for neural differentiation of mouse ES cells (Forrest et al., 2014; Lizio et al., 2015). Analysis of the 5′-terminal sequences of mRNAs produced from TSS used in differentiated neurons and ES cells suggested that a subset of genes produce substantially more 5′TOP-containing mRNAs in the differentiated neurons than in the ES cells by switching alternative TSS during neuronal development (Fig. 3F). We found 178 genes using TSSs producing mRNAs containing the 5′TOP motif in higher proportion in differentiated neurons than ES cells (Fig. 3G). By intersecting these results with our ribosome profiling results, we found only 10 genes both represent translational targets of mTOR signaling and produce 5′TOP-containing mRNAs selectively in neurons (Fig. 3H). Among them, palladin was subjected to further analysis, since it was classified as a cytoskeletal regulator in the GO term enrichment analysis. In previous reports, palladin was not detected as a translational target of mTOR signaling in other cells (Hsieh et al., 2012; Thoreen et al., 2012; Livingstone et al., 2015; Park et al., 2017), supporting that production of palladin is specifically controlled by mTOR signaling in neurons.
Palladin is translationally regulated by mTOR and localized in axons
The ribosome profiling analysis suggested that translation efficiency of palladin is impaired upon mTOR inhibition in neurons. To examine whether mTOR activity influences the levels of palladin protein, we assessed the effect of inhibition of mTOR activity on palladin expression in neurons. The neurons were treated with Torin 1 over 24 h before lysis, which allows the palladin protein levels to reflect cumulative effects of translation efficiency alterations. The levels of palladin protein were reduced in neurons upon exposure to Torin 1 (Fig. 4A), suggesting that palladin translation is controlled by mTOR activity. The palladin antibody was validated by endogenous knockdown of palladin using two distinct short hairpin RNAs (shRNAs; Fig. 4B). The RNAi effect was also confirmed by knockdown of exogenously expressed palladin in HEK293T cells (Fig. 4B). To check whether Torin 1 treatment reduces the levels of another protein that was affected by Torin 1 specifically in neurons in the ribosome profiling analyses, we performed immunoblotting analysis of Zfp36 in neurons. Consistent with our ribosome profiling result, the protein levels of Zfp36 were reduced in cortical neurons upon Torin 1 treatment (Fig. 4C). In other experiments, we assessed the effect of additional mTOR inhibitors in addition to Torin 1 on the levels of palladin in cortical neurons. We found that the mTOR inhibitors Torin 2 and PP242 reduced the levels of palladin protein (Fig. 4D), further supporting the conclusion that inhibition of mTOR reduces the translation of palladin in cortical neurons.
Figure 4.

Palladin is translationally regulated by mTOR signaling and localized in axons. A, Lysates of cortical neurons treated with control vehicle (DMSO) or Torin 1 (100 nm) for 24 h were immunoblotted with the palladin, phospho-S6, and S6 antibodies. The palladin intensity was quantified and normalized by the loading control S6. The average relative abundance of three replicates was indicated. B, Left, Lysates of cortical neurons transfected with control plasmid, palladin RNAi1, or palladin RNAi2 were immunoblotted with the palladin and ERK antibodies. The palladin intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. Right, Lysates of HEK293T cells transfected with Flag-palladin together with control plasmid, palladin RNAi1, or palladin RNAi2 were immunoblotted with the Flag and ERK antibodies. The palladin intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. C, Lysates of cortical neurons treated with control vehicle (DMSO) or Torin 1 (100 nm) for 24 h were immunoblotted with the Zfp36, phospho-S6, S6, and ERK antibodies. The Zfp36 intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. D, Lysates of cortical neurons treated with control vehicle (DMSO), Torin 1 (100 nm), Torin 2 (100 nm), or PP242 (2.5 μm) for 24 h were immunoblotted with the palladin, phospho-S6, and ERK antibodies. The palladin intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. E, Lysates of cortical neurons treated with control vehicle (DMSO), Torin 1 (100 nm), or cytochalasin D (1 μm) for 2 h followed by 30 min treatment with puromycin were immunoblotted with the puromycin, phospho-S6, S6, and ERK antibodies. Relative abundance of nascent proteins labeled with puromycin was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. F, Lysates