

Abstract
Reactive oxygen species (ROS) are key mediators in a number of inflammatory conditions, including inflammatory bowel disease (IBD). ROS, including hydrogen peroxide (H2O2), modulate intestinal epithelial ion transport and are believed to contribute to IBD-associated diarrhea. Intestinal crypt fluid secretion, driven by electrogenic Cl− secretion, hydrates and sterilizes the crypt, thus reducing bacterial adherence. Here, we show that pathophysiological concentrations of H2O2 inhibit Ca2+-dependent Cl− secretion across T84 colonic epithelial cells by elevating cytosolic Ca2+, which contributes to activation of two distinct signaling pathways. One involves recruitment of the Ca2+-responsive kinases, Src and Pyk-2, as well as extracellular signal-regulated kinase (ERK). A separate pathway recruits p38 MAP kinase and phosphoinositide 3-kinase (PI3-K) signaling. The ion transport response to Ca2+-dependent stimuli is mediated in part by K+ efflux through basolateral K+ channels and Cl− uptake by the Na+-K+-2Cl− cotransporter, NKCC1. We demonstrate that H2O2 inhibits Ca2+-dependent basolateral K+ efflux and also inhibits NKCC1 activity independently of inhibitory effects on apical Cl− conductance. Thus, we have demonstrated that H2O2 inhibits Ca2+-dependent Cl− secretion through multiple negative regulatory signaling pathways and inhibition of specific ion transporters. These findings increase our understanding of mechanisms by which inflammation disturbs intestinal epithelial function and contributes to intestinal pathophysiology.—Chappell, A. E., Bunz, M., Smoll, E., Dong, H., Lytle, C., Barrett, K. E., McCole, D. F. Hydrogen peroxide inhibits Ca2+-dependent chloride secretion across colonic epithelial cells via distinct kinase signaling pathways and ion transport proteins. FASEB J. 22, 000–000 (2008)
Keywords: reactive oxygen species, PI3-kinase, MAP kinases, NKCC1
THE GENERATION OF reactive oxygen species (ROS) is believed to play a prominent role in the mediation of injury and loss of function associated with a number of intestinal inflammatory conditions, including inflammatory bowel disease (IBD). Although the pathophysiology of IBD has not been fully elucidated, a growing body of clinical and experimental data suggests that the chronic inflammation associated with IBD may result from an inappropriate immune response to normal bacterial antigens. The dysregulated immune response results in the sustained overproduction of reactive metabolites of oxygen and nitrogen (1, 2). Data obtained from several studies indicate that chronically inflamed colonic tissue is subject to significant oxidative stress (3–6). The uncontrolled overproduction of ROS, as occurs during active episodes of IBD, can overwhelm protective antioxidant mechanisms, resulting in oxidative damage to cells and tissues. A number of studies have shown reduced levels of antioxidant enzymes, such as superoxide dismutase (SOD), in patients with Crohn’s disease (7) and increased oxidized glutathione in ulcerative colitis (UC) patients (8). In addition, there are comparatively low levels of endogenous antioxidants in the colonic mucosa, with the majority of the enzymatic antioxidant production in the colon occurring in the epithelium and little produced in the lamina propria (2). Moreover, a number of antioxidants have been shown to protect against the onset of diarrhea in mouse models of colitis (9–11).
Of the ROS believed to have a role in IBD, hydrogen peroxide (H2O2) is one that has been the subject of much investigation. H2O2 can be formed by the dismutation of superoxide or the two electron reduction of oxygen. The large number of activated phagocytes commonly found in ulcerative colitis may secrete millimolar concentrations of H2O2 in close proximity to colonocytes (12). H2O2 per se can react with lipids and sulfhydryls to inactivate key enzymes (13, 14). At physiological concentrations, H2O2 not only can cause direct damage to cultured epithelial cells and cells isolated from IBD patients, but also, along with superoxide, can interact with low-molecular-weight, redoxactive forms of iron to yield the highly reactive hydroxyl radical that also damages epithelial cells in vitro (2, 7, 15). Following activation of neutrophils and monocytes, H2O2 will also result in enhanced formation of the potent oxidant, hypochlorous acid (HOCl) from myeloperoxidase (MPO)-catalyzed oxidation of Cl− by H2O2 (13). H2O2 is therefore an important precursor of a number of very damaging ROS.
Despite evidence that many inflammatory mediators such as eicosanoids, histamine, and complement factors can activate ion transport in isolated intestine, intestinal tissues isolated from IBD patients, as well as mouse models of disease, exhibit a reduced ability to transport ions (16–20). This is believed to be a major factor in the diarrhea associated with IBD. A number of studies have investigated the effects of ROS on intestinal secretion (21). Studies in rat colonic tissue have shown that H2O2 can indirectly increase mucosal Cl− secretion by causing the release of prostaglandins. These subsequently act on submucosal cholinergic neurons to stimulate the release of neurotransmitters (i.e., acetylcholine) that evoke secretion, rather than acting directly on the epithelium (22, 23). A direct effect of H2O2 on Cl− secretion has also been investigated using T84 colonic epithelial cells (24). A high concentration of H2O2 (5.5 mmol/L) was required to evoke a small increase in short-circuit current (Isc), reflective of chloride secretion in this model. This study, and one by DuVall et al. (25), showed that H2O2 pretreatment of T84 cells for 30 min could also attenuate cAMP-dependent Cl− secretory responses. Importantly, the concentration of H2O2 used (500 μM), inhibited Cl− secretion through specific effects on the Cl− secretory pathway and did not affect paracellular permeability, nor prove toxic. The DuVall et al. study concluded that the principal effect of H2O2 on colonic cAMP-dependent Cl− secretion was inhibitory. This has been supported by the work of Walker et al. (26). Other oxidants such as HOCl and NH2Cl have been shown to increase colonic epithelial cell line and rat mucosal Cl− secretion, as well as mucosal permeability (27–29). Studies have also shown that in some cell systems H2O2 can activate mitogen-activated protein (MAP) kinases, thus modulating cell growth. Our laboratory has established that a number of signaling kinases including members of the MAP kinase family, as well as nonreceptor tyrosine kinases, including Src, can negatively regulate intestinal chloride secretion (30, 31). However, the signaling mechanisms by which ROS influence colonic ion transport have not been well characterized. Therefore, in the present study we investigated the effects of ROS, specifically H2O2, on Ca2+-dependent Cl− secretion across colonic epithelial cells and the signaling pathways activated by H2O2 that modify epithelial ion transport responses.
MATERIALS AND METHODS
Materials
Carbachol (Sigma), tyrphostin AG1478, PP2, wortmannin, LY294002, PD98059, U0126, SB203580, H2O2 (Calbiochem, San Diego, CA, USA), mouse anti-human epidermal growth factor receptor (EGFR) (clone LA1) and mouse anti-phosphotyrosine antibodies (Upstate Biotechnology, Lake Placid, NY, USA), rabbit anti-phospho-Pyk-2 antibodies (Biosource International, Camarillo, CA, USA), anti-phospho-ERK antibodies (New England Biolabs, MA, USA) and Tris-glycine electrophoresis gels (Bio-Rad, Hercules, CA, USA) were obtained from the sources noted. Rabbit polyclonal anti-human EGFR (1005) and rabbit polyclonal anti-human ERK 1 (K-23) were used to measure total EGFR and ERK, respectively (Santa Cruz Biotechnology, Santa Cruz, CA, USA). All other reagents were of analytical grade and obtained commercially.
Cell culture
Methods for maintenance of T84 cells in culture were described previously (32). Briefly, T84 cells were grown in Dulbecco’s modified Eagle’s/F-12 medium (DMEM/F12) (Mediatech Inc., Herndon, VA, USA) supplemented with 5% newborn calf serum. For Ussing chamber/voltage-clamp experiments, 5 × 105 cells were seeded onto 12-mm Millicell transwell polycarbonate filters. For Western blot analysis experiments, 106 cells were seeded onto 30-mm filters. Cells were cultured for 10–15 days prior to use. When grown on polycarbonate filters, T84 cells are known to acquire the polarized phenotype of native colonic epithelia. In accordance with muscarinic M3 receptors, distribution on intestinal epithelia, carbachol (CCh) was added basolaterally in all experiments.
HT-29.cl19A cells (33) were grown to confluence (~5 days) in DMEM supplemented with 10% FBS, l-glutamine, and streptomycin in 75 cm2 flasks. After the cells had reached confluence, they were seeded onto 12-mm round coverslips (Warner Instruments Inc., Hamden, CT, USA) and incubated for at least 24 h before use.
Physiological solutions
The composition of the Ringer’s solution used for Ussing chamber, Western blot studies, and 86Rb+ uptake studies was (in mM): 140 Na+, 120 Cl−, 5.2 K+, 25 HCO3−, 0.4 H2PO4−, 2.4 HPO42−, 1.2 Ca2+, 1.2 Mg2+, and 10 D-glucose. The physiological salt solution used in digital Ca2+ measurement contained the following (in mM): 140 Na+, 5.0 K+, 2.0 Ca2+, 147 Cl−, 10 HEPES, and 10 glucose. The solution osmolality was ~284 mosmol/kg. HEPES-buffered Hank’s balanced salt solution (HBSS) was used in 86Rb+ efflux experiments. The HBSS composition was (in mM): 137 NaCl, 5.4 KCl, 0.4 KH2PO4, 0.3 Na2HPO4, 1.0 MgCl2, 1.0 CaCl2, 15 HEPES, pH 7.2, and 10 D-glucose). The chloride-free buffer used in 86Rb+ uptake studies contained (in mM): 2.4 K2HPO4, 0.4 KH2PO4, 25 NaHCO3, 1.2 MgSO4, 1.2 mM CaSO4, 115 sodium isethionate, and 10 D-glucose.
Electrophysiological studies
T84 cells grown to confluence on permeable 12-mm filters were mounted in Ussing chambers (window area = 0.6 cm2) and bathed in oxygenated (95% O2–5% CO2) Ringer’s solution at 37°C. Monolayers were voltage-clamped to zero potential difference by the application of short-circuit current (Isc). Under these conditions, changes in Isc (ΔIsc) in response to agonists are wholly reflective of electrogenic chloride secretion (34). Transepithelial resistance (TER) was measured using a “chopstick” voltohmeter (World Precision Instruments, Sarasota, FL, USA) (35). The MAPK inhibitor, SB203580, and the MEK inhibitor, PD98059, were solubilized in dimethyl sulfoxide (DMSO). In control studies, the final concentration of DMSO (0.1%) had no effect on Isc responses to CCh in the presence or absence of H2O2 (data not shown). This is in keeping with our previous unpublished findings and with studies by other groups on MAPK phosphorylation using intestinal epithelial cell lines (36). Thus, later studies were conducted without DMSO addition to control solutions.
