

Abstract
Drug-induced abnormal heart rhythm known as Torsades de Pointes (TdP) is a potential lethal ventricular tachycardia found in many patients. Even newly released anti-arrhythmic drugs, like ivabradine with HCN channel as a primary target, block the hERG potassium current in overlapping concentration interval. Promiscuous drug block to hERG channel may potentially lead to perturbation of the action potential duration (APD) and TdP, especially when with combined with polypharmacy and/or electrolyte disturbances. The example of novel anti-arrhythmic ivabradine illustrates clinically important and ongoing deficit in drug design and warrants for better screening methods. There is an urgent need to develop new approaches for rapid and accurate assessment of how drugs with complex interactions and multiple subcellular targets can predispose or protect from drug-induced TdP. One of the unexpected outcomes of compulsory hERG screening implemented in USA and European Union resulted in large datasets of IC50 values for various molecules entering the market. The abundant data allows now to construct predictive machine-learning (ML) models. Novel ML algorithms and techniques promise better accuracy in determining IC50 values of hERG blockade that is comparable or surpassing that of the earlier QSAR or molecular modeling technique. To test the performance of modern ML techniques, we have developed a computational platform integrating various workflows for quantitative structure activity relationship (QSAR) models using data from the ChEMBL database. To establish predictive powers of ML-based algorithms we computed IC50 values for large dataset of molecules and compared it to automated patch clamp system for a large dataset of hERG blocking and non-blocking drugs, an industry gold standard in studies of cardiotoxicity. The optimal protocol with high sensitivity and predictive power is based on the novel eXtreme gradient boosting (XGBoost) algorithm. The ML-platform with XGBoost displays excellent performance with a coefficient of determination of up to R2 ~0.8 for pIC50 values in evaluation datasets, surpassing other metrics and approaches available in literature. Ultimately, the ML-based platform developed in our work is a scalable framework with automation potential to interact with other developing technologies in cardiotoxicity field, including high-throughput electrophysiology measurements delivering large datasets of profiled drugs, rapid synthesis and drug development via progress in synthetic biology.
Keywords: Drug-Induced Cardiotoxicity, hERG1 channel, Machine-Learning, Gradient-Boosting, Lead Optimization, Drug Discovery, Quantitative Structure Activity Relationship
INTRODUCTION
Abnormal cardiac electrical activity is a common side effect from unintended block of the promiscuous drug target Kv11.1 K+ channel (better known as hERG), the pore-forming domain of the delayed rectifier K+ channel in the heart. Block of the hERG channel leads to prolongation of the QT interval on the ECG, a phase of the cardiac cycle that corresponds to underlying cellular repolarization. Since 2005, the regulatory process for preclinical drug candidates includes a dedicated clinical study, primarily in healthy volunteers, the so-called “Thorough QT Study”. A drug that results in greater than 5 ms QT prolongation above normal in healthy humans indicates “regulatory concern” 1. Various drugs with unrelated structural scaffolds are known to block hERG and several drugs have been withdrawn from the market or were restricted in their usage due to their potential to block Kv11.1, lengthen the QT-interval and cause TdP. Estimates are that 40–70% of all new drug candidates bind to hERG channel and thus present a potential for cardiotoxicity 2–7. The in-vitro and animal models were developed to screen for drug blockade to hERG leading to substantial increase in cost of drug development. Therefore, development of cost-effective and efficient computational screening approaches may offer complimentary advantage to in-vitro pre-clinical studies by eliminating potentially dangerous candidates early in the development and providing information on key determinants of inadvertent hERG blockade.
One of the most successful strategies to avoid side-effects in pre-clinical drug development relies on in-silico screening with Quantitative Structure Activity Relationship (QSAR) modeling, where predictive models for desired (on-target) and undesired (anti- or off-target) drug effects are evaluated to design optimized molecules as potential lead candidates 8. The vast majority of the QSAR models reported in the literature to that point for hERG-related cardiotoxicity are ligand-based, i.e. the model builds on data derived from the ligand structures and experimental data 9–11, rather than the data derived from the molecular targets 12–14. QSAR regression models map these data to molecular activity and aim to predict the activities of novel molecules, i.e. compounds that were outside of the training sets. Popular pharmacophore models can incorporate the data of a broad variety of molecules with different structural scaffolds with the goal to generate so-called global models that aim to be predictive across various molecular scaffolds15–17. Several attempts were made to employ machine learning (ML) techniques to develop better QSAR models. Past work employed simple linear regression models such as partial least squares (PLS) 17–19 or Support Vector Machines (SVM)9,19–21 and also Bayesian approaches for classification 13,16,22,23 as well as Nearest Neighbor approaches 24. More recent papers also include ensemble based methods 9,25–27 such as Random Forest 9,25 and Gradient Boosting 9 which are widely used nowadays. Also artificial neural networks have been used 19,28,29 to study QSAR properties of various target systems. The challenge to extend regression models to pre-clinical evaluation of cardiotoxicity is related to the relatively small and diverse datasets available resulting limited success and trust in the applications to pre-clinical toxicology 9,10. Another challenge for ML methods regarding hERG-related screening is related to the limited ability in quantitative IC50 predictions. Most of the reported QSAR models with larger datasets are binary classification models, which combine compounds in groups of ‘active’ and ‘inactive’ compounds or, sometimes, provide multi-class placements for molecules such as non-active, slightly active, very active, with prediction accuracies currently between 0.8 and 0.9 on some test sets 10,30,31. In contrast to classification models, regression models allow quantitative estimations of the target value32. We found only three hERG regression models using a larger training sets (>500 compounds)17,25,33. It is not straightforward to compare the quality of these models due to different evaluation metrics used and different compositions of training and test sets which may have a considerable effect on the performance 34. To evaluate the model accuracy many publications use cross-validation, where the performance of the model is usually estimated with the cross-validated coefficient of determination, referred to as Q2. However, as other authors pointed out, high Q2 values are necessary, but not sufficient to demonstrate model generalizability. To estimate the model performance it was recommended to use multiple metrics and based on cross-validation and external data, that was not used to select or train the model 35–37.
