

Abstract
The bromodomain-containing protein BRD9, a subunit of the human BAF (SWI/SNF) nucleosome remodeling complex, has emerged as an attractive therapeutic target in cancer. Despite the development of chemical probes targeting the BRD9 bromodomain, there is a limited understanding of BRD9 function beyond acetyl-lysine recognition. We have therefore created the first BRD9-directed chemical degraders, through iterative design and testing of heterobifunctional ligands that bridge the BRD9 bromodomain and the cereblon E3 ubiquitin ligase complex. Degraders of BRD9 exhibit markedly enhanced potency compared to parental ligands (10 to 100 fold). Parallel study of degraders with divergent BRD9-binding chemotypes in models of acute myeloid leukemia resolves bromodomain polypharmacology in this emerging drug class. Together, these findings reveal the tractability of non-BET bromodomain containing proteins to chemical degradation, and highlight lead compound 6 (dBRD9) as a tool for the study of BRD9.
Keywords: Cancer, Protein Degradation, Drug Design, Bromodomain, Epigenetics
Graphical abstract
With structural guidance alongside comparative biochemical and biological assays, an iterative design strategy resulted in the development of targeted small molecule degraders that rapidly, potently, and selectively eliminate BAF complex member BRD9. These first in class non-BET bromodomain degraders offer dramatic potency improvements over existing BRD9 probes in models of acute myloid leukemia.
Small-molecule inhibitors of BRD4 established the feasibility of inhibiting acetyl-lysine recognition domains (bromodomains).[1] The broad use of the chemical probe JQ1 and other inhibitors of the bromodomain and extra-terminal domain (BET) subfamily of transcriptional co-activators has contributed an enhanced mechanistic understanding of gene control and has availed new therapeutic opportunities in cancer.[2,3] Indeed, multiple BET inhibitor candidates from several groups are now undergoing clinical trials for diverse indications in and outside of oncology.[4–6] Further, significant research effort has contributed a number of high quality chemical probes for bromodomain-containing proteins beyond the BET family.[7,8]
The bromodomain-containing protein BRD9 has garnered particular attention as a component of the human ATP-dependent chromatin remodeling BAF complex (also known as SWI/SNF). Meta-analyses of whole-genome sequencing efforts have recently identified a high frequency of recurrent somatic mutations in BAF factors in diverse human cancers.[9] Within several subsets of these genetically defined malignancies, components of the BAF complex have emerged as context-specific dependencies, either supporting growth within a residual complex following loss of function mutation, or as novel oncogenes such as the SS18-SSX fusion.[10] These observations have generated interest in therapeutic strategies to target BAF.
Beyond its presence in the BAF complex, a lack of functional annotation for BRD9 has provided incentive for development of BRD9 selective inhibitors to interrogate its biological role and to assess any therapeutic potential. Several BRD9-dircted efforts in discovery chemistry have been reported,[11–15] developing chemotypes for BRD9-specific engagement. A recent study further suggested BRD9 as a dependency in acute myeloid leukemia (AML), where bromodomain inhibition prompted a cytostatic response.[16] Beyond the bromodomain, the function of BRD9 remains unclear, and chemical tools to study other functions of BRD9 are not available. We therefore undertook to create first chemical probes that destabilize BRD9, anticipating that the study of acute BRD9 loss would offer a powerful approach to interrogate BAF complex function.
Recently, we reported a strategy to direct protein-specific degradation using bifunctional molecules to recruit the cereblon (CRBN) ubiquitin ligase complex to non-physiologic protein substrates[17], providing an all-chemical solution to prior efforts using peptides to bridge E3 ligases and ligand targets (PROTACs)[18,19]. In our prior research, we directed the degradation of BET family proteins by appending CRBN ligands to JQ1, resulting in rapid and potent degradation of BRD2, BRD3 and BRD4. These findings have been well validated, suggesting a broader utility of this strategy[20–22].