Immunoprecipitation and Western blot analysis
T84 cell monolayers were washed (3×) with Ringer’s solution, allowed to equilibrate for 30 min at 37°C and were then stimulated with agonists (±antagonists) as appropriate. The reaction was stopped by washing in ice-cold PBS, and the cells were lysed in ice-cold lysis buffer (1% Triton X-100, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml antipain, 100 μg/ml phenylmethylsulfonyl fluoride, 1 mM Na+-vanadate, 1 mM sodium fluoride, and 1 mM EDTA in phosphate buffered saline) for 45 min. Cell lysate supernatants were assayed for protein content (Bio-Rad protein assay kit) and adjusted so that each sample contained an equal amount of protein. For immunoprecipitation studies, lysates were incubated with immunoprecipitating antibody, as per the manufacturer’s instructions, for 1 h at 4°C followed by another 1 h incubation at 4°C with protein A-Sepharose. Lysates were then centrifuged for 3 min at 15,000 relative centrifugal force, and the supernatant was discarded. The pellets were washed in ice-cold phosphate-buffered saline (3×) and resuspended in 2× gel-loading buffer (50 mM Tris, pH 6.8, 2% SDS, 200 mM dithiothreitol, 40% glycerol, 0.2 bromphenol blue) and boiled prior to separation by SDS-PAGE. Resolved proteins were transferred onto polyvinylidene membranes (NEN Life Science Products Inc., Boston, MA, USA). After transfer, the membrane was preblocked with a 1% solution of blocking buffer (Upstate Biotechnology Inc.) for 30 min followed by 1 h incubation with the appropriate concentration of primary antibody in 1% blocking buffer. After washing (5×10 min) in Tris-buffered saline with 1% Tween (TBST), membranes were incubated for 30 min with a horseradish peroxidase-conjugated secondary antibody (anti-mouse or anti-rabbit IgG; BD Biosciences, San Jose, CA, USA) in 1% blocking buffer. After washing in TBST (5×10 min), immunoreactive proteins were detected using an enhanced chemiluminescence detection kit (GE Healthcare UK Ltd., Little Chalfont, UK). Densitometric analysis of Western blots was carried out using National Institutes of Health image software.
Digital Ca2+imaging
[Ca2+]cyt levels in HT-29.cl19A cells were measured by Fura-2 fluorescence ratio digital imaging, as described previously (37, 38). Briefly, HT-29.cl19A cells, grown on coverslips, were loaded with 5 μM Fura-2 acetoxymethyl ester (AM) (dissolved in 0.01% Pluronic F-127 plus 0.1% DMSO in physiological salt solution described previously) at room temperature for 50 min, then washed in normal physiological salt solution for at least 20 min. Thereafter, the coverslips were mounted in a perfusion chamber on a Nikon microscope stage. Cells were initially perfused with a normal physiological salt solution for 5 min, then perfused with the same solution containing H2O2 (500 μM). After that, H2O2 was washed out briefly, followed by treatment with CCh (100 μM) in normal physiological solution. The ratio of Fura-2 fluorescence with excitation at 340 or 380 nm (F340/380) was followed over time and captured using an intensified charge-coupled device camera (ICCD200) and a MetaFluor Imaging System (Universal Imaging, Corporation, Downingtown, PA, USA).
Basolateral 86Rb+ efflux studies
Basolateral K+ efflux was measured using 86Rb+ as a tracer in a method adapted from that described by Venglarik et al.(39). Briefly, confluent monolayers of T84 cells grown on 12-mm transwell inserts were rinsed 3× with HBSS, and incubated at 37°C for 20 min. Cells were loaded with 86Rb+ by incubating for 40 min bilaterally with 1 μCi/ml 86Rb+ in HBSS. After loading with 86Rb+, cells were washed 3× with fresh HBSS and treated bilaterally with HBSS or H2O2 (500 μM) prepared in HBSS, for 30 min. Starting at 22 min into the 30-min incubation with HBSS±H2O2 (i.e., t=8 min), inserts were transferred to new identical basolateral solutions every 2 min. At t=0, inserts were transferred to basolateral solutions of HBSS or 100 μM CCh prepared in HBSS with or without H2O2 and continued to be transferred to new identical basolateral solutions every 2 min for 14 min. At t=14 min, inserts were transferred directly into 5 ml scintillation fluid. Basolateral solutions at each time point were also transferred into 5 ml scintillation fluid, and 86Rb+ content of the inserts and each 2-min basolateral fraction was measured using standard scintillation methods. The following equation was used to calculate the apparent rate constant: r = [ln(R1) − ln(R2)]/(t1 − t2), where R1 and R2 are the percentage counts remaining in the cell monolayer at times t1 and t2, respectively. Data were expressed as rate of efflux per minute.
Basolateral 86Rb+ uptake studies
Basolateral K+ uptake was measured with 86Rb+ as a tracer, using an adaptation of a previously described method (40). Confluent monolayers of T84 cells grown on 12-mm transwell inserts were rinsed 3× with warm Ringer’s solution and incubated for 1 h at 37°C. After 1 h preincubation, Ringer’s solution was added bilaterally, or cells were treated bilaterally with H2O2 (500 μM) prepared in Ringer’s solution, for 30 min on a warming plate at 37°C. Inserts were transferred to wells containing basolateral treatment solutions of Ringer’s or 100 μM (CCh) prepared in Ringer’s solution for 1 min, and then transferred to wells with basolateral uptake solutions containing 1 μCi/ml 86Rb+ in either Ringer’s solution or CCh prepared in Ringer’s solution, and maintained at 37°C for 3 min. 86Rb+ uptake was terminated by immersing inserts several times in ice-cold 100 mM MgCl2 prepared in 10 mM Tris-HCl, pH 7.5. Filters were immediately excised from the inserts, placed directly into 5 ml scintillation fluid, and 86Rb+ content was measured by standard scintillation methods. Experiments assessing bumetanide-sensitive uptake included 10 μM bumetanide (Sigma, St. Louis, MO, USA) in the basolateral treatment and uptake solutions. In experiments with chloride-depleted cells, except for the 86Rb+ uptake solutions, Ringer’s solution was replaced in the above procedures with a chloride-free buffer (composition shown above). The 86Rb+ uptake solutions were prepared in normal Cl−-containing Ringer’s solution. 86Rb+ uptake was used to calculate K+ uptake, for which it acts as a tracer (41). K+ influx was calculated as (cpm/g protein/min)/SA, where SA is the specific activity of the uptake buffer (cpm/μmol K+). Data were expressed as μmol of K+ influx/g protein/min.
Statistical analysis
All data are expressed as means ± SE for a series of n experiments. Student’s t test or analysis of variance (ANOVA) with the Student-Newman-Keuls post-test were used to compare mean values as appropriate. Values of P<0.05 were considered to represent significant differences.
RESULTS
H2O2 inhibits CCh-stimulated Cl− secretion across T84 cells
H2O2 has previously been shown to inhibit cAMP-dependent Cl− secretion across colonic epithelial cells; however, the effects of H2O2 treatment on Ca2+-dependent Cl− secretion have not been investigated. We initially set out to determine whether H2O2 could also inhibit the ability of epithelial cells to transport Cl− in response to a calcium-dependent stimulus. Confluent monolayers of T84 colonic epithelial cells, grown on permeable supports, were mounted in Ussing chambers and pretreated bilaterally for 30 min with a range of concentrations of H2O2 (1–500 μM). The muscarinic agonist, CCh, was used as a prototypic receptor-mediated calcium-dependent secretagogue. The Cl− secretory response to CCh, expressed as change in short circuit current (ΔIsc), was not affected by H2O2 up to 100 μM. However, pretreatment with 500 μM H2O2 significantly inhibited the peak Cl− secretory response to CCh (Fig. 1A; P<0.01 vs. CCh alone, open box; n = 6). Since 500 μM was the minimum effective concentration of H2O2 capable of inhibiting CCh-stimulated secretion, this concentration was used in all subsequent experiments. Figure 1B shows a time course of H2O2 (500 μM) pretreatment on CCh-stimulated Isc. H2O2 pretreatment for 15 min caused a dramatic reduction in the peak ion transport response to CCh (P<0.05 vs. CCh alone; n = 4), while maximal inhibition was achieved with 30 min preincubation (P<0.05 vs. CCh alone; n = 4). H2O2 pretreatment for 30 min was used in all subsequent experiments. Figure 1C shows a time course of the Isc response to CCh in the presence and absence of H2O2 pretreatment. H2O2 reduced the magnitude, but not the duration, of the Cl− secretory response to CCh. To verify that the presence of H2O2 in the bathing media did not reduce the efficacy of CCh itself to stimulate ion transport through chemical modification of the ligand, we performed washout experiments in which the basolateral and apical bathing media containing H2O2, were replaced with fresh Ringer’s solution. After a brief period of stabilization (5 min), CCh was added basolaterally. Figure 1D demonstrates that there was no difference in the inhibitory effect of H2O2 on CCh-stimulated Isc in inserts that had been washed with fresh Ringer’s solution vs. inserts that still contained H2O2 in the bathing media. These data indicate that H2O2 inhibits CCh-stimulated Isc through effects on epithelial cells and not by altering the pharmacologic efficacy of CCh itself. Because H2O2 has been shown in certain studies to induce damage to epithelial monolayers, albeit at either higher concentrations, or following longer periods of incubation than used in the current studies (15, 42), we investigated whether incubation of T84 monolayers with H2O2 (500 μM) had any effect on the electrical resistance of the monolayer, as a measure of epithelial barrier integrity. Incubation of T84 monolayers with H2O2 for 30 min had no effect on TER of the monolayers when compared with Ringer’s solution alone (Fig. 1E). These data show that H2O2 inhibits Ca2+-dependent Cl− secretion across intestinal epithelial cells in the absence of any obvious defects in monolayer integrity.
Figure 1.