The compulsory hERG screening implemented in USA and European Union for all drugs entering the market resulted in large datasets of IC50 values. Therefore, there is an opportunity to validate performance of ML-based methods using large and diverse datasets. That is, the data generated by various academic and industrial consortia allows for development of quantitatively-predictive ML platforms with a potential for rapid evaluation of IC50 values comparable or surpassing that of conventional QSAR or receptor-based methods. In recent years, ensemble tree methods such as Gradient Boosting Tree Regression or Random Forest 38,39 have been applied to various QSAR learning problems with great success 9,40. To test performance of various ML techniques, we have developed a computational platform with various workflows to build QSAR models using data from the ChEMBL database. To apply ML algorithms for computational toxicology, we used the Python package for QSAR modeling that integrates RDKit, scikit-learn, pandas, XGBoost modules, among others, facilitating QSAR model prototyping. All inquiries about this program package called “pyQSAR” should be addressed to the authors of this manuscript. To illustrate predictive powers of ML algorithms we computed IC50 values for a large dataset of molecules and compared it to automated patch clamp system for a large dataset of hERG blocking and non-blocking drugs, an industry gold standard in studies of cardiotoxicity. The dataset chosen for evaluation contained all of the compounds from the Comprehensive In Vitro Proarrhythmia Assay (CiPA) 41,42. We applied cross-validation and rigorously tested the model to ensure optimal generalization properties and explored the necessary conditions for high performance in assessing large datasets available for hERG blockers.
METHODS
Overview
The aim of this study is to design an algorithm for prioritization and ranking various compounds according to the associated hERG-related cardiotoxic risks. The central approach of our study is that cardiotoxicity is assessed using the unified protocol for all compounds in the evaluation set e.g. experimental methods, protocols, cell-lines, etc are the same for all compounds. Therefore, we designed the training set accordingly ensuring that multiple compounds were screened with the same experimental condition. We used two different classes of descriptors, i.e. molecular descriptors and entries from 2D-pharmacophore fingerprints, respectively. The molecular descriptors used in ML model development are mostly real values. The 2D-pharmacophore features are binary descriptors.
Construction of the ML training set for evaluation of hERG1 blockade
The ChEMBL43 database (Dec 2015) was queried for bioactivities for the target ‘CHEMBL240’ the potassium voltage-gated channel subfamily H member 2 (the hERG channel, Kv11.1) with the Python package chembl-webresource-client (version 0.8.36). The following assay descriptions were included into the query: ‘Inhibition of human ERG’, ‘Binding affinity to human ERG’, ‘Inhibition of human ERG at 10 uM’ and ‘Inhibition of human ERG channel’. The query was restricted to the bioactivity type ‘IC50’, the assay type ‘B’ i.e. binding assays, and the target confidence ‘9’. Only items with a defined IC50 value were selected for training of ML models. 1069 compounds remained. The confidence score value reflects both the confidence that the target assigned is the correct target for that assay. The confidence scores range from 0, for as yet non-curated data entries, to 9, where a single protein target has been assigned with a high degree of confidence. Then the dataset was restricted to bioassays with at least 6 activities for different compounds leading to 822 entries. Removal of duplicates in the dataset and of compounds with a molecular masses larger than 650 atomic units lead to 729 compounds of which 29 were contained in the predefined testset 2 and were removed as well. The training set contained 700 compounds from which 100 compounds were randomly selected to serve as a test set (Test1). Histograms of descriptive properties of compounds in the dataset including training and all test sets are provided in the supplementary material.
Workflow and composition of test sets
The modeling work-flow is illustrated in Fig 1. Four different test sets were constructed to evaluate the model performance. From the 700 compounds that passed the filtering 100 compounds were selected randomly to form testset 1 (Test1). The second test set (Test2) contained 55 compounds with IC50 values reported by Kramer et al. 44. Compounds that were present in both the ChEMBL dataset and Test2 were removed manually from the ChEMBL data. This set is used as a standard evaluation toolbox for CiPA assays containing safe and cardiotoxic compounds with known activities allowing accurate calibration of the developed methods. Entries corresponding to assays that contain between 2 and 5 compounds only were combined into test set 3 (Test3) containing total 155 compounds after removal of duplicates. Finally, the remaining entries were defined as test set 4 (Test4) containing 73 compounds after removal of duplicates. Every record in Test4 has a unique ChEMBL-assay-ID. Molecules with a pIC50 > 5 were labeled as active compounds other compounds were labeled as inactive. Due to binarization of the data it is possible to characterize features and models in terms of the ROC-curve and the area under the ROC-curve (AROC). This made it possible to compare the predictive power of the model with that of the molecular logP as well as to compare our model with current classification setups.
Figure 1.

Machine Learning Platform Flowchart. For feature selection and model tuning only compounds in the training set were used. The compounds in the test sets were saved for the final evaluation of model performance. For model selection 10-fold cross-validation was used using the compounds in the training set. For the final model the complete training set was used.
Duplicate compounds in the dataset were identified based on Tanimoto similarities as implemented in RDKit 45 generated from standardized molecular representations. Duplicates were removed. Duplicate entries were observed mostly for different stereoisomers of the same chemical scaffold. To account for the most active stereoisomer the minimum of all reported IC50 value was kept. The same procedure was applied to other duplicates. Smiles codes for compounds studied were generated with MolConvert (version 6.2.1) from ChemAxon’s MarvinSketch.
Feature Preprocessing
The features referred to as molecular descriptors were calculated with RDKit. Most descriptors contain real numbers, some have binary and integer values. We removed constant feature with the same value for all compounds within the training set. Furthermore, we removed descriptors with high correlation, so no two columns had a mutual Pearson correlation coefficient larger than 0.99.
