

Abstract
Introduction:
Chronic cigarette smoking is associated with increased risk for Alzheimer’s disease (AD). The goal of this study was to determine if smoking history moderated the associations of age and APOE genotype (the most robust risk factors for AD) on brain amyloid deposition, glucose metabolism, and neurocognition in cognitively-normal elders.
Methods:
Participants (n = 264) were grouped according to their APOE ε4 carrier status (ε4 carrier: APOE4+; non-ε4 carrier: APOE4−) and smoking status (smokers: at least 1 year of smoking during lifetime; never-smokers: no history of smoking). Approximately 89% of the smoking sample was former-smokers. We specifically tested for interactions of smoking status with APOE ε4 carrier status and age on measures of cortical amyloid deposition, glucose metabolism, and neurocognition.
Results:
(1) smoking status interacted with APOE ε4 carrier status, where smoker APOE4+ showed lower glucose metabolism and poorer auditory-verbal learning and memory than never-smoking APOE4−, never-smoking APOE4+, and smoking APOE4−; (2) smoking status interacted with age on measures of semantic fluency, processing speed/set-shifting and global neurocognition; smokers, irrespective of APOE ε4 carrier status, demonstrated poorer performance with increasing age than never-smokers; and (3) smoking APOE4+ and never-smoking APOE4+ showed greater cortical amyloid deposition than never-smoking APOE4− and smoking APOE4−.
Conclusions:
The findings indicate consideration of smoking history is essential to both better understand the factors associated with neurobiological and neurocognitive abnormalities in elders, and the risk for development of AD-related neuropathology.
Introduction
The pathophysiological process promoting Alzheimer’s disease (AD) is hypothesized to begin several decades before the onset of dementia.1 During the preclinical stages,1,2 AD-related pathological makers are indicated to manifest in the following order: (1) regional cortical amyloid beta (Aβ) and tau accumulation; (2) regional synaptic dysfunction (as measured by glucose metabolism); (3) regional brain volume loss; and (4) subtle cognitive impairment. It is proposed that as the neuropathological abnormalities accrue during the preclinical stages, there is a transition from normal brain function to mild cognitive impairment (MCI), which is typified by AD-like neuropathology and clinically significant memory deficits.3,4 Although MCI may be caused by neurodegenerative processes other than AD, a large proportion of MCI patients ultimately develop dementia.4 While the pathophysiological mechanisms promoting the AD-related neurobiological abnormalities have yet to be definitively identified, increasing age, followed by inheritance of the ε4 allele of the apolipoprotein gene (APOE), are currently indicated to be the greatest risk factors for late-onset AD.5–7 Increasing age and the APOE ε4 allele are both associated with significantly increased cerebral Aβ accumulation and decreased regional glucose metabolism,8–11 which are suggested to be among the earliest appearing biomarkers of the AD pathological process.
To date, human clinical trials on disease-modifying medications for AD have yielded disappointing results.2,12 Therefore, it is of paramount importance to identify risk factors for AD that are modifiable, that is, conditions/behaviors that can be effectively targeted to reduce their prevalence prior to, or at the inception of MCI,13 in order to promote a decrease in the prevalence of AD.14 Cognitive engagement/activity, diet/nutritional supplement intake, physical activity level, vascular risk factors, alcohol consumption level, mood disorders, and cigarette smoking have been proposed as modifiable/treatable risk factors for AD.6,15,16 With respect to cigarette smoking, recent meta-analyses and cohort studies suggest cigarette smoking is robustly associated with increased risk for AD (see17 for review). Barnes and Yaffe14 estimated that smoking accounts for 574 000 (11%) of AD cases in the United States and 4.7 million (14%) cases worldwide. A 10% reduction in the total number of smokers in the United States and worldwide was projected to decrease the prevalence of AD cases by 51 000 and 412 000, respectively. Taken together, cigarette smoking has emerged as a one of the predominant modifiable risk factor for AD; however, the biological mechanisms by which cigarette smoking increases the risk for AD have not been fully delineated (see17 for review).
Age and APOE genotype have been shown to interact with other genetic and/or modifiable environmental risk factors to increase or decrease risk for AD-related neuropathology or dementia.14,18–20 Therefore, a history of chronic cigarette smoking in cognitively-normal elders may interact with age and/or APOE genotype to promote increased abnormalities in AD-related pathological makers that appear early during the preclinical stage of AD. In this study, we compared cognitively-normal elders on positron emission tomography (PET) measures of cortical Aβ deposition (via florbetapir retention) and glucose metabolism (via fluorodeoxyglucose uptake), as well as on measures of neurocognition. The relationships observed between Aβ deposition and glucose metabolism measures and neurocognitive function are inconsistent across studies of cognitively-normal elders (see17 for review). Therefore, we also examined the associations of fluorodeoxyglucose (FDG) uptake and florbetapir retention levels with neurocognition. We predicted interactions among APOE ε4 allele carrier status (carrier vs. noncarrier) and smoking status (history of smoking vs. never-smokers); these interactions will be driven primarily by significantly greater cortical Aβ deposition, decreased glucose metabolism, and poorer learning and memory in APOE ε4 carriers with a history of smoking, relative to APOE ε4 noncarrier never-smokers. Additionally, we postulated smoking status interacts with age, where smokers show greater age-related changes on measures of processing speed (ie, tasks that require that require fast and accurate responses).