Toward the elaboration of BRD9-directed degraders, we initially evaluated putative pharmacophores from reported bromodomain probes LP99 and I-BRD9, and subsequently expanded our study to a third probe, BI7273, reported during the course of this research (Figure 1 A).[12,14,15] To explore the potential of bifunctional derivatives to induce BRD9 degradation, we initially selected as our starting point a close chemical analog of I-BRD9 described by GlaxoSmithKline (GSK-39).[14] This ligand was attractive in that it offered high binding affinity (IC50 = 7.9 nM) as well as a solvent exposed methoxy substituent amenable to chemical derivatization. In our initial design strategy, we adapted this ligand by installing an ether-linked acetyl moiety as a handle for E3 ligand attachment, as exemplified by compounds 1 and 2 (Figure 1 B). Using this approach, we prepared a series of analogs that differ in linker length and composition, and explored varied attachment chemistries to CRBN or von Hippel–Lindau (VHL) E3 ligase ligands (Table S1).
Figure 1.
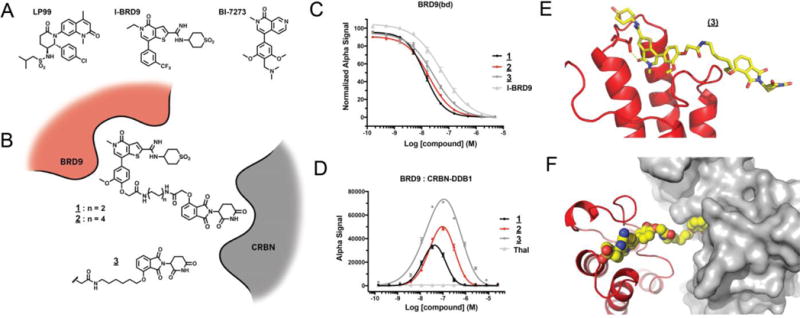
Design and characterization of thienopyridinone BRD9-targeted degraders. A) Structures of select BRD9 bromodomain probes. B) Schematic representation of degrader design. C) Vehicle-normalized BRD9(bd) displacement (AlphaScreen quadruplicate means +/− SEM). D) Compound-induced ternary complex formation of recombinant BRD9(bd) and CRBN-DDB1 (AlphaScreen quadruplicate means +/− SEM). E) Cocrystal structure of 3 with BRD9(bd) (PDB 5TWX). F) Docking of (E) into the published CRBN-DDB1 (4CI3).
To characterize these compounds biochemically, we developed competitive ligand binding assays to the BRD9 bromodomain and the co-purified CRBN-DDB1 complex. High BRD9 affinity was retained across all compounds of this series relative to the parental bromodomain ligand, as exemplified by IC50 values for compounds 1 - 3 that closely approximate the published IC50 for GSK-39 (7.9 nM; Figure 1 C, Table S1). Moderate differences were observed in CRBN-DDB1 affinity among compounds with divergently linked phthalimides (Table S1). For example, the direct alkyl ether phthalimide linkage of 3 showed slightly improved binding over acetamide ethers 1 and 2. Interestingly, measured affinities of all compounds exceeded that of unmodified thalidomide, perhaps reflecting positive affinity contributions of the pendant linker substituents.
To elicit protein degradation, bifunctional molecules must be able to efficiently associate the E3 ligase with the target. To measure this activity, we developed a homogenous luminescence assay to report on compound-induced proximity of BRD9 and CRBN. All of the bifunctional compounds in our initial series were able to significantly induce proximity of the BRD9 bromodomain and CRBN-DDB1 relative to unmodified thalidomide; an activity subsequently referred to simply as “dimerization” (Figure 1 D, Table S1). This ternary interaction exhibited a characteristic auto-inhibitory concentration dependence consistent with bimolecular interactions dominating at saturating ligand concentrations.[23]
Across a range of concentrations, the intermediate length alkyl ether analog 3 afforded robust dimerization. To understand BRD9 recognition by this compound, we solved a co-crystal structure with the BRD9 bromodomain. The pose adopted by the bromodomain warhead confirmed a conserved binding mode relative to the free probe, with the derivatized methoxy position projected to solvent as envisioned (Figure 1 E, Figure S1). In-silico modeling of the ternary assembly including CRBN-DDB1 demonstrated the steric feasibility of ternary formation, with the two ligand-binding domains brought into close assembly by compound 3 (Figure 1 F).[24]
To evaluate the ability of these compounds to degrade BRD9 in a cellular context, we treated a human AML cell line (MOLM-13) for 4 hours in dose and assessed BRD9 protein levels by immunoblot. While 1 and the extended PEG-linked S1 (see supplement) had little effect on BRD9 protein abundance, marked BRD9 loss was observed with the more potent biochemical dimerizers 2 and 3 (Figure 2 A, Table S1). Encouraged by this activity, we prepared an additional focused set of analogs exploring various molecular features. We examined the effect of liker rigidity by installing a conformationally constrained bi-piperidine linker in compound S2. This molecule showed significant improvement in both dimerization and cellular potency, possibly by enforcement of an extended ternary-competent linker conformation (Supplemental Table 1). To pursue degradation by alternate E3 ligases, we prepared the VHL-ligand conjugates S3 and S4; however, these were found to be inactive (Table S1).