Predictive features (FS1)
The remaining molecular descriptors were ranked using area under the receiver-operator-characteristic-curve(AROC) measuring the ability of each feature to distinguish between active and inactive compounds. The molecular descriptors with AROCeff = max(AROC, 1 – AROC)> 0.55 were referred to as predictive features or feature set 1 (FS1) comprising 55 descriptors all together.
Pharmacophore features (FS2)
A 2D pharmacophore is a set of chemical features with topological (2D) distances between them. Definitions from Gobbi and Poppinger 46 as implemented in RDKit were used. In RDKit these these pharmacophores can be represented as fingerprints i.e. bit-vectors with binary elements. The elements of these bit-vectors served as features for ML and referred to as pharmacophore features or feature set 2 (FS2). Only bits that were activated at least 100 times were used. The individual features were denoted as Ph2D_X, where X is an integer. The final feature set (FS3) was simply the unification of FS1 and FS2.
Metrics and scores
We used multiple scores and metrics to train and validate the performance of our model. For the fitting of the ML models the root mean squared error (RMSE) between the model prediction and the experimentally pIC50 values was used. To validate the performance comprehensively we used different metrics and scores were used: Prediction accuracy (PA), F1-score (F1), Cohen’s Kappa (CK) 47, Sensitivity (SE), Specificity (SP), False Negative Rate (FNR), False Positive Rate (FPR), True Negative Rate (TNR), True Positive Rate (TPR), Pearson’s correlation coefficient (r), as well as the coefficient of determination of the prediction vs the experiment (R2), and the experiment vs the prediction (R20), root mean squared error (RMSE), root mean absolute error (MAE), the area under the ROC curve (AROC). As well as some scores that have been suggested by Tropsha et al. 37: X0 and X1. We used the implementation in scikit-learn 48 to calculate the values for r, R2, R20 and AROC. Metrics applied to cross-validated data were noted differently for R2 (referred to as Q2) and AROC (referred to as cvAROC). To quantify similarity we used maximum similarity to compounds in the training set (MST).
MST
The the maximum Tanimoto similarity of a compound with respect to all compounds that were used to train a particular model. Therefore, the MST values are model specific, when different training sets were used. To calculate the similarity we used the Tanimoto metric as implemented in RDKit and Morgan fingerprints as bit vectors with radius 2 and 1024 bits throughout the whole study.
Feature and model selection
-
A stepwise model selection protocol was used to train an eXtreme gradient boosting model (XGBoost). After defining the feature sets FS1–3, 10-fold cross-validation 49 was used to identify the best performing unique features and parameters in the training set. The parameters and corresponding values for this grid search were: colsample_bytree: 0.2/0.3/0.4/0.5/0.6/0.7/0.8/0.9/1; subsample: 0.2/0.4/0.5/0.6/0.7/0.8/0.9/1; max_depth: 2 – 8; eta: 0.001/0.01/0.02/0.08/0.1. More information about the parameters is provided in the results section. The names of the features used here exactly match the parameter names in the python implementation of XGBoost.
For each feature set and parameter the results were projected to the parameter values and the best performing parameters were selected. For each fold, a model was trained using compounds from 9 groups and the remaining group was used for validation. The RMSE for training and validation set were monitored. When the validation error reached a plateau during first 1000 iterations the fitting stopped. The resulting data was used to calculate the cross-validated coefficient of determination Q2.
For selected parameters and features learning curves were plotted. The learning curves were generated by averaging the performance of 10 repetitions, were, random samples of the complete training set served as validation set and fractions of the remaining compounds were used to fit the model. The size of the validation set was constant and set to 10% of the training set. The number of regression trees (iterations of model fitting) was controlled for each curve and varied between 200 and 1.000. Using, learning curves were generated to characterize the dependency of the model performance on the size of the dataset, as well as to identify the optimal number of iterations for the generation of the final model. The training set was then shuffled and divided into 10 mutually exclusive groups.
The final model was trained using the complete training set and a predefined number of iterations. The non-default parameters for the final model were colsample_bytree: 0.6, eta: 0.01, max_depth: 3, subsample: 0.5. The number of iterations (number of trees) for the final model was set to 700.
Reference Models
Lasso
As a reference we used the training data to fit linear regression model using least absolute shrinkage and selection operator (lasso). Lasso is a method to build linear models which use the L1 norm as regularization term. The regularization term penalizes large feature coefficients. Lasso estimates sparse coefficients and prefers solutions with fewer parameters. Therefore, it effectively reduces the number of features the models depend on. Mathematically, it minimizes the least squares-penalty plus the regularization term:
where the regularization parameter α is a constant and ||ω||1 the L1 norm and of the parameter vector and ||…||2 is the L2 norm. The scikit-learn implementation uses coordinate descent algorithm to fit the coefficients. Here the features defined as feature set 1, i.e. the molecular descriptors, were used. Using the fingerprints was not possible due to numerical instabilities. Prior to building the model, the features were transformed to have zero mean and a standard deviation of 1. We used five-fold cross validation to identify the best value for alpha which was 0.026. 47 variables were selected in the fitting process. The R2 values for the reference model regarding the training and all test sets are shown in table 1. More details regarding the reference model are provided in the supporting information.
Table 1.
Metrics and scores used to estimate model performance of the final model with respect to the training and test sets.
| Train | Test1 | Test2 | Test3 | Test4 | |
|---|---|---|---|---|---|
| AROC | 0.93 | 0.90 | 0.93 | 0.75 | 0.51 |
| MAE | 0.31 | 0.52 | 0.80 | 0.53 | 0.69 |
| R2 | 0.88 | 0.68 | 0.61 | 0.06 | −0.64 |
| R20 | 0.82 | 0.45 | −0.24 | −2.24 | −2.21 |
| RMSE | 0.40 | 0.71 | 0.98 | 0.81 | 0.86 |
| X0 | 0.07 | 0.18 | 0.26 | 0.83 | 9.35 |
| X1 | 0.13 | 0.46 | 1.29 | 7.90 | 29.89 |
| r | 0.94 | 0.83 | 0.82 | 0.32 | 0.08 |
Random Forest models RF_FS1 and RF_FS3
Two reference models were built with the Random-Forest-Regressor class implemented in sklearn. Random forest is an ensemble estimator that fits a number of classifying decision trees on several sub-samples of the dataset. Then averaging is used to increase the predictive accuracy. We used standard parameters from sklearn to fit two models. The first model, referred to as RF_FS1, was fit using the features contained in FS1. The second model, referred to as RF_FS3, used features contained in FS3 which included the 2D-pharmacophore fingerprints.