Methods
Participants
Data used in preparation of this study were obtained from the Alzheimer Disease Neuroimaging Initiative (ADNI) database (www.loni.usc.edu/ADNI). ADNI is an ongoing, longitudinal, 50 site multicenter study (United States and Canada) designed to develop clinical, imaging, genetic, and biochemical biomarkers for the early detection and tracking of AD. ADNI is the result of the concerted effort of numerous coinvestigators from multiple academic institutions and private corporations. For current information on the ADNI project, see http://adni.loni.usc.edu/about/). Written informed consent was obtained from all participants before initiation of study procedures. The study was conducted according to the Declaration of Helsinki, and US 21 CFR Part 50—Protection of Human Subjects, and Part 56—Institutional Review Boards.
The cognitively-normal participants (n = 264) in this study were primarily Caucasian (95%) and recruited from multiple sites in the United States from 2005 to 2014 as part of the ADNI-1, ADNI-GO, and ADNI-2 study phases. Eighty-one (31%) and 183 (69%) participants were from the ADNI-1 and ADNI-GO/2 study phases, respectively. Inclusion/exclusion criteria for ADNI cognitively-normal participants are fully described in at www.adni-info.org. See Table 1 for demographic and clinical information. Briefly, all participants were between the ages of 56 and 94 years of age, had completed at least 6 years of formal education, were free of any clinically significant neurologic disease, or history of an alcohol/substance use disorder, had a Mini Mental State Examination score of at least 24, and Clinical Dementia Rating Scale score of zero. Participants were assigned as never-smokers (n = 154) if they reported no history of smoking during lifetime, and smokers if they indicated at least 1 year of smoking during lifetime (smokers; n = 110; pack-years = 26±21); 11% (n = 12) were active-smokers at the time of study; former-smokers (n = 98) quit smoking 34±15 (min = 1, max = 44; median = 37) years before study.
Table 1.
Participant Demographics and Clinical Variables
| Variable | Never-smoker APOE4− (n = 113) | Never-smoker APOE4+ (n = 41) | Smoker APOE4− (n = 79) | Smokers APOE4+ (n = 31) |
|---|---|---|---|---|
| Age | 75.8±7.2 | 73.6±7.9 | 76.8±5.6 | 74.5±5.7 |
| Education | 16.8±2.5 | 16.3±3.1 | 16.3±2.8 | 15.6±2.4 |
| Male (%) | 45 | 42 | 59 | 48 |
| Mini Mental Status Exam | 29.1±1.2 | 28.7±1.4 | 28.9±1.2 | 28.6±1.5 |
| Geriatric Depression Inventory | <1 | <1 | <1 | <1 |
| Body mass index | 27.4±4.3 | 26.8±4.9 | 27.9±5.1 | 27.3±3.7 |
| Statin/cholesterol absorption inhibitor use (%) | 36 | 27 | 37 | 32 |
| Antihypertensive use (%) | 43 | 39 | 43 | 55 |
| Triglycerides (mg/dL) | 135±71 | 136±93 | 135±84 | 168±91 |
| Total cholesterol (mg/dL) | 192±37 | 198±41 | 188±38 | 207±38 |
| Modified Hachinski score | <1 | <1 | <1 | <1 |
Florbetapir and FDG PET
Florbetapir and FDG PET data were acquired and processed as previously described.21–23 Full PET protocols and data are available online (http://adni.loni.usc.edu). In summary, quantification of florbetapir and FDG involved co-alignment of PET data with 3 Tesla, 3D T1-weighted magnetic resonance imaging (MRI) scans that were parcellated into individual cortical regions via FreeSurfer.24–27 For florbetapir, mean florbetapir uptake (standardized uptake value ratio) were extracted from gray matter within lateral and medial anterior frontal, posterior cingulate, lateral parietal, and lateral temporal regions and standardized to uptake in the whole cerebellum (white and gray matter). Florbetapir standardized uptake value ratio (SUVR) levels greater than 1.11 are considered “amyloid positive,” so a binary variable florbetapir-cutoff (≤1.11 vs. >1.11) was created based on this threshold. A florbetapir SUVR level greater than 1.11 corresponds to the upper limit of the 95% confidence interval for healthy cognitively-normal young adults and is consistent with the lower limit of a separate autopsy-validated AD sample (see28 and references therein). For FDG, mean values were extracted from the bilateral angular gyrus, posterior cingulate, and inferior temporal gyrus, and standardized to the pons/cerebellar vermis; a Composite FDG region was formed by taking the arithmetic average of the standardized mean values for the angular gyrus, posterior cingulate, and inferior temporal gyrus.