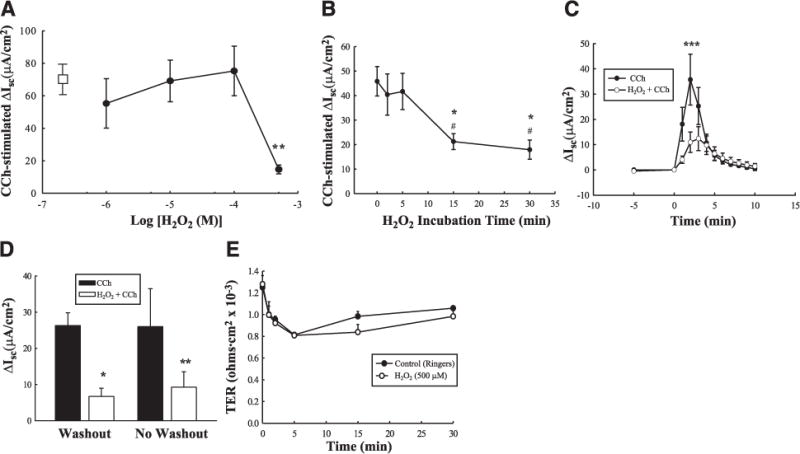
Hydrogen peroxide (H2O2) inhibits carbachol (CCh)-stimulated epithelial Isc without affecting barrier properties. A) T84 monolayers mounted in Ussing chambers were treated with H2O2 (0–500 μM; n = 6) for 30 min prior to stimulation of electrogenic Cl− secretion with CCh. The ΔIsc response to CCh alone is represented by the open rectangle. B) Time course of H2O2 (500 μM) pretreatment on peak CCh-stimulated Isc (n=4). C) Time course of the Isc response to CCh with or without pretreatment for 30 min with H2O2 (500 μM; n = 5). D) Monolayers were pretreated with H2O2 (500 μM; 30 min) and treated directly with CCh (no washout), or bathing media were replaced with fresh Ringer’s solution, and inserts were then treated with CCh (washout). E) Transepithelial resistance across T84 monolayers treated with either H2O2 (500 μM) for 30 min or Ringer’s solution, was measured using a voltohmeter (n=4). Values are presented as means ± SE. * P<0.05; **P<0.01; ***P<0.001 vs. control (CCh alone or untreated cells). #P<0.05 vs. 2 min and 5 min H2O2 pretreatment on CCh-stimulated Isc (B).
The role of MAPK signaling pathways in H2O2 inhibition of Ca2+-dependent Cl+secretion
We have previously shown important roles for MAPK signaling pathways in the negative regulation of GPCR-stimulated, Ca2+-dependent Cl− secretion across colonic epithelial cells (18, 30, 43). H2O2 has also been shown to activate MAPK signaling in other cell systems (44, 45). Consequently, we investigated whether the inhibitory effects of H2O2 on CCh-stimulated Cl− secretion incorporated established MAPK signaling pathways involved in the regulation of Ca2+-dependent Cl− secretion. Preincubation for 30 min with PD98059 (20 μM), an inhibitor of the ERK-activating kinase, MEK, significantly reduced the inhibitory effect of H2O2 on CCh-stimulated ion transport across T84 cells (Fig. 2A, peak ΔIsc, P<0.05; and Fig. 2B, time course; P<0.01; n = 7). These data were supported by an alternative MEK inhibitor, U0126 (Fig. 2C; P<0.05 vs. H2O2 CCh). To verify that H2O2 increased ERK activation and to determine the kinetics of activation, T84 monolayers were bilaterally treated with H2O2 (500 μM), and cell lysates were separated by SDS-PAGE, and resulting blots were probed for phosphorylated ERK (Thr204/Tyr 202). H2O2 transiently increased ERK phosphorylation, which was maximal at 5 min and returned to baseline by 30 min (Fig. 2D; P<0.001; n = 4).
Figure 2.
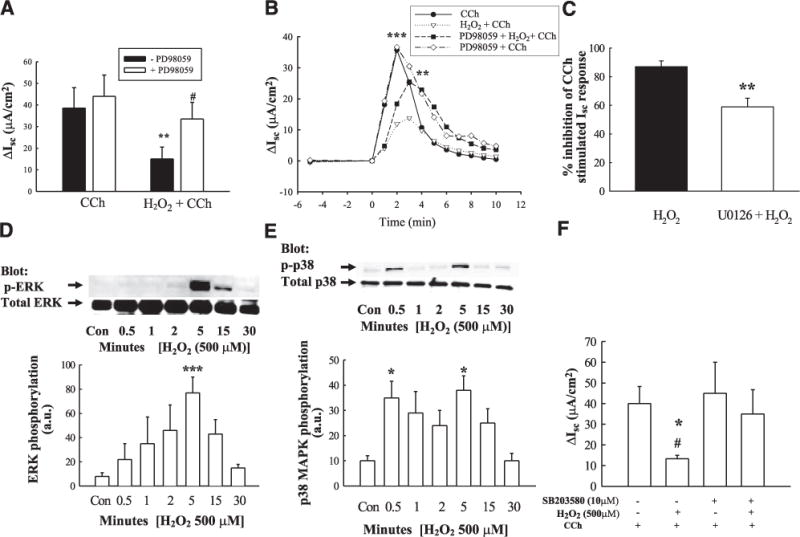
Inhibition of CCh-stimulated Cl− secretion involves ERK and p38 MAPK activation. T84 monolayers, mounted in Ussing chambers, were preincubated with the MEK inhibitor, PD98059 (A, peak ΔIsc; B, time course; 50 μM; 30 min; n = 4), the MEK inhibitor, U0126 (C; 10 μM; 30 min; n = 6; data expressed as % inhibition of peak CCh-stimulated Isc response), or the p38 inhibitor, SB203580 (F; 10 μM; 30 min; n = 5), prior to addition of H2O2 (500 μM) or Ringer’s solution for 30 min, and subsequent peak Isc responses to CCh (100 μM), and Isc response to CCh over time were measured. Error bars were removed from the time course to aid the clarity of B. H2O2 induced phosphorylation of ERK (D; n = 5), and p38 (E; n = 6) was measured by Western blot and densitometric analyses. Values are presented as means ± SE. *P<0.05; **P<0.01; ***P<0.001 vs. control (or H2O2 + CCh; B, C). #P<0.05 vs. cells treated with H2O2 + CCh (A) or H2O2 + SB203580 CCh (F).
As p38 MAPK is also involved in the negative regulation of Ca2+-dependent Cl− secretion (46), we next investigated whether p38 was involved in the inhibitory effect of H2O2 on CCh-stimulated Cl− secretion. Preincubation of T84 monolayers with the p38 inhibitor, SB203580 (10 μM), for 30 min significantly reduced the inhibitory effect of H2O2 on Cl− secretion, thus indicating a prominent role for p38 in mediating the inhibitory effect of H2O2 (Fig. 2F; P<0.05; n = 5). H2O2 actually caused a biphasic increase in p38 phosphorylation with significant increases at 0.5 and 5 min post H2O2 treatment (Fig. 2E; P<0.05; n = 6). These data indicate that the MAPKs, ERK and p38, are involved in mediating the inhibitory effect of H2O2 on Ca2+-dependent Cl− secretion.
Involvement of Src and Pyk-2 signaling pathways in H2O2 inhibition of CCh-stimulated Cl− secretion
Previous studies from our group have shown that CCh-stimulated MAPK activation occurs, at least in part, following activation of the soluble tyrosine kinase, p60Src. Therefore, we next investigated whether Src activation was involved in the inhibitory effect of H2O2 on CCh-stimulated Cl− secretion. Preincubation of T84 cells with the Src inhibitor, PP2 (20 μM), significantly reduced the inhibitory effect of H2O2 thus indicating at least a partial role for Src activation in H2O2-induced inhibition of Ca2+-dependent Cl− secretion (Fig. 3A; P<0.05; n = 5). Interestingly, H2O2 caused a rapid and sustained increase in Src phosphorylation at position Y416, as determined by Western blot analysis (Fig. 3B; P<0.05; n = 6). Another soluble tyrosine kinase involved in the negative regulation of Cl− secretion is the calcium-dependent kinase, Pyk-2. Pyk-2 and Src activation can increase each other’s phosphorylation status (30, 46, 47). Consequently, we investigated whether H2O2 also increased Pyk-2 activation, as measured by increased phosphorylation of Pyk-2 at the Y881 residue. H2O2 induced sustained Pyk-2 phosphorylation in a manner similar to Src activation, albeit with slightly delayed kinetics (Fig. 3C; P<0.001; n = 4).
Figure 3.
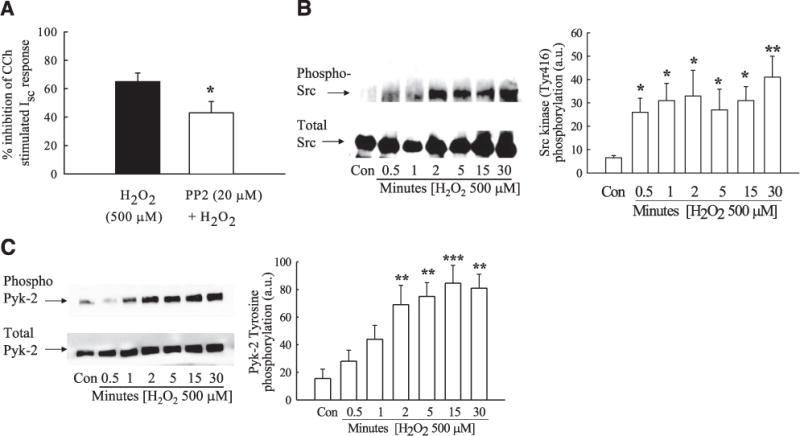
H2O2 induces activation of the Ca2+-responsive kinases, Pyk-2, and Src tyrosine kinase. A) T84 monolayers were preincubated with the Src kinase inhibitor, PP2 (20 μM; 30 min; n = 4) prior to addition of H2O2, and subsequent Isc responses to CCh (100 μM) were measured. Data are expressed as percentage inhibition of CCh-stimulated Isc response. B, C) H2O2 (500 μM)-induced phosphorylation of Src (Y416) (B; n=6) and Pyk-2 (Y881) (C; n=4), over time (0 –30 min) was determined by Western blot and densitometric analyses. Values are presented as means ± SE. *P<0.05; **P<0.01; ***P<0.001 vs. control (H2O2 + CCh in A; untreated cells in B, C).
H2O2 phosphorylates EGFR but does not recruit EGFR to mediate inhibition of Cl− secretion
As H2O2 inhibits CCh-stimulated Cl− secretion via an ERK-dependent pathway and because the EGFR is an upstream regulator of this kinase, we initially investigated whether H2O2 could activate EGFR. Bilateral H2O2 treatment increased EGFR tyrosine phosphorylation in T84 cell monolayers in both a concentration-(Fig. 4A; P<0.05–P<0.001 vs. control; n = 6) and a time-dependent (Fig. 4B; P<0.05 – P<0.01 vs. control; n = 6) manner. We next investigated the mechanisms by which H2O2 stimulated EGFR phosphorylation. Activation of EGFR by H2O2 (500 μM) was reduced by pretreatment of cells with either the EGFR kinase inhibitor, tyrphostin AG1478 (1 μM; Fig. 4C; P<0.01; n = 6), or a neutralizing anti-EGFR antibody (5 μg/ml, P<0.001) to block the EGFR ligand-binding domain. These data suggest that H2O2 phosphorylates the EGFR via the kinase activity of the EGFR itself and through the possible release and binding of an EGFR ligand. Since H2O2 inhibition of CCh-stimulated Cl− secretion is ERK and p38 dependent, we next investigated whether EGFR activation was required for stimulation of these downstream kinases. Preincubation with either tyrphostin AG1478 or a neutralizing antibody against the EGFR-ligand binding domain failed to significantly block activation of ERK or p38 by H2O2 (Fig. 4D, E). These data indicate that H2O2-induced activation of ERK and p38 occurs independently of EGFR activation. When we investigated whether EGFR activation was involved in the inhibitory effect of H2O2 on CCh-stimulated Cl− secretion, preincubation with tyrphostin AG1478 had no effect on H2O2-induced inhibition of CCh-stimulated Cl− secretion by H2O2 (Fig. 4F). These data indicate that EGFR activation is likely not involved in H2O2-induced inhibition of Ca2+-dependent Cl− secretion.