Figure 2.
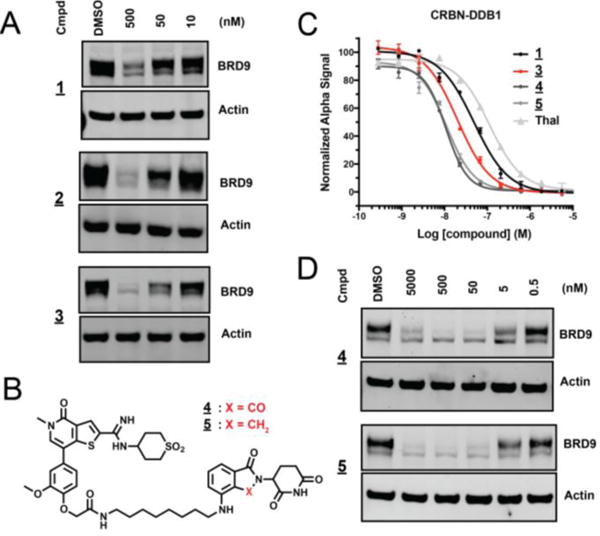
Performance of thienopyrininone degraders. A) Immunoblot for BRD9 and actin after 4 hour treatment of MOLM-13 with indicated concentrations of 1, 2, and 3. B) Chemical structures of 4 and 5. C) Vehicle-normalized CRBN-DDB1 displacement (AlphaScreen quadruplicate means +/− SEM). D) Immunoblot for BRD9 and actin after 4 hour treatment of MOLM-13 with indicated concentrations of 4 and 5.
Additional analogs explored substitution of the phenolic attachment as found in 3 for the amine type linkages found in compounds S5, 4, and 5 (Figure 2 B, Table S1). These compounds tightly bound CRBN, and effectively induced degradation of BRD9 (Figure 2 C,D). The lenalidomide-based analog 5 showed the best overall performance, effectively downregulating BRD9 protein over a broad range of concentrations. We therefore selected this molecule for further characterization.
To evaluate the kinetics of BRD9 degradation, we exposed MOLM-13 cells to 5 at a fixed concentration (100 nM) and assessed BRD9 abundance over time by immunoblot. Near complete BRD9 loss was observed within 1 hour, with no detectable return observed for the duration of the 24-hour treatment period (Figure 3 A). This profile is appropriate to enable study of primary consequences of acute BRD9 loss, as well as viability defects manifested over longer periods.
Figure 3.
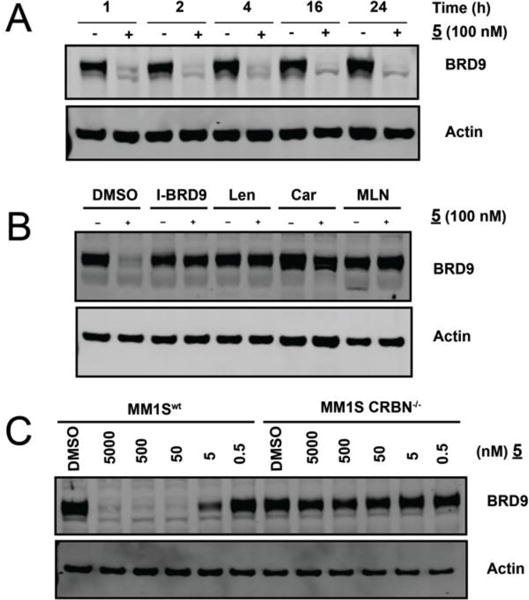
Temporal and mechanistic characterization of BRD9 degradation by 5. A) Immunoblot for BRD9 and actin after treatment of MOLM-13 Cells with 100nM 5 for the indicated times. B) Immunoblot for BRD9 and actin after a 4hr pre-treatment of MM.1S cells with vehicle, I-BRD9, Lenalidomide,Carfilzomib*, or MLN-4924, followed by a 2-hour treatment with 5 (100 nM). * Car pretreatment 30 min. C) Immunoblot for BRD9 and actin after 4 hour treatment with 5 at the indicated doses in MM.1Swt or MM.1SCRBN−/− cells.