Neurocognitive, Behavioral, and Clinical Measures
From the ADNI neurocognitive battery, the following measures were selected: Rey Auditory Verbal Learning Test (AVLT): immediate and delayed recall; Wechsler Memory Scale-Revised Logical Memory: immediate and delayed recall; Category Fluency (Animals), Trail Making Test (Parts A and B). Numbers of correct words recalled for trails 1–5 were summed from the AVLT to form the immediate recall measure. The foregoing measures were selected because they are sensitive to the effects of chronic smoking29 and polymorphisms of the APOE gene.30 Additionally, the Alzheimer’s Disease Assessment Scale-Cognitive (13-item modification) subscale, and the Mini Mental Status Examination were selected as measures of global cognitive function, and the Geriatric Depression Scale (GDI) assessed for self-reported unipolar depressive symptomatology. Total score for the Modified Hachinski Scale was calculated. Raw scores for the above neurocognitive measures were standardized to never-smoking APOE noncarriers to form t-scores. Measures of total plasma triglycerides and cholesterol were also obtained. See www.adni-info.org/scientists/CognitiveTesting.aspx for corresponding references and administration procedure for the foregoing measures.
Design and Statistics
ADNI-1 participants completed their PET scans 55±7 months after baseline assessment and ADNI-GO/ADNI-2 participants underwent PET scans at baseline assessment. Neurocognitive, behavioral, and clinical measures were obtained at the time of PET scans for all participants. To adjust for previous exposure to neurocognitive testing for ADNI-1 participants, ADNI study phase (ADNI-1 vs. ADNI-GO/2) served as a binary factor in all analyses.
Generalized linear models were used in all analyses. Generalized linear modeling employs maximum likelihood parameter estimation; a chi-square statistic (Wald), and corresponding P value, is generated for each parameter estimate. The following variables served as predictors of Florbetapir SUVR and Composite FDG dependent measures: sex, ADNI study phase, education, smoking status (smokers vs. nonsmokers) and APOE ε4 carrier status (carrier [APOE4−] vs. noncarrier [APOE4+]), as well as interactions among smoking status and age, and APOE ε4 carrier status and age. The following variables served as predictors of all neurocognitive dependent measures: sex, ADNI study phase, education, composite FDG and florbetapir levels, smoking status and APOE ε4 carrier status, as well as interactions among smoking status and age, and APOE ε4 carrier status and age. All main effects, interactions, and t tests for a priori predictions were considered statistically significant at P < .05. Significant main effects and/or interactions (among categorical predictors) were followed-up with pairwise t tests (two-tailed). Effect sizes (ES) for mean differences on dependent measures among the four groups were calculated with Cohen’s d; magnitudes for Cohen’s d are as follows: weak ≤ 0.49; moderate 0.50–0.79; strong ≥ 0.80.31 Comparisons of never-smoker APOE4−, never-smoker APOE4+, smoker APOE4−, and smoker APOE4+ on frequency of amyloid positivity (ie, florbetapir levels > 1.11) were conducted with the chi-square test and z test for proportions of independent groups. In smokers, associations of pack-years and years of smoking cessation with florbetapir level, FDG, and neurocognitive measures were examined; all associations were controlled for multiple comparisons via false discovery rate.32
Results
Participants
Smoking APOE4+ had significantly lower education than never-smoking APOE4− (P = .02), and higher cholesterol than never-smoker APOE4− and smoker APOE4− (both P < .04). No significant differences among groups were observed for age, triglycerides, modified Hachinski total score or use of statins/cholesterol absorption blocking, and antihypertensive medications (Table 1). There were no significant differences between smoking APOE4+ and smoking APOE4− on the frequency of active smokers, pack-years, and the number of years of smoking cessation (all P > .70).
Group Comparisons on Florbetapir, FDG, and Neurocognitive Measures
Florbetapir
Main effects for smoking status [χ2(1) = 7.77, P = .005], APOE4 carrier status [χ2(1) = 32.36, P < .001], and sex [χ2(1) = 3.91, P = .048] were observed. Follow-up pairwise comparisons indicated smoking APOE4+ (ES = 1.18) and never-smoking APOE4+ (ES = 0.74) showed higher florbetapir retention level than never-smoking APOE4− (all P < .015; see Figure 1). Similarly, smoking APOE4+ (ES = 0.91) and never-smoking APOE4+ (ES = 0.48) showed higher florbetapir level than smoking APOE4− (both P < .015). Smoking APOE4− demonstrated a trend for a higher level than never-smoking APOE4− (P = .082; ES = 0.26), and smoking APOE4+ showed a trend for higher florbetapir level than never-smoking APOE4+ (P = .067; ES = 0.44). Females showed a higher florbetapir level than males. The frequency of amyloid positive (ie, above the 1.11 cutoff) cases for each group, based on predicted scores from the above analyses, were significantly different [χ2(3) = 204.2, P < .001] and as follows: 0% of never-smoking APOE4−, 85% of never-smoking APOE4+, 13% of smoking APOE4−, and 94% of smoking APOE4+. Approximately 28% of the total sample was amyloid positive. Never-smoking APOE4− had a significantly lower proportion of amyloid positive cases than all other groups; smoking APOE4− lower proportion of amyloid positive cases than never-smoking APOE4+ and smoking APOE4+ (all P < .05), and never-smoking APOE4+ and smoking APOE4+ showed an equivalent proportion of amyloid positive cases.
Figure 1.