XGB simple
A boosting tree model using feature set FS1 with default configuration using 10 trees.
Software versions
For the presented publication Python 3.5.2 on a 64 bit Linux environment was used with the following packages: Pandas (0.19.1), Numpy (1.11.2), scikit-learn (0.18.1), RDKit (2016.03.1), molvs (0.0.5), XGBoost (0.4).
Software availability
The ML software will be available to all academic users for a nominal fee of $50 CAD or for evaluation purposes with a permission from the developers. Please note, the software is now part of an integrated toxicology platform developed by Achlys Inc (Edmonton) and licensed through TEC Edmonton (Government of Alberta) for commercial users.
RESULTS
Defining chemical underpinnings of hERG blockade is one of the major goals in rapid screening drug candidates. Such a QSAR model would allow for rapid and accurate evaluation of drug candidates, reducing potential risks in drug development. Regression models based on established machine learning (ML) techniques may help to identify critical factors of the torsadogenic activity. Here we present a model based on the eXtreme gradient boosting algorithm for the prediction of small chemicals pIC50 values with respect to hERG. Input vectors were molecular descriptors and 2D pharmacophore features and experimental data from the ChEMBL database.
The ChEMBL database contained 15741 entries for the target ID CHEMBL240. However, the database contained data from diverse sources and types of assays. The bioactivity type IC50 (8033 entries) was the most common followed by Inhibition (3549 entries) and Ki values (2344 entries). However, the IC50 data contained 405 different assay descriptions and 835 assay IDs, many of them unique. To increase the consistency of experimental data in our training set we restricted the data as described in the methods section.
Three different sets of features (FS1–3), as described in the methods section, were tested in combination with different sets of model parameters using 10-fold cross validation. The results of the grid search are summarized in fig. 3. For a broad set of parameters feature set FS3 was superior to FS1 and FS2 (fig. 3A). The boxplots in fig. 3B show that FS3 performed best with an median Q2 above 0.65. The final set of parameters was based on the the best mean Q2 values in figures 3C–F. The parameter colsample_by_tree defines the fraction of columns that is used in each iteration to build the regression tree. For all feature sets this parameter had a small impact on the performance. The learning rate eta showed a clear preference of smaller values around 0.01 consistently for all feature sets. Interestingly, the parameter maximum_depth was optimal at values of 2 and 3, favoring less complex models. The final parameter subsample was optimal at values between 0.4 and 0.6 on average. Based on this results we selected the parameters for the final model.
Figure 3.

A) Q2 values from 10-fold cross-validation for the three feature sets FS1 (●), FS2 (▲), FS3 (■). B) Boxplots summarizing Q2 values. C–F) mean Q2 projected on the C) fraction of columns used to build each decision tree, D) the learning rate eta, E) the maximal depth of each tree and F) the size of the sample used for each tree. G) Training (■) and validation (●) RMSE over the size of the training set for different numbers of trees using the final parameters. Here fractions (between 0.2 and 1) of the training set were used to analyse the dependency of the predictive power on the size of the training set.
The optimal number of trees was estimated based on the learning curves shown in figure 3F. With more than 700 trees the test RMSE stopped decreasing while the training RMSE decreased, indicating that the model started to overfit the training data. Therefore, we chose 700 as the number of trees for the final model. So far exclusively compounds from the training set were used.
The fitted model was applied to pre-defined test sets to estimate the model’s ability to generalize and to estimate the off-sample performance. All test sets were combined to analyze dependency of the model performance on the ligand maximum similarity to compounds in the training set (fig 4A–F). Figures 4A–B show the performance on the union of all test sets in terms of absolute agreement and ranking performance (ROC). The model allows a better separation of active and inactive compounds as compared to the molecular logP feature. Furthermore, the ROC-curve shows that the model performs significantly better than a random prediction as indicated by the black area in the plot. As indicated by fig 4C the compounds basically fall into two groups of similar compounds (MST > 0.5) and dissimilar compounds (MST < 0.5). The model performed very differently on Test1-4 (fig 4D). The model can confidently predict values in Test1 and Test2, but fails for Test3-4. A threshold analysis revealed a dependency of the accuracy with the MST values of the compounds fig 4E–F. Subsets with higher MST values also gained higher accuracies. Figure 4G shows the location of compounds with range violations, i.e. features with values outside the range defined by the according features in the training set. The separation of similar and dissimilar compounds suggests a possible way to define an applicability domain. Using both criteria, MST > 5 and no range violations led to figures 4H–I.
Figure 4.

Model performance for all test sets combined. A) Correlation to experimental data. B) ROC curve using same class criteria. C) Error over the distance to the training set for each compound. Color codes illustrate the MST values (blue: MST > 0.5, red: MST < 0.5). Model performance for the combined test sets (D) and dependences on different similarity thresholds (E and F). G) Location of range violations. Number or range violations indicated by color and size of spheres. Cross-symbols indicate compounds with no range violations. H) and I) model performance for compounds within the recommended applicability domain.
Y-randomization 50 was used to check rule out chance correlation. The target variable was shuffled and the complete model selection and fitting procedure was repeated. The XGBoost was able to fit y-randomized training data, however, none of the Y-randomized datasets was able to gain statistically significant values for Q2 that could be distinguished from a random prediction.
