

Abstract
The IFN gamma receptor 1 (IFNGR1) binds IFN-γ and activates gene transcription path ways crucial for controlling bacterial and viral infections. Although decreases in IFNGR1 surface levels have been demonstrated to inhibit IFN-γ signaling, little is known regarding the molecular mechanisms controlling receptor stability. Here, we show in epithelial and monocytic cell lines that IFNGR1 displays K48 polyubiquitination, is proteasomally degraded, and harbors three ubiquitin acceptor sites at K277, K279, and K285. Inhibition of glycogen synthase kinase 3 beta (GSK3β) destabilized IFNGR1 while overexpression of GSK3β increased receptor stability. We identified critical serine and threonine residues juxtaposed to ubiquitin acceptor sites that impacted IFNGR1 stability. In CRISPR–Cas9 IFNGR1 generated knockout cell lines, cellular expression of IFNGR1 plasmids encoding ubiquitin acceptor site mutations demonstrated significantly impaired STAT1 phosphoryl ation and decreased STAT1-dependent gene induction. Thus, IFNGR1 undergoes rapid site-specific polyubiquitination, a process modulated by GSK3β. Ubiquitination appears to be necessary for efficient IFNGR1-dependent gamma gene induction and represents a relatively uncharacterized regulatory mechanism for this receptor.
Introduction
There are three types of interferons: Class I; IFNα & β, Class II; IFN-γ, and Class III; IFN-λ1, 2 & 3 (also called IL29, IL28A, and IL28B). The cytokines IL-2, IL-12, IL-18 drive the production of IFN-γ mostly by natural killer, natural killer T, CD4 Th1, and CD8 cytotoxic T lymphocyte effector T cells. IFN-γ binds to cells expressing the two subunits of the interferon gamma receptor, interferon gamma receptor 1 (IFNGR1) and interferon gamma receptor 2 (IFNGR2). JAK1 (Janus kinase 1) and JAK2 (Janus kinase 2) are constitutively associated with the cytosolic domains of IFNGR1 and IFNGR2 receptor, respectively, and become activated after IFN-γ binding [1]. STAT1 is then recruited to the complex, is phosphorylated, and transported to the nucleus [2]. IFN-γ complex targets gamma-activating sequences within key promoters of genes 15–30 min after binding. Hundreds of genes are up-regulated either ubiquitously or in a tissue or cell type-specific manner. Approximately two-thirds of IFN-stimulated genes (ISGs) are STAT1-dependent [2]. IFN-γ signaling also increases the respiratory burst in macrophages by up-regulating NADPH-oxidase components, the inducible nitric oxide synthase, and antigen processing and presentation through enhanced expression of both MHC class I and II, and activation of the immunoproteasome. IFN-γ also skews the adaptive immune response by increasing TH1 cytokine production and enhancing apoptosis of TH2 cells [2]. Thus, IFN-γ signaling through its cognate receptors represents an indispensable mode of activity in innate immunity in response to a variety of virulent pathogens.
Human IFNGR1 is a single pass, 489 amino acid membrane receptor (mouse 477 AA) that is expressed in nearly all tissues. Impaired or inhibited expression in individuals with IFNGR1 mutations results in deficiencies in host responses to bacterial, parasitic, and viral infections [3,4]. These deficiencies have been replicated in IFNGR−/− mice. Several studies have demonstrated that degradation of IFNGR1 is enhanced in response to various stimuli including TLR2 stimulation and viral infection, resulting in decreased IFN-γ-mediated STAT1 phosphorylation and inhibited ISG transcription. Both lysosomal and proteasomal degradation have been implicated in IFNGR1 degradation [5–7]. However, the precise molecular mechanism whereby the IFNGR1 is degraded in cells remains relatively unknown. Identification of the regulatory pathways for IFNGR1 cellular disposal might lead to identification of molecular targets for therapeutics that could modulate surface receptor availability to alter immune responses to pathogens.
Ubiquitin is an 8.5 kDa protein that is transferred to target proteins by E1-activating enzymes, E2-conjugating enzymes, and E3 ligases, which ultimately generates an isopeptide bond between the substrate protein and ubiquitin. Ubiquitination can serve as the signal for protein degradation either via the proteasome or lysosome, or alter protein trafficking and/or signaling [8]. The ubiquitination and degradation of membrane proteins is well established and has been demonstrated in receptors such as the interferon alpha receptor, epidermal growth factor receptor (EGFR), Toll-like receptor 2, CFTR, Notch, E-Cadherin, etc. [9–12]. Of the post-translational modifications known to alter protein ubiquitination, phosphorylation is the most widely studied. Phosphorylation of substrates serves as a recognition signal to recruit some E3 ubiquitin ligases leading to substrate ubiquitination of proteins [13].
In the present study, we investigated the mechanisms that control the post-translation stability of the IFNGR1. Our data suggest that glycogen synthase kinase 3 beta (GSK3β) phosphorylates IFNGR1 thereby enhancing receptor protein stability by limiting ubiquitin proteasomal degradation. We identified the lysine ubiquitin acceptor sites and have uncovered adjacent GSK3β phosphorylation sites that modify IFNGR1 stability. Finally, we determined that lysine ubiquitin acceptor sites in IFNGR1 are necessary for efficient IFN-γ signaling. The molecular processes that dictate IFNGR1 lifespan in cells by co-ordinate actions of GSK3β and the ubiquitin-proteasome apparatus has important implications in the control of IFN-γ signaling.
Experimental
Cells
HEK 293FT cells were cultured with Dulbecco’s modified eagle medium containing 10% fetal bovine serum (FBS) and antibiotics. THP-1 and A549 cells were cultured in Roswell Park Memorial Institute medium supplemented with 10% FBS. To generate THP-1 macrophages, THP-1 monocytes were incubated with 20 nM PMA for 48 h. PMA media were then removed and replaced with fresh media for 24 h before treatment.
Chemicals used
The following reagents were purchased: bortezomib (Santa Cruz Biotech), cycloheximide (CHX) (FisherThermo Scientific), benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (BADGP) (Sigma), leupeptin (Sigma), PMA (Sigma), tunicamycin (Sigma), recombinant IFN-γ (Biolegend), TWS119 (Cayman Chemical). From CalBiochem: MG132, protein kinase G inhibitor, KN-93, bisindolylmaleimide I, ML-7, staurosporine, H-89. From TOCRIS Biosciences: U0126, SB 202190, SP 600125.
Plasmids, cloning, and transfection
Human IFNGR1 in a pLX304 backbone was ordered from the plasmid repository, DNASU. IFNGR1 mutants were created using the QuickChange site-directed mutagenesis kits (Agilent). Constructs were validated via sequencing. Plasmids were transfected into cells using XtremeGene transfection reagent (Roche) according to manufacturer’s protocol.
RNAi
DsiRNA against GSK3β was obtained from IDT oligonucleotides. DsiRNA #2: sense 5′-rGrArArUrCrUrGrCr CrArUrCrGrGrGrArUrArUrUrArArACC-3′, antisense 5′-rGrGrUrUrUrArArUrArUrCrCrCrGrArUrGrGrCr ArGrArUrUrCrCrA-3′; DsiRNA #3: sense 5′-rArArCrArUrGrCrUrCrArGrUrCrArArArCrCrArArArUrCAA-3′ antisense 5′–rUrUrGrArUrUrUrGrGrUrUrUrGrArCrUrGrArGrCrArUrGrUrUrUrC-3′; THP-1 and HEK were transfected with 30 nM DsiRNA using the GeneMute siRNA transfection reagent (SignaGen) according to manufacturer’s instructions.
Immunoblotting
Cell lysis and immunoblotting was performed as previously described [14]. Briefly, cells were lysed in PBS supplemented with 0.25% triton X and cOmplete, Mini Protease Inhibitor Cocktail (Roche). Lysates were then sonicated, and then centrifuged at 500×g to remove debris. An aliquot of sample containing total cellular protein was frozen at −80°C. Total cellular proteins, soluble, and membrane proteins were loaded on SDS–PAGE gels (Invitrogen) and processed for immunoblotting using the following antibodies: anti-E-Cadherin (Life Technologies, 13-5700), anti-hemagglutinin, anti-ubiquitin (LifeSensors, VU101), anti-K48-linked ubiquitin (Cell Signaling, 8081S), anti-K63-linked ubiquitin (Cell Signaling, 5621S), anti-CD172a (Millipore, 566310), anti-IFNGR1(Santa Cruz, sc-28363), anti-GSK3β (Cell Signaling, D613M), anti-V5 (Cell Signaling, 13202S), anti-GAPDH (Sigma), anti-STAT-1 (Cell Signaling, 9172P), anti-pSTAT-1 (Cell Signaling, 7649P), anti-EGFR (Cell Signaling, 4267S), and anti-β-actin mouse monoclonal antibody (Sigma). Immunoblots were exposed to Supersignal West Femto chemiluminescent substrate (Thermo Scientific).