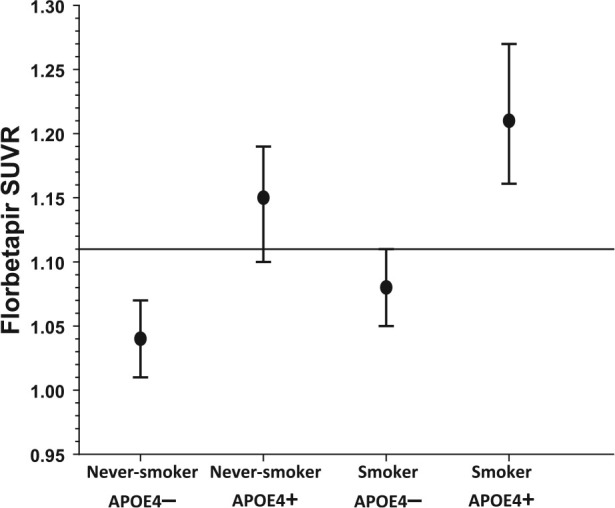
Florbetapir retention level across groups. Higher values indicate greater amyloid level. Levels above the horizontal line indicate amyloid positivity. Mean ± standard error of the mean.
FDG
A smoking status × APOE4 carrier status interaction was observed [χ2(1) = 4.46, P < .001; Figure 2], where smoking APOE4+ had significantly lower FDG uptake than never-smoker APOE4− (P < .001; ES = 0.77), never-smoker APOE4+ (P = .005; ES = 0.68), and smoker APOE4− (P = .003; ES = 0.62). Main effects were yielded for smoking status [χ2(1) = 10.59, P = .001], APOE4 carrier status, [χ2(1) = 4.55, P = .033], and age [χ2(1) = 13.42, P < .001]; smokers showed lower FDG uptake than never-smokers, APOE4 carriers had lower FDG uptake than non-carriers, and increasing age was associated with lower FDG uptake.
Figure 2.

Composite glucose uptake level across groups. Higher values indicate greater glucose metabolism. Mean ± standard error of the mean.
Neurocognition
AVLT Immediate Recall:
A smoking status × APOE4 carrier status interaction was observed [χ2(1) = 4.43, P = .035], where smoking APOE4+ had significantly lower immediate recall than never-smoker APOE4− (P = .032; ES = 0.48), never-smoker APOE4+ (P = .023; ES = 0.60), and smoker APOE4+ (P = .022; ES = 0.53). Main effects were yielded for sex [χ2(1) = 18.07, P < .001], age [χ2(1) = 20.07, P < .001], and education [χ2(1) = 4.43, P = .003]; males performed significantly worse than females, greater age was associated with lower immediate recall, and lower education was associated with poorer performance.
AVLT Delayed Recall:
A smoking status × APOE4 carrier status interaction was observed [χ2(1) = 7.42, P = .006], where smoking APOE4+ had significantly lower delayed recall than never-smoker APOE4+ (P = .019; ES = 0.62). Main effects were yielded for sex [χ2(1) = 10.52, P = .001] and age [χ2(1) = 10.46, P = .001]; males performed worse than females, and greater age was associated with poorer performance.
Logical Memory Immediate Recall:
Main effects were observed for smoking status [χ2(1) = 3.86, P = .049] and education [χ2(1) = 6.33, P = .012]; smokers performed significantly worse than never-smokers, and lower education was associated with poorer performance. Pairwise comparisons showed smoking APOE4+ had significantly poorer immediate recall than never-smoking APOE4− (P = .011; ES = 0.57), and smoking APOE4− (P = .045; ES = 0.46), with a trend for lower recall than never-smoking APOE4+ (P = .059; ES = 0.50).
Logical Memory Delayed Recall:
A main effect was found for smoking status [χ2(1) = 3.86, P = .032], where smokers performed significantly worse than never-smokers. Pairwise comparisons showed smoking APOE4+ had significantly poorer delayed recall than never-smoking APOE4− (P = .02; ES = 0.52), and smoking APOE4− (P = .026; ES = 0.59), with a trend for lower recall than never-smoking APOE4+ (P = .051; ES = 0.45).
Semantic Fluency:
A smoking status × age interaction was observed [χ2(1) = 4.66, P = .031], where smokers showed significantly poorer semantic fluency with increasing age compared with never-smokers. Main effects were yielded for age [χ2(1) = 17.57, P < .001] and education [χ2(1) = 14.49, P < .001]; greater age was associated with worse performance, and lower education was associated with poorer performance.
Trails A
: A main effect for education was yielded [χ2(1) = 3.86, P = .032], where lower education was associated with worse performance.
Trails B:
A smoking status × age interaction was observed [χ2(1) = 7.72, P = .005; see Figure 3], where smokers showed significantly poorer performance with increasing age compared with never-smokers. A main effects was seen for education [χ2(1) = 3.99, P < .001]; lower education was associated with poorer performance.
Figure 3.