To interrogate the mechanism of degradation by compound 5 in a cellular context, we assessed the requirement for target binding, proteasome activity, and activated cullin E3 ligases via chemical and genetic perturbations (Figure 3 B,C). Pretreatment with excess I-BRD9 or lenalidomide competed with 5 for binding to BRD9 or CRBN, respectively, and prevented degradation, consistent with a requirement for intracellular engagement of both targets (Figure 3 B).
Degradation was abolished by the co-treatment with the proteasome inhibitor carfilzomib, confirming a requirement for proteasome function. Pretreatment using a mechanism-based inhibitor of neddylation also rescued BRD9 levels, as expected given the requirement for neddylation of CRL E3 ligases for activity.[25,26] We further established a requirement for CRBN by examining the effects of compound 5 treatment in cells rendered CRBN deficient by CRISPR/Cas9 (CRBN−/−). While treatment of wild type MM.1S cells resulted in marked dose-dependent BRD9 loss, treatment of the paired MM.1S CRBN−/− line failed to induce BRD9 degradation (Figure 3 C).[27] These data support CRBN- and proteasome-dependent degradation of BRD9 by 5.
We aimed to further characterize 5 by establishing the biochemical selectivity profile among 32 representative members of the human bromodomain family. While the results of this analysis confirmed potent engagement of BRD9, we also observed substantial off-target binding activity, notably including BET bromodomains (Figure 4 A). Because of the confounding transcriptional and anti-proliferative effects associated with BET inhibition or loss, we felt selectivity over this family to be an important concern. At this stage of research, the concurrent publication of BI-7273, a highly selective BRD9 probe from Boehringer Ingelheim[14,15], inspired exploration of a novel chemical series of bifunctional degraders (Supplemental Table 2).
Figure 4.
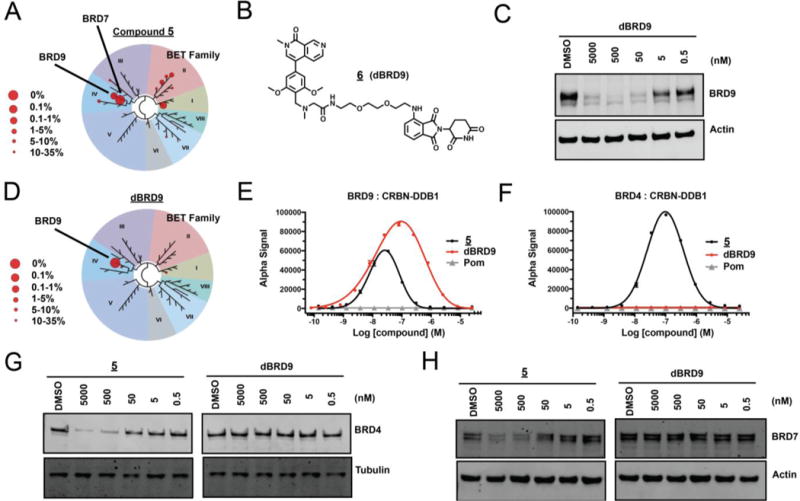
Napthiridinone degrader 6 (dBRD9) offers improved biochemical and cellular selectivity. A) Selectivity of phage-displayed bromodomain displacement by 5 (Bromoscan). B) Chemical structure of dBRD9. C) Immunoblot of BRD9 and actin after 4hr treatment of MOLM-13 cells with indicated concentrations of dBRD9. D) Selectivity of phage-displayed bromodomain displacement by dBRD9 (Bromoscan). E) Compound-induced ternary complex formation of recombinant BRD9(bd) and CRBN-DDB1 (AlphaScreen quadruplicate means +/− SEM). F) Compound-induced ternary complex formation of recombinant BRD4(1) and CRBN-DDB1 as in (E). G) Immunoblot for BRD7 and actin after 4hr treatment of MOLM-13 cells with indicated concentrations of 5 or dBRD9. H) Immunoblot for BRD4 following treatment as in (G).