Biotinylation of plasma membrane proteins
As previously described [9], cells were washed three times with PBS pH 8.0, and incubated with EZ-Link Sulfo-NHS-SS-biotin (Thermo Scientific) in PBS at pH 8.0 per manufacturer’s instructions. Cells were then incubated on ice for 15 min, and quenched three times with 50 mM Tris buffer. After washing with PBS, cells were lysed with RIPA buffer. Biotinylated proteins were captured with Neutravidin-coated Sepharose beads (Thermo Scientific) for 1 h at 4°C. Beads were then washed five times with PBS supplemented with 0.25% triton X to remove unbound protein and protein eluted with SDS sample buffer at 95°C for 5 min.
Immunoprecipitation
For ubiquitin experiments cells were isolated in lysis buffer supplemented with 10 mM of the deubiquitinase inhibitor N-ethylmaleimide (ThermoFisher Scientific). Between 500 and 1000 mg of total protein from cell lysates was precleared with protein G beads (ThermoFisher Scientific) for 30 min. Primary antibody was added for 2 h at room temperature. Protein G beads were then added for an additional 1 h of incubation. Beads were magnetically sorted and washed five times. The beads were heated at 95°C for 5 min with protein sample buffer prior to SDS–PAGE and immunoblotting. For TUBES (tandem ubiquitin binding entities) pull-down, pre-cleared samples were incubated with TUBES reagent (LifeSensors) for 2 h at 4°C, followed by washing, heating, and preparation as above.
Fluorescent immunostaining
As described previously [15], briefly, A549 cells were inoculated into glass-bottomed 35-mm plates and transiently transfected with indicated plasmids for 24 h. Cells were treated as indicated and washed with cold PBS twice and fixed with 4% paraformaldehyde for 30 min prior to incubation with permeabilization solution (0.1% Triton X-100 in PBS) for 15 min. The cells were then blocked in PBS + 2% BSA for 45 min and probed with a primary antibody in PBS + 0.5% BSA solution for 2 h or overnight. Plates were washed five times and incubated with fluorescence-conjugated secondary antibodies in PBS + 0.5% BSA solution for 1 h. Plates were then washed five times and stained with DAPI (1:5000) for 3 min. Images were acquired by a combination laser-scanning microscope system [Nikon A1, Nikon (Melville, NY)], and the results were analyzed using the Nikon NIS-Elements software.
qRT-PCR
RNA was isolated from cells using the RNeasy mini kits (Qiagen) according to the manufacturer’s protocol. Isolated RNA was immediately converted to cDNA using the high-capacity RNA-to-cDNA kits (Life Technologies). Real-time PCR assays were performed using SYBR® Select Master Mix for CFX (2×, Life Technologies) per the protocol provided in a C1000 Thermal Cycler (Bio-Rad). The following primers were used: IFNGR1 — sense 5′-GCTCTCCTCTTTCTCCTACC-3′, antisense 5′-AGTTGGTGTAGGCACTGAG-3′, IRF1, sense — 5′-GCTCATCTGGATTAATAAAGAGGAG-3′, antisense — 5′-CATCCTTGTTGATGTCCCAG-3′, UBE3A — sense 5′-GAAGCCGGAATCTAGATTTCC-3′, antisense 5′-TAATCAGAACAGAGTCCCTGG-3′; CXCL10 — sense 5′-ACGTGTTGAGATCATTGCT-3′, antisense 5′-GTAAATTCTTGATGGCCTTCG-3′; ICAM1 — sense 5′-ACCATCTACAGCTTTCCGG-3′; antisense 5′-ACACTTCACTGTCACCTCG-3′; ISG15 — sense 5′-GTCACGAATCGCTTAATTCTG-3′, antisense 5′-GACACCTGGAATTCGTTGC-3′; MX1 — sense 5′-TAATAAAGCCCAGAATGCCA-3′, antisense 5′-TTAGAGTCAGATCCGGGAC-3′; LMP7 — sense 5′-CTACATTAGTGCCTTACGGGT-3′, antisense 5′-TACTGACAGTCTGCTGCAC-3′; IFIT3 — sense 5′-AACAGCCATCATGAGTGAG-3′, antisense 5′-AAGTTCCAGGTGAAATGGC-3′.
CRISPR–Cas9
HEK cells were transfected with recombinant cas9, crRNA targeted against the IFNGR1 receptor gene locus (#1 AUUGUACACCCUAAUGUAACGUUUUAGAGCUAUGCU, #2 ACAUGAACCCUAUCGUAUAUG UUUUAGAGCUAUGCU) and trans-activating crRNA from IDT oligonucleotides. Cell were co-transfected with homology directed repair (HDR) plasmids (Santa Cruz) containing a puromycin resistance gene, an RFP fluorescent marker, and 5′ and 3′sequences homologous to three separate regions of IFNGR1. HDR integration resulted in the integration of puromycin resistance and RFP expression in the targeted sites. Cells were then treated with puromycin (Santa Cruz) (2 μg/ml) for 1–2 weeks for stable selection, and seeded into a 96-well plate for selection of single colonies. Individual clones were tested for IFNGR1 expression and for activation of STAT1 after IFN-γ treatment to confirm knockout.
Statistics
All Student’s t-test was used for statistical analysis for comparison of two groups. The comparison of statistical significance among three or more groups was determined by one-way analysis of variance followed by pairwise comparisons using Tukey’s t-test.
Results
IFNGR1 is proteasomally degraded through K48 mediated ubiquitination in epithelial and monocytic cells
To determine IFNGR1 stability, we examined endogenous expression of IFNGR1 in alveolar epithelial cell lines and measured its degradation in the presence of proteasomal and lysosomal inhibitors after the inhibition of new protein synthesis with CHX. Of note, distinct bands were not always observed on immunoblots for IFNGR1 due to considerable post-translational modification by glycosylation [16]. Proteasomal inhibition with MG132 significantly extended the half-life of IFNGR1 while lysosomal inhibitors had no significant effect on IFNGR1 stability (Figure 1a). To confirm that the lysosomal inhibitors were effective in our system, we treated A549 cells with epidermal growth factor (EGF) to stimulate lysosomal degradation of EGFR. Both bafilomycin, and to a lesser degree, leupeptin, prevented the degradation of EGFR at the concentrations used in (Figure 1a,b). To confirm proteasomal specificity, we treated cells with both MG132 and the more specific inhibitor bortezomib. Both inhibitors increased IFNGR1 expression in a dose-dependent manner (Figure 1c). Because mature membrane-associated receptors are frequently associated with lysosomal degradation, we directly examined proteasomal degradation of plasma membrane-associated IFNGR1. We biotinylated plasma membrane-associated proteins and measured the degradation of biotin labeled IFNGR1 in the presence or absence of MG132. Plasma membrane-associated IFNGR1 had a similar half-life to total IFNGR1 (Figure 1a) and was also stabilized in the presence of MG132 (Figure 1d). In contrast, the plasma membrane-associated protein E-cadherin (E-Cad) was stable over the same time frame. Degradation via the proteasome is preceded by ubiquitination. Thus, when cells were overexpressed with HA-ubiquitin in our system we observed that IFNGR1 protein half-life was decreased (Figure 1e). To confirm receptor ubiquitination, we overexpressed a plasmid encoding IFNGR1 in HEK cells and immunoprecipitated proteins in the presence of MG132. Ubiquitinated IFNGR1 increased in a time-dependent manner after blocking proteasomal degradation (Figure 1f). In addition, we observed lysine 48 (K48) ubiquitin linkages that are characteristic of proteins processed via proteasomal degradation. To confirm K48-linked ubiquitination of IFNGR1, we transfected HEK cells with plasmids encoding wild-type (Wt) ubiquitin, lysine-less ubiquitin (K-R), and ubiquitin mutants retaining only lysine 48 (R48K) or 63 (R63K). Both Wt ubiquitin and R48K permitted the formation of a poly-ubiquitin chain (Figure 1g). However, K-R ubiquitin and R63K ubiquitin did not allow the formation of a high-molecular-weight poly-ubiquitin smear suggesting that lysine 48 is necessary and sufficient for the polyubiquitination of IFNGR1. Although K48 ubiquitination was observed, it appeared that the level of ubiquitination was higher in cells expressing Wt ubiquitin. These data suggest that other lysine linkages (K6, K11, K27, K29, K33, linear) might be involved in the degradation of the receptor. Overall, these data suggest that IFNGR1 has a short half-life due to K48 (and possibly other lysine) linked polyubiquitination and proteasomal degradation. The results do not exclude other modes for IFNGR1 degradation including the lysosome.