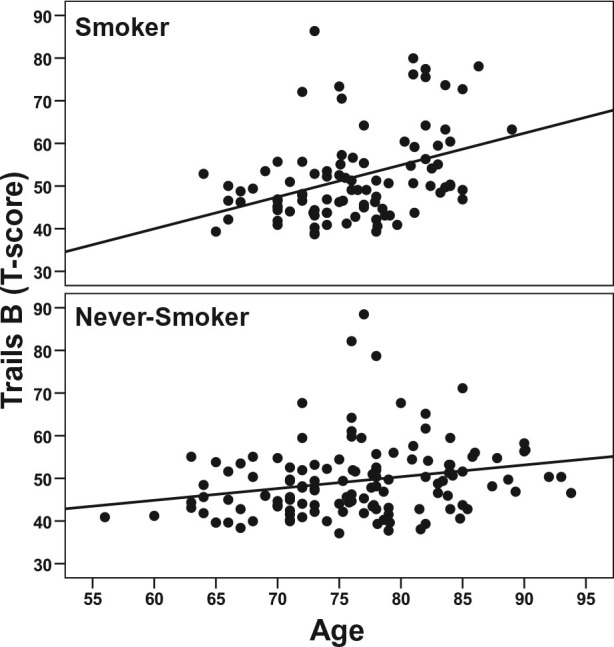
Age-related effects on Trails B performance in smokers and never-smokers. Higher scores reflect poorer performance.
Alzheimer’s Disease Assessment Scale-Cognitive:
A smoking status × age interaction was detected [χ2(1) = 5.23, P = .022], where smokers showed significantly poorer performance with increasing age compared with never-smokers. Main effects were seen for sex [χ2(1) = 11.82, P = .001] and age [χ2(1) = 28.85, P < .001]; males were inferior to females, and greater age was associated with poorer performance.
There were no significant interactions between smoking status and sex, or of APOE4 carrier status with age or sex for any of the above dependent measures. Florbetapir (also as a binary variable, ie, amyloid positive vs. amyloid negative, data not shown) and FDG measures were not significant predictors of any of the above neurocognitive tests. Additionally, vascular risk factors (triglyceride and total cholesterol levels, Modified Hachinski score) were not significant predictors of any dependent measure. The reported findings were essentially unchanged when analyses were repeated after removing active-smokers (approximately 11% of smoking sample) from the smoking sample.
Associations of Smoking Severity With Florbetapir, FDG, and Neurocognitive Measures in Smokers
After false discovery rate correction, pack-years, lifetime years of smoking, and years of smoking were not significantly related to florbetapir, FDG, or neurocognitive measures in smoking APOE4+ and smoking APOE4−.
Conclusions
The main findings from this cohort of cognitively-normal elders were as follows: (1) Smoking status interacted with APOE4 carrier status, where APOE4+ showed lower glucose metabolism and poorer performance on a auditory-verbal learning and memory list learning task (ie, AVLT) than never-smoking APOE4−, never-smoking APOE4+ and smoking APOE4−; (2) On measures of semantic fluency, processing speed/set-shifting and global neurocognition, smoking status interacted with age; irrespective of APOE4 carrier status, smokers demonstrated poorer performance with increasing age than never-smokers on these measures; (3) Smokers were inferior to never-smokers on an auditory-verbal and learning and memory story recall task (ie, Logical Memory); (4) Smoking APOE4+ and never-smoking APOE4+ showed greater cortical amyloid deposition than never-smoking APOE4− and smoking APOE4−; and (5) Across the sample, cortical amyloid deposition and glucose metabolism levels were not related to any neurocognitive measure. No significant relationships of smoking severity/cessation with cortical amyloid deposition, glucose metabolism or neurocognitive measures were observed.
The APOE ε4 allele and increasing age are currently indicated to be the greatest risk factors for late onset AD. In this cohort of cognitively-normal elders, smoking status interacted with APOE4 carrier status on measures of glucose metabolism and auditory-verbal learning and memory, and with age on semantic fluency, processing speed/set-shifting and global neurocognition measures. On measures of glucose metabolism and auditory-verbal learning and memory (based on a list learning task), smoking APOE4+ generally showed the moderate magnitude differences from never-smokers APOE4− and never-smoker APOE4+, followed by smoker APOE4−; there were no significant differences between never-smoker APOE4−, never-smoker APOE4+, and smoker APOE4− on these measures. In middle-aged and elder adults, both chronic smokers29 and APOE4+ 33 demonstrated poorer performance on measures of learning and memory, processing speed and global cognition. In some studies, former-smokers across adulthood performed intermediate to active and never-smokers.29 Cognitively-normal middle-aged and elder APOE4 carriers showed lower regional glucose metabolism than non-APOE4 carriers11,33 and lower regional cortical blood flow, which is closely coupled with cortical glucose metabolism, was reported in adult smokers.29 However, in the foregoing studies, the effects of smoking status, APOE4 carrier status and their interaction were not concurrently evaluated. In the current study, smoking status also interacted with age, where smokers, independent of APOE4 carrier status, showed poorer performance with increasing age than never-smokers on measures of semantic fluency, processing speed/set-shifting and global neurocognition. Although no significant mean differences were observed among the four groups on measures of semantic fluency, processing speed/set-shifting, and global neurocognition, the findings indicated that both APOE4− and APOE4+ smokers showed significantly greater age-related changes on these measures. Increasing age34,35 and chronic smoking29,36 are independently associated with poorer performance on measures of processing speed and global cognitive function in middle-aged and elder adults. The findings from this study are consistent with reports suggesting chronic smoking is associated with greater adverse age-related effects on neurocognition and regional brain atrophy in cognitively-normal adults.29,37,38
For cortical amyloid deposition, additive main effects for smoking status and APOE4 carrier status were apparent, but the effect size for APOE4 carrier status was substantially larger than for smoking status (data not shown). The largest magnitude difference (ES = 1.18) was between smoking APOE4+ and never-smoking APOE4−. We previously found that cognitively-normal elders with a history of smoking demonstrated significantly greater amyloid deposition in the cingulate, parietal, and temporal gray matter than never-smokers;17 however, specific comparisons among groups as a function of smoking status and APOE carrier status were not conducted. It is noteworthy that none of the never-smoking APOE4− were amyloid positive (ie, above the 1.11 cutoff), while 85% of never-smoking APOE4+, 13% of smoking APOE4−, and 94% of smoking APOE4+ were amyloid positive. Approximately 28% of the total sample was amyloid positive, which was primarily driven by smoking and nonsmoking APOE4+. The frequency of amyloid positive participants observed across this cohort is consistent with studies indicating approximately 30% of cognitively-normal elders exhibit amyloid deposition levels that are observed in AD cases,28,39,40 but the frequencies observed in never-smoking and smoking APOE4+ are markedly higher than expected. Our findings are commensurate with the strong APOE4 carrier status effect typically observed on cortical amyloid deposition.8 However, previous human postmortem studies reported a history of chronic cigarette smoking was associated with higher cerebral amyloid levels (see17 for review).
