

SUMMARY
Granulocyte colony stimulating factor (G-CSF) is a multi-modal hematopoietic growth factor, which also has profound effects on the diseased central nervous system. G-CSF has been shown to enhance recovery from neurologic deficits in rodent models of ischemia. G-CSF appears to facilitate neuroplastic changes by both mobilization of bone marrow-derived cells and by its direct actions on CNS cells. The overall objective of the study was to determine if G-CSF administration in a mouse model of AD (Tg APP/PS1) would impact hippocampal-dependent learning by modifying the underlying disease pathology. A course of sub-cutaneous administration of G-CSF for a period of less than three weeks significantly improved cognitive performance, decreased β-amyloid deposition in hippocampus and entorhinal cortex and augmented total microglial activity. Additionally, G-CSF reduced systemic inflammation indicated by suppression of the production or activity of major pro-inflammatory cytokines in plasma. Improved cognition in AD mice was associated with increased synaptophysin immunostaining in hippocamal CA1 and CA3 regions and augmented neurogenesis, evidenced by increased numbers of calretinin-expressing cells in dentate gyrus. Given that G-CSF is already utilized clinically to safely stimulate hematopoietic stem cell production, these basic research findings will be readily translated into clinical trials to reverse or forestall the progression of dementia in AD.
Keywords: hippocampus, microglia, bone marrow, β-amyloid, synaptophysin, neurogenesis
Granulocyte colony stimulating factor (G-CSF; Filgastrim) is one of several hematopoietic growth factors (HGF)s or colony stimulating factors (CSFs) that control the production of circulating blood cells by the bone marrow. G-CSF is produced by endothelium, macrophages, and a number of other immune cells. G-CSF and its receptor have also been reported to be expressed in neurons and neural progenitor cells that reside in the hippocampal neurogenic niche (Schneider et al., 2005). Although G-CSF has been used primarily as an agent to treat leukopenia, the agent has been studied in animal models of stroke where it has been reported to reduce brain damage and improve outcome (Schabitz et al., 2003, Six et al., 2003, Shyu et al., 2006, Solaroglu et al., 2006). Several ongoing clinical trials are evaluating the effectiveness and safety of G-CSF for treatment of ischemic stroke, and results are still pending (Schabitz and Schneider, 2006). It has also been used experimentally to treat myocardial infarction in humans (Takano et al., 2003, Kuethe et al., 2004, Nienaber et al., 2006, Ripa et al., 2006, Suzuki et al., 2006). In addition, G-CSF has putative neuroprotective actions (e.g. anti-apoptotic and/or neurogenic) independent of its regulation of granulocyte differentiation and stem cell mobilization (Schabitz et al., 2003, Gibson et al., 2005, Solaroglu et al., 2006). G-CSF has also been shown to modulate systemic immune responses by inhibiting pro-inflammatory cytokines. Animal, volunteer, and patient studies have all shown that G-CSF reduces inflammatory activity by inhibiting the production or activity of the main inflammatory mediators interleukin-1 (IL-1), tumor necrosis factor-alpha (TNF-α), and interferon gamma (IFγ) (Hartung, 1998). Importantly, decreased plasma levels of G-CSF (and other hematopoietic factors) have been linked to Alzheimer’s disease (AD) and are in fact predictive of conversion from mild cognitive impairment (MCI) to AD (Ray et al., 2007).
One of the mechanisms by which G-CSF hastens recovery from ischemic neurologic deficits in animal models of stroke is related to increased activation of resident microglia but not to increased mobilization of marrow-derived microglia (Komine-Kobayashi et al., 2006). However, many of the microglia that infiltrate the core of beta-amyloid (Aβ) plaques in transgenic mouse models of AD have been shown to originate from the bone marrow (Malm et al., 2005, Simard et al., 2006). Both Aβ1-40 and Aβ1-42 are effective in triggering this chemo attraction. Although resident microglia are clearly activated in response to Aβ deposition, the bone marrow-derived microglia appear to be very efficient in reabsorption of these Aβ deposits (Simard et al., 2006). Furthermore, penetration of bone marrow-derived cells into the brain can be boosted by microinjection of lipopolysaccharide into hippocampus of AD mice (Malm et al., 2005). This procedure significantly increased the number of bone marrow-derived cells in aged AD mice and resulted in decreased hippocampal Aβ burden (Malm et al., 2005). Administration of G-CSF has been reported to improve cognitive performance in Tg2576 mice without reduction of total brain amyloid burden (Tsai et al., 2007). That study also reported an increased proliferation of cells around the amyloid plaques, based on BrdU labeling. The authors indicated that the BrdU-labeled cells co-expressed neuronal markers and suggested hematopoietic stem/progenitor cells trans-differentiated into neurons around the plaques (Tsai et al., 2007). In this context, our prior review of the literature has raised interesting and paradoxical issues that warrant the systematic study of G-CSF and other hematopoietic growth factors (HGFs) for treatment of AD (Sanchez-Ramos et al., 2008).
The primary objective of the present study was to determine whether a short course of G-CSF administration would have an impact on the pathological hallmark of AD, the age-dependent accumulation of Aβ deposits, in a transgenic mouse model of AD (APP+PS1; Tg). A second objective was to determine whether such treatment would impact cognitive performance in a hippocampal-dependent memory paradigm. To explain the G-CSF triggered amyloid reduction and associated reversal of cognitive impairment, several mechanisms of action were explored. 1) G-CSF was hypothesized to increase activation of resident microglia and to increase mobilization of marrow-derived microglia. The effect of G-CSF on microglial activation was examined by quantitative measurements of total microglial burden. To determine if G-CSF increased trafficking of marrow-derived microglia into brain, bone marrow-derived green fluorescent protein-expressing (GFP+) microglia were visualized in the brains of chimeric AD mice. 2) To assess the role of immune-modulation in mediating G-CSF effects, a panel of cytokines was measured in both plasma and brain. 3) To test the hypothesis that reduction of Aβ deposits can affect synaptic area, quantitative measurement of synaptophysin immunoreactivity in hippocampal CA1 and CA3 sectors was undertaken. 4) To learn whether enhanced hippocampal neurogenesis was induced by G-CSF treatment, numbers of calretinin-expressing cells were determined in dentate gyrus.
MATERIALS AND METHODS
Animals
Mice were generated from a cross between F6 generation mice heterozygous for the mutant APPK670N, M671L gene (i.e., the APPsw, Swedish mutation derived from Tg2576 mice), and littermate mutant PS1 (6.2 line) mice bearing the M146V mutation. (All mice contained a mixed background of C57B6, B6D2F1, SJL, and SW). Mice were initially genotyped at the time of weaning and then had a confirmatory genotyping at 4 months of age. A total of 52 mice in 2 age-based cohorts comprised the behavioral studies. Cohort 1 (n=25) ranged from 13-15 months old and cohort 2 (n=27) ranged from 7 to 9 months old. The behavioral study initially involved only Cohort 1 mice. However, upon seeing a strong cognitive-enhancing effect of G-CSF in this cohort, we decided to repeat the study in a second cohort of aged mice (Cohort 2) to confirm our behavioral results. A total of 24 APP+PS1 double transgenic mice (e.g., mice bearing both APPsw and PS1 mutations) and 28 non-transgenic littermates were distributed between the two cohorts. A third cohort of APP+PS1 mice (n=16) was utilized to generate chimeric mice with GFP-expressing bone marrow. Mice were housed and maintained in a specific pathogen-free facility under a 12/12 hrs light–dark cycle, with ad libitum access to rodent chow and water. The animal research protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of South Florida.
Chimeric Tg APP/PS1 Mice With Bone Marrow Derived From Tg “Green Mice”
Sixteen APP+PS1 mice from the same colony (2M old) were lethally irradiated with 10 Gy total body irradiation (delivered in two fractions of 5 Gy at dose rate of 1.03 Gy/min in a Gammacell 40 Extractor (Furuya et al., 2003). This was followed by rescue with a bone marrow transplant (8 × 106 mononuclear cells in 0.2 ml) from Tg GFP mice (C57BL/6-Tg [ACTB-EGFP] 1Osb/J, 003291; The Jackson Laboratory, Bar Harbor, ME) infused via tail vein. Bone marrow-derived cells in the rescued mice were readily tracked by virtue of their green fluorescence. Examination of blood smears from tail clippings for the presence of green monocytes confirmed successful engraftment. An average of 75% of the irradiated mice had successful engraftment and exhibited GFP+ mononuclear cells in their peripheral blood.
G-CSF Dose and Administration Schedule
G-CSF, a natural human glycoprotein, exists in two forms of a 174- and 180-amino-acid-long protein of molecular weight 19,600 grams per mole. The more-abundant and more-active 174-amino acid form has been used in the development of pharmaceutical products by recombinant human DNA (rhDNA) technology. Filgrastim (Neupogen®, Amgen, Inc. Thousand Oaks, CA), one of three proprietary G-CSF compounds, was utilized in the present study. The dose of G-CSF (250 μg/kg X 6) administered in the present study was lower than that reported to be effective in mobilizing bone marrow in a rat model of stroke (300 μg/kg X 10 days) but higher than that utilized by others in rodent models of stroke (Schabitz et al., 2003, Six et al., 2003, Solaroglu et al., 2006) and in a recently published report on the treatment of memory impairment in a mouse model of AD (50 μg/kg s.c for 5 days) (Tsai et al., 2007). The schedule of administration of G-CSF was one injection sub-cutaneously (s.c.) every other day for 3 weeks. In the experiments in which behavioral testing was performed before and after administration of G-CSF, the drug continued to be given on alternate days during the final behavioral testing. Dilution of G-CSF was in 5% dextrose as recommended by the manufacturer (Amgen, Inc. Thousand Oaks, CA). Vehicle (5% dextrose) was administered to control transgenic and non-transgenic groups.
Behavioral Testing
Spatial working memory was assessed in a ‘‘win-stay’’ version of the radial arm water maze (RAWM) task. Animals were pre-treatment tested for 8 consecutive days in the RAWM task, and then each genotype (NT, Tg) was divided into two groups behaviorally balanced in pre-treatment RAWM performance. For Tg mice, blood Aβ levels following pre-treatment testing were also utilized to balance treatment and control Tg groups. Beginning at two weeks into treatment, all mice were re-tested in the RAWM for 4 consecutive days. For RAWM testing, an aluminum insert was introduced into a 100 cm pool in order to divide the pool into six equally spaced swim arms (30.5 cm length 19 cm width) radiating from a central circular swim area (40 cm diameter). The insert extended 5 cm above the surface of the water, allowing the mice to easily view surrounding visual cues, which were generously placed outside of the pool. Visual/spatial cues consisted of large brightly colored 2D and 3D objects, including a beach ball, poster, and inflatable pool toys. During testing, the pool water was maintained at 23–27 °C. In one of the arms, a transparent 9 cm submerged escape platform was placed 1.5 cm below the water near the wall end. Each mouse was given five 1-min trials per day. The last of the four consecutive acquisition trials (Trial 4, T4) and a 30-min delayed retention trial (Trial 5, T5) are indices of working memory. On any given day, the escape platform location was placed at the end of one of the six arms, with the platform moved to a different arm in a semi-random fashion for each day of testing. In contrast to the stationary platform of Morris water maze, moving the escape platform forced the animal to learn a new platform location daily, therefore evaluating working memory. On each day, different start arms for each of the five daily trials were selected from the remaining five swim arms in a semi-random sequence that involved all five arms. For any given trial, the mouse was placed into that trial’s start arm, facing the center swim area, and given 60 s to find the platform. When the mouse made an incorrect choice, it was gently pulled back to that trial’s start arm and an error was recorded. An error was also recorded if the mouse failed to make a choice in 20 s (in which case it was returned to that trails start arm), or if the animal entered the platform-containing arm, but failed to locate the platform. A 30-s stay was given once the mouse had found the platform. If the mouse did not find the platform within a 60-s trial, it was guided by the experimenter to the platform, allowed to stay for 30 s, and was assigned a latency of 60 s. Both errors (incorrect arm choices) and escape latency were recorded for each daily trial.