of cortical neurons treated with control vehicle (DMSO), Torin 1 (100 nm), or cytochalasin D (1 μm) for 24 h were immunoblotted with the palladin and ERK antibodies. The palladin intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. G, Lysates of TSC1 mutant Neuro2A cells or control cells were immunoblotted with palladin, TSC1, phospho-S6, S6, and ERK antibodies. The schematic of mouse TSC1 gene indicates sites of mutations introduced with the CRISPR–Cas9 system. The palladin intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. H, Lysates of cortical neurons from different time points were immunoblotted with the palladin and ERK antibodies. The palladin intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated. I, Hippocampal neurons cultured for 5 d in vitro were subjected to immunocytochemistry with the palladin and βIII Tubulin antibodies. Scale bar, 5 μm. J, Sections of E16 mouse cerebral cortex were subjected to immunohistochemistry with the palladin antibody (green), Tau-1 antibody (red) and stained with Hoechst (blue). Regions in the boxes 1–4 are enlarged in the bottom panels. The palladin immunoreactivity is higher in the intermediate zone where axons reside compared with other parts of the cerebral cortex. Scale bar, 100 μm. K, Lysates of whole cells or isolated neurites of cortical neurons treated with control vehicle (DMSO) or Torin 1 (100 nm) for 12 h were immunoblotted with the palladin, phospho-S6, S6, ERK, and nucleoporin antibodies. The palladin intensity was quantified and normalized by the loading control ERK. The average relative abundance of three replicates was indicated in the graphs. Asterisks indicate p < 0.05 in the graphs.
Torin 1 inhibits the two distinct mTOR-containing protein complexes mTORC1 and mTORC2 (Thoreen et al., 2009). Although mTORC1 directly controls protein synthesis, mTORC2 controls other cellular activities including actin cytoskeleton. To reveal overall activity of protein synthesis within neurons, we performed SUnSET analysis (Schmidt et al., 2009) upon treatment with Torin 1 or actin polymerization inhibitor cytochalasin D. We found that Torin 1, but not cytochalasin D, reduced the amount of newly synthesized proteins (Fig. 4E), suggesting that actin polymerization had little or no effect on protein synthesis. In other analyses, we found that cytochalasin D did not reduce palladin protein levels in cortical neurons (Fig. 4F). These data suggest that the effect of Torin 1 treatment on palladin protein translation was not caused by actin regulation through mTORC2, and hence presumably via regulation of protein synthesis through mTORC1.
We next mutated the gene encoding the mTOR inhibitor protein TSC1 using the CRISPR/Cas9 system (Ran et al., 2013) in neural cells. We tested the effect of mTOR activation upon mutation of TSC1 on the levels of palladin in cells. We confirmed that mutation of the TSC1 gene downregulated the levels of TSC1 protein and increased phosphorylation of the mTOR downstream signaling protein S6 in cells (Fig. 4G). Importantly, the levels of palladin protein were significantly increased in the TSC1 mutant cells (Fig. 4G), supporting the conclusion that palladin translation is regulated by the mTOR signaling cascade.
Palladin is a cytoskeletal regulator; however, its contribution to neuronal morphogenesis is still unclear (Boukhelifa et al., 2001; Shu et al., 2009). In temporal analyses of palladin in mouse cortical neurons, palladin was expressed during early developmental phases of neurons coinciding with axon extension (Fig. 4H). In immunocytochemical analyses in cortical neurons, palladin immunoreactivity was present in axon terminals and cell bodies (Fig. 4I). In immunohistochemical analyses of the mouse cerebral cortex, palladin-immunoreactivity was enriched in the intermediate zone where axons extend from neurons in the cortical plate (Fig. 4J). Thus, the temporal and spatial expression of palladin suggest a function in axon morphogenesis.
To examine local synthesis of palladin in neurites, we isolated neurites and treated them with Torin 1 (Fig. 4K). Neurons were cultured on a filter bearing pores that accommodate neurites to grow on the opposite side of their soma, and the neurites were isolated by removing soma on the filter as described previously (Vidaki et al., 2017). We found that Torin 1-treatment of isolated neurites significantly reduced the protein level of palladin. Palladin protein level was similarly reduced by Torin 1-treatment in the whole cells, suggesting that palladin is synthesized within neurites as well as in the soma.