Compared to males, females showed greater amyloid deposition despite better auditory-verbal learning and memory performance and global neurocognitive function. These findings are consistent with previous studies that reported higher amyloid levels (via PET)41 and better learning and memory in cognitively-normal elder females relative to males.29 On the AVLT, a list learning task, smoking status interacted with APOE carrier status, but on Logical Memory, a story recall task, only main effects for smoking status were observed. Although both the AVLT and Logical Memory are measures of auditory-verbal learning and memory, performance on story tasks benefits from the intrinsic semantic organization of the material, while word-list tasks necessitates the self-generation of organizational strategies.
Overall, the greatest neurobiological and neurocognitive abnormalities were observed in smoking APOE4+. Except for cortical amyloid level, smoking APOE4− were generally intermediate to smoking APOE4+ and never-smoking APOE4− and APOE4+ across measures. Based on recently suggested criteria2 proposed to operationalize and stage the severity of the neuropathological correlates of preclinical AD, smoking APOE4+ showed elevated amyloid, neuronal injury (as measured with FDG PET), and subtle cognitive dysfunction; this corresponds to the most advanced preclinical AD stage (so called stage 3). Eighty-nine percent of smokers in this sample were former smokers with 34±15 of smoking cessation; therefore, the greater cortical amyloid deposition, lower glucose metabolism, and poorer neurocognition exhibited by smoking APOE4+ may reflect the extended residual consequences of chronic smoking into middle-age. The observed findings were not likely fully mediated by vascular risk factors, given the equivalence of groups on triglyceride level, modified Hachinski total score, and use of statins/cholesterol absorption blocking and antihypertensive medications. Additionally, the foregoing variables and cholesterol level were not significant predictors of any dependent measure. See17 for review of potential mechanisms by which chronic smoking may increase risk for AD-related neuropathology and neurocognitive decline.
This study has limitations that may influence the generalizability of the findings. The sample was composed of predominately well-educated Caucasians. Cigarette smoking in the United States is associated with at least a 10-year reduction in life expectancy,42 which may create a survivor bias. In other words, the study of the neurobiological and neurocognitive effects of smoking in elders will tend to be biased toward the healthiest smokers—individuals who survived or did not experience significant smoking-related morbidity.43,44 Consequently, the effects of smoking status on the dependent measures in this elder sample may be underestimated due to survivor bias. Given the lack of associations between cigarette consumption variables (eg, pack-years), years of smoking cessation in the smoking groups, and dependent measures in this study, the observed group differences may have been influenced by comorbid/premorbid conditions (eg, subclinical pulmonary, cerebrovascular disease) not accounted for in this research.
Results indicated that chronic smoking, even with several decades of smoking cessation, was associated with brain injury and cognitive deficiencies, primarily in the presence of APOE ε4 or increasing age. Notably, the observed group differences did not appear to be attributable to smoking-related morbidity such as vascular disease risk factors. Smoking status had a more limited effect on the level of amyloid pathology than APOE ε4. This suggests that smoking may increase the risk for AD-related brain injury and dysfunction through non-amyloid pathways.45 However, the results support the supposition that modifiable environmental factors, such as chronic smoking, may interact with genetic predispositions to increase the risk for AD-related neuropathology.17,46 Despite markedly elevated cortical amyloid deposition and decreased glucose metabolism, smoking APOE4+ were cognitively-normal, which, at least partially, may be related to survivor bias44 and/or high cognitive reserve47 in this highly educated Caucasian sample. Amyloid deposition and glucose metabolism were not significant predictors of neurocognition in this sample of cognitively-normal elders, which is consistent with several previous reports (see17 for review). However, increased rate of amyloid accumulation and decreased glucose metabolism are associated with heightened risk for cognitive decline in cognitively-normal elders, as well as conversion from MCI to AD;48–50 therefore, longitudinal tracking of this sample of APOE ε4 carriers with a history of cigarette smoking may advance our understanding of the factors promoting increased risk for, as well as resiliency to, AD-dementia, particularly in the rapidly growing numbers of oldest-old (ie, ≥90 years of age) in the United States.51 In conclusion, results from this study provide additional novel evidence of the adverse effects of chronic smoking on the brain and its functions, particularly in the presence of AD-related biological risk factors. The findings indicate that, even with extended smoking cessation, chronic smoking may be associated with potentially enduring deleterious effects on neurobiology and neurocognition in elder adults.