The frequencies of the features used in the final model were evaluated with the f-score method implemented in XGBoost. The score reflects how often a particular feature was used in the model. The most frequently used feature was the molecular logP value (fig 5A). The top ranks are dominated by molecular descriptors. Only one 2D-pharmacophore was under the top 20 ranks. Nevertheless, the model gained performance from using the pharmacophore features (fig. 3).
Figure 5.

The number of times features have been used in the model: A) the top 20 features and B) the following features. Molecular descriptors (blue) and pharmacophore features (black). Only scores of the top 150 features (of ~400) are shown.
The linear model (Lasso) performs worst in terms of fitting the training set as well as predicting the values in the test sets. Only for Test2 the Random Forest model RF_FS1 archives lower values. Interestingly, for Test1 and Test4 the performance of all the ensemble based models, including the final model, is comparable to each other. The performance of the final model comparable to the ensemble based reference models on Test1 (Fig. 6). On Test2 the final model showed increased ability to predict the experimental values indicated by an R2 of 0.61 compared to values 0.46 by the reference models at maximum. As well for the Random Forest models, RF_FS1 and RF_FS3, using the molecular fingerprint based features seem to enhance model performance on Test1 and Test2. All models perform poorly on Test3 and Test4 with R2 values around zero or even negative values respectively.
Figure 6.
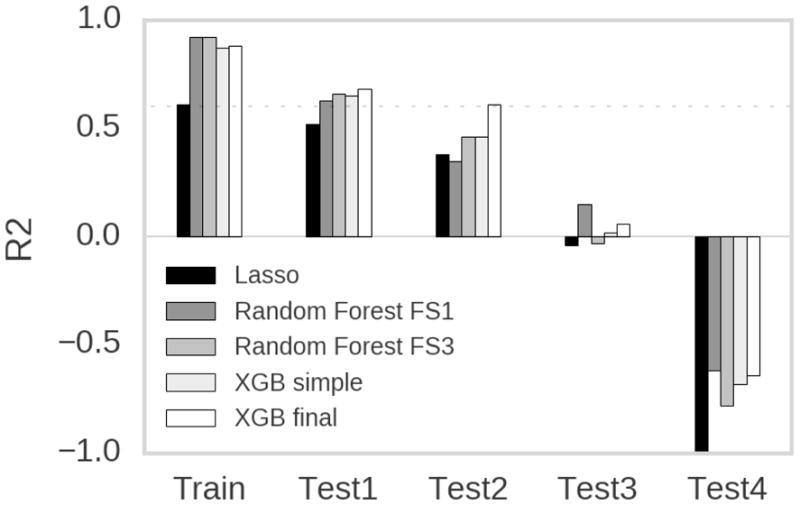
Comparison of the final model with reference models. Only the range between -1 and 1 is shown. The value for the Lasso model for test set 4 (Test4) was - 1.22. The dotted line marks R2 = 0.6.
4. DISCUSSIONS
We have developed a statistically significant QSAR machine learning (ML) model in agreement with OECD guidelines for the prediction of pIC50 values regarding the potassium channel Kv11.1, also known as hERG channel. We provide a detailed protocol of the model selection and parameterization procedure. The presented framework can be used to model data for other biological targets when sufficient experimental data is available. Our results show that quantitative estimations for new compounds are accurate if they are similar to compounds in the training set. The higher the mutual similarities the more accurate the predictions. Optimally, the model should be used to predict compounds with small derivations from the training set. As soon as new data is available, the model should be re-trained incorporating the new data. This observation is in line with that of Gavaghan et al17 who monitored the performance of their model over a period of 15 months after implementation and found that more and more compounds were outside the applicability domain making it necessary to update the model on a regular basis. Such a model is useful in the drug optimization process to identify cardiac safe or risky derivatives of already tested substances. As it is difficult to derive molecular parameters that can be optimized from an ensemble based model, we suggest to generate possible derivatives of compounds which are subsequently scored with our model. Then derivatives that have lower risk of blocking hERG can be selected.
The ML model failed to predict pIC50 values from Test3 and Test4 (fig. 4D) clearly rendering application boundaries. The low performance is a related to several important limitations central to other methods as well. First, the compounds are rather dissimilar to compounds in the training set (fig 2B). Secondly, the experimental data origins from different assays as the data in the training set and Test1. And finally, especially for Test4, the data comes from diverse sources. In Test4 every item had a unique ChEMBL-Assay-ID, indicating the extreme diversity of experimental values. In contrast, the model was able to predict the values in Test2. Fig. 4D shows that that classification of active and inactive compounds based on the prediction works for almost 100% of the compounds. However, so does the classification based on logP values. Interestingly, for the RMSE is highest for Test2 compared to all other test sets. Our interpretation of these findings is that inconsistent experimental conditions add substantial noise to the target values. For example, different temperatures used in different assays can have complex influence on the kinetics and thermodynamics of the hERG block 51. Such parameters, therefore, should be recorded in databases like ChEMBL.
Figure 2.

A) Approximated density of pIC50 values in the Training and Test sets. B) Approximated density of the maximum similarities to compounds in the training set for all test sets. For the training set the similarity to the next most similar compound is shown. The curves are scaled so that the area under the curve is 1.
To highlight the dependency of the performance on the ligand similarity we combined all four test sets and performed a threshold analysis. We found a clear trend towards better performance for compounds with high MST values. For subsets of less similar compounds the performance dropped. However, even for the least similar subset the result was still better than random. Notably, even for the subset of most dissimilar compounds the performance was better as for Test4 as mentioned above. The distributions of MST of all test compounds, showed two maxima around 0.3 and 0.8 and a minimum around 0.5. Accurate estimations were possible for compounds with higher MST, suggesting a natural applicability domain for the model (MST > 0.5). For compounds with lower MST the performance converged to that of the baseline model, i.e. the best molecular descriptor (MolLogP). For all thresholds the model performs significantly better than random, however, for very dissimilar subsets of compounds the predictive power is almost identical to the baseline model. Therefore, the model should be used or compounds with higher MST values. Correlation of hERG affinities with the molecular solubility have been observed before and features that account for solubility or lipophilicity like the logP and logD frequently turned out to be the most influential factors 52.