Figure 4.
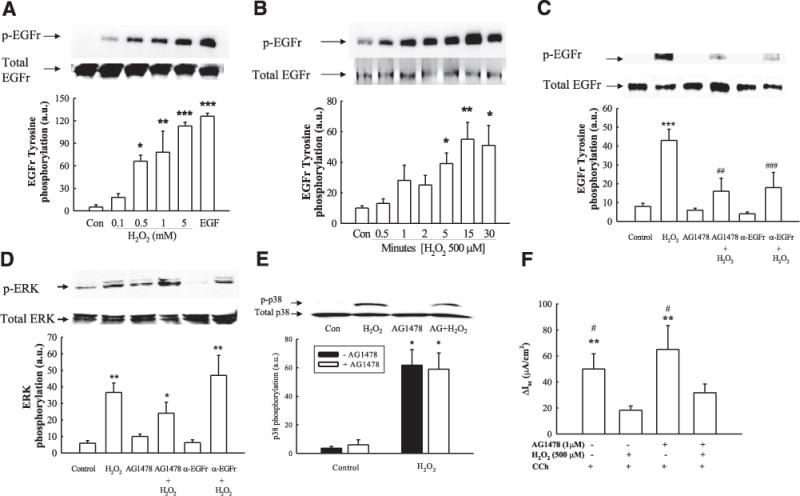
H2O2 stimulates EGFR tyrosine phosphorylation. A, B) T84 monolayers were bilaterally treated with a range of concentrations of H2O2 for 5 min (A; n=6) or with 500 μM H2O2 over different time points (B; n=6). EGFR immunoprecipitates were probed for tyrosine phosphorylation levels by Western blot analysis. Bands were quantified by densitometric analysis. C, D) Monolayers were preincubated with either the EGFR kinase inhibitor, tyrphostin AG1478 (1 μM), or a neutralizing antibody against the ligand-binding domain of the EGFR (5 μg/ml) to inhibit EGFR activation prior to treatment with H2O2. H2O2-induced EGFR (C; n=6), and ERK phosphorylation (D; n=6) was determined by Western blot analysis and densitometry. E) T84 monolayers were preincubated with tyrphostin AG1478 (1 μM), and H2O2-induced p38 phosphorylation was determined by Western blot analysis and densitometry (histogram; n=3). F) T84 monolayers mounted in Ussing chambers were pretreated with tyrphostin AG1478 (1 μM) prior to addition of H2O2, and subsequent Isc responses to CCh (100 μM) were measured (n=6). Results are presented as means ± SE for levels of phosphorylation (a.u., arbitrary units). *P<0.05; **P< 0.01; ***P<0.001 vs. control (untreated cells in A–E; H2O2 + CCh in F). #P<0.05; ##P<0.01; ###P<0.001vs. cells treated with H2O2 (C) or H2O2 + AG1478 + CCh (F).
Role of cytosolic calcium ([Ca2+]cyt) in signaling events mediated by H2O2
An elevation in [Ca2+]cyt levels is one of the major second messengers involved in the prosecretory actions of stimuli such as CCh and other GqPCR agonists. However, [Ca2+]cyt is also involved in the recruitment of antisecretory signaling pathways, including ERK and p38 (27, 41). H2O2 has been shown to cause an increase in [Ca2+]cyt levels in some cell systems. In addition, we have shown that H2O2 increases activation of the Ca2+-dependent kinase, Pyk-2, which translates elevations in [Ca2+]cyt to a tyrosine kinase-dependent signal. Consequently, we investigated whether H2O2 recruitment of signaling pathways involved in the negative regulation of Cl− secretion required elevations in [Ca2+]cyt. In fact, pretreatment of T84 monolayers with the [Ca2+]cyt chelator, BAPTA-AM (20 μM), reduced H2O2 activation of Src (Fig. 5A; P<0.05; n = 5) and ERK MAPK (Fig. 5B; P<0.001; n = 4). In addition, BAPTA-AM also partially reduced p38 activation (Fig. 5C; P<0.001; n = 7) and blocked Pyk-2 activation (Fig. 5D; P<0.01; n = 5).
Figure 5.
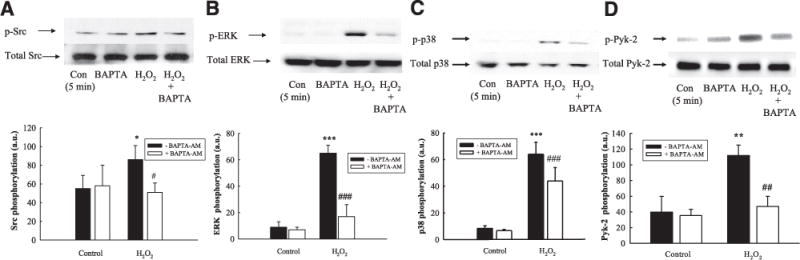
H2O2 increases Src, ERK, and p38 activation in a Ca2+-dependent manner. T84 monolayers were preincubated with the Ca2+ chelator, BAPTA-AM (20 μM) for 30 min prior to treatment with H2O2 (500 μM; 5 min). H2O2-induced phosphorylation of Src (Y416) (A; n=5), ERK (T202/Y204) (B; n=5), p38 (T180/Y182) (C; n=7), and Pyk-2 (Y881) (D) was assessed by Western blot analysis and subsequent densitometric analysis. Results are presented as means ± SE for levels of phosphorylation (a.u., arbitrary units). *P<0.05; **P<0.01; ***P<0.001 vs. control (untreated cells). #P<0.05; ##P<0.01; ###P 0.001 vs. cells treated with H2O2 alone.
To directly investigate whether H2O2 treatment alters [Ca2+]cyt levels, we performed digital calcium imaging by preincubating HT-29.cl19a colonic epithelial cells, grown on coverslips, for 30 min with the Ca2+-dye, Fura-2 (5 μM). HT-29.cl19a cells were used because they gave a robust and stable response to CCh, whereas repeated attempts with T84 cells grown on coverslips failed to show a stable response to CCh, possibly because of an inability to access muscarinic receptors that are restricted to the basolateral membrane. In complementary Ussing chamber studies, the Isc response to CCh in HT-29.cl19a cell monolayers pretreated with H2O2 (500 μM) was reduced to 39±20% that of the response seen in monolayers treated with CCh alone (4.0±0.5 vs. 1.7±1.0 μA/cm2; P<0.05; n = 3), a similar level of inhibition as seen in T84 cells. HT-29.cl19a cells grown on coverslips were mounted on an imaging stage. H2O2 (500 μM) caused a gradual increase in [Ca2+]cyt levels, which reached a plateau (Fig. 6A, B; P<0.001; n = 51). Since CCh stimulation of ion transport is Ca2+dependent, we then investigated whether H2O2 inhibited the ability of CCh to evoke a Ca2+ response. Cells grown on coverslips, preloaded with Fura-2, were incubated with H2O2 or HBSS for 30 min prior to treatment with CCh. The [Ca2+]cyt response to CCh (Fig. 6C) was partially reduced by H2O2 pretreatment (Fig. 6D; P<0.001; n = 51), but CCh still induced a robust calcium signal, as the level of inhibition by H2O2 was only 18±6% (Fig. 6E, P<0.001; n=51). These data suggest that H2O2 likely does not deplete intracellular Ca2+ stores sufficiently to block CCh-stimulated increases in [Ca2+]cyt levels that are adequate to drive a chloride secretory response.
Figure 6.
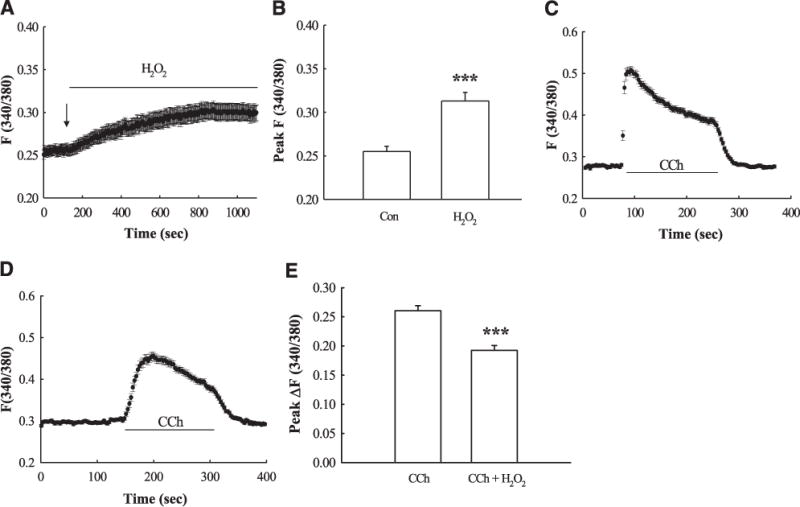
H2O2 increases [Ca2+]cyt levels. HT-29.cl19a cells grown on coverslips were preloaded with Fura-2 (5 μM; 30 min) followed by a 60-min wash in HBSS. A) Cumulative record of the change in [Ca2+]cyt following addition (signified by the arrow) of H2O2 (500 μM; n=51). B) Peak change in [Ca2+]cyt caused by H2O2 expressed as the fluorescence ratio, F340/380 (n=51). C) Cumulative trace of [Ca2+]cyt elevation caused by CCh (100 μM; n=51). D) Effect of pretreatment of H2O2 (30 min) on CCh-stimulated [Ca2+]cyt (n=51). E) Peak change in CCh-stimulated [Ca2+]cyt with or without H2O2 pretreatment (n=51). Results are presented as means ± SE. ***P 0.001<vs. control or CCh-treated cells.