Funding
The study sponsors had no role in the study design, in the collection, analysis, and interpretation of data, in the writing of the report, and in the decision to submit the article for publication. This work was supported by the National Institutes of Health (NIH DA24136 to TCD) and by the use of resources and facilities at the San Francisco Veterans Administration Medical Center. All data collection and sharing for this project was supported by ADNI. ADNI is funded by the National Institute on Aging (U01 AG024904 to MWW), the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co, Medpace, Inc, Merck and Co, Inc, Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc, and Wyeth, as well as nonprofit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the US Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University Southern California. This research was also supported by NIH grants P30 AG010129, K01 AG030514, R01 AG010897, R01 AG012435, and The Dana Foundation. The above funding agencies had no role in the design and conduct of the study, collection, management, analysis, and interpretation of the data, preparation, review, or approval of the manuscript. Original data used in preparation of this article were obtained from the ADNI database (www.loni.usc.edu/ADNI). As such, the investigators within ADNI contributed to the design and implementation of ADNI and/or provided data, but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Authorship_List.pdf.
Delaration of Interests
None declared.
References
- 1. Jack CR, Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet neurol. 2013;12(2):207–216. doi:10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi:S1552-5260(11)00099-9/10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen RC, Roberts RO, Knopman DS, et al. Mild cognitive impairment: ten years later. Arch Neurol. 2009;66(12):1447–1455. doi:66/12/1447 10.1001/archneurol.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drago V, Babiloni C, Bartres-Faz D, et al. Disease tracking markers for Alzheimer’s disease at the prodromal (MCI) stage. J Alzheimers Dis. 2011;26(suppl 3):159–199. doi:10.3233/JAD-2011-0043 K8245641Q214J660. [DOI] [PubMed] [Google Scholar]
- 5. Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012;8(2):131–168. doi:S1552-5260(12)00032-5 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 6. Daviglus ML, Plassman BL, Pirzada A, et al. Risk factors and preventive interventions for Alzheimer Disease: State of the science. Arch Neurol. 2011;68 (9):1185–1190. doi:archneurol.2011.100 10.1001/archneurol.2011.100. [DOI] [PubMed] [Google Scholar]
- 7. Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25(5):641–650. doi:10.1016/j.neurobiolaging.2003.12.023 S0197458004001009. [DOI] [PubMed] [Google Scholar]
- 8. Vemuri P, Wiste HJ, Weigand SD, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol. 2010;67(3):308–316. doi:10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–969. doi:10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 10. Risacher SL, Kim S, Shen L, et al. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E-MCI). Front Aging Neurosci. 2013;5:11. doi:10.3389/fnagi.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jagust WJ, Landau SM. Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. 2012;32(50):18227–18233. doi:10.1523/JNEUROSCI.3266-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schneider LS, Insel PS, Weiner MW. Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer’s Disease Neuroimaging Initiative. Arch Neurol. 2011;68(1):58–66. doi:68/1/58 10.1001/archneurol.2010.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jagust W. APolipoprotein e, neurodegeneration, and alzheimer disease. JAMA neurol. 2013;70(3):299–300. doi:10.1001/jamaneurol.2013.726. [DOI] [PubMed] [Google Scholar]
- 14. Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819–828. doi:10.1016/S1474-4422(11)70072-2 S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ersche KD, Barnes A, Jones PS, Morein-Zamir S, Robbins TW, Bullmore ET. Abnormal structure of frontostriatal brain systems is associated with aspects of impulsivity and compulsivity in cocaine dependence. Brain. 2011;134(pt 7):2013–2024. doi:awr138 10.1093/brain/awr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Etgen T, Sander D, Bickel H, Forstl H. Mild cognitive impairment and dementia: the importance of modifiable risk factors. Dtsch Arztebl Int. 2011;108(44):743–750. doi:10.3238/arztebl.2011.0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durazzo TC, Mattsson N, Weiner MW. Smoking and increased Alzheimer’s disease risk: a review of potential mechanisms. Alzheimers Dement. 2014;10(3):S122–S145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kivipelto M, Rovio S, Ngandu T, et al. Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med. 2008;12(6B):2762–2771. doi:10.1111/j.1582-4934.2008.00296.x JCMM296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67(1):122–131. doi:10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol. 2010;67(12):1473–1484. doi:archneurol.2010.201 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132(pt 5):1310–1323. doi:awn320 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jagust WJ, Landau SM, Shaw LM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73(15):1193–1199. doi:10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32 (7):1207–1218. doi: 10.1016/j.neurobiolaging.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9(2):179–194. [DOI] [PubMed] [Google Scholar]