Figure 1. IFNGR1 is ubiquitinated and degraded by the proteasome.
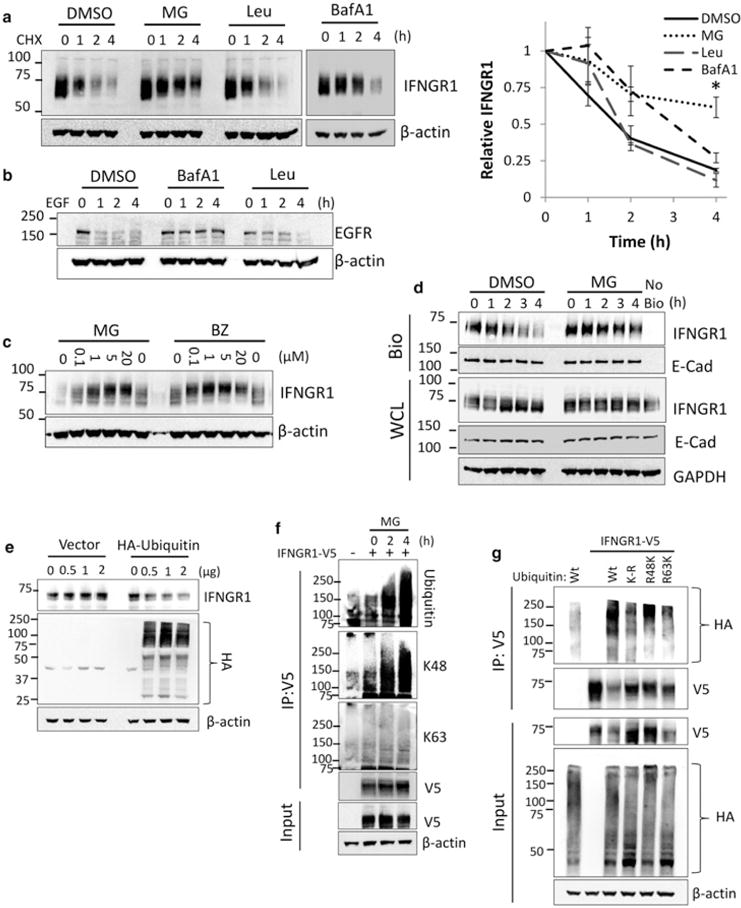
(a) A549 cells were treated with CHX for the indicated times in the presence of DMSO, MG132 (10 μM), bafilomycin A1 (BafA1) (100 nM) or leupeptin (Leu) (100 μM). Cells were then harvested and probed for IFNGR1 expression. Right: IFNGR1 protein densitometry normalized to β-actin and CHX 0 h to determine protein half-life. n = 3–4 independent experiments, ±SE. *P < 0.05 vs. control by ANOVA and Tukey’s t-test. (b) A549 cells were treated with EGF (100 μM) for the indicated times in the presence of DMSO, BafA1, or Leu at the concentrations above. Cells were then harvested and probed for EGFR expression. (c) A549 cells were treated with indicated concentrations of MG132 or bortezomib (BZ) for 4 h, harvested, and probed for IFNGR1 expression. (d) A549 cells were incubated with membrane impermeable biotin at 4°C, returned to the incubator and treated with DMSO or MG132 for indicated times. Cells were probed for plasma membrane (biotinylated, [Bio]) and whole cellular lysates (WCL) IFNGR1. E-cadherin was included as a plasma membrane marker, no bio = no biotinylation control. n = 2 independent experiments (e) HEK cells were transfected with HA-ubiquitin or an empty vector control. At 24 h, cells were harvested and probed for endogenous IFNGR1. The middle blot below shows levels of total cellular ubiquitination. (f) HEK cells were transfected with V5-tagged IFNGR1. At 2 and 4 h prior to harvest cells were treated with 10 μM MG132. 48 h post-transfection cell lysates were boiled, immunoprecipitated (IP) for V5, and probed for total ubiquitin, K48-linked ubiquitin, and K63-linked ubiquitin; n = 3 independent experiments. (g) HEK cells were transfected with V5-tagged IFNGR1 and Wt, lysine-less (K-R), lysine 48 only (R48K), and lysine 63 only (R63K) HA-tagged ubiquitin plasmids. At 4 h prior to harvest cells were treated with 10 μM MG132. 48 h post-transfection cell lysates were boiled, subjected to IP for V5, and probed for HA; n = 2 independent experiments.
Ligand stimulation with IFN-γ does not alter receptor degradation
Frequently, ligand binding leads to rapidly increased receptor degradation via post-translation modifications [17]. We therefore examined IFNGR1 stability in response to IFN-γ treatment. To determine whether ligand stimulation altered IFNGR1 mRNA stability in differentiated THP-1, we measured IFNGR1 mRNA via qRT-PCR at 2 and 6 h post IFN-γ treatment. We observed no differences in IFNGR1 stability (Figure 2a). As a positive control, we treated THP-1 with IFN-β which has been previously shown to decrease IFNGR1 mRNA [18]. As expected, in contrast with IFN-γ treatment, treatment with IFN-β led to a dramatic decrease in IFNGR1 mRNA. Next, to examine post-translational changes in IFNGR1, we treated THP-1 with IFN-γ and measured changes in IFNGR1 stability over a 2-h period (Figure 2b). No significant alteration of total IFNGR1 expression was observed. To confirm that this effect was consistent in multiple cell types, we treated A549 treated with varying concentrations of IFN-γ over 4 h and also showed no changes in IFNGR1 expression (Figure 2c). To determine whether the plasma membrane pool might be preferentially altered, we examined cell surface IFNGR1 via biotinylation (Figure 2d). We did not observe significant differences in plasma membrane IFNGR1 after ligand stimulation. Finally, we examined IFNGR1 half-life via cycloheximide chase in differentiated THP-1. IFNGR1 stability was not altered over a 4 h treatment with IFN-γ (Figure 2e). These data suggest that IFNGR1 protein expression is not significantly altered by ligand stimulation.
Figure 2. IFN-γ treatment does not alter IFNGR1 expression.
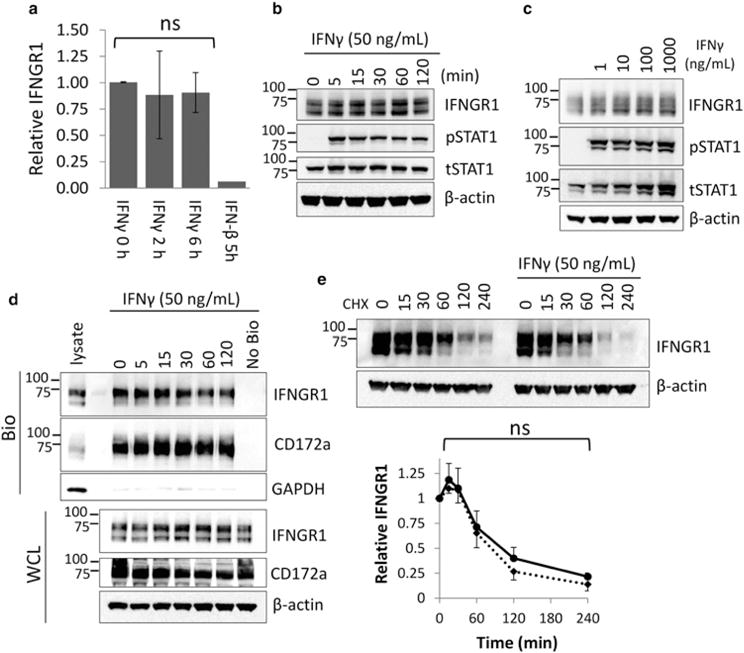
(a) Differentiated THP-1 cells were treated with 50 ng/ml IFN-γ for 2 and 6 h or 50 ng/ml IFN-β. Total RNA was harvested, converted to cDNA, and probed for the indicated genes via qRT-PCR using IFNGR1-specific primers. n = 3 independent experiments, ±SE. ns, not significant to 0 h control by Student’s t-test. (b) THP-1 differentiated macrophages were treated with IFN-γ at 50 ng/ml for the indicated times, harvested, and immunoblotted for IFNGR1, phosphorylated STAT1, and total STAT1. (c) A549 cells were treated with indicated concentrations of IFN-γ for 4 h. Cells were harvested and lysates immunoblotted for indicated proteins. (d) THP-1 differentiated macrophages were treated with IFN-γ at 50 ng/ml for the indicated times prior to incubation with membrane impermeable biotin. Whole cell lysates (WCL) and biotinylated plasma membrane proteins (biotinylated, [Bio]) were probed for IFNGR1 and CD172a as a membrane control. (e) THP-1 differentiated macrophages were treated with CHX alone or CHX + IFN-γ for indicated times. Cells were harvested and lysates immunoblotted for IFNGR1. IFNGR1 densitometry normalized to β-actin expression; n = 3 independent experiments, ±SE. ns, not significant vs. CHX only control by Student’s t-test.