Recruitment of multiple regulatory signaling pathways by H2O2
The recruitment of signaling molecules involved in the negative regulation of Ca2+-dependent Cl− secretion also involves stimulus-selective recruitment of specific signaling pathways. One example of this signaling divergence is the recruitment of phosphoinositide 3-kinase (PI3-K) signaling following activation of the EGFR by native ligand, but a lack of PI3-K recruitment following CCh-stimulated EGFR transactivation (48, 49). When we investigated whether PI3-K activation plays a role in mediating the inhibitory effects of H2O2 on CCh-stimulated Cl− secretion, preincubation with the PI3-K inhibitor, wortmannin (50 nM), significantly reduced the inhibitory effect of H2O2, thus indicating a prominent role for PI3-K activation in the inhibitory effect of H2O2 on Ca2+-dependent Cl− secretion (Fig. 7A; P<0.01; n = 4). These data were supported by studies using an alternative PI3-K inhibitor, LY294002 (20 μM). Pre-incubation of T84 cells with LY294002 significantly reduced the inhibitory effect of H2O2 on CCh-stimulated Cl− secretion (Fig. 7B; P<0.05; n = 5). H2O2 treatment also activated PI3-K signaling, as measured by increased phosphorylation of the downstream signaling target, Akt1 (Thr 308). This effect was shown to occur, at least in part, in a Ca2+-dependent manner as pretreatment with BAPTA significantly reduced Akt1 phosphorylation (Fig. 7C; P<0.05; n = 4). However, H2O2-stimulated Akt1 activation occurred independently of EGFR kinase activity as the EGFR inhibitor, tyrphostin AG1478, did not significantly inhibit Akt1 activation [7±2arbitrary units in untreated; 20±3 in H2O2 treated (P<0.01 vs. untreated); 9±3 in AG1478 treated; 15±2 in AG1478+H2O2 treated cells (P<0.01 vs. untreated); n = 5].
Figure 7.
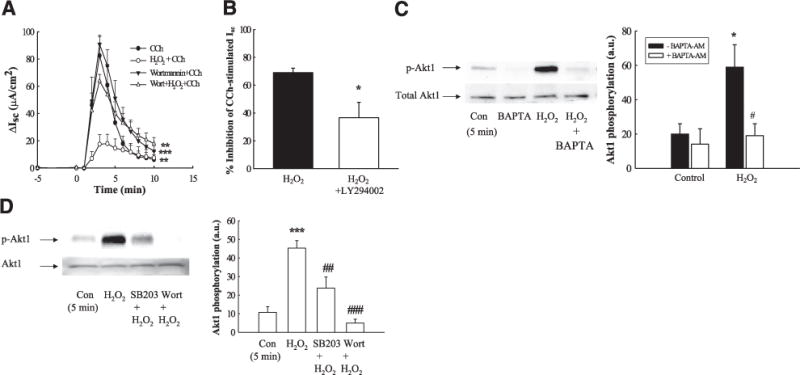
H2O2 inhibition of Isc involves PI3-K activation downstream of p38 activation. A) T84 monolayers mounted in Ussing chambers were treated with the PI3-K inhibitor, wortmannin (50 nM), for 30 min prior to addition of H2O2, and subsequent Isc responses to CCh (100 μM) were measured (n=4). B) T84 monolayers mounted in Ussing chambers were treated with the PI3-K inhibitor, LY294002 (20 μM), for 30 min prior to addition of H2O2, and subsequent Isc responses to CCh (100 μM) were measured (n=4). Data are expressed as percentage inhibition of CCh-stimulated Isc response. C) Monolayers were pretreated with BAPTA-AM (20 μM; 30 min) prior to incubation with H2O2 (500 μM; 5 min). Lysates were blotted for phosphorylation of the downstream PI3-K target, Akt1 (T308; n=4). D) T84 monolayers were preincubated for 30 min with the p38 inhibitor, SB203580 (10 μM; n=5), or the PI3-K inhibitor, wortmannin (50 nM; 30 min; n=5), and H2O2-stimulated Akt1 phosphorylation was measured. Results are means ± SE for phosphorylation (a.u., arbitrary units). *P<0.05; ***P<0.001 vs. H2O2 + CCh (A, B) or control (untreated cells; C, D). #P<0.05; ##P<0.01; ###P<0.001 vs. cells treated with H2O2 alone.
Because both PI3-K and p38 appeared to play prominent roles in mediating the inhibitory effect of H2O2 on CCh-stimulated Cl− secretion, we next investigated whether PI3-K and p38 activation were components of one, or separate, signaling pathways. T84 cells were pretreated with either the PI3-K inhibitor, wortmannin, or the p38 inhibitor, SB203580, for 30 min prior to treatment with H2O2 for 5 min, a time point where significant phosphorylation of both p38 (cf. Fig. 2E) and Akt1 (cf. Fig. 7C) had been observed. Wortmannin pretreatment exerted no inhibitory effect on H2O2-induced p38 phosphorylation (Fig. 8B; n = 4) but did inhibit phosphorylation of the PI3-K downstream target, Akt1, while SB203580 significantly reduced phosphorylation of Akt1 (Fig. 7D; P<0.01; n = 5). These data suggest that p38 activation in response to H2O2 treatment lies upstream of PI3-K.
Figure 8.
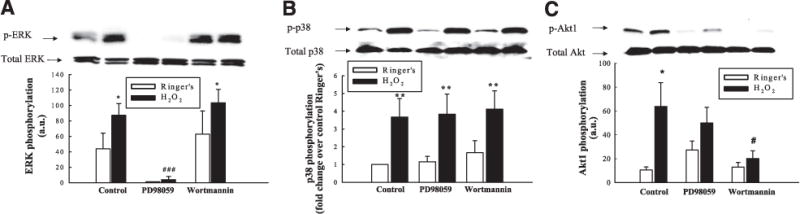
H2O2 activates multiple regulatory signaling pathways. T84 monolayers were preincubated with the MEK inhibitor, PD98059 (50 μM), or the PI3-K inhibitor, wortmannin (50 nM), for 30 min prior to treatment with H2O2 (500 μM; 5 min). Cell lysates were probed by Western blotting for ERK phosphorylation (A; n=3), p38 phosphorylation (B; n=4), and Akt1 phosphorylation (C; n=5). Results are presented as means ± SE for levels of phosphorylation (a.u., arbitrary units), or fold change over control for p38 phosphorylation (B). *P<0.05; **P<0.01 vs. control (untreated cells). #P<0.05; ###P<0.001 vs. cells treated with H2O2 alone.
We next investigated whether H2O2-induced ERK activation occurred in sequence or in parallel with the p38-PI3-K activation pathway. Preincubation with the MEK inhibitor, PD98059 (50 μM), significantly blocked H2O2-induced ERK phosphorylation (Fig. 8A; P<0.001; n = 3). However, PD98059 had no significant effect on H2O2-induced p38 (Fig. 8B; n = 4) or Akt1 phosphorylation (Fig. 8C; n = 5). These data suggest that H2O2-induced ERK activation does not lie upstream of p38 or PI3-K. Furthermore, the PI3-K inhibitor, wortmannin, which blocked Akt1 phosphorylation (Fig. 8C; P<0.05; n = 5), had no effect on ERK phosphorylation in response to H2O2 (Fig. 8A). This suggests that PI3-K signaling does not influence ERK activation. In addition, wortmannin had no effect on p38 phosphorylation (Fig. 8B), thus supporting our earlier findings that PI3-K lies downstream of p38 (cf. Fig. 7D). Collectively, these data demonstrate that H2O2 activates two separate signaling pathways that contribute to negative regulation of Ca2+-dependent ion transport responses.
Identity of ion transport proteins targeted by H2O2
We next set out to identify specific ion transport proteins that are involved in the inhibitory effect of H2O2. The secretory response to CCh is dependent on the activity of a number of ion transport proteins. A basolateral K+ efflux is essential to maintain the electrochemical gradient required to drive active Cl− secretion by CCh (50, 51). We therefore investigated whether CCh-stimulated opening of basolateral K+ channels is affected by H2O2 pretreatment. T84 monolayers were preloaded with 86Rb+, a surrogate for K+, prior to incubation with H2O2 (500 μM) or Ringer’s solution for 30 min. Cells were then treated with CCh (100 μM), and 86Rb+ efflux was determined at 2-min time intervals. H2O2 pretreatment significantly reduced CCh-stimulated 86Rb+ efflux, by 31 7% (Fig. 9A; P<0.05; n = 4). Treatment of control monolayers with H2O2 alone had no effect on basal 86Rb+ efflux (Fig. 9A). These data imply that H2O2 reduces CCh-stimulated opening of basolateral K+ channels.
Figure 9.
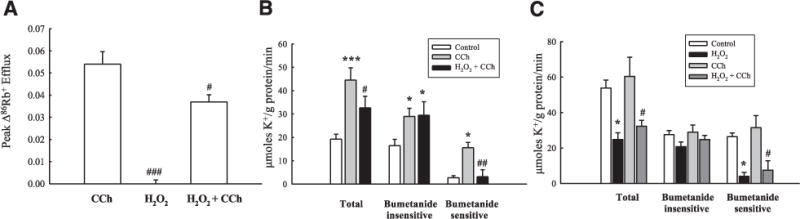
H2O2 inhibits basolateral K+ efflux and NKCC1 activity. A) T84 monolayers were preloaded with the K+ tracer 86Rb+ and treated with H2O2 (500 μM) bilaterally for 30 min prior to basolateral treatment with CCh. The efflux of 86Rb+ into the basolateral bathing solution was measured by scintillation counting. Data were expressed as mean ± SE peak rate of 86Rb+ efflux. B) T84 monolayers were treated with H2O2 for 30 min prior to basolateral incubation in CCh for 1 min and transfer to a basolateral 86Rb+-containing buffer for 3 min (n=8). The bumetanide insensitive and sensitive components of the response were assessed by incubating inserts in bumetanide (10 μM) concurrently with CCh treatment (n=5). C) T84 monolayers were depleted of intracellular Cl− for 60 min prior to treatment as in B, but with H2O2 and CCh solutions prepared in Cl−-free buffer. Cells were then transferred to basolateral 86Rb+ buffer containing Cl− (n=9). Data are presented as mean ± SE rate of K+ uptake (μmol K+/g protein/min). *P<0.05; ***P<0.001 vs. control (untreated cells). #P<0.05; ##P<0.01; ###P<0.001 vs. cells treated with CCh alone.