Most regression models published to date were formulated from small sets of compounds. We found only three hERG regression models using a larger training sets (>500 compounds). It is not straightforward to compare the quality of the models due to different evaluation metrics used and different compositions of training and test sets which may have a considerable effect on the performance 34. The compulsory safety screening for all drugs in preclinical development finally led to large publicly available datasets of compounds with known QSAR properties. In addition, high-performance training algorithms for supervised learning have been developed and utilized for computational toxicology 53. For example, complex models like deep neural-nets 29 and gradient boosting have been under the winning contributions at competitions like the Merck’s drug discovery effort at kaggle.com. To evaluate the model accuracy many publications use cross-validation, where the performance of the model is usually estimated with the cross-validated coefficient of determination, referred to as Q2. However, high Q2 values 35–37 are necessary, but not sufficient condition to demonstrate model generalizability and transferability. The better strategy has to utilize multiple metrics and as well as cross-validation and input from external data, that was not used to select or train the model 35–37.
Cianchetta et. al33 trained a regression model based on a dataset of 885 molecules. They reported an off-sample R2 of 0.9 or greater. However, the test set only contained 16 molecules uniformly spanning the activity range of the dataset! The compounds were manually selected and removed from the training set. Such a test set invariably lead to an overestimation of the off-sample accuracy or the accuracy on data that has not been used to train or select the model. Gavaghan et. al17 used a dataset of 1312 molecules with IC50 values that were all measured with the same protocol (IonWorks High Throughput Electrophysiology Assay) to fit a PLS model. The test set contained 7520 compounds. Neither the training set nor the test set are publicly available. The authors were particularly interested in the validity of external and internal validation techniques. The reported R2 values estimated with the cross-validation displayed R2 coefficients ranging between 0.31 and 0.64. Hansen et al25 used multiple models (Ridge Regression, Gaussian Processes, SVM and Random Forest) to set standards for building consensus models. Their training set contained around 660 compounds. The model performance was not evaluated with a test set from an independent source. The reported cross-validation RMSE values were between 0.57 and 0.73 comparing predicted pIC50 against available experimental data.
How could your model be improved? The shape of the learning curves (Fig. 3G) indicates that more training data would improve the performance as with increased size of the training set the validation RMSE decreases. Furthermore, improved features may increase the accuracy. The use of 2D-pharmacophore features increased the mean Q2 value from around 0.6 to 0.65. We used morgan fingerprints with radius 2 and 1024 bit length. Other configurations or types of fingerprints might be able to improve the model performance. We speculate that features that integrate 3D information, or even 4D features that incorporate motion, may be a promising route for future development of rapid toxicology screening based on ML approaches. However, the generation of useful 3D and 4D features is not trivial and their purpose has been questioned 8.
Supplementary Material
Machine-Learning Platform (MLP) were developed to predict cardiotoxicity with large training dataset of compounds
MLP allows for fast and accurate predictions of IC-50 values for hERG blockade
MLP provides an opportunity to determine key molecular characteristics responsible for high-affinity hERG blockade
Acknowledgments
This work was supported by the Heart and Stroke Foundation of Alberta (2011HSF-2014NOSKOV) and the National Institutes of Health NHLBI R01HL128170-01 to Sergei Y. Noskov. Soren Wacker was partially supported by funding from the Alberta Innovates Technical Futures Strategic Chair in (Bio)Molecular Simulation and funds available through the Li Ka Shing Applied Virology Institute, University of Alberta. Useful discussion with Professors Colleen Clancy, Michael Houghton, Henry J. Duff and Jack Tuszynski are greatly acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) Channels: Structure, Function, and Clinical Significance. Physiol Rev. 2012;92(3):1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- 2.Witchel HJ. The hERG Potassium Channel as a Therapeutic Target. Expert Opin Ther Targets. 2007;11(3):321–336. doi: 10.1517/14728222.11.3.321. [DOI] [PubMed] [Google Scholar]
- 3.Witchel HJ. Drug-Induced hERG Block and Long QT Syndrome. Cardiovasc Ther. 2011;29(4):251–259. doi: 10.1111/j.1755-5922.2010.00154.x. [DOI] [PubMed] [Google Scholar]
- 4.Di Veroli GY, Davies MR, Zhang H, Abi-Gerges N, Boyett MR. High-Throughput Screening of Drug-Binding Dynamics to HERG Improves Early Drug Safety Assessment. Am J Physiol Heart Circ Physiol. 2013;304(1):H104–H117. doi: 10.1152/ajpheart.00511.2012. [DOI] [PubMed] [Google Scholar]
- 5.Cocco G, Jerie P. Torsades de Pointes Induced by the Concomitant Use of Ivabradine and Azithromycin: An Unexpected Dangerous Interaction. Cardiovasc Toxicol. 2015;15(1):104–106. doi: 10.1007/s12012-014-9274-y. [DOI] [PubMed] [Google Scholar]