Palladin regulates axonal morphogenesis downstream of the mTOR signaling pathway
We next asked whether palladin plays a role in neuronal morphogenesis downstream of mTOR signaling. To assess the function of palladin in neuronal morphogenesis, we first expressed palladin in primary mouse hippocampal neurons. We found that significantly higher percentage of palladin-expressing neurons possessed multiple axons compared with control neurons (Fig. 5A), resembling the phenotype induced by activation of the mTOR pathway (Li et al., 2008; Mejia et al., 2013). The exogenous expression of palladin in axons was confirmed by immunocytochemistry analysis with palladin antibody (Fig. 5B). The phenotype of multiple axons was consistently observed when palladin was expressed as a GFP fusion protein (Fig. 5C).
Figure 5.
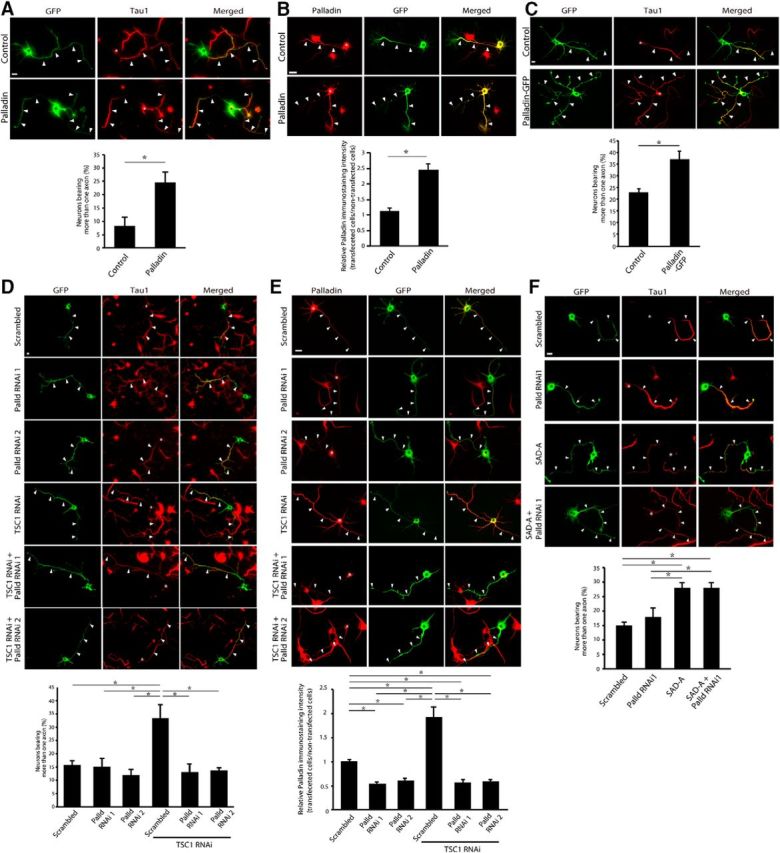
Palladin controls axonal morphology downstream of mTOR signaling. A, Hippocampal neurons were transfected with the palladin expression plasmid or control plasmid together with a GFP expression plasmid 2–6 h after plating. Five days after transfection, neurons were subjected to immunocytochemistry using the GFP (green) and Tau1 (red) antibodies. Palladin-expressing neurons had a higher percentage of neurons bearing more than one axon than control-transfected neurons. Arrowheads and asterisks indicate axons and cell bodies, respectively. B, Hippocampal neurons were transfected with the palladin expression plasmid or control plasmid together with a GFP expression plasmid, and cultured for 3 d. The neurons were subjected to immunocytochemistry using the palladin (red) and GFP (green) antibodies. The palladin intensity was significantly increased by the exogenous expression of palladin in axons. Arrowheads and asterisks indicate axons and cell bodies, respectively. C, Hippocampal neurons were transfected with the GFP-fused palladin expression plasmid or control GFP plasmid. Five days after transfection, neurons were subjected to immunocytochemistry using the GFP (green) and Tau1 (red) antibodies. Palladin-GFP-expressing neurons had a higher percentage of neurons bearing more than one axon than control-transfected neurons. Arrowheads and asterisks indicate axons and cell bodies, respectively. D, Hippocampal neurons transfected with the TSC1 RNAi or control scrambled RNAi together with the palladin RNAi 1, palladin RNAi 2, or the control plasmid were analyzed. Palladin RNAi 1 and palladin RNAi 2 target distinct regions of palladin mRNA. The neurons were subjected to immunocytochemistry using the GFP (green) and Tau1 (red) antibodies. TSC1 RNAi increased the percentage of neurons bearing multiple axons compared with control-transfected neurons, whereas palladin RNAi 1 or 2 did not alter the percentage of neurons bearing multiple axons. Palladin knockdown with both RNAi 1 and 2 significantly reduced the percentage of neurons bearing multiple axons in the background of TSC1 knockdown. E, Hippocampal neurons transfected with the TSC1 RNAi or control scrambled plasmid together with the palladin RNAi 1, palladin RNAi 2 or control scrambled plasmid were analyzed by immunocytochemistry with palladin (red) and GFP (green) antibodies. F, Hippocampal neurons transfected with the palladin RNAi 1 or control scrambled plasmid together with the SAD-A expression plasmid or control plasmid were analyzed. The neurons were subjected to immunocytochemistry using the GFP (green) and Tau1 (red) antibodies. The SAD-A expression significantly induced supernumerary axons, and the palladin RNAi1 did not suppress the supernumerary axons induced by the SAD-A expression. Scale bars, 10 μm. Asterisks indicate p < 0.05 in the all graphs.