Sample Preparation and Immunohistochemistry
At the conclusion of the behavioral testing (approximately 3 weeks into treatment and at 14-15 months of age for the first cohort of mice and at 8-9 months for the second cohort), mice were anesthetized under deep chloral hydrate (10%) anesthesia. Blood was collected intracardially and a plasma sample was stored at -80°C for later analysis of Aβ levels. After transcardial perfusion of the brain with 0.1 M phosphate buffered saline (PBS), the brain was removed and bisected parasagitally. Further, the right hemisphere was dissected into two pieces (frontal and caudal brains). These brain pieces were immediately frozen on dry ice and stored at -80°C for determination of Aβ levels by ELISA. The left hemisphere was placed in 4% paraformaldehyde in 0.1M phosphate buffer (PB, pH 7.4) over night. Like the right hemisphere, the brain was dissected into two pieces (frontal and caudal brains) and the frontal piece was transferred to a graded series of sucrose solutions (10%, 20%, and finally 30%). Quarter brains that had been stored in 30% sucrose solution were removed, rinsed in dH2O, and frozen on the stage of a sliding microtome.
The caudal part of the left hemisphere was routinely embedded in paraffin with 24 hrs processing. For paraffin sectioning, five coronal sections (per set) with a 150 μm interval were cut at a thickness of 5 μm in hippocampus (H) and entorhinal cortex (EC), bregma -2.92 mm to – 3.64 mm (Paxinos and Franklin, 2001). Four sets of five sections from H and EC were prepared for analyses of Aβ and Iba1 (ionized calcium-binding adapter molecule 1). Immunohistochemical staining was performed following the manufacturer’s protocol using a Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA) coupled with the diaminobenzidine reaction, except that the biothinylated secondary antibody step was omitted for Aβ immunohistochemical staining. The following primary antibodies were used for immunohistochemical staining: a biothinylated human amyloid-β monoclonal antibody (clone 4G8; 1:200, Covance Research Products, Emeryville, CA), an Iba-1 polyclonal antibody (1:1000, Wako, Osaka, Japan), and rabbit synaptophysin polyclonal antibody (undiluted, DAKO, Carpinteria, CA).
For the experiments using chimeric Tg APP+PS1 mice, no behavioral analysis was performed. At the end of 2 weeks of G-CSF treatment, animals were anesthetized and perfused as above. Brains were bisected parasagitally; with one half for Aβ level determinations by ELISA and the other half for fluorescence immunohistochemistry. The left hemisphere was placed in 4% paraformaldehyde in 0.1M phosphate buffer (PB, pH 7.4) over night. The hemisphere was dissected into two pieces (frontal and caudal brains) and the frontal piece was transferred to a graded series of sucrose solutions (10%, 20%, and finally 30%). Quarter brains that had been stored in 30% sucrose solution were removed, rinsed in dH2O, and frozen on the stage of a sliding microtome. Antibodies to GFP (Chicken anti-GFP 1:1250, Chemicon International, Temecula, CA) were used to enhance the GFP+ signal from bone marrow derived cells that accumulated around amyloid deposits. The secondary antibody was Alexa Fluor 488 labeled goat anti-chicken IgG, 1:400 (Invitrogen, Invitrogen Corp., Carlsbad, CA). For calretinin immunofluroescence, sections were blocked in PBS+ (PBS, 10% normal goat serum, 1% TX-100, 1M Lysine) for 1 hr at 4°C and incubated overnight at 4°C in rabbit anti-calretinin 1:2500 (Swant, International, Bellinzona, Switzerland) in PBS. Sections were washed in PBS and incubated in goat anti-rabbit IgG Alexa Fluor 488 (Invitrogen Corp., Carlsbad, CA) and coated with Vectorshield mounting medium (Invitrogen Corp.). Labeled cells were visualized with fluorescence microscopy (IX2 inverted microscope; Olympus, Tokyo, Japan) using appropriate filters or with a confocal microscope (LSM 510; Carl Zeiss, Inc., Thornwood, NY). Digital images were captured with either the DP-70 digital camera system (Olympus) or by collecting Z-stacks of the confocal images (Carl Zeiss, Inc., Thornwood, NY).
Proliferation Assay for Hippocampal Neural Stem/Progenitor cells (NSC) in vitro
NSC from adult hippocampus of C57BL mice were isolated and maintained in cell culture in the presence of EGF and bFGF using standard methods as described previously in our laboratory (Sava et al., 2007). G-CSF was added to hippocampal NSC in basal media containing DMEM+10% FBS for 48 hrs, (in absence of EGF and bFGF), and incorporation of [3H]-thymidine was measured as previously described (Sava et al., 2007). Briefly, at various intervals after G-CSF addition (0, 24, 48, and 72 h), 37,000 Bq/ml (500 pmol/m;) [3H]-thymidine (PerkinElmer Life and Analytical Sciences, Inc., MA) was added to the cultures for 4 h, then cells were harvested, and [3H]-thymidine incorporated into DNA was determined. The specific activity of [3H]-thymidine was 74 Bq/pmol. Uptake was normalized and expressed as percent change from control.
Estimates of Hippocamal Neurogenesis in vivo
Unbiased estimates of the number of immature neurons in dentate gyrus (DG) were made by counting calretinin-immunoreactive cells in serially sectioned hippocampus according to the method previously described (Shors et al., 2001, Shors et al., 2002). Briefly, positively labeled cells were counted in every 6th section (each section separated by 180 μm) using a modification to the optical dissector method; cells on the upper and lower planes were not counted to avoid counting partial cells. The number of calretinin+ cells counted in every 6th section was multiplied by 6 to get the total number of calretinin+ cells in the DG.
Aβ Burden, Iba1 (microgliosis) Burden, and Synaptophysin Immunostaining By Quantitative Image Analyses
Images were acquired as digitized tagged-image format files to retain maximum resolution using an Olympus BX60 microscope with an attached digital camera system (DP-70, Olympus, Tokyo, Japan), and digital images were routed into a Windows PC for quantitative analyses using SimplePCI software (Compix, Inc. Imaging Systems, Cranberry Township, PA). Images of five sections (each 5 μm thick and 150 μm apart) were captured from serially sectioned EC and H and a threshold optical density was obtained that discriminated staining from background. Each anatomic region of interest was manually edited to eliminate artifacts. For Aβ and Iba1 (microgliosis) burden analyses, data are reported as the percentage of labeled area captured (positive pixels) divided by the full area captured (total pixels). Bias was eliminated by analyzing each entire region of interest (H and EC) represented by the sampling of 5 sections per region. Stereological equipment was not used. To evaluate synaptophysin immunostaining, after the mode of all images was converted to gray scale, the average optical density of positive signals from each image was quantified in the CA1 and CA3 regions of hippocampus as a relative number from zero (white) to 255 (black) and expressed as mean intensity of synpatophysin immunoreactivity. Each analysis was done by a single examiner blinded to sample identities.
Aβ ELISA
Hippocampal and cortical levels of soluble Aβ1–40 and Aβ1–42 were measured by ELISA. Briefly, 30 mg brain tissues were homogenized in 400 μl RIPA buffer (100 mM Tris [pH8.0], 150 mM NaCl, 0.5% DOC, 1% NP-40, 0.2% SDS, and 1 tablet proteinase inhibitor per 100 ml (S8820, Sigma, St. Louis, MO), and sonicated for 20 s on ice. Samples were then centrifuged for 30 min at 27,000 g at 4°C, and supernatants were transferred into new screw cap tubes. The supernatants obtained from this protocol were then stored at -80°C for determination of soluble Aβ levels using ELISA kits (KHB3482 for 40, KHB3442 for 42, Invitrogen). Standards and samples were mixed with detection antibody and loaded on the antibody pre-coated plate as the designated wells. HRP-conjugated antibody was added after washing, substrates were added for colorimetric reaction, and then stopped with sulfuric acid. Optical density was obtained and concentrations were calculated according to the standard curve. Plasma Aβ1-40 and 42 levels were determined with the same protocol and using the same ELISA kits.
Cytokine Analyses
For blood samples taken at euthanasia from Cohort 1 of the behavioral study, plasma levels of 23 cytokines were measured using Bio-Rad Bio-Plex kits (Bio-Rad, catalogue # 171F11181). Hippocampal levels of these 23 cytokines were also determined with the same protocol. Samples and standards were prepared using company protocols with the initial concentration of standards ranging from 32 ng/ml to 1.95 pg/ml. Plasma samples were prepared for analysis by diluting 1 volume of the serum sample with 3 volumes of the Bio-Plex mouse sample diluent. Using the microplate readout, each cytokine level was calculated based on its own standard curve.
Statistical Analysis
Behavioral performance was statistically evaluated (Statistica) to determine group differences based on genotype and treatment with G-CSF compared to placebo. The RAWM data was analyzed using both one-way ANOVAs and two-way repeated measure ANOVAs. Prior to analysis, the RAWM behavioral data was divided into 2-day blocks to aid in data presentation and analysis. After ANOVA analysis, post hoc pair-by-pair differences between groups (planned comparisons) were resolved using the Fisher’s LSD test. Pre- vs. post-GCSF behavioral measures were statistically evaluated using paired Student’s t-test, with Bonferroni’s correction for multiple comparisons, as appropriate. Neurohistologic and neurochemical analyses were performed using ANOVA. All group data are presented as mean +/- SEM.group and all comparisons were considered significant at p < 0.05.
RESULTS
G-CSF Reverses Memory Impairment in Aged AD Transgenic Mice
The APP+PS1 (Tg) mice from the colony used in this study have been previously shown to be cognitively impaired in a variety of tasks by 5 months of age (Jensen et al., 2005, Costa et al., 2007). To confirm that the 13-14 month old and the 8 month old Tg mice to be treated with G-CSF were impaired in working memory, Tg and non-transgenic (NT) littermates were tested in the RAWM task. Since there were no differences in RAWM performance for either Tg or NT mice between the two cohorts of mice, behavioral data from each genotype was combined for all statistical analysis. With both cohorts of mice thus combined, performance during the last block of pre-treatment testing showed clear impairment of Tg mice during working memory trials T4 and/or T5 for both errors and latency measures (Figure 1A). Immediately thereafter, Tg mice (n=24) and NT mice (n=28) were each divided into two groups (n=12-14 per group) balanced in behavioral performance and blood Aβ levels. For each group, G-CSF (or vehicle) was administered for two weeks, with injections continued for RAWM re-testing during the third week. The large number of mice being behaviorally evaluated required two separate cohorts of mice (the 13-14 month old and 8 month old mice), with each cohort a complete study separately since all 4 groups were included. Though data presentation in this report involves combined cohorts 1 and 2, it is important to note that separate statistical analysis from each group yielded identical transgenic and G-CSF treatment effects, thus, the beneficial effects of GCSF that we currently report were duplicated in two independent cohorts of mice that differed in age by 5 months.
Figure 1. G-CSF Treatment Reverses Working Memory Impairment of Tg Mice.