Mutation of specific lysines increases IFNGR1 stability
Because we observed that ubiquitination played a role in IFNGR1 stability, we made a series of lysine to arginine point mutations within putative ubiquitin acceptor sites (Figure 3a and Supplementary Figure S1). We then measured the half-life of overexpressed IFNGR1 in HEK cells after CHX treatment. Mutation of membrane proximal lysines 277, 279, and 285 had the most substantial effect on receptor protein stability (Figure 3b). We, therefore, mutated all three lysines and measured IFNGR1 half-life. Expression of a plasmid encoding a triple mutant of all three lysines led to robust increased receptor expression of this IFNGR1 variant. Although we were unable to see differences among the receptor variants in protein half-life in the short-time course initially measured (Figure 3c), at later time points, the triple mutant was more stable than either the single or double mutants (Figure 3d,e). We then transfected HEK cells with V5-tagged IFNGR1 K-R mutants and immunoprecipitated proteins from lysates using the TUBEs reagent to capture all ubiquitinated proteins. The IFNGR1 277, 279, and 285 triple mutant plasmid showed a decreased level of ubiquitination in comparison with the Wt protein or the 271, 272 double mutant (Figure 3f).
Figure 3. Lysine ubiquitin acceptor sites impact IFNGR1 stability.
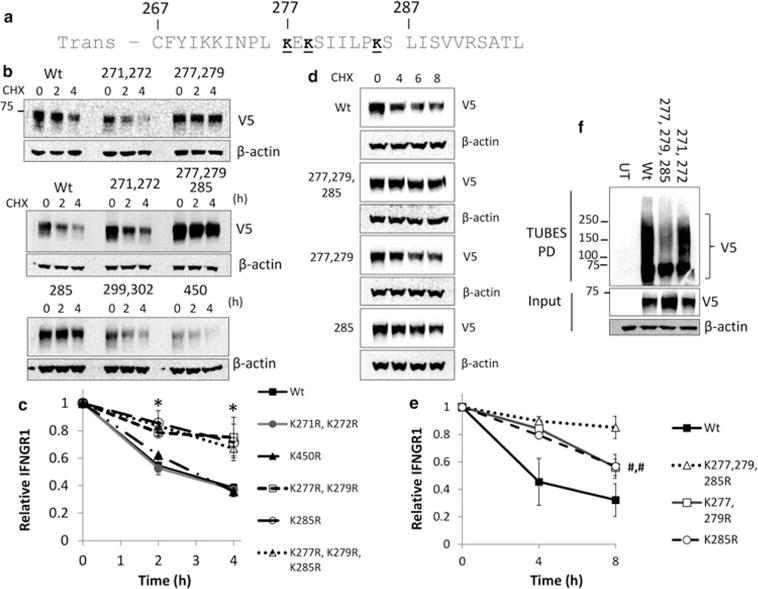
(a) Primary sequence of the IFNGR1 cytoplasmic juxtamembrane region. Putative residues necessary for protein stability and ubiquitination are underlined. Trans, transmembrane domain. (b–e) HEK Cells were transfected with Wt or various IFNGR1 V5 lysine to arginine mutant plasmids to determine the role of putative ubiquitin acceptor sites on IFNGR1 degradation. At 24–48 h post-transfection, cells were treated with CHX for indicated times and immunoblotted for V5 to determine protein half-life. (c) IFNGR1 protein densitometry normalized to β-actin and CHX 0 h to determine protein half-life; At least n = 3 independent experiments per group, ±SE. *P < 0.05 vs. control by ANOVA and Tukey’s t-test. (e) IFNGR1 protein densitometry normalized to β-actin and CHX 0 h to determine protein half-life; At least n = 3 independent experiments per group, ±SE. #,#P < 0.05 vs. K277,279,285R by Student’s t-test. (f) HEK cells were transfected with V5-tagged IFNGR1. At 48 h post-transfection, cell lysates were boiled and immunoprecipitated using the TUBEs reagent (Lifesensors) to pull down total ubiquitinated proteins. Immunoprecipitated lysates were probed for exogenous IFNGR1 with a V5 antibody. Input was probed for V5 and actin as a loading control. UT, untransfected. n = 3 independent experiments per group.
Inhibition of GSK3β decreases IFNGR1 stability
To assess the role of serine or threonine kinases in the stabilization of IFNGR1, we first treated HEK cells with varying concentrations of kinase inhibitors and measured endogenous IFNGR1 expression. While most kinase inhibitors had no effect on IFNGR1 stability in the time-frame tested, several kinase inhibitors decreased IFNGR1 expression including the broad spectrum kinase inhibitor staurosporine, the JNK inhibitor SP600125, and the GSK3β inhibitor TWS119 (data not shown). These results suggest that phosphorylation enhances IFNGR1 stability. While our results with the JNK inhibitor were inconsistent between cells, we observed that TWS119 significantly decreased the stability of IFNGR1 in both HEK (Figure 4a) and THP-1 cells (Figure 4b). To confirm that GSK3β is responsible for the observed changes, we knocked down endogenous GSK3β using RNAi and measured IFNGR1 half-life after CHX treatment. In both HEK (Figure 4c) and differentiated THP-1 (Figure 4d), GSK3β cellular depletion significantly decreased IFNGR1 stability. In addition, overexpression of exogenous GSK3β increased the stability of IFNGR1 (Figure 4e). Finally, we examined the role of receptor phosphorylation in IFN-γ signaling. Inhibition of GSK3β with TWS119 decreased the phosphorylation of STAT1 in THP-1 (Figure 4f).
Figure 4. Inhibition of GSK3β decreases IFNGR1 stability.
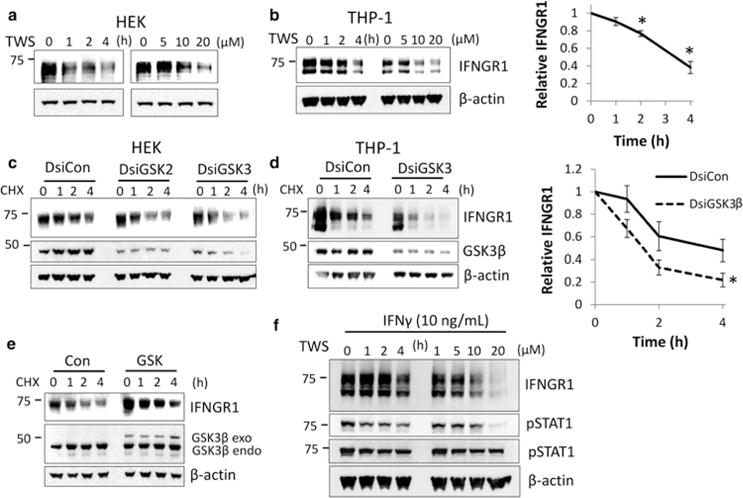
(a) HEK and (b) THP-1 cells were treated with TWS119 (10 μM) for indicated times (left) or with TWS119 for 4 h at the indicated concentrations (right); n = 3 independent experiments. Right: THP-1 IFNGR1 protein densitometry normalized to β-actin and TWS-119 0 h to determine protein stability. *P < 0.05 vs. control by ANOVA and Tukey’s t-test. (c) HEK and (d) THP-1 cells were transfected with dicer substrate short interfering RNA (DsiRNA) against GSK3β (Dsi GSK2/GSK3) or a dicer control RNA (DsiCon) for 72 h and then treated with CHX for the indicated times prior to harvest. n = 3 independent experiments for each group. Right: THP-1 IFNGR1 protein densitometry normalized to β-actin and TWS-119 0 h to determine protein stability. *P < 0.05 vs. DsiCon by ANOVA. (e) HEK cells were transfected with a plasmid expressing GSK3β or an empty vector control for 48 h and then treated with CHX for the indicated times prior to harvest. GSK3β endo (endogenous), exo (exogenous). Con, control plasmid. n = 3 independent experiments. (f) THP-1 cells were treated with TWS119 (10 μM) for indicated times (left) or with TWS119 for 4 h at the indicated concentrations (right). Cells were then treated with IFN-γ 15 min prior to harvesting and immunoblotted for IFNGR1 protein and phosphorylated STAT1 (pSTAT1) to measure IFN-γ activity. n = 3 independent experiments.