Compound 6 (dBRD9), a PEG-linked pomalidomide conjugate, was found to prompt rapid BRD9 degradation over a broad range of concentrations (Figure 4 B,C). Gratifyingly, dBRD9 also showed an improved bromodomain engagement profile, with reduced binding activity across the BET family (Figure 4 D). A comparison of biochemical affinity of 5 and dBRD9 for the BET bromodomain, BRD4, by competitive ligand displacement confirmed this result (Supplemental Figure 2). Moreover, while 5 was able to effectively induce biochemical association of CRBN-DDB1 with either BRD9 or BRD4, dBRD9 lost all ability to dimerize BRD4 with CRBN-DDB1 above background levels, but retained robust dimerization of BRD9 (Figure 4 E,F). Consistent with this result, dBRD9 showed improved cellular selectivity. Off target degradation activity on BRD4 and BRD7, observed at high concentrations of 5, was not detectable by western blot following dBRD9 treatment (Figure 4 G,H).
To assess the cellular selectivity for BRD9 degradation in an unbiased, quantitative manner, we measured effects of dBRD9 (100 nM for 2 hours) versus vehicle (DMSO) on all cellular proteins in MOLM-13 cells detected by isobaric tagging and mass spectrometry.[28] Strikingly, of the 7326 proteins quantified in this experiment, BRD9 was the singular protein showing a marked and statistically significant difference in abundance, showing a 5.5 median fold lower abundance in dBRD9 treated samples (FDR corrected q-value < .01) (Figure 5). Levels of other proteins were remarkably static between treatments with 99% of proteins differing less than 0.30 fold. Consistent with quantification by western blot, no significant change in BRD4 or BRD7 levels was observed.
Figure 5.
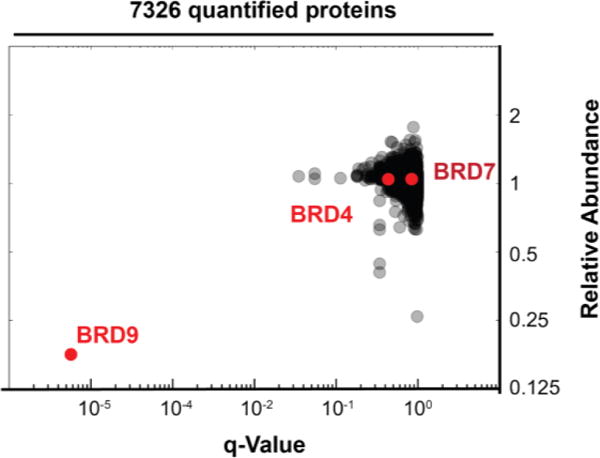
dBRD9 selectivity established by whole-cell lysate proteomics. Fold change in relative abundance of 7326 proteins quantified from MOML-13 cells treated for two hours with dBRD9 (100nM) or vehicle (DMSO), versus q-value for quintuplicate replicates.
Having characterized two potent and pharmacologically distinct degraders of BRD9, we next sought to evaluate the anti-proliferative activity of these molecules in comparison to the parental bromodomain inhibitors. In the context of human AML lines (EOL-1, MOLM-13, MV4;11), compound 5 and dBRD9 both exerted a potent anti-proliferative effect, exceeding non-degrading probe potencies in excesses of 10 to 100 fold (Figure 6 A, Figure S3). Interestingly, although these two compounds exhibited comparable low nanomolar half-maximal anti-proliferative concentrations, the maximal effect (Emax) of 5 exceeded that of dBRD9, likely owing to the polympharmacology associated with 5, particularly activity on BRD4, a well described AML dependency.[17] These data argue that polypharmacologic degraders are a viable chemical strategy, and further demonstrate the ability of cereblon ligand conjugation to reveal relevant cellular off-target activities of chemical probes.
Figure 6.