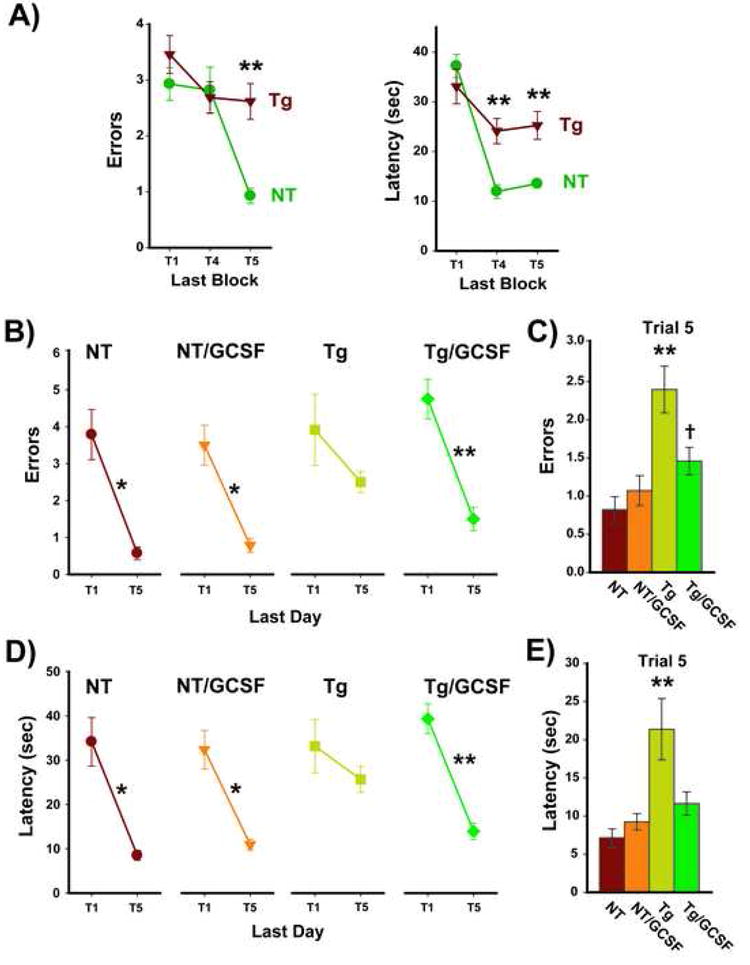
(A) Tg mice were impaired in working (short-term) memory performance prior to G-CSF treatment. Performance during the last block of pre-testing in the RAWM task revealed clear impairment of Tg mice during working memory trials T4 and/or T5, and for both Errors and Latency. **p<0.0001 between groups for that trial. Tg and NT mice were then divided into two groups balanced in behavioral performance and blood Aβ levels. (B,D) On the last of 4 test days, and at 2½ weeks into G-CSF treatment, Tg control mice could not improve their error (B) and escape latency (D) performance from naïve T1 to the T5 memory retention trial. By contrast, Tg/GCSF mice showed a significant reduction in errors from T1 to T5. NT mice also showed highly significant T1 vs. T5 decreases in both errors and latency, with G-CSF unable to improve upon the already excellent performance level of NT mice. *p<0.001, **p<0.00005 for T1 vs. T5. (C,E) For the final 2-day block of testing concurrent with treatment, Tg control mice were impaired versus all other groups in both errors (C) and latency (E). By contrast, Tg/GCSF mice performed similar to NT mice and substantially better the Tg controls. **p<0.005 or higher level of significance versus other 3 groups; †p<0.05 vs. NT group).
At RAWM re-testing, G-CSF treatment had no effect on the already excellent performance of NT mice across multiple RAWM measures, while the same G-CSF treatment resulted in marked and consistent improvement in the RAWM performance of Tg mice (Figures 1 and 2). During the final day of RAWM testing, control Tg mice could not reduce their number of errors or escape latency between Trial 1 (T1; the naïve trial) and working memory T5 (Figure 1B,C). By contrast, Tg mice treated with G-CSF (Tg/GCSF) showed a highly significant reduction in both errors and latency between T1 and T5. Performance of Tg/GCSF mice was in fact comparable to that of both NT groups. When T5 memory retention was evaluated for the last block of testing (Figure 1C,E), Tg control mice once again exhibited significant memory impairment compared to all other groups, while Tg/GCSF mice performed much better and comparable to both NT groups.
Figure 2. G-CSF Treatment Significantly Improves Upon Pre-treatment RAWM Performance of Tg Mice.
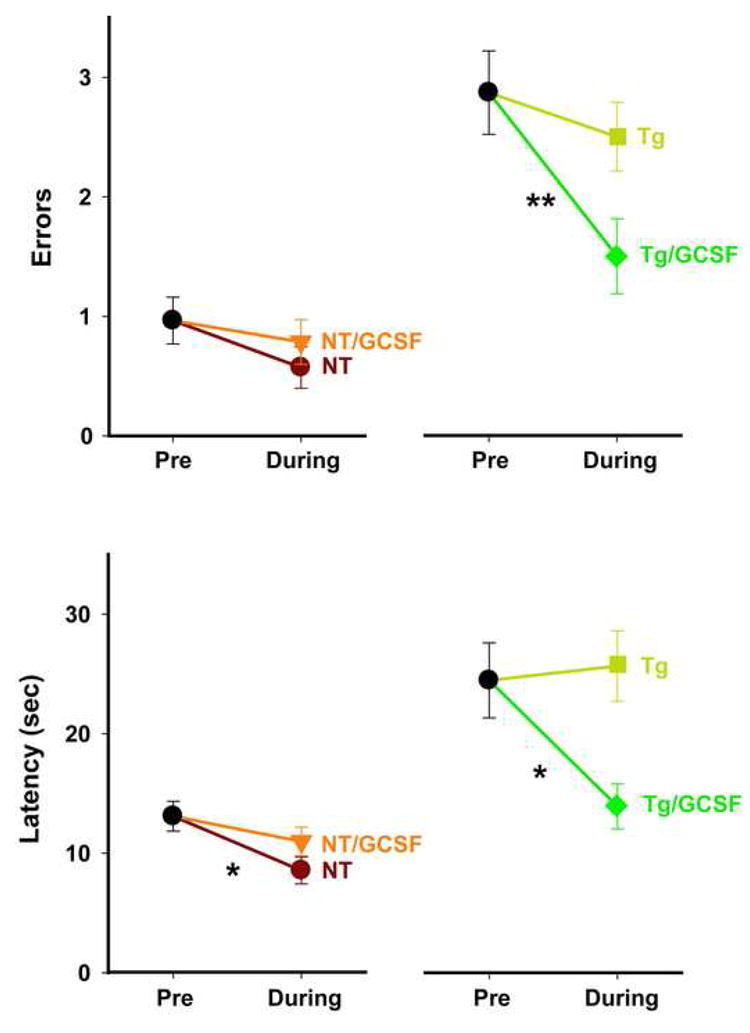
Both Errors (upper) and Latency (lower) measures are presented. Comparing the last day of pre-testing to the last day of testing with concurrent G-CSF administration, mice in both NT groups showed continuing excellent performance irrespective of GCSF treatment. Tg control mice exhibited consistently poor performance for both pre- and during-treatment testing. By sharp contrast, Tg mice treated with GCSF showed significant improvements in both errors and latency during GCSF treatment. Pre-treatment mean ± SEM values are for all animals in that group. *p<0.05, **p<0.01 for pre-treatment versus during-treatment performance (paired t-test).
As another index of memory performance, pre-treatment versus during-treatment T5 working memory was evaluated for each group. Irrespective of whether errors (Figure 2, upper) or escape latency (Figure 2, lower) was assessed, GCSF treatment did not further improve upon the already excellent memory performance of NT mice seen during pre-treatment testing. Tg controls also could not improve upon the impaired pre-treatment working memory performance of Tg mice when tested during ensuing placebo treatment (Figure 2). By sharp contrast, Tg/GCSF mice showed significantly improved memory performance during GCSF treatment compared to the high pre-treatment level of errors and escape latencies in Tg mice (Figure 2).
G-CSF Reduces Brain Aβ Deposition and Aβ Levels in Tg Mice
After completion of the behavioral testing, animals were euthanized, a blood sample taken, then half of each brain was processed for quantitative immunohistochemical determination of total amyloid deposition in situ and the other half processed for assessment of soluble Aβ levels. Compared to controls, Tg mice of cohort 1 (14.5M old) treated with G-CSF exhibited highly significant reductions of 39% and 42% in Aβ deposits within hippocampus and entorhinal cortex, respectively (Figure 3A). Photomicrographic examples of Aβ immunostaining from both of these brain areas are presented in Figure 3B. A reduction in size and extent of brain Aβ deposits is qualitatively evident in G-CSF treated Tg mice compared to Tg controls. The younger Cohort 2 (8.5M age) also exhibited a highly significant 36% reduction of Aβ deposits in both brain areas (Figure 3A), though the Aβ burden in saline-control Tg mice was less than in the older mice of Cohort 1.
Figure 3. G-CSF Treatment Reduces Aβ Deposition in Both Hippocampus and Entorhinal Cortex, While Also Reducing Soluble Aβ Levels in Hippocampus.
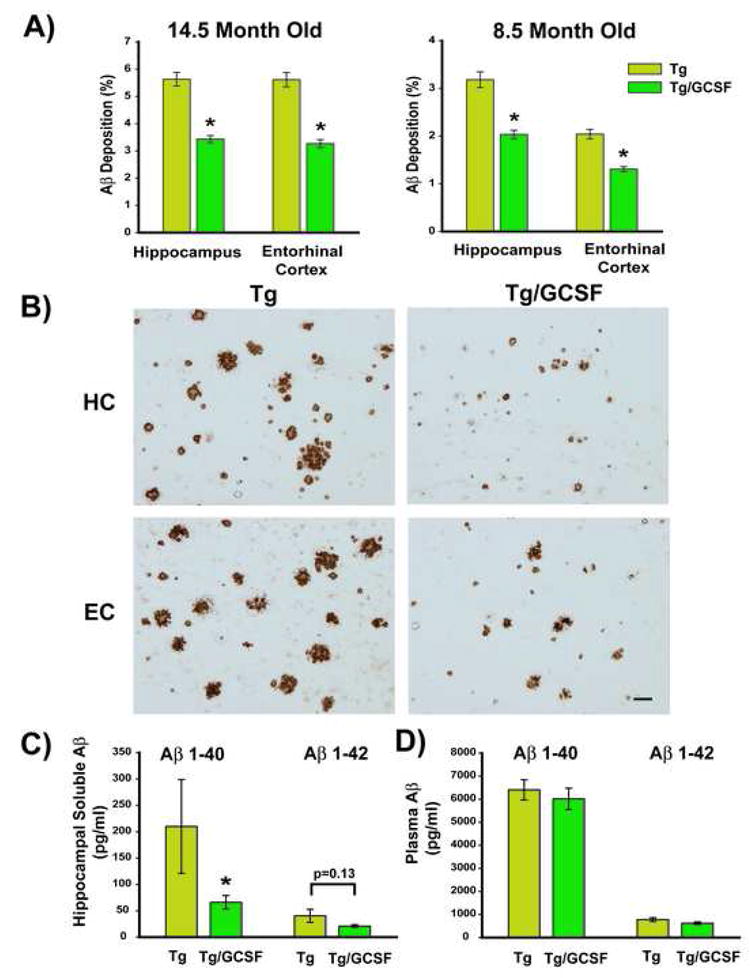
(A) Quantification of Aβ immunostaining in hippocampus (HC) and entorhinal cortex (EC) in 14.5 and 8.5 month old cohorts revealed very significant reductions in both brain regions for Tg mice treated with G/CSF compared to Tg controls. *p<0.0005 versus Tg controls. (B) Examples of Aβ immunohistochemical staining in HC and EC for G-CSF and saline treated Tg mice from 14.5 month old cohort. The reduction in both size and extent of Aβ deposition is evident in both brain regions. Scale bar = 20 μm. (C) For both cohorts of Tg mice combined, hippocampal levels of soluble Aβ1-40 were significantly reduced in comparison to Tg controls, while the reduction in hippocampal Aβ1-42 levels was nearly significant. *p<0.05 (Kruskal-Wallis test). (D) Also for both cohorts of Tg mice combined, plasma levels of both Aβ1-40 and Aβ1-42 were unaffected by G-CSF treatment.