Consensus phosphorylation sites modulate IFNGR1 stability
We tested the hypothesis that phosphorylation of residues near the lysine ubiquitin acceptor site modifies protein stability. Because the phosphorylation of serine/threonine residues by GSK3β was important for IFNGR1 stability, we made mutations in serine and threonine residues near the membrane proximal, ubiquitin adjacent domain which contained a GSK3β-binding motif. We initially made a quadruple mutant in which serine 286, serine 289, serine 293, and threonine 295 were mutated to either alanine (S/T-A) or the phosphomimetic aspartic acid (S/T-D) (Figure 5a). Consistent with a role for phosphorylation in increasing membrane stability, the S/T-A mutant when expressed displayed decreased receptor stability while the mimicking phosphorylation variant (S/T-D) exhibited increased stability (Figure 5b). We proceeded to express constructs harboring individual point mutations in the residues at or near this domain. While IFNGR1 variants that expressed individual mutations at residues S280, S286, S289 had little overall impact on membrane stability, expression of phosphomimetic constructs with mutation of residues S293 and T295 to aspartic acid resulted in a protein with increased stability (Figure 5b). To examine how the modification of adjacent serine/threonine alters ubiquitination of IFNGR1, we overexpressed V5-tagged Wt, S/T-A, and S/T-D plasmids in HEK cells and pulled downed the IFNGR1 variant proteins. We observed that in comparison with Wt IFNGR1, the S/T-A mutant was similarly enriched for ubiquitination despite lower expression. In contrast, the S/T-D phosphorylation mimic showed less overall ubiquitination (Figure 5c). Finally, to test whether phosphorylation and ubiquitination were necessary for GSK3β-mediated destabilization, we overexpressed Wt, K-R, and S/T-A mutant IFNGR1 in HEK cells in the presence or absence of TWS119 and measured protein expression. We found that while Wt IFNGR1 was degraded shortly after TWS119 treatment, S/T-A and K-R mutant IFNGR1 were significantly protected from degradation in the presence of TWS119 (Figure 5d). These results suggest that IFNGR1 phosphorylation at key serine and threonine residues in the membrane proximal domain by GSK3β stabilizes IFNGR1 by decreasing receptor ubiquitination.
Figure 5. Consensus phosphorylation sites modulate IFNGR1 stability.
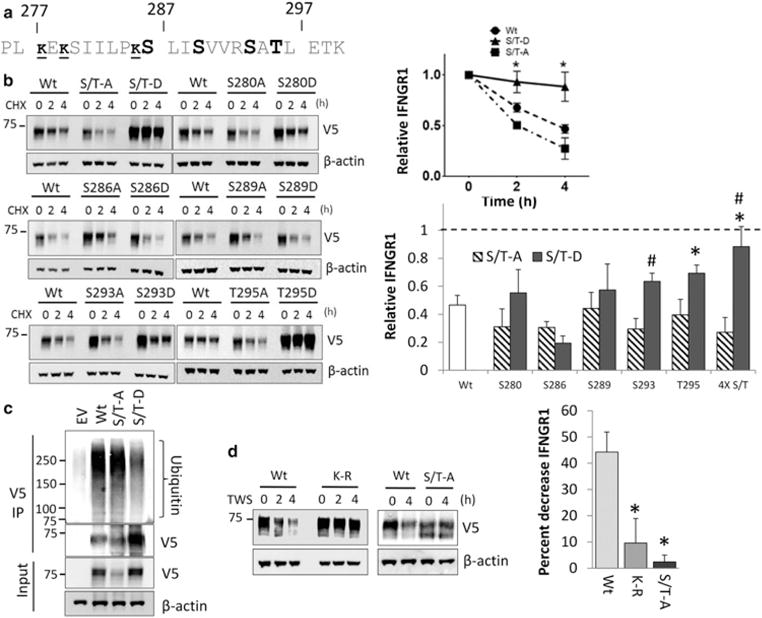
(a) Primary sequence of the IFNGR1 cytoplasmic juxtamembrane region. Serine and threonine residues mutated to either alanine and aspartic acid in the quadruple mutant are in bold. The sites of ubiquitination described in Figure 3 are indicated in small font and underlined. (b) HEK cells were transfected with various IFNGR1 V5-tagged serine or threonine point mutants. At 24 h post-transfection, cells were treated with CHX for indicated times and immunoblotted to determine protein half-life. Top right: Wt IFNGR1 and quadruple mutant serine/threonine mutant (S/T-A, S/T-D) densitometry normalized to β actin expression; n = 3 independent experiments per group, ±SE. *P < 0.05 S/T-D vs. Wt by ANOVA and Tukey’s t-test. Bottom right: IFNGR1 remaining at 4 h post-CHX treatment normalized to 0 h. n = 3 independent experiments per group, ±SE. *P < 0.05 mutant vs. Wt by ANOVA and Tukey’s t-test, #P < 0.05 aspartate vs. alanine mutant by Student’s t-test. (c) HEK cells were transfected with plasmids encoding V5-tagged IFNGR1 Wt, and the S/T-A, S/T-D mutants for 48 h followed by treatment with MG132 for 2 h to stabilize ubiquitinated proteins prior to harvest. Cells were then harvested, boiled, and subjected to IP for V5, and probed for ubiquitin. n = 2 independent experiments. (d) HEK cells were transfected with plasmids encoding V5-tagged IFNGR1 Wt, K-R (277, 279, 285R) or the S/T-A quadruple mutants for 48 h followed by treatment with DMSO or TWS-119 (10 μM) for 2 or 4 h. Cells were then harvested and probed for IFNGR1 expression. Right: percent decrease in IFNGR1 expression after 4 h treatment with TWS119. n = 3 independent experiments ± SE. *P < 0.05 mutant vs. Wt by ANOVA and Tukey’s t-test.
To test the trafficking of the ubiquitin and phosphorylation mutants, we overexpressed IFNGR1 Wt, K-R, S/T-A, and S/T-D constructs in HEK cells. We then biotinylated plasma membrane proteins and measured the association of each mutant with the plasma membrane. As previously observed, total K-R and S/T-D protein expression was enhanced over the Wt protein, while the S-A protein was less stable. However, despite differences in expression, the ratio of total vs. plasma membrane-associated IFNGR1 appeared similar in all the constructs (Figure 6a). In addition, we overexpressed IFNGR1 mutants in A549 cells and measured localization via immunofluorescence microscopy. Wt, K-R, S/T-A, and S/T-D mutant IFNGR1 all localize to the plasma membrane and appear to have a similar cellular distribution (Figure 6b).
Figure 6. Localization of IFNGR1 mutant proteins.