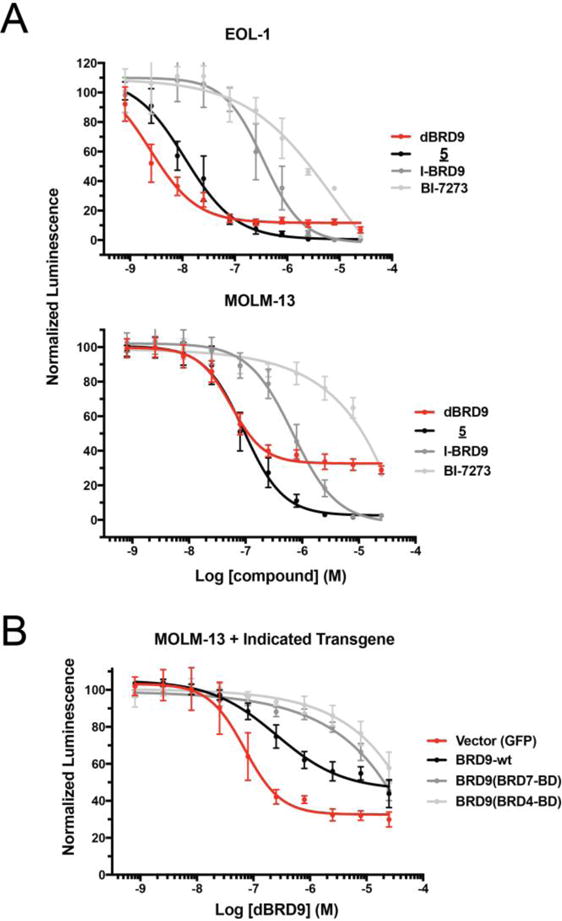
Impact of BRD9 degradation on cultured human leukemia lines. A) Viability of EOL-1 and MOML-13 cell lines treated for 7 days with the indicated compounds (ATP-Lite quadruplicate means +/− SEM). B) Viability of MOLM-13 AML measured as in (A) following transduction with recombinant BRD9 alleles or vector control.
To further scrutinize the conclusion that the observed growth defect results from on-target activity of dBRD9, we extended the bromodomain-swap strategy of the Vakoc laboratory, who found that substitution of BRD4 or BRD7 bromodomains for the endogenous BRD9 domain produced recombinant BRD9 alleles that lost affinity to BI-7273, but could functionally substitute for the wild type protein. We therefore stably transduced MOLM-13 cells with wild type BRD9, bromodomain subsituted BRD9 alleles, or a GFP vector control, and re-evaluated sensitivity to dBRD9 in the presence of each transgene. In both lines expressing domain-swap alleles, the antiproliferative affect of dBRD9 was dramatically rescued relative to vector control. Viral expression of exogenous wild type BRD9 also shifted sensitivity, but to an intermediate degree, consistent with retained succeptibility to dBRD9 degradation. These responses are fully congruent with the observed activity of dBRD9 in each line by western blot (Supplimental Figure 4 A). Consistent with it’s polypharmacology, the activity of compound 5 was only partially shifted by domain-swapped alleles, while the BRD9 independent activity of BET inhibitor JQ1 was wholly unaffected (Supplimental Figure 4 B). Together with expression proteomics, these data provide extensive support that the observed sensitivity in MOLM-13 is a BRD9 specific effect.
In the course of our on-target validation work, we prepared a non-targeting control analog (compound S10), which lacks a key hydrogen bonding moiety while being otherwise structurally identical to dBRD9 (Supplimental Figure 5 A). As expected, this molecule lost biochemical BRD9 bromdomain affinity, the ability to degrade BRD9, and accordingly, lost antiproliferative activity in MOLM-13 (MOLM-13 IC50 > 10 uM) (Supplimental Figure 5 B–D). Unexpectedly however, S10 retained considerable activity in the multiple myelome derived MM.1S line, which we had identified as dBRD9 sensitive in studies of activity outside AML (Supplimental Figure 5 E). Contemplating the known activity of IMiDs in MM.1S,[27] we were lead to discover that S10 and dBRD9 retain activity against the IKZF family of linage specific transcription factors; activity not previously observed with dBET-1 (Supplimental Figure 5 F).[17] We therefore caution that in select cell lines of lymphoid origin that express and depend on IKZF family proteins, researchers should control for this feature. We suspect that published molecules featuring similar CRBN targeting chemistry may also retain activity.