Analysis of soluble Aβ levels from hippocampus and plasma of Tg mice from both the 14.5M and 8.5M cohorts revealed no group differences between cohorts. As such, data from Tg and Tg/GCSF mice in both cohorts was combined for presentation. Tg mice treated with G-CSF exhibited a significant 69% reduction in hippocampal Aβ1-40 levels (Figure 3C). Similarly, soluble Aβ1-42 levels in hippocampus were reduced 49% (nearly significant) by GCSF treatment. Soluble Aβ levels in frontal cortex of Tg mice were unaffected by G-CSF treatment (data not shown). Plasma levels of both Aβ1-40 and Aβ1-42 were also not affected by G-CSF treatment (Figure 3D).
Plasma and brain cytokine levels were analyzed from the animals in Cohort 1 (aged 14.5M at euthanasia). In NT mice, no effect of G-CSF on any of the 23 plasma cytokines was observed (data not shown). Twelve out of the 23 cytokines measured were significantly elevated in Tg control mice (Table 1). However, Tg mice receiving G-CSF showed normal levels in all but one of the 12 plasma cytokines which were elevated in untreated animals (Table 1). By contrast, hippocampal levels of inflammatory cytokines were not impacted by G-CSF treatment (data not shown); this was true for both NT and Tg mice treated with G-CSF. Thus, GCSF treatment did not result in a global inflammatory response but rather, may have selectively attenuated the elevated plasma cytokine levels occurring in untreated APP+PS1 mice. Interestingly, hG-CSF treatment reduced levels of endogenous G-CSF in plasma of NT and Tg mice collectively, comparing pre-treatment vs. post-treatment levels (521±93 vs. 321±37 ng/ml p<0.05, paired t-test). This likely reflects the functionality of hG-CSF in mice and a consequent down-regulation of endogenous G-CSF production. A separate determination of hG-CSF in hippocampal tissue from Tg/GCSF mice revealed measurable levels, indicating that administered hG-CSF had indeed entered the brain.
Table 1.
Plasma cytokine levels (ng/ml) in APP+PS1 transgenic mice Following three weeks of G-CSF treatment
| Cytokine | NT | Tg | Tg/GCSF | GCSF Effect |
|---|---|---|---|---|
| IL-1α | 29±6 | 371±47 | 282±39 | |
| IL1β | 44±4 | 66±16* | 51±7 | ↓ |
| IL-2 | 43±8 | 53±9 | 50±14 | |
| IL-3 | 57±7 | 139±53* | 70±15 | ↓ |
| IL-4 | 26±2 | 76±37** | 25±2 | ↓↓ |
| IL-5 | 70±11 | 161±37* | 123±30 | ↓ |
| IL-6 | 54±6 | 103±36** | 47±12 | ↓↓ |
| IL-9 | 65±19 | 156±71 | 181±89 | |
| IL-10 | 119±17 | 191±38* | 115±23 | ↓ |
| IL-12(p70) | 2093±419 | 1945±405 | 969±89 | |
| IL-12(p40) | 243±44 | 409±118 | 340±120 | |
| IL-13 | 34±3 | 111±48** | 45±13 | ↓↓ |
| IL-17 | 819±252 | 1461±558 | 1085±496 | |
| TNF-α | 95±10 | 183±34** | 114±22 | ↓↓ |
| Eotaxin | 121±6 | 161±16* | 143±8 | ↓ |
| GCSF | 432±80 | 516±116 | 289±30 | |
| GMCSF | 89±5 | 120±15 | 121±23 | |
| IFN-γ | 195±42 | 323±100 | 301±75 | |
| KC | 402±65 | 713±120* | 436±110 | ↓ |
| MCP-1 | 62±9 | 81±20 | 72±19 | |
| MIP-1α | 512±36 | 669±31* | 632±19* | Ø |
| MIP-1β | 59±3 | 128±47** | 65±4 | ↓↓ |
| Rantes | 150±23 | 226±39 | 173±24 |
p<0.05 or higher level of significance vs. NT group
p<0.05 or higher level of significance vs. NT and Tg/GCSF groups
G-CSF Increases Microglial Burden in Tg Mice
It was hypothesized that the most likely mechanism responsible for the marked reduction of brain Aβ deposits was related to the extent and distribution of microglial activity. Therefore, measurement of the total microglial burden was performed by quantitative immunohistochemical studies of Iba1 expression in hippocampus and entorhinal cortex from animals in Cohort 2 (8-9M old at euthanasia). Iba1 is a macrophage/microglia-specific calcium-binding protein expressed in both ramified and resting microglia as well as in activated microglia and macrophages (Imai and Kohsaka, 2002). G-CSF treatment to NT mice had no effect on Iba-1 burdens in hippocampus and entorhinal cortex (Figure 4A). Compared to both groups of NT mice, Tg control mice exhibited a substantial elevation in microglial burdens. G-CSF administration to Tg mice resulted in a further increase in microglial burdens above Tg control levels, as indicated by quantitative measures of Iba1 immunoreactivity in both the hippocampus (↑17%) and entorhinal cortex (↑20%) (Figure 4A). Qualitative analysis of brain sections revealed the expected microgliosis in Tg compared to NT mice (Figure 4B). The Iba1 immunoreactive cells were distributed around amyloid plaques in both hippocampus and entorhinal cortex of Tg mice (hippocampus is shown in Figure 4B), with G-CSF treatment resulting in an observable increase in microglial burdens.
Figure 4. G-CSF Increases Total Microglial Activity Selectively in Tg APP+PS1 Mice.
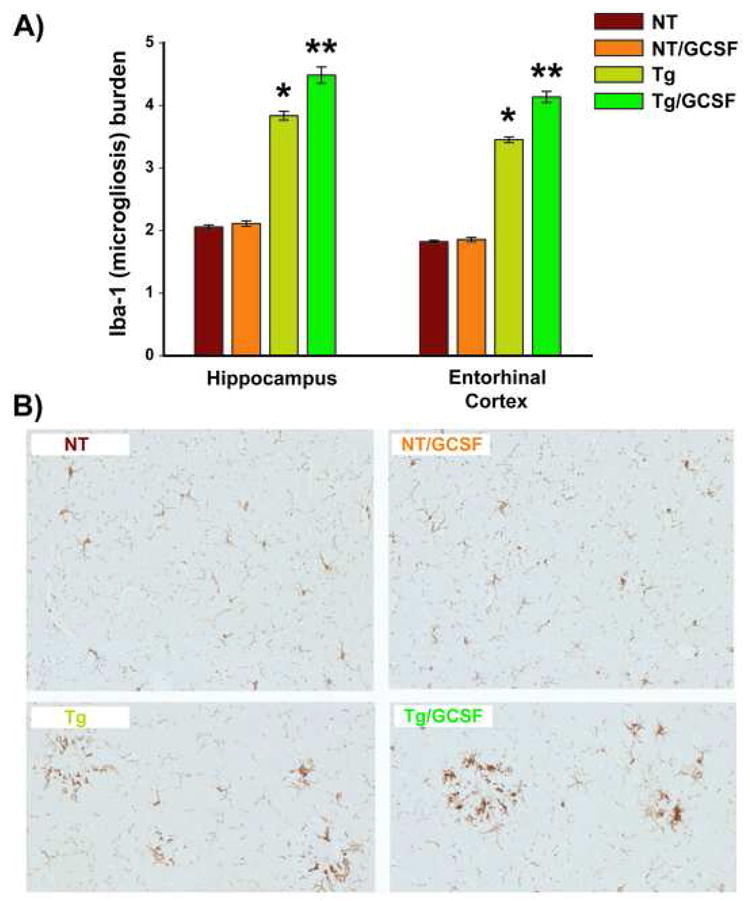
(A) Increased microgliosis was evident by quantitative Iba1 immunostaining in both hippocampus and entorhinal cortex of Tg/GCSF mice, but not NT/GCSF mice; * p < 0.00001 versus both NT groups, **p<0.00001 versus Tg group. (B) Saline-treated Tg APP+PS1 mice exhibited clusters of microglia surrounding amyloid, while G-CSF treated Tg mice revealed a significant increase in Iba1 immunoreactivity around and within amyloid deposits in hippocampus (B) and entorhinal cortex (not shown). Scale bar = 50 μm.
Effects of G-CSF in Chimeric Tg APP/PS1 Mice
To determine whether infiltration of bone-marrow derived cells into brain contributed to the microgliosis, a chimeric Tg APP+PS1 mouse was generated. Following whole body irradiation and intravenous infusion of fresh suspensions of mononuclear cells from GFP+ bone marrow at 2 months of age, 11 of 16 transgenic APP+PS1 mice were successfully rescued by the bone marrow transplantation. Examination of tail vein blood at age 4 and 6 months revealed that all mononuclear cells expressed GFP+, indicating successful engraftment of the marrow by the GFP+ stem/progenitor cells. These mice were then allowed to mature to the age when brain amyloid levels/deposits interfere with cognitive function (6-8 months). At this time, blood levels of soluble Aβ1-40 were measured and it was discovered that 3 of the mice did not express Aβ. The eight surviving chimeric AD mice were divided into two groups (n=4 per group), one of which received G-CSF injections sub-cutaneously for 2 weeks and the other group received vehicle control (saline). Levels of blood Aβ were closely matched in the two groups prior to G-CSF or vehicle treatment.
Evaluation of the histology in non-transgenic chimeric mice treated with G-CSF revealed GFP+ cells in meningeal and cortical blood vessels, but these circulating bone marrow-derived cells rarely infiltrated brain parenchyma (Figure 5C). In the chimeric Tg mice, GFP+ cells decorated amyloid deposits in entorhinal cortex in both G-CSF treated and saline control mice, and visual inspection showed an increased abundance of GFP+ cells surrounding amyloid deposits in the G-CSF group (Figure 5A). Similarly, GFP+ cells surrounded and infiltrated amyloid deposits in hippocampus in both the G-CSF group and saline treated Tg mice (Figure 5B). GFP+ cells surrounding and infiltrating β-amyloid plaques were observed to be transformed into amyloid-ingesting macrophages (Figure 5C). The actual numbers of GFP+ cells that penetrated brain parenchyma were not counted in these immunostained sections because of difficulty distinguishing individual GFP+ cells around A-β plaques and a limited number of mice per condition that survived bone marrow grafting for this experiment. Quantitative estimates of the number of GFP+ cells that infiltrate the brain may be done in the future using fluorescence activated cell sorting (FACS) as described by others (Cardona et al., 2006). Despite the absence of quantitation, these immunofluorescent images from chimeric mice, taken in the context of the significantly increased microgliosis evidenced by Iba1 immunoreactivity, suggests that G-CSF increases microglial activity in part by recruitment of bone-marrow derived microglia.