Basolateral entry of Cl− and K+ into colonic epithelial cells is mediated by the Na+-K+-2Cl− cotransporter, NKCC1, while 3 K+ ions also enter the cell via the energy-consuming Na+-K+-ATPase pump in exchange for 2 Na+ ions (52). The coordinated activation of both of these transporters is required to create the concentration gradient necessary for electrogenic Cl− secretion to occur. Therefore, we investigated whether H2O2 attenuated the secretory response to CCh by inhibiting the influx of K+ ions. H2O2 pretreatment (500 μM; 30 min) had no effect on baseline K+ influx, as measured by influx of the K+ surrogate, 86Rb+, while acute treatment with H2O2 for 1 min also had no effect on K+ influx (data not shown). However, H2O2 did significantly reduce CCh-stimulated K+ influx by 51±14% (Fig. 9B; P<0.05; n=4). To investigate the role of NKCC1 in CCh-stimulated K+ influx, we used the NKCC1 inhibitor, bumetanide (10 μM) to determine the ability of H2O2 to inhibit the bumetanide-sensitive component of CCh-stimulated K+ influx. In the presence of bumetanide, residual uptake of K+ (bumetanide-insensitive uptake) in CCh-treated tissues was identical in the presence or absence of H2O2. When the bumetanide-insensitive component was subtracted from the total K+ uptake to yield the bumetanide-sensitive K+ uptake, CCh-stimulated uptake was completely blocked in H2O2 pretreated cells. These data indicate that H2O2 strongly inhibits CCh-stimulated NKCC1 activity. During the transition to the secreting state, activation of NKCC1 via phosphorylation depends critically on the decrease in intracellular Cl− concentration that accompanies regulated apical Cl− exit (53). To determine whether the inhibitory effect of H2O2 on NKCC1 activity occurred secondary to inhibition of apical Cl− channels, we measured K+ influx in Cl−-depleted T84 cells. This was accomplished by preincubating T84 cells in a Cl−-free isethionate medium prior to treatment with H2O2. In unstimulated Cl−-depleted cells, K+ influx was 2.8-fold higher than in control cells containing normal Cl− (cf. Fig. 9B), and CCh did not induce a further increase in K+ uptake. The elevated basal K+ influx presumably reflected a stimulation of NKCC1 in response to Cl− depletion alone (Fig. 9C). The elevated uptake of K+ was inhibited by H2O2 pretreatment (P<0.05; n=9). In the presence of bumetanide, the bumetanide-insensitive uptake was equivalent under all four treatment conditions. The bumetanide-sensitive uptake of K+ in control and CCh-treated cells was dramatically inhibited by H2O2 pretreatment (P<0.05; n=9). This demonstrates that when the contribution of apical Cl− efflux to NKCC1 activation is removed, H2O2 still inhibits NKCC1 activity. Therefore, these data strongly suggest that H2O2 inhibits NKCC1 independently of possible alterations in intracellular Cl− concentration.
DISCUSSION
In the present study, we have demonstrated that H2O2 inhibits Ca2+-dependent epithelial Cl− secretion by activating specific signaling pathways that inhibit membrane ion transport proteins. This inhibitory effect supports previous findings of H2O2 inhibition of cAMP-dependent Cl− secretion (25). Moreover, in agreement with these studies, H2O2 (500 μM) had no effect on T84 transepithelial resistance, in contrast to the effects of incubation for longer durations or with higher concentrations of H2O2 (15, 24). This indicates that our observed effects of H2O2 on ion transport regulation are not due to nonspecific damage to the epithelial monolayer. Other groups have observed stimulatory effects of H2O2 on epithelial Isc. We did not detect a significant stimulatory effect of H2O2 on epithelial ion transport (not shown). Our findings and those of other groups suggest that at lower experimental concentrations (i.e., 500 μM), H2O2 exerts an inhibitory effect on Isc, whereas at higher concentrations (i.e., 2.2–5.5 mM), a small stimulatory response can occur (24).
H2O2 has emerged as an important transducer of cellular signals, including mediating responses to growth factors such as EGF (54), as well as participating in the activation of MAPK and NF-κB signaling pathways (55, 56). In addition, we have previously established the involvement of EGFR and MAPK pathways in the regulation of Ca2+-dependent Cl− secretion in intestinal epithelial cells (31, 43, 46, 48, 57). Here, we observed that H2O2 activated a number of discreet signaling pathways that occurred, at least in part, downstream of elevations in cytosolic Ca2+levels. H2O2 has been shown in a number of systems to modify [Ca2+]cyt concentrations (58, 59). Indeed, the pattern of a slow and sustained elevation in [Ca2+]cyt, similar to that seen in Fig. 6A, has been described in H2O2-treated pancreatic acinar cells (60). At this stage, we have not identified the H2O2-responsive Ca2+ pool that was activated in these studies, or whether H2O2 increases the influx of extracellular Ca2+.
The elevation in [Ca2+]cyt was not sufficient to prevent CCh from mounting a robust Ca2+ response. This fact is important from the perspective of continued Ca2+-dependent processes in the face of exposure to an inflammatory mediator. Moreover, H2O2-induced elevations in [Ca2+]cyt were able to activate two separate signaling pathways. We have previously established a role for the Ca2+-responsive soluble kinases, Pyk-2, and Src in the regulation of CCh-stimulated Cl− secretion (30, 43). In this current study, we similarly observed a role for Ca2+ in the activation of ERK MAPK, downstream of Src and Pyk-2, and a separate pathway involving p38 MAPK activation and downstream activation of PI3-K signaling. The kinetics of activation of these signaling kinases correlates with their upstream role in mediating H2O2 inhibition of CCh-stimulated Isc, which was highly significant after 15 min pretreatment (cf. Fig. 1B). The transient nature of the activation of certain kinases, i.e., ERK, suggests a discrete level of control is in operation to prevent consequences arising from sustained activation of these mediators. We have previously reported the potentiating effect of inhibitors of ERK, p38, PI3-K, and Src, on CCh-stimulated Cl− secretion. However, in order to clearly demonstrate the role of Src and PI3-K in H2O2-mediated inhibition of CCh-stimulated Isc, these data were presented as the % inhibition of CCh-stimulated Isc, consequently masking the definite potentiating effect of PP2 and LY294002 on the CCh response. Although the ERK and p38 inhibitors did not show a clear potentiating effect on CCh-stimulated Isc (in monolayers not treated with H2O2), this may have been due to the lower experimental number than used in our previous studies to specifically demonstrate a potentiating effect of ERK and p38 inhibition (31, 46). Interestingly, EGFR, which lies upstream of MAPK signaling in response to a number of stimuli, was not involved in the inhibitory effect of H2O2 on CCh-stimulated Cl− secretion, even though H2O2 did increase EGFR phosphorylation. Furthermore, EGFR activation appears to occur via the release of an EGFR ligand, as H2O2-induced EGFR phosphorylation was dramatically inhibited by a neutralizing antibody against the ligand-binding domain of the EGFR. On the basis of our previous studies of EGFR transactivation in these cells in response to a Ca2+-generating agonist (CCh), the ligand responsible for H2O2-induced EGFR transactivation is likely transforming growth factor-α (TGF-α), although this remains to be confirmed (38). The significance of H2O2-induced EGFR activation in these experiments is unclear, although there is conflicting evidence for a protective role of EGFR activation against H2O2-induced increases in the permeability of Caco-2 intestinal epithelial monolayers (61, 62). Moreover, the reason why H2O2-induced EGFR phosphorylation becomes uncoupled from downstream signaling pathways is unknown but is worthy of additional study.
Two key events involved in the Cl− secretory response to CCh are the efflux of K+ ions through basolateral K+ channels and the NKCC1-mediated ba-solateral uptake of Cl− ions (in conjunction with Na+ and K+) that replenishes intracellular Cl− lost through apical Cl− channels. Our data indicate that H2O2 inhibits both of these processes. Although DuVall et al. (25) demonstrated that acute treatment with H2O2 could increase apical to basolateral K+ transport, we did not observe an increase in basolateral 86Rb+ efflux in response to H2O2. However, our studies were not conducted in the presence of the Na+-K+-ATPase inhibitor, ouabain, which potentiates the basolateral K+ conductance response by inhibiting reuptake of 86Rb+ through the Na+ pump. Previous studies have demonstrated that H2O2 can inhibit cAMP-stimulated apical Cl− efflux (25, 26). Nevertheless, by depleting T84 monolayers of Cl−, we were able to demonstrate that H2O2 inhibits NKCC1 activity independent of effects on apical Cl− conductance. NKCC1 regulation is mediated in part by phosphorylation of threonine residues on the N terminus of NKCC1 (63–66). Furthermore, internalization of NKCC1 plays an important role in terminating the involvement of NKCC1 in CCh-stimulated ion transport in both cultured colonic epithelial cells and isolated human colonic crypts (67, 68). Interestingly, in Calu-3 airway cells, basolateral application of H2O2 potentiated forskolin-stimulated Isc responses by increasing NKCC1 activity, while apical application of H2O2 inhibited Isc responses (69). In addition to differences between cAMP- and Ca2+-dependent ion transport, this observation likely reflects inherent differences between airway and intestinal epithelial ion transport processes, as we observed identical levels of inhibition of CCh-stimulated Isc in T84 cells exposed solely on the apical or basolateral surface to H2O2 (data not shown). The exact nature of NKCC1 regulation by H2O2, and the signaling events involved, are the focus of our current studies.
In conclusion, we have demonstrated that pathophysiologically relevant concentrations of H2O2 can inhibit Ca2+-dependent Cl− secretion by activating kinase signaling pathways that negatively regulate ion transport. In addition, H2O2 inhibits basolateral K+ efflux and the uptake of K+ and Cl− via NKCC1 (Fig. 10). In vivo, Ca2+-dependent Cl− secretion is a critical response to neuronal reflexes activated by changes in luminal contents, distension, and physical contact between the epithelium and luminal contents (70). Reduced Cl− secretion leads to reduced flushing of the crypt to remove noxious substances and pathogenic bacteria. Therefore, fluid secretion is an important component of the epithelial barrier and host protection, and when secretion is compromised, this likely contributes to a decrease in overall barrier effectiveness. Indeed, the importance of Cl− secretion was emphasized in a recent study demonstrating that inhibition of the CFTR Cl− channel in isolated airway epithelial cells induces an inflammatory profile similar to that observed in cystic fibrosis (71). Therefore, dysregulation of ion transport can strongly influence the inflammatory status of the mucosa. The diarrhea associated with IBD is believed to be due primarily to defective solute and fluid absorption, thus leading to retention of fluid in the lumen, rather than an excess of active secretion (19, 72, 73). Although most studies on ion transport defects in human and murine colitis have focused on altered expression of ion transport proteins as the primary cause of defective transport, we, and others have previously demonstrated that the ion-transporting properties of isolated tissues from mouse models of colitis can be restored, at least in part, by acute modification of regulatory signaling pathways, including those investigated in this study (26, 74). Therefore, in addition to defects in the structural integrity of the epithelial barrier itself (28, 75), inhibition of fluid secretion, through altered expression and/or regulation of ion transport proteins, might play an important role in facilitating bacterial colonization of the intestinal crypt and the consequent excessive inflammatory response associated with IBD.
Figure 10.