Because our biochemical analyses suggest that palladin expression is regulated by mTOR activity, we next tested whether palladin operates downstream of mTOR kinase by assessing the percentage of cells with multiple axons upon knockdown of TSC1 and knockdown of palladin simultaneously. As reported, suppression of TSC1 induced formation of more than one axon per neuron (Fig. 5D). Although knockdown of palladin did not change the percentage of cells bearing multiple axons, palladin knockdown reversed the multiple axon phenotype in TSC1 knockdown neurons (Fig. 5D). Immunocytochemistry analyses revealed that palladin immunoreactivity signal was reduced upon knockdown of palladin, validating the palladin antibody for immunocytochemistry (Fig. 5E). Knockdown of TSC1 upregulated palladin immunoreactivity, while knockdown of palladin together with TSC1 knockdown reduced the palladin immunoreactivity (Fig. 5E), suggesting that palladin protein level is controlled by the mTOR signaling pathway. These results demonstrate that palladin regulates neuronal morphogenesis downstream of the mTOR signaling pathway.
Because SAD kinases are known to regulate axon morphology downstream of mTOR signaling pathway (Choi et al., 2008), we also characterized the functional relationship between SAD kinase and palladin in axon morphogenesis. The expression of SAD-A kinase-induced supernumerary axons, and knockdown of palladin failed to suppress the supernumerary axons induced by SAD-A (Fig. 5F). These results suggest that SAD kinases operate downstream of or in parallel with palladin in the control of axon morphology induced by activation of mTOR signaling.
Collectively, we have found that mTOR signaling regulates the translation of a specific set of proteins in neurons including the cytoskeletal regulator palladin and thereby controls axon morphogenesis.
Discussion
In this study, we have discovered that the mTOR signaling pathway regulates the translation of specific proteins in neurons. We have identified the cytoskeletal regulator palladin as a neuron-specific translational target of mTOR signaling, which mediates mTOR-regulation of axon morphogenesis. Ribosome profiling analyses revealed that translation efficiency of palladin is modified upon acute inhibition of mTOR specifically in neurons, while codon-specific ribosome occupancy and ribosomal stalling were not altered significantly. Consistent with this finding, palladin protein amount was modulated by alterations of mTOR activity. Palladin was localized in axons, and expression of palladin-induced neurons to form multiple axons. Supernumerary axons induced by knockdown of the mTOR-regulator TSC1 were suppressed by simultaneous knockdown of palladin, suggesting that palladin functions downstream of mTOR. Our findings define a mechanism that regulates neuronal morphology via mTOR-mediated control of protein translation.
The mTOR signaling pathway controls diverse aspects of protein synthesis, including initiation and elongation of translation. Consequently, we assessed changes of translation landscape upon Torin 1 treatment from multiple perspectives. We observed limited differences in codon-specific ribosome occupancy and translational pausing in cells upon exposure to the mTOR inhibitor Torin 1. However, comparison of neurons and ES cells exposed differential codon ribosome occupancy and ribosome pausing. Recent studies suggest that the availability of aminoacyl-tRNA varies in different cell types, leading to changes in codon optimality and codon ribosome occupancy at A-site (Plotkin and Kudla, 2011; Kirchner and Ignatova, 2015; Bazzini et al., 2016). Quantitative understanding of the differences in codon optimality between neurons and ES cells might reveal the function and importance of codon optimality during development and differentiation. How P-site codon ribosome occupancy is differentially regulated in neurons and ES cells requires further study. Proline codons were significantly more occupied by ribosomes in ES cells than in neurons, raising the possibility that eIF5A, a regulator of proline elongation (Gutierrez et al., 2013; Schuller et al., 2017), may be regulated in a cell-type-specific manner. Likewise, differences in ribosome stalling were more pronounced between neurons and ES cells than between neurons treated with Torin 1 and control neurons. It will be interesting to determine in future studies how ribosome stalling during translation elongation occurs differentially in distinct cells.