Figure 5. Bone Marrow-Derived (GFP+) Cells Contribute To Brain Microglial Activity and Are Increased in G-CSF Treated Tg Mice.
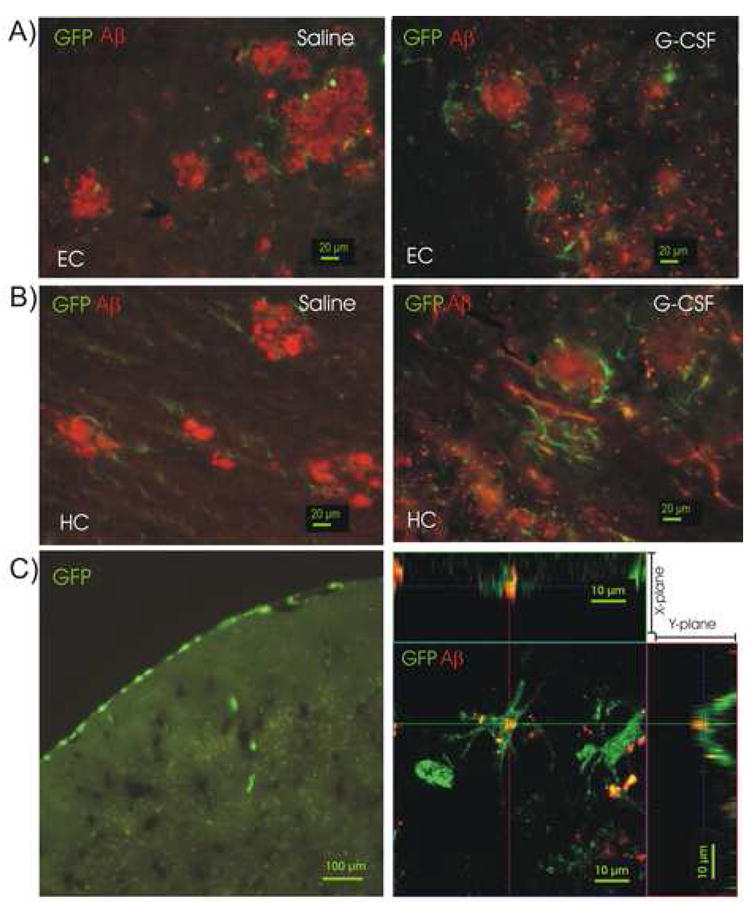
(A) GFP+ cells are seen surrounding amyloid plaques (red) in entorhinal cortex (EC) of a saline-treated Tg mouse (left panel) and in a G-CSF-treated Tg mouse (right panel). Scale bar = 20 μm. (B) GFP+ cells surrounding amyloid in hippocampus (HC) of saline-treated Tg mouse (left panel) and in a G-CSF-treated Tg mouse (right panel). In both HC and EC, visual inspection suggested a greater number of GFP+ cells surrounding the Aβ deposits in G-CSF-treated mice. (C) Low power fluorescence photomicrograph of bone marrow-derived (GFP+) cells in a saline-control non-tg mouse (Left panel). GFP+ cells are seen migrating in meningeal vessels (scale bar = 100 μm). An activated GFP+ macrophage, derived from bone marrow of a chimeric Tg mouse is shown engulfing amyloid (red) in a confocal photomicrograph (Zeiss LSM510) with views along X- and Y-planes, (right panel). Scale bar = 10 μm.
G-CSF Increases Synaptophysin Immunostaining in Hippocampus of Both Normal and Tg Mice
The loss of synapses, particularly the associated presynaptic vesicle protein synaptophysin in the hippocampus and association cortices, is considered to be a robust correlate of Alzheimer’s disease-associated cognitive decline. As a molecular marker for the presynaptic vesicle membrane, synaptophysin has been widely used in both animal models and human patients. Therefore, the relationship between amyloid reduction and synaptophysin immunostaining was explored. Brain sections from the CA1 and CA3 region of the hippocampus were quantitatively analyzed for synaptophysin immunostaining using optical densitometry. This method has been previously reported to reflect the number of synaptophysin immunoreactive nerve terminal boutons in the hippocampal formation (Li et al., 2002). The optical density of synaptophysin immunohistochemical labeling was reported to be linearly correlated with number of synaptophysin-immunoreactive terminals (R2=0.99) (Li et al., 2002). As shown in Figure 6, G-CSF treatment enhanced the optical density of synaptophysin immunostaining in hippocampus for both NT and Tg mice, and in both 14.5 month (A) and 8.5 month old (B) cohorts. Interestingly, increased synaptophysin immunostaining was evident between NT and Tg controls, which we have previously reported and determined to be due to aberrant synaptic terminal staining in Tg mice, particularly on the periphery of Aβ deposits (Cracchiolo et al., 2007). Photomicrographic examples depicting G-CSF’s enhancement of hippocampal synaptic area in the CA1 regions of hippocampus are presented in Figure 6C.
Figure 6. G- CSF Treatment Enhances Synaptophysin Immunostaining in Hippocampus for Both NT and Tg Mice.
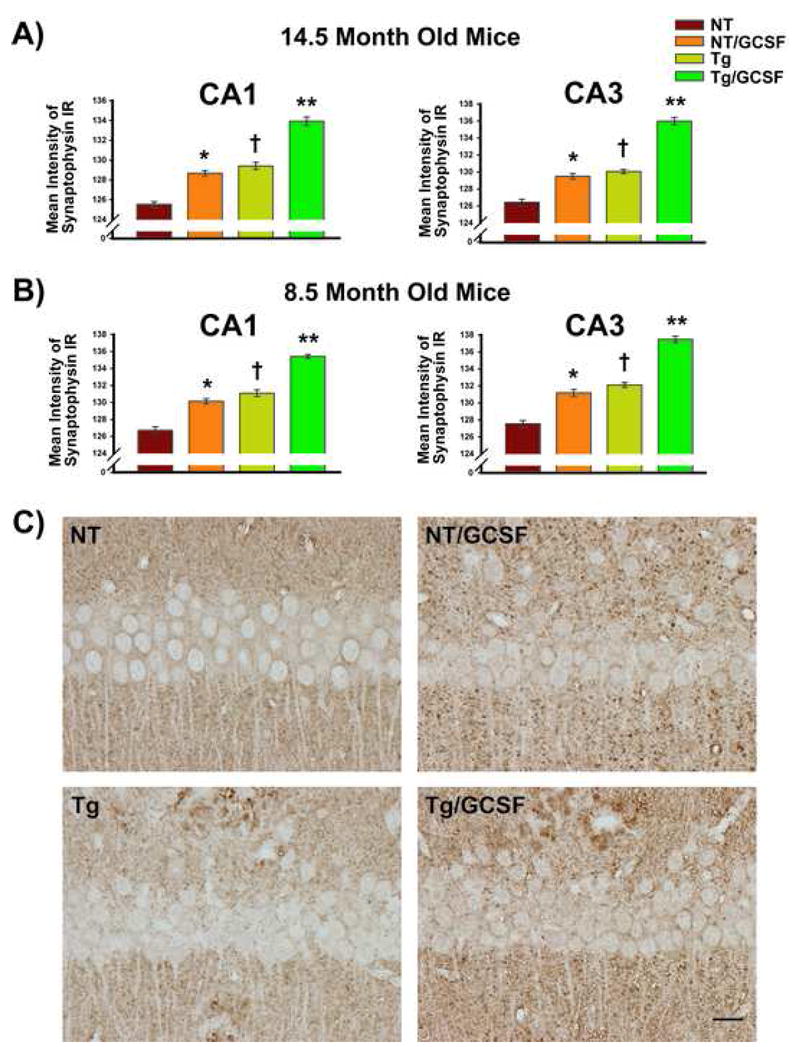
(A and B) Increased staining was evident in both CA1 and CA3 regions of the hippocampus of mice given G-CSF treatment in both 14.5 month and 8.5 month old cohorts. Interestingly, increased immunostaining was evident between NT and Tg controls due to aberrant synaptic terminal staining of Tg mice, particularly on the periphery of Aβ deposits. (C) Photomicrographic examples depicting G-CSF’s enhancement of synaptophysin immunostaining in the CA1 regions of hippocampus are presented for both NT and Tg mice. Scale bar = 50 μm.
G-CSF Administration Increases Hippocampal Neurogenesis in Tg Mice
The ligand and receptor for G-CSF is widely expressed in neurons and by adult neural stem/progenitor cells (Schneider et al., 2005, Zhao et al., 2007). It is reasonable to expect that G-CSF will have direct actions on those cells, especially since it is known that G-CSF is actively transported into the CNS (Zhao et al., 2007) and in view of our finding measurable levels of hG-CSF in brains from mice of these studies. Adult hippocampal neural stem/progenitor cells isolated from normal adult mice and grown in cell culture express the G-CSF receptor. (Figure 7A). Addition of G-CSF to neural stem cell cultures resulted in a dose-dependent increase in DNA synthesis (Figure 7B). To determine if G-CSF had an effect on neurogenesis in Tg mice in vivo, hippocampal sections from Cohort 2 (8.5 months old at euthanasia) were examined for the presence of calretinin-expressing cells, a marker of immature neurons. Unbiased estimates of the number of calretinin-expressing cells in the dentate gyrus showed no significant difference between control NT and vehicle-treated Tg mice. However, there was a significant increase (by 36.5%) in calretin-expressing cells in G-CSF treated Tg mice compared to the vehicle-treated Tg mice (Figure 8).
Figure 7.
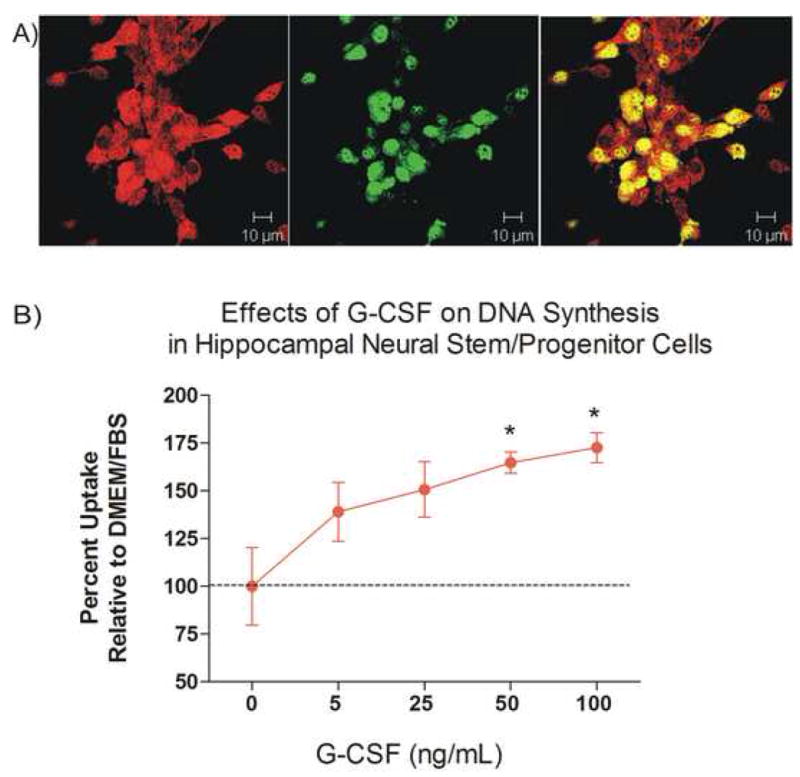
(A) Neural stem/progenitor cells (NSCs) co-express G-CSF receptor and nestin. Polyclonal rabbit anti-G-CSF-R 1:500 and goat anti-nestin antibody 1/200 (both fromSanta Cruz Biotechnology; Santa Cruz, CA) were incubated for 24 hrs at 4 C, washed with PBS, then incubated with secondary antibodies for 1 hr (anti-rabbit-rhodamine and anti-goat FITC). Image taken with a Zeiss LSM510 confocal microscope. Red= G-CSF receptor; green= nestin. Scale= 10 μm.