- 25. Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: Inflation, flattening, and a surface-based coordinate system. Neuroimage. 1999;9(2):195–207. [DOI] [PubMed] [Google Scholar]
- 26. Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97(20):11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fischl B, Destrieux C, Halgren E, et al. Automatic parcellation of the human cerebral cortex. Cereb Cort. 2004;14(1):11–22. [DOI] [PubMed] [Google Scholar]
- 28. Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72(4):578–586. doi:10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durazzo TC, Meyerhoff DJ, Nixon SJ. Chronic cigarette smoking: implications for neurocognition and brain neurobiology. Int J Environ Res Public Health. 2010;7(10):3760–3791. doi:10.3390/ijerph7103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu L, Boyle PA, Leurgans S, Schneider JA, Bennett DA. Disentangling the effects of age and APOE on neuropathology and late life cognitive decline. Neurobiol Aging. 2014;35(4):819–826. doi:10.1016/j.neurobiolaging.2013.10.074 S0197-4580(13)00526-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen J. Statistical Power Analysis for the Behavioral Sciences (rev. ed.). New York, NY: Academic Press; 1977. [Google Scholar]
- 32. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc. 1995;57(Series B):289–300. [Google Scholar]
- 33. Langbaum JB, Chen K, Caselli RJ, et al. Hypometabolism in Alzheimer-affected brain regions in cognitively healthy Latino individuals carrying the apolipoprotein E epsilon4 allele. Arch Neurol. 2010;67(4):462–468. doi:10.1001/archneurol.2010.3067/4/462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salthouse TA. Aging and measures of processing speed. Biol Psychol. 2000;54(1–3):35–54. [DOI] [PubMed] [Google Scholar]
- 35. Salthouse TA. The processing-speed theory of adult age differences in cognition. Psychol Rev. 1996;103(3):403–428. [DOI] [PubMed] [Google Scholar]
- 36. Durazzo TC, Meyerhoff DJ, Nixon SJ. A comprehensive assessment of neurocognition in middle-aged chronic cigarette smokers. Drug Alcohol Depend. 2012;122(1–2):105–111. doi:S0376-8716(11)00407-8.10.1016/j.drugalcdep.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Durazzo T, Meyerhoff DJ, Nixon SJ. Interactive effects of chronic cigarette smoking and age on hippocampal volumes. Drug Alcohol Depend. 2013;133 (2):704–711. doi:http://dx.doi.org/10.1016/j.drugalcdep.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Durazzo TC, Mon A, Pennington D, Abe C, Gazdzinski S, Meyerhoff DJ. Interactive effects of chronic cigarette smoking and age on brain volumes in controls and alcohol-dependent individuals in early abstinence. Addict Biol. 2014;19(1):132–143. doi:10.1111/j.1369-1600.2012.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. [DOI] [PubMed] [Google Scholar]
- 40. Price JL, Morris JC. Tangles and plaques in nondemented aging and "preclinical" Alzheimer’s disease. Ann Neurol. 1999;45(3):358–368. [DOI] [PubMed] [Google Scholar]
- 41. Johnson SC, Christian BT, Okonkwo OC, et al. Amyloid burden and neural function in people at risk for Alzheimer’s Disease. Neurobiol Aging. 2014;35(3):576–584. doi:10.1016/j.neurobiolaging.2013.09.028S0197-4580(13)00430-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jha P, Ramasundarahettige C, Landsman V, et al. 21st-century hazards of smoking and benefits of cessation in the United States. N Engl J Med. 2013;368(4):341–350. doi:10.1056/NEJMsa1211128. [DOI] [PubMed] [Google Scholar]
- 43. Kukull WA. The association between smoking and Alzheimer’s disease: effects of study design and bias. Biol Psychiatry. 2001;49(3):194–199. doi:S0006322300010775. [DOI] [PubMed] [Google Scholar]
- 44. Chang CC, Zhao Y, Lee CW, Ganguli M. Smoking, death, and Alzheimer disease: a case of competing risks. Alzheimer Dis Assoc Disord. 2012;26(4):300–306. doi:10.1097/WAD.0b013e3182420b6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jack CR, Jr, Wiste HJ, Weigand SD, et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology. 2013;81(20):1732–1740. doi:10.1212/01.wnl.0000435556.21319.e4 01.wnl.0000435556.21319.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wirth M, Villeneuve S, La Joie R, Marks SM, Jagust WJ. Gene-environment interactions: lifetime cognitive activity, APOE genotype, and β-amyloid burden. J Neurosci. 2014;34(25):8612–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012;11(11):1006–1012. doi:10.1016/S1474-4422(12)70191-6S1474-4422(12)70191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Small GW, Siddarth P, Kepe V, et al. Prediction of cognitive decline by positron emission tomography of brain amyloid and tau. Arch Neurol. 2012;69(2):215–222. doi:10.1001/archneurol.2011.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192. doi:10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mosconi L, Brys M, Glodzik-Sobanska L, De Santi S, Rusinek H, de Leon MJ. Early detection of Alzheimer’s disease using neuroimaging. Exp Gerontol. 2007;42(1–2):129–138. [DOI] [PubMed] [Google Scholar]
- 51. Corrada MM, Brookmeyer R, Paganini-Hill A, Berlau D, Kawas CH. Dementia incidence continues to increase with age in the oldest old: the 90+ study. Ann Neurol. 2010;67(1):114–121. doi:10.1002/ana.21915. [DOI] [PMC free article] [PubMed] [Google Scholar]