Through the analysis of translation efficiency, we found that mRNAs of 431 genes acutely responded to mTOR inhibition at the level of translation specifically in neurons. Using GO term enrichment analysis of the identified genes, we found that a significant portion of the genes is associated with cytoskeletal regulation, whereas energy and mitochondria-related genes were enriched in the ES cell-specific mTOR-responsive genes. The difference in the function of the observed genes suggests that subsets of genes are programmed to respond at the level of protein translation to adapt cells specifically to their differentiation status. It is noteworthy that translation machinery proteins were commonly downregulated by inhibition of mTOR, which may exhibit different effects in different cell types. It will be interesting to explore how newly synthesized ribosomes might regulate protein synthesis in distinct cell types.
Palladin was enriched in axon terminals. Recent evidence shows that protein synthesis occurs in axons, and local translation in axons enables neurons to control axon growth and regeneration (Jung et al., 2012; Korsak et al., 2016; Twiss et al., 2016; Spaulding and Burgess, 2017). We observed that palladin protein level in isolated neurites responded to Torin 1-treatment, suggesting that palladin may be locally synthesized in axons. To understand the precise localization and dynamics of palladin synthesis in axons, further detailed analyses of palladin synthesis in axons are required. Neuronal compartment-specific ribosome profiling might provide further insights into local regulation of protein synthesis in neurons.
Although knockdown of palladin rescued the supernumerary axons induced by knockdown of TSC1, knockdown of palladin itself had little or no effect on the morphogenesis of axons (Fig. 5D). TSC1/2 overexpression has been reported to suppress axon formation (Choi et al., 2008), suggesting that other proteins may mediate the control of axon morphogenesis upon inactivation of mTOR signaling. Knockdown of palladin also failed to suppress supernumerary axons induced by SAD-A expression, whereas it suppressed supernumerary axons induced by knockdown of TSC1. These results suggest that palladin operates downstream of TSC1, but upstream or in parallel with SAD kinases.
The identification of a function of palladin as a neuronal morphogenesis effector protein of mTOR signaling may shed light on pathogenesis of mTOR-related neurological disorders including TSC. Among diverse clinical symptoms caused by TSC, neurological symptoms including intellectual disability, epilepsy, and ASD may be caused through dysregulation of neuron-specific mechanisms. Recent findings suggest that dysregulation of mTOR signaling and protein synthesis might underlie broader neurological disorders including autism and schizophrenia (Kelleher and Bear, 2008; Gururajan and van den Buuse, 2014).
Footnotes
This work was supported by Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research 26890008, 15K14311, 15H01528, and 17H05661 (Y.I.), Uehara Memorial Foundation (Y.I.), Mochida Memorial Foundation (Y.I.), Mathers Foundation (A.B.), and NIH Grant NS051255 (A.B.). We thank Shintaro Iwasaki and Jagat Chhipishrestha for critical discussion and providing PP242; Luis de la Torre-Ubieta, Luis Mejia, and Anna Oldenborg for providing support for plasmids used in the study; the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with sequencing analyses; computing resource was provided by Human Genome Center, the Institute of Medical Science, the University of Tokyo; pSpCas9(BB)-2A-Puro (pX459) V2.0 was a gift from Feng Zhang (Addgene plasmid # 62988); and the SAD-A expression plasmid was a gift from Josh Sanes.
The authors declare no competing financial interests.