(B) Addition of G-CSF to hippocampal NSC in basal media containing DMEM+10% FBS for 48 hrs, (in absence of EGF and bFGF), resulted in a dose-dependent increase in 3H-thymidine uptake expressed as percent change from control conditions (DMEM+FCS). * p <0.05 compared to control uptake.
Figure 8. G-CSF Treatment Increased Neurogenesis in Dentate Gyrus.
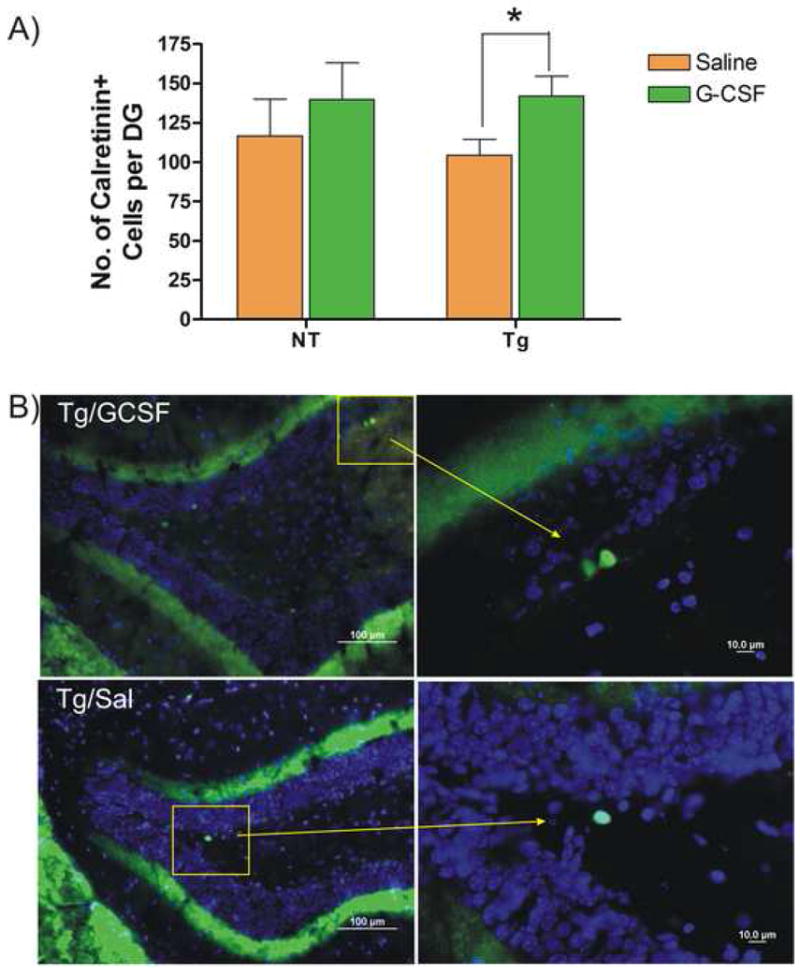
(A) Mean number of calretinin+ cells was estimated in NT and Tg mice from cohort 2 mice (8.5 months of age) treated with G-CSF or saline. G-CSF significantly increased numbers of calretinin+ cells in Tg mice by 36.5%. (*p < 0.05; two-tailed t-test). Mean number of calretinin+ cells in the Tg mice was slightly lower than in NT mice, but this result did not reach statistical significance. (B) Immunofluorescent images of calretinin+ cells in dentate gyrus of hippocampus. Upper panels illustrate calretinin+ cells in a G-CSF treated Tg mouse and lower panels show cells from a saline-treated Tg mice. (Green = Calretinin-immunoreactivity; Blue = DAPI). Panels on the right are magnifications of the indicated regions, showing immature neurons expressing calretinin in the subgranular zone of the dentate gyrus (scale bar = 100 μm in left panels and 10 μm in right panels).
DISCUSSION
This study provides clear evidence that G-CSF administration can reverse cognitive impairment in a commonly used AD mouse model (APP+PS1 transgenic mice; Tg). Moreover, GCSF-induced cognitive benefits appear to involve both Aβ-dependent and Aβ independent mechanisms, including activation of brain microglia, recruitment of bone marrow-derived microglia, peripheral anti-inflammatory actions, enhancement of synaptophysin immunoreactivity and increased neurogenesis. With only a short 2½ week course of treatment, Tg mice markedly improved performance in a challenging test of hippocampal-dependent working memory (the radial arm water maze; RAWM). This nootropic effect was associated with a marked and rapid reversal of brain Aβ pathology, as evidenced by both decreased Aβ deposition in hippocampus and entorrhinal cortex, as well as-significantly decreased soluble Aβ within hippocampus. The robust reduction in Aβ deposits was associated with increased microgliosis as measured by quantitative Iba1 immunostaining. This microglial activation occurred without induction of a CNS or global pro-inflammatory response – indeed, GCSF provided strong anti-inflammatory actions in plasma of Tg mice.
The accumulation of brain Aβ with time is clearly associated with cognitive decline. The APP+PS1 mice used in this study have been shown previously to become cognitively impaired by age 5 to 6 months of age in a variety of tasks spanning multiple cognitive domains, including the RAWM task of working (short-term) memory utilized in the present study (Arendash et al., 2001a, Jensen et al., 2005). Accompanying this cognitive impairment in APP+PS1 mice are increasing brain levels of both soluble and insoluble Aβ1-40 and Aβ1-42 beginning in young adulthood. Early β-amyloid plaque formation is evident in both cerebral cortex and hippocampus by 5-6 months of age and is prominent by 10-11 months of age in these brain areas critical to cognitive function (Cracchiolo et al., 2006, Ethell et al., 2006). Thus, G-CSF administration to 8 and 14 month old APP+PS1 mice in the present study was administered at an age wherein Aβ deposition is marked and cognitive impairment is widespread across tasks.
The RAWM task is particularly sensitive to brain Aβ deposition/levels, with strong correlations consistently being present between brain Aβ deposition/levels and RAWM performance in APP+PS1 mice at the ages evaluated in the present study (Arendash et al., 2001a, Ethell et al., 2006). It was therefore no surprise that the substantial reduction in brain Aβ deposition and Aβ levels was associated with cognitive improvement in these Tg mice. This improvement was seen across multiple measures of working memory in the RAWM task. It should be underscored that Aβ aggregation involves a continuum from soluble (dimeric, oligomeric) through aggregated (diffuse, compact) forms. In this context, we have found correlations between cognitive impairment and both soluble and deposited forms of Aβ in multiple prior studies (Arendash et al., 2001b, Arendash et al., 2004, Costa et al., 2007, Cao et al., 2009) leading us to conclude that the entire process of Aβ aggregation impacts cognitive function. Nonetheless, recent studies have suggested that soluble forms of Aβ are more toxic than deposited Aβ forms (Walsh and Selkoe, 2004). In addition, brain injections of medium containing soluble Aβ moieties inhibit long-term potentiation in rats (Walsh et al., 2002).
The ability of G-CSF to decrease both brain Aβ deposition and soluble Aβ is best appreciated in the context of the dynamic equilibrium between deposited Aβ and soluble Aβ in the brain (Figure 9). As such, the effect of G-CSF treatment to reduce soluble Aβ levels in Tg mouse brains would appear to be secondary to the primary action of G-CSF to reduce Aβ deposition, resulting in a flux of Aβ from the soluble to the deposited brain pool. This could result in less uni-directional flux of soluble Aβ from brain to blood, although not reflected as an actual decrease in blood Aβ levels chronically.
Figure 9. Diagrams depicting proposed effects of G-CSF treatment on the dynamic equilibrium between soluble and deposited Aβ in the brain.
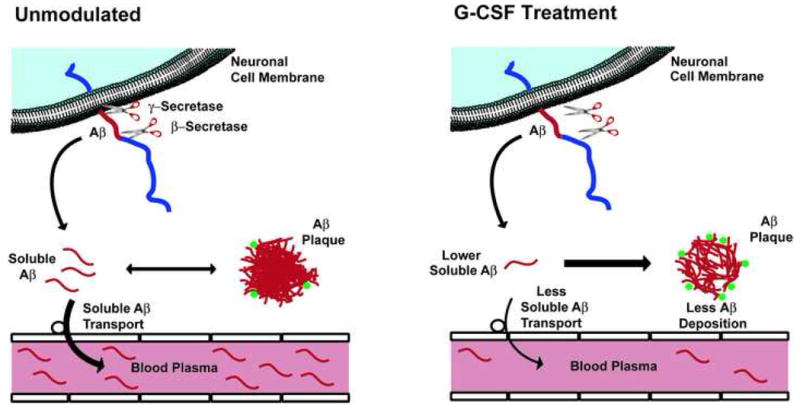
(Unmodulated) Newly produced Aβ, resulting from the actions of both β- and γ secretase on APP, enters an equilibrium between soluble and deposited (insoluble) Aβ in the brain. Some bone marrow-derived microglia surround Aβ plaques (green), continually engulfing deposited Aβ. Transport of soluble Aβ from brain to blood plasma is concentration-dependent on levels of soluble Aβ in the brain. (G-CSF Treatment) A relatively short course of G-CSF treatment increases the penetration of bone marrow-derived microglia (green) into the brain and increases activation of reside brain microglia, thus enhancing Aβ engulfment (removal) from plaques. This causes a shift “to the right” in the equilibrium between soluble Aβ ⇔ deposited Aβ, decreasing soluble levels of brain Aβ and resulting in less soluble Aβ transport into the blood.
Other therapeutics that decrease brain Aβ deposition/levels have also been shown to improve cognitive function. Active and passive immunization with different forms of Aβ using various protocols have been shown to decrease brain Aβ levels/deposition and to improve cognitive performance in various AD transgenic mouse lines (Bard et al., 2000); review by (Zhang and Ma, 2003). Leuprolide acetate (a gonadotropin-releasing hormone analogue) administration mediated a reduction of Aβ and this effect correlated with improved cognition in a Tg model of AD (Casadesus et al., 2006). Moreover, long-term caffeine administration reduces brain Aβ levels and protects against otherwise inevitable cognitive impairment in the Alzheimer’s Tg mice (Arendash et al., 2006).
The GCSF-induced microglial activation that we presently report appears to be a result of direct stimulation of resident microglia which are known to express G-CSF receptor (Hasselblatt et al., 2007) and to some extent, mobilization of microglia from bone marrow-derived cells. Microglia surround and infiltrate amyloid plaques in Tg mouse models of AD (Meda et al., 1996, Ishizuka et al., 1997, Janelsins et al., 2005, Bolmont et al., 2008). Indeed, perhaps as many as 10% of microglial cells associated with amyloid plaques originate from the bone marrow, with both Aβ1-40 and Aβ1-42 isoforms triggering this chemo attraction (Simard et al., 2006). These newly recruited cells also exhibit a specific immune reaction to both exogenous and endogenous Aβ in the brain. (Malm et al., 2005, Simard et al., 2006). In the present study, G-CSF treatment appeared to enhance microglial activity, even in the Tg APP+PS1 mice that did not receive irradiation or bone marrow transplantation. The ability of G-CSF to enhance mobilization/recruitment of “new” bone marrow-derived microglia may be particularly important in view of a recent study showing that, as APP+PS1 mice age, their microglia become dysfunctional by exhibiting significantly reduced expression of Aβ-binding receptors and Aβ-degrading enzymes (Hickman et al., 2008). Since these microglia from aged APP+PS1 mice nonetheless maintain their ability to produce pro-inflammatory cytokines, it is important to underscore that the augmented microglial activity triggered by G-CSF administration was not associated with a global inflammatory response.