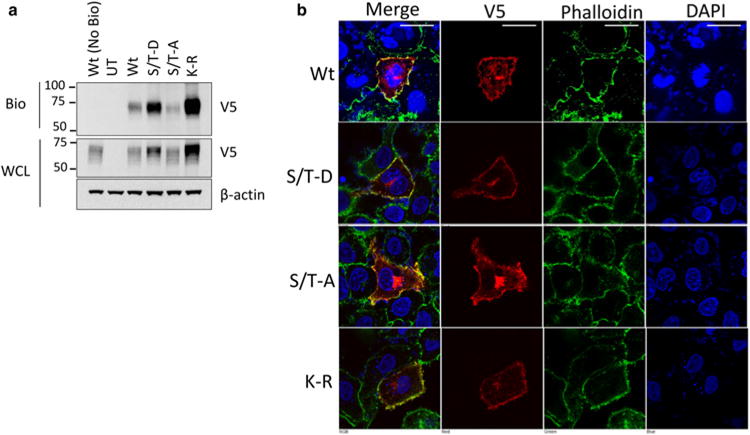
(a) HEK cells were transfected with plasmids encoding V5-tagged IFNGR1 Wt, K-R, or the S/T-A, S/T-D mutants for 48 h. Cells were then treated with membrane impermeable biotin, harvested, and probed for IFNGR1 to determine plasma membrane localization. Wt (No Bio) = transfected, no biotin control, UT, untransfected. n = 2 independent experiments. (b) A549 cells were transfected with plasmids encoding V5-tagged IFNGR1 Wt, K-R, or the S/T-A, S/T-D mutants on coverslips for 48 h. Cells were then fixed, permeabilized and stained for DAPI for nuclear staining, phalloidin to visualize the plasma membrane, and V5 to examine IFNGR. Scale bar = 25 μm.
Ubiquitination acceptor sites are necessary for efficient IFNGR1 signaling
To determine whether ubiquitination of IFNGR1 modifies signaling through the receptor, we developed several IFNGR1 knockout cell lines using the CRISPR–Cas9 system. After selecting single cell lines completely deficient for IFNGR1 expression, we rescued IFN-γ mediated STAT1 signaling by ectopically expressing IFNGR1 plasmids. All knockout cell lines showed no STAT1 phosphorylation in response to IFN-γ (Figure 7a). As expected, overexpression of Wt IFNGR1 plasmid rescued pSTAT1 signaling, demonstrating an intact signaling platform. Having confirmed that exogenous IFNGR1 overexpression was sufficient to restore IFN-γ responses, we next tested IFNGR1 ubiquitin mutant signaling in knockout cells. As previously observed, expression of constructs containing mutations of lysine 277 and 279 or lysine 285 individually only modestly preserved IFNGR1 stability and did not result in noticeable changes in pSTAT1 (Figure 7b). However, although mutation of all three lysine residues robustly increased receptor expression, induction of pSTAT1 was decreased in comparison with the Wt receptor when considering levels of plasmid expression. To directly compare signaling between the Wt and K-R mutant receptors, we overexpressed various concentrations of the IFNGR1 receptor and stimulated cells with IFN-γ. At all concentrations, despite exhibiting higher expression, the IFNGR1 K-R mutant showed reduced STAT1 activation (Figure 7c,d). When we normalized the phosphorylation of STAT1 to total IFNGR1 expression, we found that signaling was reduced by ~50–70% depending on the amount of plasmid expressed (Figure 7d, below). When we normalized for plasma membrane expression of IFNGR1 obtained via biotinylation experiments, signaling also remained significantly reduced (Figure 7e). Finally, we measured the induction of several genes known to be up-regulated in response to IFN-γ in HEK cells (Supplementary Figure S2a) [19,20]. As an additional control, we measured IFNGR1 mRNA after IFNGR1 Wt and K-R plasmid transfection in Cas9 IFNGR1 knockout cells. As expected, our transfection was comparable despite the differences in protein expression further suggesting stability changes accounting for the increased expression observed (Supplementary Figure S2b). We then selected highly induced genes for comparison in control HEK cells and IFNGR1 Cas9 knockout HEK cells overexpressing Wt or mutant receptors. As expected, knockout cells displayed no gene induction in response to IFN-γ. Wt receptor expression was able to partially rescue gene induction (Figure 7f). Despite dramatically higher expression of the IFNGR1 K-R mutant, IFN-γ stimulated gene induction in transfected knockout cell was significantly reduced compared with cells transfected with the Wt IFNGR1 receptor (Figure 7g). These data are consistent with attenuation of STAT1 by the expression of the K-R mutant proteins and suggest that either ubiquitination is involved in signal transduction or that other elements that link receptor protein stability to signal transduction are involved that limit downstream gene expression in response to IFN-γ.
Figure 7. Ubiquitination is necessary for efficient IFNGR1 signaling.
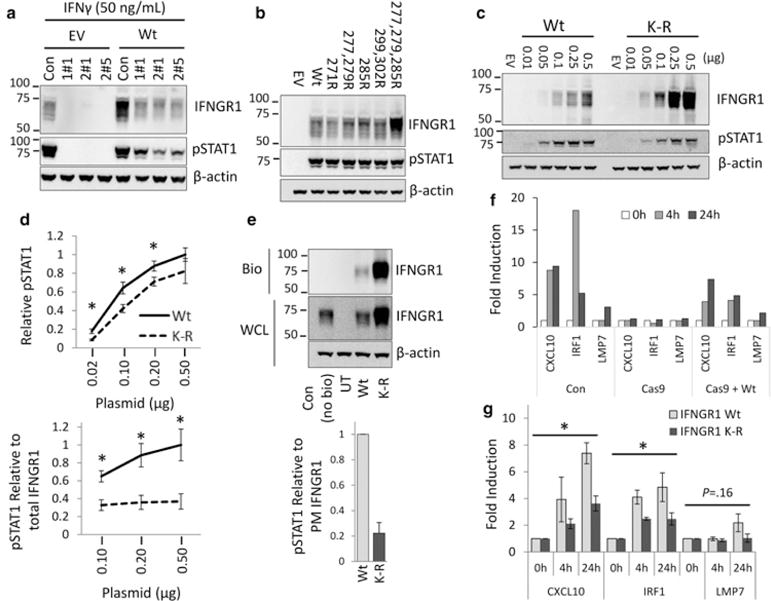
(a) Several HEK cell lines with IFNGR1 knocked out by CRISPR–Cas9 (HEK Cas9) were transfected with IFNGR1 to rescue IFNGR1 signaling. Cells were transfected with empty vector or IFNGR1 for 48 h and treated with 50 ng/ml IFN-γ 15 min prior to harvest. Cells were then harvested and probed for IFNGR1, and pSTAT1. HEK Cas9 IFNGR1 KO cell line 1#1 was used in subsequent experiments and cells were treated as described above. (b) HEK Cas9 cells were transfected with various IFNGR1 lysine to arginine mutant plasmids and probed for IFNGR1 levels and phosphorylated STAT1 (pSTAT1). (c) HEK cells were transfected with indicated amounts of Wt IFNGR1 or the K-R (277, 279, 285R) mutant plasmids for 48 h, harvested and probed for IFNGR1 and pSTAT1 by immunoblotting. (d) Densitometry of pSTAT1 in HEK transfected with 0.02, 0.1, 0.20, 0.5 μg/ml of Wt or K-R mutant IFNGR1. *P < 0.05 Wt vs. K-R by Student’s t-test. Below: pSTAT1 signaling normalized to total protein expression at 0.10, 0.20, 0.50 μg/ml DNA transfection. *P < 0.05 Wt vs. K-R by Student’s t-test. (e) HEK cells were transfected with 0.5 μg/ml plasmids encoding V5-tagged IFNGR1 Wt or K-R, for 48 h. Cells were then treated with membrane impermeable biotin, harvested, and probed for IFNGR1 to determine plasma membrane localization. Wt (No Bio) = transfected, no biotin control, UT, untransfected. Below: pSTAT1 normalized to plasma membrane IFNGR1 expression in cells transfected with Wt or K-R mutant IFNGR1. (f) IFN-γ stimulated gene induction in HEK cells. HEK cells and HEK IFNGR1 Cas9 KO cells were seeded into 6-well plates. HEK IFNGR1 Cas9 KO cells were transfected with empty vector, Wt IFNGR1 0.5 μg/ml, for 24–48 h. Cells were then treated with IFN-γ (50 ng/ml) for indicated times, harvested for RNA and cDNA, and probed for IFN-γ stimulated gene induction by qRT-PCR. (g) HEK IFNGR1 Cas9 KO cells were transfected with Wt IFNGR1 or K-R mutant as described above. n = 3–4 independent experiments ± SE. *P < 0.05 K-R vs. Wt by ANOVA.