- 6.Chi KR. Revolution Dawning in Cardiotoxicity Testing. Nat Rev Drug Discov. 2013;12(8):565–567. doi: 10.1038/nrd4083. [DOI] [PubMed] [Google Scholar]
- 7.Beltrame JF. Ivabradine and the SIGNIFY Conundrum. Eur Heart J. 2015;36(46):3297– 3299. doi: 10.1093/eurheartj/ehv368. [DOI] [PubMed] [Google Scholar]
- 8.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. QSAR Modeling: Where Have You Been? Where Are You Going To? J Med Chem. 2014;57(12):4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Tropsha A, Andrade CH. Tuning HERG out: Antitarget QSAR Models for Drug Development. Curr Top Med Chem. 2014;14(11):1399–1415. doi: 10.2174/1568026614666140506124442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villoutreix BO, Taboureau O. Computational Investigations of hERG Channel Blockers: New Insights and Current Predictive Models. Adv Drug Deliv Rev. 2015;86:72–82. doi: 10.1016/j.addr.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Taboureau O, Jorgensen FS. In Silico Predictions of hERG Channel Blockers in Drug Discovery: From Ligand-Based and Target-Based Approaches to Systems Chemical Biology. Comb Chem High Throughput Screen. 2011;14(5):375–387. doi: 10.2174/138620711795508322. [DOI] [PubMed] [Google Scholar]
- 12.Durdagi S, Duff HJ, Noskov SY. Combined Receptor and Ligand-Based Approach to the Universal Pharmacophore Model Development for Studies of Drug Blockade to the hERG1 Pore Domain. J Chem Inf Model. 2011;51(2):463–474. doi: 10.1021/ci100409y. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Sun H, Liu H, Li D, Li Y, Hou T. ADMET Evaluation in Drug Discovery. 16. Predicting hERG Blockers by Combining Multiple Pharmacophores and Machine Learning Approaches. Mol Pharm. 2016 doi: 10.1021/acs.molpharmaceut.6b00471. [DOI] [PubMed] [Google Scholar]
- 14.Du-Cuny L, Chen L, Zhang S. A Critical Assessment of Combined Ligand- and Structure-Based Approaches to HERG Channel Blocker Modeling. J Chem Inf Model. 2011;51(11):2948–2960. doi: 10.1021/ci200271d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waring MJ, Johnstone C. A Quantitative Assessment of hERG Liability as a Function of Lipophilicity. Bioorg Med Chem Lett. 2007;17(6):1759–1764. doi: 10.1016/j.bmcl.2006.12.061. [DOI] [PubMed] [Google Scholar]
- 16.Sun H. An Accurate and Interpretable Bayesian Classification Model for Prediction of HERG Liability. ChemMedChem. 2006;1(3):315–322. doi: 10.1002/cmdc.200500047. [DOI] [PubMed] [Google Scholar]
- 17.Gavaghan CL, Arnby CH, Blomberg N, Strandlund G, Boyer S. Development, Interpretation and Temporal Evaluation of a Global QSAR of hERG Electrophysiology Screening Data. J Comput Aided Mol Des. 2007;21(4):189–206. doi: 10.1007/s10822-006-9095-6. [DOI] [PubMed] [Google Scholar]
- 18.Su B-H, Shen M-Y, Esposito EX, Hopfinger AJ, Tseng YJ. In Silico Binary Classification QSAR Models Based on 4D-Fingerprints and MOE Descriptors for Prediction of hERG Blockage. J Chem Inf Model. 2010;50(7):1304–1318. doi: 10.1021/ci100081j. [DOI] [PubMed] [Google Scholar]
- 19.Roche O, Trube G, Zuegge J, Pflimlin P, Alanine A, Schneider G. A Virtual Screening Method for Prediction of the HERG Potassium Channel Liability of Compound Libraries. Chembiochem. 2002;3(5):455–459. doi: 10.1002/1439-7633(20020503)3:5<455::AID-CBIC455>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20.Jia L, Sun H. Support Vector Machines Classification of hERG Liabilities Based on Atom Types. Bioorg Med Chem. 2008;16(11):6252–6260. doi: 10.1016/j.bmc.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 21.Doddareddy MR, Klaasse EC, Shagufta, Ijzerman AP, Bender A. Prospective Validation of a Comprehensive in Silico hERG Model and Its Applications to Commercial Compound and Drug Databases. ChemMedChem. 2010;5(5):716–729. doi: 10.1002/cmdc.201000024. [DOI] [PubMed] [Google Scholar]
- 22.Liu L-L, Lu J, Lu Y, Zheng M-Y, Luo X-M, Zhu W-L, Jiang H-L, Chen K-X. Novel Bayesian Classification Models for Predicting Compounds Blocking hERG Potassium Channels. Acta Pharmacol Sin. 2014;35(8):1093–1102. doi: 10.1038/aps.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obrezanova O, Csanyi G, Gola JMR, Segall MD. Gaussian Processes: A Method for Automatic QSAR Modeling of ADME Properties. J Chem Inf Model. 2007;47(5):1847– 1857. doi: 10.1021/ci7000633. [DOI] [PubMed] [Google Scholar]
- 24.Kireeva NV, Ovchinnikova SI, Kuznetsov SL, Kazennov AM, Tsivadze AY. Impact of Distance-Based Metric Learning on Classification and Visualization Model Performance and Structure-Activity Landscapes. J Comput Aided Mol Des. 2014;28(2):61–73. doi: 10.1007/s10822-014-9719-1. [DOI] [PubMed] [Google Scholar]
- 25.Hansen K, Rathke F, Schroeter T, Rast G, Fox T, Kriegl JM, Mika S. Bias- Correction of Regression Models: A Case Study on hERG Inhibition. J Chem Inf Model. 2009;49(6):1486–1496. doi: 10.1021/ci9000794. [DOI] [PubMed] [Google Scholar]
- 26.Riniker S, Landrum GA. Similarity Maps - a Visualization Strategy for Molecular Fingerprints and Machine-Learning Methods. J Cheminform. 2013;5(1):43. doi: 10.1186/1758-2946-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schierz AC. Virtual Screening of Bioassay Data. J Cheminform. 2009;1:21. doi: 10.1186/1758-2946-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polak S, Wiśniowska B, Glinka A, Fijorek K, Mendyk A. Slow Delayed Rectifying Potassium Current (IKs) – Analysis of the in Vitro Inhibition Data and Predictive Model Development. J Appl Toxicol. 2013;33(8):723–739. doi: 10.1002/jat.2719. [DOI] [PubMed] [Google Scholar]