G-CSF, which is known to increase neutrophil differentiation in the periphery, modulates the immune response by inhibiting the production or activity of the main pro-inflammatory mediators (Hartung, 1998, Boneberg et al., 2000). The functional activity of G-CSF receptors in peripheral blood cells has been shown by challenging isolated leukocyte populations to release cytokines with endotoxin in the presence of G-CSF (Boneberg et al., 2000). For example, G-CSF treatment attenuated the release of the proinflammatory cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-12, IL-1β, and interferon (IFN)- γ in ex vivo lipopolysaccharide (LPS)-stimulated whole blood. Hence the potential pro-inflammatory effect of increasing the circulating neutrophil population by G-CSF is modulated by inhibition of pro-inflammatory cytokine release.
Through a mechanism that may have both Aβ dependent and Aβ-independent components, G-CSF’s ability to increase synaptic area, as reflected by increased optical density of synaptophysin immunostaining, could be critical for the recovery of Tg mice from cognitive dysfunction. Even NT mice treated with G-CSF showed an increase in hippocampal synaptophysin immunostaining. This novel observation of increased synaptic area with G-CSF treatment is important for several reasons. Synapse pathology is considered by many experts to be a major correlate of cognitive decline not only in AD patients, but also in other dementia syndromes (DeKosky and Scheff, 1990, Terry et al., 1991, Dickson et al., 1995, Terry, 1996, Scheff and Price, 2003). The loss of synapses, and particularly the associated presynaptic vesicle protein synaptophysin in the hippocampus and association cortices is considered a consequence of Aβ deposition. However, loss of synaptophysin and impaired synaptic function has been reported to occur before the onset of Aβ deposition in Tg mice (Hsia et al., 1999, Mucke et al., 2000, Oddo et al., 2003).
On the other hand, Aβ plaques/deposition have been reported to disrupt neuronal circuitry and therefore may indirectly affect global synaptic function (Stern et al., 2004) or cognitive function (Chen et al., 2000). Support for the hypothesis that Aβ deposition results in synaptic dysfunction was provided by reports that reduction in amyloid load by immunization protected against progressive loss of synaptophysin in the hippocampal molecular layer and frontal neocortex of a transgenic mouse model of Alzheimer’s disease (Buttini et al., 2005). These results were substantiated by quantitative electron microscopic analysis of synaptic density and strongly support a direct causative role of Aβ in the synaptic degeneration seen in AD. In addition, amyloid plaques and neuritic lesions were reversed within a short period of time after administration of a single dose of Aβ antibody to Tg APP mice(Lombardo et al., 2003). Amyloid clearance and recovery of normal neuronal geometries were observed as early as 4 days and lasted at least 32 days after a single treatment. These results suggest that neuronal and synaptic morphology is self-correcting after amyloid is cleared (Lombardo et al., 2003). Our future studies will determine whether GCSF’s ability to increase synaptic immunoreactivity in Tg mice within several weeks is a reflection of increased synaptic number (e.g., synaptogenesis) or some other mechanism of synaptic action (e.g., enlargement of existing synapses). In any event, effects of G-CSF on hippocampal LTP are critical and are currently being studied in our laboratories.
It is important to underscore that G-CSF does more than activate resident microglia and possibly increase trafficking of microglia from marrow to brain to presumably decrease brain Aβ deposition through enhanced phagocytotic activity. G-CSF readily passes through the blood-brain barrier (Zhao et al., 2007) and has been shown to act directly on neural tissue via interaction with G-CSF receptors expressed by neurons and neural stem cells (Schneider et al., 2005). Both the G-CSF ligand and its receptor are widely expressed by brain neurons (as well as microglia) and their expression is induced by ischemia. In the present study, we verified that administered hG-CSF does indeed enter the brain. Through CNS actions, administered hG-CSF thus increased the generation of new neurons in hippocampus, as indicated by increased numbers of calretinin-expressing cells in the dentate gyrus.
Increased neurogenesis and associated synaptogenesis promoted by G-CSF may be the neural substrate for the improved cognition. However, the relationship between cognitive performance and neurogenesis in the Tg APP/PS1 mouse is uncertain. Tg APP/PS1 mice raised in enriched environments exhibited increased cognitive performance and an increased rate of hippocampal NSC proliferation when compared to Tg APP/PS1 mice raised in an “impoverished” environment (Catlow et al., 2009). However, the number of BrdU birth-dated cells and calretinin-expressing cells that survived two weeks after BrdU injection was not different between the groups of Tg mice (Catlow et al., 2009). A recent review of neurogenesis in various Tg mouse models of AD reveals conflicting results in the literature, with reports of increases, no effect and decreases in hippocampal neurogenesis depending on the mouse model, age, temporal parameters and assay methods (Kuhn et al., 2007). Given the uncertainty regarding the relationship between neurogenesis, cognitive performance and amyloid deposition in Tg AD models, increased neurogenesis may play a role, but cannot be cited as the critical mechanism at this time, for improved cognitive function produced by G-CSF administration.
Of direct relevance to the present study, G-CSF administration was recently reported to have pro-cognitive effects without reducing either soluble or deposited forms of Aβ in the brains of AD transgenic mice (Tsai et al., 2007). A 5-day regime of G-CSF administered to Tg2576 mice was reported to result in improved learning (acquisition) in the Morris Water Maze task (Tsai et al., 2007). In their study, Tsai et al. (2007) behaviorally evaluated mice after stopping G-CSF treatment and only a single reference learning measure was evaluated. In contrast, the present study utilized multiple measures/endpoints in a very challenging working memory task (the radial arm water maze) that is highly sensitive to both soluble and deposited brain Aβ. Moreover, G-CSF treatment was continued throughout behavioral testing and until mice were euthanitized. This methodology, in addition to higher doses of G-CSF administered over a longer period of time, is probably why we were able to demonstrate G-CSF induced decreases in brain Aβ levels/deposition, while Tsai et al (2007) reported no effects of G-CSF treatment on brain Aβ. Despite these differences in effects of G-CSF on total plaque burden, both reports found improvement in cognitive performance, underscoring the uncertain relationship between cognitive function and amyloid burden. An interesting finding in Tsai et al. (2007) report was an increase in the number of BrdU-labeled neurons located around Aβ plaques in cerebral cortex and hippocampus. The authors suggested that these new neurons were derived from hematopoietic stem cells that had infiltrated the brain and perhaps the behavioral improvement was related to increased neurogenesis. “Transdifferentiation” of bone-marrow derived cells has been reported, but is considered to be a rare phenomenon and may represent fusion of bone marrow-derived cells with injured neurons (Alvarez-Dorado et al., 2003).
Concluding Remarks
To summarize results of the present study, G-CSF administration impacts both the bone marrow and the CNS, resulting in reversal of cognitive deficits associated with brain Aβ reduction, increased synaptophysin immunostaining, and neurogenesis. Additionally, G-CSF reduces inflammation by suppressing production/activity of major pro-inflammatory cytokines in plasma, consistent with previous reports not involving AD transgenic mice (Hartung, 1998, Solaroglu et al., 2007). As underscored previously, the cognitive benefits of G-CSF may involve not only induction of Aβ-reducing phagocytotic activity, but also multiple mechanisms of “neuroprotection” and neuroplasticity (e.g., reduction of inflammation, stimulation of neurogenesis and synaptogenesis). Indeed, G-CSF has been shown to be protective against loss of dopaminergic neurons in an animal model of Parkinson’s Disease — the MPTP-treated mouse (Meuer et al., 2006). Of direct importance to AD, decreased blood levels of G-CSF is one of 18 plasma biomarkers found to identify MCI patients who progressed to AD within a few years (Ray et al., 2007). In characterizing the physiologic categories of these 18 biomarkers, the authors point to “systemic dysregulation” of hematopoiesis, apoptosis, neuroprotection, and inflammation as key indicators of pre-symptomatic AD. Not co-incidentally, G-CSF would appear to favorably impact all four of these physiologic categories. Additional research is underway in our laboratories to clarify the pleiotropic actions of G-CSF upon brain and peripheral tissues, specifically as they relate to AD.
The multiplicity of beneficial mechanisms provided by GCSF, in combination with its inherent safety through use in other medical conditions such as leukopenia, make this hematopoietic growth factor a most attractive candidate for clinical trials in AD.
Acknowledgments
This work was supported by Grants from Elan-ADDF (J.S.R.), the Helen Ellis Endowment (J.S.R.), The Florida Alzheimer’s Disease Research Center (G.W.A.), and The Byrd Alzheimer’s Disease Research Center (G.W.A.).