Discussion
As the receptor that directly binds IFN-γ, IFNGR1 is essential for the transduction of IFN-γ signaling. Modification of IFNGR1 degradation has been shown to alter IFN-γ activity in several systems [7,21]. Our studies examine the role of ubiquitination on the stability of the IFNGR1 receptor and the resulting modification of IFN-γ-mediated signaling. We initially found that IFNGR1 was degraded through the proteasome via K48-linked polyubiquitination at specific lysine ubiquitin acceptor sites. We also demonstrate that serine and threonine residues adjacent to ubiquitin acceptor were essential for GSK3β-mediated stabilization of IFNGR1. Finally, we examined IFNGR1 signaling in IFNGR1 knockout HEK cells and found significantly reduced pSTAT1-dependent gene induction after expressing plasmids that encode for IFNGR1 stable mutants. These latter observations suggest that polyubiquitination is an important signature that elicits downstream IFN-γ signaling or that enhanced IFNGR1 receptor stability in cells might trigger feedback inhibitory signals that limit innate immune responses in cells. Overall, these findings provide new insight into the molecular events that control IFNGR1 cell surface protein abundance and may impact inflammatory gene responses after microbial infection.
Prior studies implicated both proteasomal and lysosomal degradation in IFNGR1 turnover. Curry et al. [7] demonstrated that blocking the proteasome prevented degradation of IFNGR1 through TLR2 stimulation. In contrast, Shah et al. measured the half-life of IFNGR1 and determined the degradation was slowed by MG132 and chloroquine. However, another lysosomal inhibitor, ammonium chloride, had no effect on degradation [5]. In our hands neither the lysosomal protease-specific inhibitor leupeptin nor the v-ATPase inhibitor bafilomycin A1 (Figure 1a) altered stability. However, the proteasome inhibitors MG132 and bortezomib dose dependently enhanced IFNGR1 stability and led to an accumulation of the ubiquitinated receptor. Interestingly, chloroquine has been previously demonstrated to inhibit proteasome activity suggesting that the chloroquine effect on IFNGR1 may be through the proteasome as well [22]. In combination with our data demonstrating K48-linked ubiquitination, which normally results in proteasomal degradation, but failing to show K63-linked ubiquitination, frequently important in lysosomal targeting, these results provide significant evidence supporting IFNGR1 proteasomal degradation as the predominant mechanism in our system [23].
Transmembrane proteins are translated in the endoplasmic reticulum and transported through the Golgi apparatus where complex glycosylation occurs. Maturely glycosylated IFNGR1 is transported to the plasma membrane to participate in signaling events. ER-associated degradation of newly synthesized, improperly folded proteins occurs primarily through the proteasomal pathway [24]. We therefore sought to specifically examine the mature receptor and to determine whether it was subject to proteasomal degradation. Interestingly, degradation of mature, plasma membrane associated, IFNGR1 was substantially slowed by proteasomal inhibition. Several mature receptors have been demonstrated to be subject to proteasomal degradation by various mechanisms including metalloproteinase cleavage and internalization followed by membrane extraction [25,26]. Glycosylation, in some circumstances, appeared to be required for ubiquitination and proteasomal degradation [27]. In separate studies, we demonstrate that IFNGR1 glycosylation does not impact stability of the receptor (Supplementary Figure S3). The exact mechanism by which IFNGR1 is targeted and degraded remains to be explored.
Owing to the established role of ligand binding in the ubiquitination and degradation of membrane receptors, we examined whether ligand stimulation altered IFNGR1 stability [10,17,28,29]. Several studies have previously investigated the role of IFN-γ binding on receptor stability with conflicting results [30–32]. In our system, IFN-γ treatment did not acutely alter IFNGR1 expression or stability in either epithelial cells or macrophage differentiated monocytes (Figure 2).
Although there are several post-translational modifications that have been demonstrated to modify protein ubiquitination, phosphorylation is the most studied mechanism [33–35]. The role of phosphorylation by serine or threonine kinases on the stability of IFNGR1 had not been explored. Our observations that GSK3β-enhanced IFNGR1 stability is consistent with several studies showing ability of GSK3β to modulate IFN-γ signaling [36,37]. Our laboratory has previously implicated GSK3β in the modification of receptor protein stability. Zhao et al. [29] showed that phosphorylation at Ser442 by the kinase GSK3β enhanced the degradation of the IL-33 receptor STL2. Similar to the mechanism we describe, GSK-3β overexpression results in accumulation of IL-22R protein through a decrease in the receptor’s phosphorylation and degradation [38]. Whether IFNGR1 is directly targeted by GSK3β was not determined in this study. In this regard, it is known that GSK3β often co-ordinates with priming phosphorylation events on substrates mediated by other kinases. The precise interplay between GSK3β and other priming events will also require additional studies.
Finally, we tested the downstream signaling after ectopic expression of IFNGR1 ubiquitin mutant constructs in CRISPR–Cas9 knockout cells devoid of the receptor. Interestingly in cells expressing a protein variant harboring K-R mutations signaling was decreased in comparison with the Wt receptor, despite greater expression. There are several explanations for these observations. The simplest explanation is that the mutations modify the interactions with binding partners leading to decreased signaling. Although the potential site of ubiquitination is near the JAK1 binding motif, previous research suggests that only proline 284 is necessary for JAK1 binding with mutation of the adjacent lysine having no effect on signaling [39]. We also find that a single point mutation in this domain did not dramatically alter phosphorylation of STAT1 (Figure 7b). Interactions with downstream modulators of signaling are complicated by the fact that alterations in ubiquitination itself might change receptor localization or affinity. Transition to lipid microdomains is necessary for IFNGR2 binding and STAT1 phosphorylation [40]. Several studies have also suggested that IFNGR1 is necessary for nuclear transport of IFN-γ and STAT1 [41–43]. All these steps may be modified by ubiquitination. Another possibility is that extended life span of an IFNGR1 protein at the cell surface incites feedback control mechanisms to restrict IFNGR1-dependent pro-inflammatory gene expression as a protective mechanism. The role of ubiquitin in receptor trafficking and internalization will be explored in future studies.
Supplementary Material
Acknowledgments
Funding
This work was supported, in part, by National Institutes of Health Grants HL096376, HL097376, HL098174, HL081784, P01 HL114453, and 4T32HL007563-29 to R.K.M., National Institutes of Health Grants HL116472 and HL132862 to B.B.C., and American Heart Grant 17POST33410945 to J.D.L. This work was also supported in part by the United States Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, and a Merit Review Award from the United States Department of Veterans Affairs (R.K.M.).