References
- Bazzini AA, Del Viso F, Moreno-Mateos MA, Johnstone TG, Vejnar CE, Qin Y, Yao J, Khokha MK, Giraldez AJ (2016) Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J 35:2087–2103. 10.15252/embj.201694699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhelifa M, Parast MM, Valtschanoff JG, LaMantia AS, Meeker RB, Otey CA (2001) A role for the cytoskeleton-associated protein palladin in neurite outgrowth. Mol Biol Cell 12:2721–2729. 10.1091/mbc.12.9.2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nat Rev Neurosci 8:776–789. 10.1038/nrn2150 [DOI] [PubMed] [Google Scholar]
- Brittis PA, Lu Q, Flanagan JG (2002) Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell 110:223–235. 10.1016/S0092-8674(02)00813-9 [DOI] [PubMed] [Google Scholar]
- Browne GJ, Proud CG (2004) A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol 24:2986–2997. 10.1128/MCB.24.7.2986-2997.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Boutros PC (2011) VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. 10.1186/1471-2105-12-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X (2008) Tuberous sclerosis complex proteins control axon formation. Genes Dev 22:2485–2495. 10.1101/gad.1685008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Monteggia LM (2013) mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci 16:1537–1543. 10.1038/nn.3546 [DOI] [PubMed] [Google Scholar]
- Darnell JC, Klann E (2013) The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 16:1530–1536. 10.1038/nn.3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deglincerti A, Jaffrey SR (2012) Insights into the roles of local translation from the axonal transcriptome. Open Biol 2:120079. 10.1098/rsob.120079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, Weissman JS (2016) Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17:958. 10.1186/s12864-016-3278-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller WJ, Jackson TJ, Knight JR, Ridgway RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB, Dudek KM, Casey HA, Scopelliti A, Cordero JB, Vidal M, Pende M, Ryazanov AG, Sonenberg N, Meyuhas O, Hall MN, et al. (2015) mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517:497–500. 10.1038/nature13896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, Andersson R, Mungall CJ, Meehan TF, Schmeier S, Bertin N, Jørgensen M, Dimont E, Arner E, Schmidl C, Schaefer U, et al.; FANTOM Consortium and the RIKEN PMI and CLST (2014) A promoter-level mammalian expression atlas. Nature 507:462–470. 10.1038/nature13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A (2002) RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem 277:46442–46446. 10.1074/jbc.M206653200 [DOI] [PubMed] [Google Scholar]
- Gentilella A, Kozma SC, Thomas G (2015) A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim Biophys Acta 1849:812–820. 10.1016/j.bbagrm.2015.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gururajan A, van den Buuse M (2014) Is the mTOR-signalling cascade disrupted in schizophrenia? J Neurochem 129:377–387. 10.1111/jnc.12622 [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, Dever TE (2013) eIF5A promotes translation of polyproline motifs. Mol Cell 51:35–45. 10.1016/j.molcel.2013.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485:55–61. 10.1038/nature10912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WF, Chahrour MH, Walsh CA (2014) The diverse genetic landscape of neurodevelopmental disorders. Annu Rev Genomics Hum Genet 15:195–213. 10.1146/annurev-genom-090413-025600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS (2012) The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7:1534–1550. 10.1038/nprot.2012.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies HB, Reinhard C, Kozma SC, Thomas G (1994) Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc Natl Acad Sci U S A 91:4441–4445. 10.1073/pnas.91.10.4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Yoon BC, Holt CE (2012) Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13:308–324. 10.1038/nrn3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ (2004) The UCSC table browser data retrieval tool. Nucleic Acids Res 32:D493–D496. 10.1093/nar/gkh103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Bear MF (2008) The autistic neuron: troubled translation? Cell 135:401–406. 10.1016/j.cell.2008.10.017 [DOI] [PubMed] [Google Scholar]
- Kenney JW, Moore CE, Wang X, Proud CG (2014) Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv Biol Regul 55:15–27. 10.1016/j.jbior.2014.04.003 [DOI] [PubMed] [Google Scholar]
- Kirchner S, Ignatova Z (2015) Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet 16:98–112. 10.1038/nrg3861 [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A (2004) Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303:1026–1030. 10.1126/science.1093712 [DOI] [PubMed] [Google Scholar]