Abbreviations
- AD
Alzheimer’s disease
- Aβ
beta Amyloid
- CNS
central nervous system
- G-CSF
granulocyte colony stimulating factor
- HGF
hematopoietic growth factor
- IL-1
interleukin-1
- IFγ
interferon gamma
- RAWM
radial arm water maze
- rhDNA
recombinant human DNA
- s.c
sub-cutaneous
- TNF-α
tumor necrosis factor-alpha
- Tg
transgenic
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K, Lois C, Morrison SJ, Alvarez-Buylla A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–973. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- Arendash GW, Garcia MF, Costa DA, Cracchiolo JR, Wefes IM, Potter H. Environmental enrichment improves cognition in aged Alzheimer’s transgenic mice despite stable beta-amyloid deposition. Neuroreport. 2004;15:1751–1754. doi: 10.1097/01.wnr.0000137183.68847.4e. [DOI] [PubMed] [Google Scholar]
- Arendash GW, Gordon MN, Diamond DM, Austin LA, Hatcher JM, Jantzen P, DiCarlo G, Wilcock D, Morgan D. Behavioral assessment of Alzheimer’s transgenic mice following long-term Abeta vaccination: task specificity and correlations between Abeta deposition and spatial memory. DNA Cell Biol. 2001a;20:737–744. doi: 10.1089/10445490152717604. [DOI] [PubMed] [Google Scholar]
- Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain research. 2001b;891:42–53. doi: 10.1016/s0006-8993(00)03186-3. [DOI] [PubMed] [Google Scholar]
- Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience. 2006;142:941–952. doi: 10.1016/j.neuroscience.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the Microglial/Amyloid Interaction Indicate a Role in Plaque Maintenance. Neurobiology of Disease. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneberg EM, Hareng L, Gantner F, Wendel A, Hartung T. Human monocytes express functional receptors for granulocyte colony-stimulating factor that mediate suppression of monokines and interferon-gamma. Blood. 2000;95:270–276. [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Arendash GW, Dickson A, Mamcarz MB, Lin X, Ethell DW. Abeta-specific Th2 cells provide cognitive and pathological benefits to Alzheimer’s mice without infiltrating the CNS. Neurobiol Dis. 2009;34:63–70. doi: 10.1016/j.nbd.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Huang D, Sasse ME, Ransohoff RM. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nature protocols. 2006;1:1947–1951. doi: 10.1038/nprot.2006.327. [DOI] [PubMed] [Google Scholar]
- Casadesus G, Webber KM, Atwood CS, Pappolla MA, Perry G, Bowen RL, Smith MA. Luteinizing hormone modulates cognition and amyloid-beta deposition in Alzheimer APP transgenic mice. Biochim Biophys Acta. 2006;1762:447–452. doi: 10.1016/j.bbadis.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Catlow BJ, Rowe AR, Clearwater CR, Mamcarz M, Arendash GW, Sanchez-Ramos J. Effects of environmental enrichment and physical activity on neurogenesis in transgenic PS1/APP mice. Brain research. 2009;1256:173–179. doi: 10.1016/j.brainres.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Costa DA, Cracchiolo JR, Bachstetter AD, Hughes TF, Bales KR, Paul SM, Mervis RF, Arendash GW, Potter H. Enrichment improves cognition in AD mice by amyloid-related and unrelated mechanisms. Neurobiology of aging. 2007;28:831–844. doi: 10.1016/j.neurobiolaging.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Cracchiolo J, Mori T, Bachstetter A, Mervis R, Nazian S, Tan J, Potter H, Arendash G. Enhanced cognitive activity - over and above social or physical activity - is required to protect Alzhiemer’s mice against cognitive impairment, reduce A deposition, and increase synaptic area. 2006 doi: 10.1016/j.nlm.2007.07.007. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracchiolo JR, Mori T, Nazian SJ, Tan J, Potter H, Arendash GW. Enhanced cognitive activity--over and above social or physical activity--is required to protect Alzheimer’s mice against cognitive impairment, reduce Abeta deposition, and increase synaptic immunoreactivity. Neurobiol Learn Mem. 2007;88:277–294. doi: 10.1016/j.nlm.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiology of aging. 1995;16:285–298. doi: 10.1016/0197-4580(95)00013-5. discussion 298-304. [DOI] [PubMed] [Google Scholar]
- Ethell DW, Shippy D, Cao C, Cracchiolo JR, Runfeldt M, Blake B, Arendash GW. Abeta-specific T-cells reverse cognitive decline and synaptic loss in Alzheimer’s mice. Neurobiol Dis. 2006;23:351–361. doi: 10.1016/j.nbd.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Furuya T, Tanaka R, Urabe T, Hayakawa J, Migita M, Shimada T, Mizuno Y, Mochizuki H. Establishment of modified chimeric mice using GFP bone marrow as a model for neurological disorders. Neuroreport. 2003;14:629–631. doi: 10.1097/00001756-200303240-00021. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Jones NC, Prior MJ, Bath PM, Murphy SP. G-CSF suppresses edema formation and reduces interleukin-1beta expression after cerebral ischemia in mice. J Neuropathol Exp Neurol. 2005;64:763–769. doi: 10.1097/01.jnen.0000179196.10032.dd. [DOI] [PubMed] [Google Scholar]
- Hartung T. Anti-inflammatory effects of granulocyte colony-stimulating factor. Current opinion in hematology. 1998;5:221–225. doi: 10.1097/00062752-199805000-00013. [DOI] [PubMed] [Google Scholar]
- Hasselblatt M, Jeibmann A, Riesmeier B, Maintz D, Schabitz WR. Granulocyte-colony stimulating factor (G-CSF) and G-CSF receptor expression in human ischemic stroke. Acta neuropathologica. 2007;113:45–51. doi: 10.1007/s00401-006-0152-y. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kohsaka S. Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia. 2002;40:164–174. doi: 10.1002/glia.10149. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry and clinical neurosciences. 1997;51:135–138. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. Journal of neuroinflammation. 2005;2:23. doi: 10.1186/1742-2094-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW. Lifelong immunization with human beta-amyloid (1-42) protects Alzheimer’s transgenic mice against cognitive impairment throughout aging. Neuroscience. 2005;130:667–684. doi: 10.1016/j.neuroscience.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Komine-Kobayashi M, Zhang N, Liu M, Tanaka R, Hara H, Osaka A, Mochizuki H, Mizuno Y, Urabe T. Neuroprotective effect of recombinant human granulocyte colony-stimulating factor in transient focal ischemia of mice. Journal of Cerebral Blood Flow and Metabolism. 2006;26:402–413. doi: 10.1038/sj.jcbfm.9600195. [DOI] [PubMed] [Google Scholar]
- Kuethe F, Figulla HR, Voth M, Richartz BM, Opfermann T, Sayer HG, Krack A, Fritzenwanger M, Hoffken K, Gottschild D, Werner GS. [Mobilization of stem cells by granulocyte colony-stimulating factor for the regeneration of myocardial tissue after myocardial infarction] Dtsch Med Wochenschr. 2004;129:424–428. doi: 10.1055/s-2004-820061. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Cooper-Kuhn CM, Boekhoorn K, Lucassen PJ. Changes in neurogenesis in dementia and Alzheimer mouse models: are they functionally relevant? European archives of psychiatry and clinical neuroscience. 2007;257:281–289. doi: 10.1007/s00406-007-0732-4. [DOI] [PubMed] [Google Scholar]
- Li S, Reinprecht I, Fahnestock M, Racine RJ. Activity-dependent changes in synaptophysin immunoreactivity in hippocampus, piriform cortex, and entorhinal cortex of the rat. Neuroscience. 2002;115:1221–1229. doi: 10.1016/s0306-4522(02)00485-2. [DOI] [PubMed] [Google Scholar]
- Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT. Amyloid-{beta} Antibody Treatment Leads to Rapid Normalization of Plaque-Induced. Neuritic Alterations vol. 2003;23:10879–10883. doi: 10.1523/JNEUROSCI.23-34-10879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Meda L, Bernasconi S, Bonaiuto C, Sozzani S, Zhou D, Otvos L, Jr, Mantovani A, Rossi F, Cassatella MA. Beta-amyloid (25-35) peptide and IFN-gamma synergistically induce the production of the chemotactic cytokine MCP-1/JE in monocytes and microglial cells. J Immunol. 1996;157:1213–1218. [PubMed] [Google Scholar]
- Meuer K, Pitzer C, Teismann P, Krüger C, Göricke B, Laage R, Lingor P, Peters K, Schlachetzki JCM, Kobayashi K, Dietz GPH, Weber D, Ferger B, Schäbitz WR, Bach A, Chulz JB, Bähr M, Schneider A, Weishaupt JH. Granulocyte-colony stimulating factor is neuroprotective in a model of Parkinson’s disease. Journal of Neurochemistry. 2006;97:675–686. doi: 10.1111/j.1471-4159.2006.03727.x. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienaber CA, Petzsch M, Kleine HD, Eckard H, Freund M, Ince H. Effects of granulocyte-colony-stimulating factor on mobilization of bone-marrow-derived stem cells after myocardial infarction in humans. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S73–77. doi: 10.1038/ncpcardio0443. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego, Calif.: London: Academic Press; 2001. [Google Scholar]
- Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- Ripa RS, Jorgensen E, Wang Y, Thune JJ, Nilsson JC, Sondergaard L, Johnsen HE, Kober L, Grande P, Kastrup J. Stem cell mobilization induced by subcutaneous granulocyte-colony stimulating factor to improve cardiac regeneration after acute ST-elevation myocardial infarction: result of the double-blind, randomized, placebo-controlled stem cells in myocardial infarction (STEMMI) trial. Circulation. 2006;113:1983–1992. doi: 10.1161/CIRCULATIONAHA.105.610469. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ramos J, Song S, Cao C, Arendash G. The potential of hematopoietic growth factors for treatment of Alzheimer’s disease: a mini-review. BMC neuroscience. 2008;9(Suppl 2):S3. doi: 10.1186/1471-2202-9-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sava V, Velasquez A, Song S, Sanchez-Ramos J. Adult hippocampal neural stem/progenitor cells in vitro are vulnerable to the mycotoxin ochratoxin-A. Toxicological Sciences. 2007;98:187–197. doi: 10.1093/toxsci/kfm093. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Kollmar R, Schwaninger M, Juettler E, Bardutzky J, Scholzke MN, Sommer C, Schwab S. Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2003;34:745–751. doi: 10.1161/01.STR.0000057814.70180.17. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Schneider A. Developing granulocyte-colony stimulating factor for the treatment of stroke: current status of clinical trials. Stroke; a journal of cerebral circulation. 2006;37:1654. doi: 10.1161/01.STR.0000227299.62106.0e. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiology of aging. 2003;24:1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Schneider A, Kruger C, Steigleder T, Weber D, Pitzer C, Laage R, Aronowski J, Maurer MH, Gassler N, Mier W, Hasselblatt M, Kollmar R, Schwab S, Sommer C, Bach A, Kuhn HG, Schabitz WR. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. Journal of Clinical Investigation. 2005;115:2083–2098. doi: 10.1172/JCI23559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu WC, Lin SZ, Lee CC, Liu DD, Li H. Granulocyte colony-stimulating factor for acute ischemic stroke: a randomized controlled trial. Cmaj. 2006;174:927–933. doi: 10.1503/cmaj.051322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Six I, Gasan G, Mura E, Bordet R. Beneficial effect of pharmacological mobilization of bone marrow in experimental cerebral ischemia. Eur J Pharmacol. 2003;458:327–328. doi: 10.1016/s0014-2999(02)02785-1. [DOI] [PubMed] [Google Scholar]
- Solaroglu I, Jadhav V, Zhang JH. Neuroprotective effect of granulocyte-colony stimulating factor. Frontiers in Bioscience. 2007;12:712–724. doi: 10.2741/2095. [DOI] [PubMed] [Google Scholar]
- Solaroglu I, Tsubokawa T, Cahill J, Zhang JH. Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience. 2006;143:965–974. doi: 10.1016/j.neuroscience.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Bacskai BJ, Hickey GA, Attenello FJ, Lombardo JA, Hyman BT. Cortical synaptic integration in vivo is disrupted by amyloid-beta plaques. J Neurosci. 2004;24:4535–4540. doi: 10.1523/JNEUROSCI.0462-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Nagashima K, Arai M, Uno Y, Misao Y, Takemura G, Nishigaki K, Minatoguchi S, Watanabe S, Tei C, Fujiwara H. Effect of granulocyte colony-stimulating factor treatment at a low dose but for a long duration in patients with coronary heart disease. Circ J. 2006;70:430–437. doi: 10.1253/circj.70.430. [DOI] [PubMed] [Google Scholar]
- Takano H, Ohtsuka M, Akazawa H, Toko H, Harada M, Hasegawa H, Nagai T, Komuro I. Pleiotropic effects of cytokines on acute myocardial infarction: G-CSF as a novel therapy for acute myocardial infarction. Curr Pharm Des. 2003;9:1121–1127. doi: 10.2174/1381612033455008. [DOI] [PubMed] [Google Scholar]
- Terry RD. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1996;55:1023–1025. [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tsai KJ, Tsai YC, Shen CK. G-CSF rescues the memory impairment of animal models of Alzheimer’s disease. The Journal of experimental medicine. 2007;204:1273–1280. doi: 10.1084/jem.20062481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Ma QJ. [Prophylactic and therapeutic vaccines against Alzheimer’s disease] Sheng Wu Gong Cheng Xue Bao. 2003;19:641–645. [PubMed] [Google Scholar]
- Zhao LR, Navalitloha Y, Singhal S, Mehta J, Piao CS, Guo WP, Kessler JA, Groothuis DR. Hematopoietic growth factors pass through the blood-brain barrier in intact rats. Experimental Neurology. 2007;204:569–573. doi: 10.1016/j.expneurol.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]