In summary, the present work describes the design and characterization of first in class chemical degraders of BRD9. These studies demonstrate the utility of the targeted degradation strategy to bromodomain-containing proteins beyond the BET family. This is particularly relevant, as competitive bromodomain inhibition has failed to phenocopy the effects of protein knock-down (shRNA) or knock-out (CRISPR-Cas9) for non-BET bromodomain proteins.[29]. Thus, using comparative biochemical and biological assays, we have qualified a lead BRD9 chemical degrader, dBRD9, as a selective probe useful for the study of BAF complex biology. The rapid and potent activity of this compound render it ideally suited to the study of fast biological responses such as transcriptional effects and nucleosome positioning. Finally, potent activity of dBRD9 in cellular models of human AML is confirmed to be on target through expression proteomics, alongside chemical and genetic controls within the exemplar MOLM-13 line.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jennifer Perry for critical reading of the manuscript, Eben McCue for assistance with images and N. Gray for technical discussions. We thank Mette Isohey for her work on conditions for linker synthesis, and Stephen DeAngelo for technical contributions to protein production. D.L.B. is a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2196-14). G.L.B. is supported by an EMBO Long-Term Fellowship (ALTF 1235–2015) with additional support from Marie Curie Actions. M.S. was supported by NIH grant F31 GM095450. This was supported by the Claudia Adams Barr Program for Innovative Cancer Research (to D.L.B.), and by NIH/NCI grant P01-CA066996, to J.E.B. and S.A.A. J.E.B. is now an executive and shareholder in Novartis AG.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Nature. 2011;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fu LL, Tian M, Li X, Li JJ, Huang J, Ouyang L, Zhang Y, Liu B. Oncotarget. 2015;6:5501–5516. doi: 10.18632/oncotarget.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicodème E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Nature. 2011;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaidos A, Caputo V, Karadimitris A. Ther Adv Hematol. 2015;6:128–141. doi: 10.1177/2040620715576662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wadhwa E, Nicolaides T. Cureus. 2016;8:e620. doi: 10.7759/cureus.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theodoulou NH, Tomkinson NC, Prinjha RK, Humphreys PG. Current Opinion in Chemical Biology. 2016;33:58–66. doi: 10.1016/j.cbpa.2016.05.028. [DOI] [PubMed] [Google Scholar]
- 7.Filippakopoulos P, Knapp S. Nature reviews Drug discovery. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 8.Smith SG, Zhou MM. ACS Chemical Biology. 2016;11:598–608. doi: 10.1021/acschembio.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Nature Genetics. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadoch C, Crabtree GR. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guetzoyan L, Ingham RJ, Nikbin N, Rossignol J, Wolling M, Baumert M, Burgess-Brown NA, Strain-Damerell CM, Shrestha L, Brennan PE, et al. Med Chem Commun. 2014;5:540–7. [Google Scholar]
- 12.Clark PGK, Vieira LCC, Tallant C, Fedorov O, Singleton DC, Rogers CM, Monteiro OP, Bennett JM, Baronio R, Müller S, et al. Angewandte Chemie. 2015;127:6315–6319. doi: 10.1002/ange.201501394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Picaud S, Strocchia M, Terracciano S, Lauro G, Méndez J, Daniels DL, Riccio R, Bifulco G, Bruno I, Filippakopoulos P. Journal of Medicinal Chemistry. 2015;58:2718–2736. doi: 10.1021/jm501893k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Theodoulou NH, Bamborough P, Bannister AJ, Becher I, Bit RA, Che KH, Chung CW, Dittmann A, Drewes G, Drewry DH, et al. Journal of Medicinal Chemistry. 2016;59:1425–1439. doi: 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin LJ, Koegl M, Bader G, Cockcroft XL, Fedorov O, Fiegen D, Gerstberger T, Hofmann MH, Hohmann AF, Kessler D, et al. Journal of Medicinal Chemistry. 2016;59:4462–4475. doi: 10.1021/acs.jmedchem.5b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hohmann AF, Martin LJ, Minder JL, Roe JS, Shi J, Steurer S, Bader G, Mcconnell D, Pearson M, Gerstberger T, et al. Nature chemical biology. 2016;12:672–679. doi: 10.1038/nchembio.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Proceedings of the National Academy of Sciences. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. Journal of the American Chemical Society. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 20.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. Chemistry and Biology. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zengerle M, Chan KH, Ciulli A. ACS chemical biology. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AMK, Wang J, Chen X, Dong H, Siu K, et al. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Douglass EF, Miller CJ, Sparer G, Shapiro H, Spiegel DA. Journal of the American Chemical Society. 2013;135:6092–6099. doi: 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, et al. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 27.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. Analytical Chemistry. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS, Protopopov A, et al. Cancer Research. 2015;75:3865–3878. doi: 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.