- Korsak LI, Mitchell ME, Shepard KA, Akins MR (2016) Regulation of neuronal gene expression by local axonal translation. Curr Genet Med Rep 4:16–25. 10.1007/s40142-016-0085-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9:e1003118. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Werner H, Püschel AW (2008) Rheb and mTOR regulate neuronal polarity through Rap1B. J Biol Chem 283:33784–33792. 10.1074/jbc.M802431200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone M, Sikström K, Robert PA, Uzé G, Larsson O, Pellegrini S (2015) Assessment of mTOR-dependent translational regulation of interferon stimulated genes. PLoS One 10:e0133482. 10.1371/journal.pone.0133482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F, Ishikawa-Kato S, Mungall CJ, Arner E, Baillie JK, Bertin N, Bono H, de Hoon M, Diehl AD, Dimont E, Freeman TC, Fujieda K, et al. (2015) Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol 16:22. 10.1186/s13059-014-0560-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia LA, Litterman N, Ikeuchi Y, de la Torre-Ubieta L, Bennett EJ, Zhang C, Harper JW, Bonni A (2013) A novel Hap1–Tsc1 interaction regulates neuronal mTORC1 signaling and morphogenesis in the brain. J Neurosci 33:18015–18021. 10.1523/JNEUROSCI.2290-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge JM, Harrow J (2015) Creating reference gene annotation for the mouse C57BL/6J genome assembly. Mamm Genome 26:366–378. 10.1007/s00335-015-9583-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkova DD, Leidel SA (2015) Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161:1606–1618. 10.1016/j.cell.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Masui S, Chambers I, Smith AG, Miyazaki J (2002) Phenotypic complementation establishes requirements for specific POU domain and generic transactivation function of oct-3/4 in embryonic stem cells. Mol Cell Biol 22:1526–1536. 10.1128/MCB.22.5.1526-1536.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Matsui H, Ohtsuka S, Niwa H (2004) A novel mechanism for regulating clonal propagation of mouse ES cells. Genes Cells 9:471–477. 10.1111/j.1356-9597.2004.00736.x [DOI] [PubMed] [Google Scholar]
- Olshen AB, Hsieh AC, Stumpf CR, Olshen RA, Ruggero D, Taylor BS (2013) Assessing gene-level translational control from ribosome profiling. Bioinformatics 29:2995–3002. 10.1093/bioinformatics/btt533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, Huganir RL, Linden DJ, Worley PF (2008) Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59:70–83. 10.1016/j.neuron.2008.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Reyna-Neyra A, Philippe L, Thoreen CC (2017) mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep 19:1083–1090. 10.1016/j.celrep.2017.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12:32–42. 10.1038/nrg2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET: a nonradioactive method to monitor protein synthesis. Nat Methods 6:275–277. 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- Schuller AP, Wu CC, Dever TE, Buskirk AR, Green R (2017) eIF5A functions globally in translation elongation and termination. Mol Cell 66:194–205.e5. 10.1016/j.molcel.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF (2011) New views of arc: a master regulator of synaptic plasticity. Nat Neurosci 14:279–284. 10.1038/nn.2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu RZ, Zhang F, Liu XS, Li CL, Wang L, Tai YL, Wu XL, Yang X, Liao XD, Jin Y, Gu MM, Huang L, Pang XF, Wang ZG (2009) Target deletion of the cytoskeleton-associated protein palladin does not impair neurite outgrowth in mice. PLoS One 4:e6916. 10.1371/journal.pone.0006916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding EL, Burgess RW (2017) Accumulating evidence for axonal translation in neuronal homeostasis. Front Neurosci 11:312. 10.3389/fnins.2017.00312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Nawa H (2014) mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci 7:28. 10.3389/fnmol.2014.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. 10.1038/nn1566 [DOI] [PubMed] [Google Scholar]
- The RNAcentral Consortium (2017) RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res 45:D128–D134. 10.1093/nar/gkw1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284:8023–8032. 10.1074/jbc.M900301200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113. 10.1038/nature11083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiss JL, Kalinski AL, Sachdeva R, Houle JD (2016) Intra-axonal protein synthesis: a new target for neural repair? Neural Regen Res 11:1365–1367. 10.4103/1673-5374.191193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpelli C, Piccoli G, Zibetti C, Zanchi A, Gardoni F, Huang K, Brambilla D, Di Luca M, Battaglioli E, Sala C (2010) Synaptic activity controls dendritic spine morphology by modulating eEF2-dependent BDNF synthesis. J Neurosci 30:5830–5842. 10.1523/JNEUROSCI.0119-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidaki M, Drees F, Saxena T, Lanslots E, Taliaferro MJ, Tatarakis A, Burge CB, Wang ET, Gertler FB (2017) A requirement for Mena, an actin regulator, in local mRNA translation in developing neurons. Neuron 95:608–622.e5. 10.1016/j.neuron.2017.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DN, Arenz S, Beckmann R (2016) Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr Opin Struct Biol 37:123–133. 10.1016/j.sbi.2016.01.008 [DOI] [PubMed] [Google Scholar]