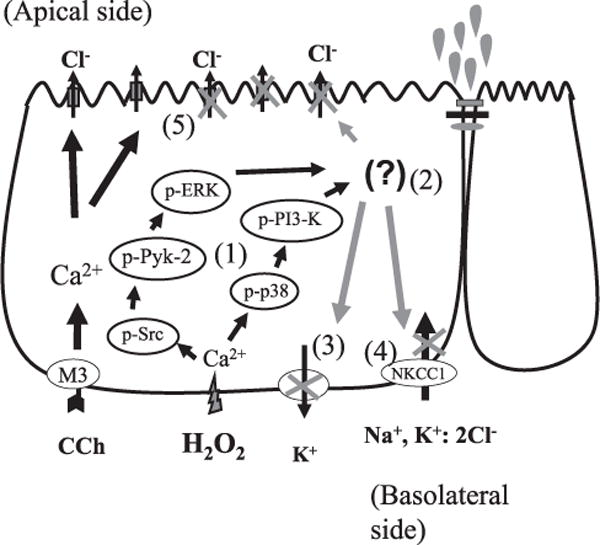
Integration of H2O2-activated regulatory signaling pathways. 1). H2O2 stimulates an increase in cytosolic Ca2+ concentration, which triggers two separate kinase pathways that are required for the overall inhibitory effect of H2O2 on Ca2+-dependent Cl− secretion. 2). Whether these pathways influence different downstream transport processes or converge via a downstream signal is unknown. 3). H2O2 also inhibits basolateral Ca2+-responsive K+ efflux. 4). H2O2 inhibits NKCC1 activity independently of possible effects on apical Cl− channels. 5). The inhibition of K+ efflux and NKCC1 activity likely contributes to the overall inhibitory effect of H2O2 on Ca2+-dependent ion transport in colonic epithelial cells.
Acknowledgments
We gratefully acknowledge the technical assistance of Linh Pham, Anahita Dashtei, and Sameer Gupta (University of California, San Diego). This research was funded by a Crohn’s and Colitis Career Development Award (to D.F.M.) and U.S. National Institutes of Health grant DK-28305 (to K.E.B.).
References
- 1.Grisham MB, Yamada T. Neutrophils, nitrogen oxides, and inflammatory bowel disease. Ann N Y Acad Sci. 1992;664:103–115. doi: 10.1111/j.1749-6632.1992.tb39753.x. [DOI] [PubMed] [Google Scholar]
- 2.Pavlick KP, Laroux FS, Fuseler J, Wolf RE, Gray L, Hoffman J, Grisham MB. Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease. Free Radic Biol Med. 2002;33:311–322. doi: 10.1016/s0891-5849(02)00853-5. [DOI] [PubMed] [Google Scholar]
- 3.Conner EM, Grisham MB. Inflammation, free radicals, and antioxidants. Nutrition. 1996;12:274–277. doi: 10.1016/s0899-9007(96)00000-8. [DOI] [PubMed] [Google Scholar]
- 4.Grisham MB. Oxidants and free radicals in inflammatory bowel disease. Lancet. 1994;344:859–861. doi: 10.1016/s0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 5.Harris ML, Schiller HJ, Reilly PM, Donowitz M, Grisham MB, Bulkley GB. Free radicals and other reactive oxygen metabolites in inflammatory bowel disease: cause, consequence or epiphenomenon. Pharmacol Ther. 1992;53:375–408. doi: 10.1016/0163-7258(92)90057-7. [DOI] [PubMed] [Google Scholar]
- 6.Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, Weissman GS, Katz S, Floyd RA, McKinley MJ, Fisher SE, Mullin GE. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci. 1996;41:2078–2086. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 7.Kruidenier L, Kuiper I, Van Duijn W, Mieremet-Ooms MA, van Hogezand RA, Lamers CB, Verspaget HW. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003;201:17–27. doi: 10.1002/path.1408. [DOI] [PubMed] [Google Scholar]
- 8.Holmes EW, Yong SL, Eiznhamer D, Keshavarzian A. Glutathione content of colonic mucosa: evidence for oxidative damage in active ulcerative colitis. Dig Dis Sci. 1998;43:1088–1095. doi: 10.1023/a:1018899222258. [DOI] [PubMed] [Google Scholar]
- 9.Keshavarzian A, Haydek J, Zabihi R, Doria M, D’Astice M, Sorenson JR. Agents capable of eliminating reactive oxygen species. Catalase, WR-2721, or Cu(II)2(3,5-DIPS)4 decrease experimental colitis. Dig Dis Sci. 1992;37:1866–1873. doi: 10.1007/BF01308081. [DOI] [PubMed] [Google Scholar]
- 10.Choudhary S, Keshavarzian A, Yong S, Wade M, Bocckino S, Day BJ, Banan A. Novel antioxidants zolimid and AEOL11201 ameliorate colitis in rats. Dig Dis Sci. 2001;46:2222–2230. doi: 10.1023/a:1011975218006. [DOI] [PubMed] [Google Scholar]
- 11.Oz HS, Chen TS, McClain CJ, de Villiers WJ. Antioxidants as novel therapy in a murine model of colitis. J Nutr Biochem. 2005;16:297–304. doi: 10.1016/j.jnutbio.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Test ST, Weiss SJ. Quantitative and temporal characterization of the extracellular H2O2 pool generated by human neutrophils. J Biol Chem. 1984;259:399–405. [PubMed] [Google Scholar]
- 13.Klebanoff SJ. Oxygen metabolites from phagocytes. In: Gallin JI, Goldstein IM, Snyderman R, editors. Inflammation: Basic Principles and Clinical Correlates. Raven Press; New York: 1992. pp. 541–588. [Google Scholar]
- 14.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 15.Watson AJ, Askew JN, Sandle GI. Characterisation of oxidative injury to an intestinal cell line (HT-29) by hydrogen peroxide. Gut. 1994;35:1575–1581. doi: 10.1136/gut.35.11.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kachur JF, Won-Kim S, Anglin C, Gaginella TS. Eicosanoids and histamine mediate C5a-induced electrolyte secretion in guinea pig ileal mucosa. Inflammation. 1995;19:717–725. doi: 10.1007/BF01534574. [DOI] [PubMed] [Google Scholar]
- 17.Keely SJ, Stack WA, O’Donoghue DP, Baird AW. Regulation of ion transport by histamine in human colon. Eur J Pharmacol. 1995;279:203–209. doi: 10.1016/0014-2999(95)00156-f. [DOI] [PubMed] [Google Scholar]
- 18.McCole DF, Otti B, Newsholme P, Baird AW. Complement activation of electrogenic ion transport in isolated rat colon. Biochem Pharmacol. 1997;54:1133–1137. doi: 10.1016/s0006-2952(97)00266-9. [DOI] [PubMed] [Google Scholar]
- 19.Sandle GI, Higgs N, Crowe P, Marsh MN, Venkatesan S, Peters TJ. Cellular basis for defective electrolyte transport in inflamed human colon. Gastroenterology. 1990;99:97–105. doi: 10.1016/0016-5085(90)91235-x. [DOI] [PubMed] [Google Scholar]
- 20.Kachur JF, Keshavarzian A, Sundaresan R, Doria M, Walsh R, de las Alas MM, Gaginella TS. Colitis reduces short-circuit current response to inflammatory mediators in rat colonic mucosa. Inflammation. 1995;19:245–259. doi: 10.1007/BF01534465. [DOI] [PubMed] [Google Scholar]
- 21.Gaginella TS, Kachur JF, Tamai H, Keshavarzian A. Reactive oxygen and nitrogen metabolites as mediators of secretory diarrhea. Gastroenterology. 1995;109:2019–2028. doi: 10.1016/0016-5085(95)90772-6. [DOI] [PubMed] [Google Scholar]
- 22.Karayalcin SS, Sturbaum CW, Wachsman JT, Cha JH, Powell DW. Hydrogen peroxide stimulates rat colonic prostaglandin production and alters electrolyte transport. J Clin Invest. 1990;86:60–68. doi: 10.1172/JCI114715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamai H, Kachur JF, Baron DA, Grisham MB, Gaginella TS. Monochloramine, a neutrophil-derived oxidant, stimulates rat colonic secretion. J Pharmacol Exp Ther. 1991;257:887–894. [PubMed] [Google Scholar]
- 24.Nguyen TD, Canada AT. Modulation of human colonic T84 cell secretion by hydrogen peroxide. Biochem Pharmacol. 1994;47:403–410. doi: 10.1016/0006-2952(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 25.DuVall MD, Guo Y, Matalon S. Hydrogen peroxide inhibits cAMP-induced Cl− secretion across colonic epithelial cells. Am J Physiol Cell Physiol. 1998;275:C1313–C1322. doi: 10.1152/ajpcell.1998.275.5.C1313. [DOI] [PubMed] [Google Scholar]
- 26.Walker J, Jijon HB, Churchill T, Kulka M, Madsen KL. Activation of AMP-activated protein kinase reduces cAMP-mediated epithelial chloride secretion. Am J Physiol Gastrointest Liver Physiol. 2003;285:G850–G860. doi: 10.1152/ajpgi.00077.2003. [DOI] [PubMed] [Google Scholar]
- 27.Grisham MB, Gaginella TS, von Ritter C, Tamai H, Be RM, Granger DN. Effects of neutrophil-derived oxidants on intestinal permeability, electrolyte transport, and epithelial cell viability. Inflammation. 1990;14:531–542. doi: 10.1007/BF00914274. [DOI] [PubMed] [Google Scholar]
- 28.Söderholm JD, Olaison G, Peterson KH, Franzén LE, Lindmark T, Wirén M, Tagesson C, Sjödahl R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut. 2002;50:307–313. doi: 10.1136/gut.50.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamai H, Gaginella TS, Kachur JF, Musch MW, Chang EB. Ca-mediated stimulation of Cl secretion by reactive oxygen metabolites in human colonic T84 cells. J Clin Invest. 1992;89:301–307. doi: 10.1172/JCI115576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keely SJ, Calandrella SO, Barrett KE. Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T(84) cells is mediated by intracellular Ca2+, PYK-2, and p60(src) J Biol Chem. 2000;275:12619–12625. doi: 10.1074/jbc.275.17.12619. [DOI] [PubMed] [Google Scholar]
- 31.Keely SJ, Uribe JM, Barrett KE. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells. Implications for carbachol-stimulated chloride secretion. J Biol Chem. 1998;273:27111–27117. doi: 10.1074/jbc.273.42.27111. [DOI] [PubMed] [Google Scholar]
- 32.Weymer A, Huott P, Liu W, McRoberts JA, Dharmsathaphorn K. Chloride secretory mechanism induced by prostaglandin E1 in a colonic epithelial cell line. J Clin Invest. 1985;76:1828–1836. doi: 10.1172/JCI112175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Augeron C, Laboisse CL. Emergence of permanently differentiated cell clones in a human colonic cancer cell line in culture after treatment with sodium butyrate. Cancer Res. 1984;44:3961–3969. [PubMed] [Google Scholar]
- 34.Cartwright CA, McRoberts JA, Mandel KG, Dharmsathaphorn K. Synergistic action of cyclic adenosine monophosphate- and calcium-mediated chloride secretion in a colonic epithelial cell line. J Clin Invest. 1985;76:1837–1842. doi: 10.1172/JCI112176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Resta-Lenert S, Barrett KE. Enteroinvasive bacteria alter barrier and transport properties of human intestinal epithelium: role of iNOS and COX-2. Gastroenterology. 2002;122:1070–1087. doi: 10.1053/gast.2002.32372. [DOI] [PubMed] [Google Scholar]