- 29.Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J Chem Inf Model. 2015;55(2):263–274. doi: 10.1021/ci500747n. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Mussa HY, Lowe R, Glen RC, Yan A. Probability Based hERG Blocker Classifiers. Mol Inform. 2012;31(9):679–685. doi: 10.1002/minf.201200011. [DOI] [PubMed] [Google Scholar]
- 31.Aronov AM, Goldman BB. A Model for Identifying HERG K+ Channel Blockers. Bioorg Med Chem. 2004;12(9):2307–2315. doi: 10.1016/j.bmc.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Johnson SR, Yue H, Conder ML, Shi H, Doweyko AM, Lloyd J, Levesque P. Estimation of hERG Inhibition of Drug Candidates Using Multivariate Property and Pharmacophore SAR. Bioorg Med Chem. 2007;15(18):6182–6192. doi: 10.1016/j.bmc.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 33.Cianchetta G, Li Y, Kang J, Rampe D, Fravolini A, Cruciani G, Vaz RJ. Predictive Models for hERG Potassium Channel Blockers. Bioorg Med Chem Lett. 2005;15(15):3637–3642. doi: 10.1016/j.bmcl.2005.03.062. [DOI] [PubMed] [Google Scholar]
- 34.Marchese Robinson RL, Glen RC, Mitchell JBO. Development and Comparison of hERG Blocker Classifiers: Assessment on Different Datasets Yields Markedly Different Results. Mol Inform. 2011;30(5):443–458. doi: 10.1002/minf.201000159. [DOI] [PubMed] [Google Scholar]
- 35.Golbraikh A, Tropsha A. Beware of q2! J Mol Graph Model. 2002;20(4):269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 36.Fourches D, Muratov E, Tropsha A. Trust, but Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J Chem Inf Model. 2010;50(7):1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tropsha A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol Inform. 2010;29(6–7):476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 38.Breiman L. Random Forests. Mach Learn. 45(1):5–32. [Google Scholar]
- 39.Breiman L. Classification and Regression Trees. Wadsworth International Group; 1984. [Google Scholar]
- 40.Yang P, Hwa Yang Y, Zhou BB, Zomaya YA. A Review of Ensemble Methods in Bioinformatics. Curr Bioinform. 2010;5(4):296–308. [Google Scholar]
- 41.Cavero I, Holzgrefe H. Comprehensive in Vitro Proarrhythmia Assay, a Novel in Vitro/in Silico Paradigm to Detect Ventricular Proarrhythmic Liability: A Visionary 21st Century Initiative. Expert Opin Drug Saf. 2014;13(6):745–758. doi: 10.1517/14740338.2014.915311. [DOI] [PubMed] [Google Scholar]
- 42.Fermini B, Hancox JC, Abi-Gerges N, Bridgland-Taylor M, Chaudhary KW, Colatsky T, Correll K, Crumb W, Damiano B, Erdemli G, Gintant G, Imredy J, Koerner J, Kramer J, Levesque P, Li Z, Lindqvist A, Obejero-Paz CA, Rampe D, Sawada K, Strauss DG, Vandenberg JI. A New Perspective in the Field of Cardiac Safety Testing through the Comprehensive In Vitro Proarrhythmia Assay Paradigm. J Biomol Screen. 2016;21(1):1–11. doi: 10.1177/1087057115594589. [DOI] [PubMed] [Google Scholar]
- 43.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012;40(Database issue):D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kramer J, Obejero-Paz CA, Myatt G, Kuryshev YA, Bruening-Wright A, Verducci JS, Brown AM. MICE Models: Superior to the HERG Model in Predicting Torsade de Pointes. Sci Rep. 2013;3:2100. doi: 10.1038/srep02100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landrum G. RDKit: Open-source cheminformatics. http://www.rdkit.org.
- 46.Gobbi A, Poppinger D. Genetic Optimization of Combinatorial Libraries. Biotechnol Bioeng. 1998;61(1):47–54. doi: 10.1002/(sici)1097-0290(199824)61:1<47::aid-bit9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 47.Cohen J. A Coefficient of Agreement for Nominal Scales. Educ Psychol Meas. 1960;20(1):37–46. [Google Scholar]
- 48.Buitinck L, Louppe G, Blondel M, Pedregosa F, Mueller A, Grisel O, Niculae V, Prettenhofer P, Gramfort A, Grobler J, Layton R, Vanderplas J, Joly A, Holt B, Varoquaux G. API Design for Machine Learning Software: Experiences from the Scikit-Learn Project. 2013 arXiv [cs.LG] [Google Scholar]
- 49.Kohavi R. A Study of Cross-Validation and Bootstrap for Accuracy Estimation and Model Selection. Proceedings of the 14th International Joint Conference on Artificial Intelligence - Volume 2; IJCAI’95; San Francisco, CA, USA: Morgan Kaufmann Publishers Inc; 1995. pp. 1137–1143. [Google Scholar]
- 50.Rucker C, Rucker G, Meringer M. Y-Randomization and Its Variants in QSPR/QSAR. J Chem Inf Model. 2007;47(6):2345–2357. doi: 10.1021/ci700157b. [DOI] [PubMed] [Google Scholar]
- 51.Windley MJ, Mann SA, Vandenberg JI, Hill A. Temperature Effects on Kinetics of Kv11.1 Drug Block Have Important Consequences for in Silico Proarrhythmic Risk Prediction. Mol Pharmacol. 2016 doi: 10.1124/mol.115.103127. [DOI] [PubMed] [Google Scholar]
- 52.Zachariae U, Giordanetto F, Leach AG. Side Chain Flexibilities in the Human Ether-a-Go-Go Related Gene Potassium Channel (hERG) Together with Matched-Pair Binding Studies Suggest a New Binding Mode for Channel Blockers. J Med Chem. 2009;52(14):4266–4276. doi: 10.1021/jm900002x. [DOI] [PubMed] [Google Scholar]
- 53.Cramer RD. The Inevitable QSAR Renaissance. J Comput Aided Mol Des. 2012;26(1):35–38. doi: 10.1007/s10822-011-9495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.