- 36.Shiratsuchi H, Basson MD. Activation of p38 MAP-Kalpha by extracellular pressure mediates the stimulation of macrophage phagocytosis by pressure. Am J Physiol Cell Physiol. 2005;288:C1083–C1093. doi: 10.1152/ajpcell.00543.2004. [DOI] [PubMed] [Google Scholar]
- 37.Dong H, Sellers ZM, Smith A, Chow JY, Barrett KE. Na+/Ca2+ exchange regulates Ca2+-dependent duodenal mucosal ion transport and HCO3− secretion in mice. Am J Physiol Gastrointest Liver Physiol. 2005;288:G457–G465. doi: 10.1152/ajpgi.00381.2004. [DOI] [PubMed] [Google Scholar]
- 38.Smith AJ, Chappell AE, Buret AG, Barrett KE, Dong H. 5-Hydroxytryptamine contributes significantly to a reflex pathway by which the duodenal mucosa protects itself from gastric acid injury. FASEB J. 2006;20:2486–2495. doi: 10.1096/fj.06-6391com. [DOI] [PubMed] [Google Scholar]
- 39.Venglarik CJ, Bridges RJ, Frizzell RA. A simple assay for agonist-regulated Cl− and K conductances in salt-secreting epithelial cells. Am J Physiol Cell Physiol. 1990;259:C358–C364. doi: 10.1152/ajpcell.1990.259.2.C358. [DOI] [PubMed] [Google Scholar]
- 40.Matthews JB, Awtrey CS, Madara JL. Microfilament-dependent activation of Na+/K+/2Cl-cotransport by cAMP in intestinal epithelial monolayers. J Clin Invest. 1992;90:1608–1613. doi: 10.1172/JCI116030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owen NE, Prastein ML. Na/K/Cl cotransport in cultured human fibroblasts. J Biol Chem. 1985;260:1445–1451. [PubMed] [Google Scholar]
- 42.Rao R, Baker RD, Baker SS. Inhibition of oxidant-induced barrier disruption and protein tyrosine phosphorylation in Caco-2 cell monolayers by epidermal growth factor. Biochem Pharmacol. 1999;57:685–695. doi: 10.1016/s0006-2952(98)00333-5. [DOI] [PubMed] [Google Scholar]
- 43.McCole DF, Keely SJ, Coffey RJ, Barrett KE. Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor-alpha. J Biol Chem. 2002;277:42603–42612. doi: 10.1074/jbc.M206487200. [DOI] [PubMed] [Google Scholar]
- 44.Basuroy S, Seth A, Elias B, Naren AP, Rao R. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J. 2006;393:69–77. doi: 10.1042/BJ20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim YM, Song EJ, Seo J, Kim HJ, Lee KJ. Proteomic analysis of tyrosine phosphorylations in vascular endothelial growth factor- and reactive oxygen species-mediated signaling pathway. J Proteome Res. 2007;6:593–601. doi: 10.1021/pr060326s. [DOI] [PubMed] [Google Scholar]
- 46.Keely SJ, Barrett KE. p38 mitogen-activated protein kinase inhibits calcium-dependent chloride secretion in T84 colonic epithelial cells. Am J Physiol Cell Physiol. 2003;284:C339–C348. doi: 10.1152/ajpcell.00144.2002. [DOI] [PubMed] [Google Scholar]
- 47.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 48.McCole DF, Truong A, Bunz M, Barrett KE. Consequences of direct versus indirect activation of epidermal growth factor receptor in intestinal epithelial cells are dictated by protein-tyrosine phosphatase 1B. J Biol Chem. 2007;282:13303–13315. doi: 10.1074/jbc.M700424200. [DOI] [PubMed] [Google Scholar]
- 49.Uribe JM, Keely SJ, Traynor-Kaplan AE, Barrett KE. Phosphatidylinositol 3-kinase mediates the inhibitory effect of epidermal growth factor on calcium-dependent chloride secretion. J Biol Chem. 1996;271:26588–26595. doi: 10.1074/jbc.271.43.26588. [DOI] [PubMed] [Google Scholar]
- 50.Dharmsathaphorn K, Pandol SJ. Mechanism of chloride secretion induced by carbachol in a colonic epithelial cell line. J Clin Invest. 1986;77:348–354. doi: 10.1172/JCI112311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirota CL, McKay DM. Cholinergic regulation of epithelial ion transport in the mammalian intestine. Br J Pharmacol. 2006;149:463–479. doi: 10.1038/sj.bjp.0706889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- 53.Lytle C, Forbush B. Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am J Physiol Cell Physiol. 1996;270:C437–C448. doi: 10.1152/ajpcell.1996.270.2.C437. [DOI] [PubMed] [Google Scholar]
- 54.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 55.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 56.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κB transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uribe JM, Gelbmann CM, Traynor-Kaplan AE, Barrett KE. Epidermal growth factor inhibits Ca2+-dependent Cl− transport in T84 human colonic epithelial cells. Am J Physiol Cell Physiol. 1996;271:C914–C922. doi: 10.1152/ajpcell.1996.271.3.C914. [DOI] [PubMed] [Google Scholar]
- 58.Zheng Y, Shen X. H2O2 directly activates inositol 1,4,5-trisphosphate receptors in endothelial cells. Redox Rep. 2005;10:29–36. doi: 10.1179/135100005X21660. [DOI] [PubMed] [Google Scholar]
- 59.González A, Granados MP, Pariente JA, Salido GM. H2O2 mobilizes Ca2+ from agonist- and thapsigargin-sensitive and insensitive intracellular stores and stimulates glutamate secretion in rat hippocampal astrocytes. Neurochem Res. 2006;31:741–750. doi: 10.1007/s11064-006-9078-y. [DOI] [PubMed] [Google Scholar]
- 60.Granados MP, Salido GM, González A, Pariente JA. Dose-dependent effect of hydrogen peroxide on calcium mobilization in mouse pancreatic acinar cells. Biochem Cell Biol. 2006;84:39–48. doi: 10.1139/o05-150. [DOI] [PubMed] [Google Scholar]
- 61.Banan A, Choudhary S, Zhang Y, Fields JZ, Keshavarzian A. Oxidant-induced intestinal barrier disruption and its prevention by growth factors in a human colonic cell line: role of the microtubule cytoskeleton. Free Radic Biol Med. 2000;28:727–738. doi: 10.1016/s0891-5849(00)00160-x. [DOI] [PubMed] [Google Scholar]
- 62.Forsyth CB, Banan A, Farhadi A, Fields JZ, Tang Y, Shaikh M, Zhang LJ, Engen PA, Keshavarzian A. Regulation of oxidant-induced intestinal permeability by metalloprotease-dependent epidermal growth factor receptor signaling. J Pharmacol Exp Ther. 2007;321:84–97. doi: 10.1124/jpet.106.113019. [DOI] [PubMed] [Google Scholar]
- 63.Payne JA, Xu JC, Haas M, Lytle CY, Ward D, Forbush B., 3rd Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J Biol Chem. 1995;270:17977–17985. doi: 10.1074/jbc.270.30.17977. [DOI] [PubMed] [Google Scholar]
- 64.Lytle C. Activation of the avian erythrocyte Na-K-Cl cotransport protein by cell shrinkage, cAMP, fluoride, and calyculin-A involves phosphorylation at common sites. J Biol Chem. 1997;272:15069–15077. doi: 10.1074/jbc.272.24.15069. [DOI] [PubMed] [Google Scholar]
- 65.Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem. 2002;277:37542–37550. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- 66.Dowd BF, Forbush B. PASK (prolinealanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2003;278:27347–27353. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- 67.Del Castillo IC, Fedor-Chaiken M, Song JC, Starlinger V, Yoo J, Matlin KS, Matthews JB. Dynamic regulation of Na+-K+-2Cl− cotransporter surface expression by PKC-å in Cl−–secretory epithelia. Am J Physiol Cell Physiol. 2005;289:C1332–C1342. doi: 10.1152/ajpcell.00580.2004. [DOI] [PubMed] [Google Scholar]
- 68.Reynolds A, Parris A, Evans LA, Lindqvist S, Sharp P, Lewis M, Tighe R, Williams MR. Dynamic and differential regulation of NKCC1 by calcium and cAMP in the native human colonic epithelium. J Physiol. 582:507–524. doi: 10.1113/jphysiol.2007.129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohashi T, Ito Y, Matsuno T, Sato S, Shimokata K, Kume H. Paradoxical effects of hydrogen peroxide on human airway anion secretion. J Pharmacol Exp Ther. 2006;318:296–303. doi: 10.1124/jpet.106.102541. [DOI] [PubMed] [Google Scholar]
- 70.Cooke HJ. Enteric tears: Chloride secretion and its neural regulation. News Physiol Sci. 1998;13:269–274. [PubMed] [Google Scholar]
- 71.Perez A, Issler AC, Cotton CU, Kelley TJ, Verkman AS, Davis PB. CFTR inhibition mimics the cystic fibrosis inflammatory profile. Am J Physiol Lung Cell Mol Physiol. 2007;292:L383–L395. doi: 10.1152/ajplung.00403.2005. [DOI] [PubMed] [Google Scholar]
- 72.Greig E, Sandle GI. Diarrhea in ulcerative colitis. The role of altered colonic sodium transport. Ann N Y Acad Sci. 2000;915:327–332. doi: 10.1111/j.1749-6632.2000.tb05260.x. [DOI] [PubMed] [Google Scholar]
- 73.Schmitz H, Barmeyer C, Gitter AH, Wullstein F, Bentzel CJ, Fromm M, Riecken EO, Schulzke JD. Epithelial barrier and transport function of the colon in ulcerative colitis. Ann N Y Acad Sci. 2000;915:312–326. doi: 10.1111/j.1749-6632.2000.tb05259.x. [DOI] [PubMed] [Google Scholar]
- 74.McCole DF, Rogler G, Varki N, Barrett KE. Epidermal growth factor partially restores colonic ion transport responses in mouse models of chronic colitis. Gastroenterology. 2005;129:591–608. doi: 10.1016/j.gastro.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 75.Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermüller N, Otto HF, Autschbach F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281:G216–G228. doi: 10.1152/ajpgi.2001.281.1.G216. [DOI] [PubMed] [Google Scholar]