Abbreviations
- CHX
cycloheximide
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- GAS
gamma-activated sequence
- GSK3β
glycogen synthase kinase 3 beta
- HDR
homology directed repair
- IFNGR1
interferon gamma receptor 1
- ISG
interferon stimulated genes
- JAK
Janus kinase
- TUBES
tandem ubiquitin binding entities
- Wt
wild-type
Footnotes
Author Contribution
J.D.L. designed the studies, performed experiments, analyzed the data, wrote and edited the manuscript; D.L.G., L.S.M. and T.B.L. performed experiments. N.M.W., T.L.S. and B.B.C. assisted with experiments and edited the manuscript. R.K.M. designed the studies, analyzed the data and edited the manuscript.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Pollard KM, Cauvi DM, Toomey CB, Morris KV, Kono DH. Interferon-γ and systemic autoimmunity. Discov Med. 2013;16:123–131. [PMC free article] [PubMed] [Google Scholar]
- 2.Haverkamp MH, van Dissel JT, Holland SM. Human host genetic factors in nontuberculous mycobacterial infection: lessons from single gene disorders affecting innate and adaptive immunity and lessons from molecular defects in interferon-γ-dependent signaling. Microbes Infect. 2006;8:1157–1166. doi: 10.1016/j.micinf.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 3.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 4.de Vor IC, van der Meulen PM, Bekker V, Verhard EM, Breuning MH, Harnisch E, et al. Deletion of the entire interferon-γ receptor 1 gene causing complete deficiency in three related patients. J Clin Immunol. 2016;36:195–203. doi: 10.1007/s10875-016-0244-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah KM, Stewart SE, Wei W, Woodman CB, O’Neil JD, Dawson CW, et al. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene. 2009;28:3903–3914. doi: 10.1038/onc.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q, Means R, Lang S, Jung JU. Downregulation of γ interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J Virol. 2007;81:2117–2127. doi: 10.1128/JVI.01961-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curry H, Alvarez GR, Zwilling BS, Lafuse WP. Toll-like receptor 2 stimulation decreases IFN-γ receptor expression in mouse RAW264.7 macrophages. J Interferon Cytokine Res. 2004;24:699–710. doi: 10.1089/jir.2004.24.699. [DOI] [PubMed] [Google Scholar]
- 8.Weathington NM, Sznajder JI, Mallampalli RK. The emerging role of the ubiquitin proteasome in pulmonary biology and disease. Am J Respir Crit Care Med. 2013;188:530–537. doi: 10.1164/rccm.201304-0754PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Londino JD, Lazrak A, Noah JW, Aggarwal S, Bali V, Woodworth BA, et al. Influenza virus M2 targets cystic fibrosis transmembrane conductance regulator for lysosomal degradation during viral infection. FASEB J. 2015;29:2712–2725. doi: 10.1096/fj.14-268755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKelvey AC, Lear TB, Dunn SR, Evankovich J, Londino JD, Bednash JS, et al. RING finger E3 ligase PPP1R11 regulates TLR2 signaling and innate immunity. eLife. 2016;5:e18496. doi: 10.7554/eLife.18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuchs SY. Ubiquitination-mediated regulation of interferon responses. Growth Factors. 2012;30:141–148. doi: 10.3109/08977194.2012.669382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Bras S, Loyer N, Le Borgne R. The multiple facets of ubiquitination in the regulation of notch signaling pathway. Traffic. 2011;12:149–161. doi: 10.1111/j.1600-0854.2010.01126.x. [DOI] [PubMed] [Google Scholar]
- 13.Weathington NM, Mallampalli RK. New insights on the function of SCF ubiquitin E3 ligases in the lung. Cell Signal. 2013;25:1792–1798. doi: 10.1016/j.cellsig.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen BB, Mallampalli RK. Masking of a nuclear signal motif by monoubiquitination leads to mislocalization and degradation of the regulatory enzyme cytidylyltransferase. Mol Cell Biol. 2009;29:3062–3075. doi: 10.1128/MCB.01824-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Londino JD, Gulick D, Isenberg JS, Mallampalli RK. Cleavage of signal regulatory protein α (SIRPα) enhances inflammatory signaling. J Biol Chem. 2015;290:31113–31125. doi: 10.1074/jbc.M115.682914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hershey GK, Schreiber RD. Biosynthetic analysis of the human interferon-γ receptor. Identification of N-linked glycosylation intermediates. J Biol Chem. 1989;264:11981–11988. [PubMed] [Google Scholar]
- 17.Kumar KGS, Krolewski JJ, Fuchs SY. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem. 2004;279:46614–46620. doi: 10.1074/jbc.M407082200. [DOI] [PubMed] [Google Scholar]
- 18.Kearney SJ, Delgado C, Eshleman EM, Hill KK, O’Connor BP, Lenz LL. Type I IFNs downregulate myeloid cell IFN-γ receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences ifngr1 transcription. J Immunol. 2013;191:3384–3392. doi: 10.4049/jimmunol.1203510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pawliczak R, Logun C, Madara P, Barb J, Suffredini AF, Munson PJ, et al. Influence of IFN-γ on gene expression in normal human bronchial epithelial cells: modulation of IFN-γ effects by dexamethasone. Physiol Genomics. 2005;23:28–45. doi: 10.1152/physiolgenomics.00011.2005. [DOI] [PubMed] [Google Scholar]
- 20.Waddell SJ, Popper SJ, Rubins KH, Griffiths MJ, Brown PO, Levin M, et al. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS ONE. 2010;5:e9753. doi: 10.1371/journal.pone.0009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uetani K, Hiroi M, Meguro T, Ogawa H, Kamisako T, Ohmori Y, et al. Influenza A virus abrogates IFN-γ response in respiratory epithelial cells by disruption of the Jak/Stat pathway. Eur J Immunol. 2008;38:1559–1573. doi: 10.1002/eji.200737045. [DOI] [PubMed] [Google Scholar]
- 22.Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286:22426–22440. doi: 10.1074/jbc.M110.149252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bednash JS, Mallampalli RK. Regulation of inflammasomes by ubiquitination. Cell Mol Immunol. 2016;13:722–728. doi: 10.1038/cmi.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lippincott-Schwartz J, Roberts TH, Hirschberg K. Secretory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol. 2000;16:557–589. doi: 10.1146/annurev.cellbio.16.1.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeffers M, Taylor GA, Weidner KM, Omura S, Vande Woude GF. Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol Cell Biol. 1997;17:799–808. doi: 10.1128/MCB.17.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vecchi M, Carpenter G. Constitutive proteolysis of the ErbB-4 receptor tyrosine kinase by a unique, sequential mechanism. J Cell Biol. 1997;139:995–1003. doi: 10.1083/jcb.139.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao N, Zhang AS, Worthen C, Knutson MD, Enns CA. An iron-regulated and glycosylation-dependent proteasomal degradation pathway for the plasma membrane metal transporter ZIP14. Proc Natl Acad Sci USA. 2014;111:9175–9180. doi: 10.1073/pnas.1405355111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokouchi M, Kondo T, Houghton A, Bartkiewicz M, Horne WC, Zhang H, et al. Ligand-induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c-Cbl RING finger and UbcH7. J Biol Chem. 1999;274:31707–31712. doi: 10.1074/jbc.274.44.31707. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Wei J, Mialki RK, Mallampalli DF, Chen BB, Coon T, et al. F-box protein FBXL19–mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat Immunol. 2012;13:651–658. doi: 10.1038/ni.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skrenta H, Yang Y, Pestka S, Fathman CG. Ligand-independent down-regulation of IFN-γ receptor 1 following TCR engagement. J Immunol. 2000;164:3506–3511. doi: 10.4049/jimmunol.164.7.3506. [DOI] [PubMed] [Google Scholar]
- 31.Fischer DG, Novick D, Orchansky P, Rubinstein M. Two molecular forms of the human interferon-γ receptor. Ligand binding, internalization, and down-regulation. J Biol Chem. 1988;263:2632–2637. [PubMed] [Google Scholar]
- 32.Bader T, Wietzerbin J. Modulation of murine and human interferon-γ receptor expression by their ligands or phorbol ester. Cytokine. 1994;6:70–78. doi: 10.1016/1043-4666(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 33.Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y, et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol. 2013;14:470–479. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W, Xiong S, Li J, Li X, Liu Y, Zou C, et al. The ubiquitin E3 ligase SCF-FBXO24 recognizes deacetylated nucleoside diphosphate kinase A to enhance its degradation. Mol Cell Biol. 2015;35:1001–1013. doi: 10.1128/MCB.01185-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guinez C, Mir AM, Dehennaut V, Cacan R, Harduin-Lepers A, Michalski JC, et al. Protein ubiquitination is modulated by O-GlcNAc glycosylation. FASEB J. 2008;22:2901–2911. doi: 10.1096/fj.07-102509. [DOI] [PubMed] [Google Scholar]
- 36.Beurel E, Jope RS. Glycogen synthase kinase-3 promotes the synergistic action of interferon-γ on lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell Signal. 2009;21:978–985. doi: 10.1016/j.cellsig.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai CC, Kai JI, Huang WC, Wang CY, Wang Y, Chen CL, et al. Glycogen synthase kinase-3β facilitates IFN-γ-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J Immunol. 2009;183:856–864. doi: 10.4049/jimmunol.0804033. [DOI] [PubMed] [Google Scholar]
- 38.Weathington NM, Snavely CA, Chen BB, Zhao J, Zhao Y, Mallampalli RK. Glycogen synthase kinase-3β stabilizes the interleukin (IL)-22 receptor from proteasomal degradation in murine lung epithelia. J Biol Chem. 2014;289:17610–17619. doi: 10.1074/jbc.M114.551747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplan DH, Greenlund AC, Tanner JW, Shaw AS, Schreiber RD. Identification of an interferon-γ receptor α chain sequence required for JAK-1 binding. J Biol Chem. 1996;271:9–12. doi: 10.1074/jbc.271.1.9. [DOI] [PubMed] [Google Scholar]
- 40.Marchetti M, Monier MN, Fradagrada A, Mitchell K, Baychelier F, Eid P, et al. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol Biol Cell. 2006;17:2896–2909. doi: 10.1091/mbc.E06-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larkin J, 3rd, Johnson HM, Subramaniam PS. Differential nuclear localization of the IFNGR-1 and IFNGR-2 subunits of the IFN-γ receptor complex following activation by IFN-γ. J Interferon Cytokine Res. 2000;20:565–576. doi: 10.1089/10799900050044769. [DOI] [PubMed] [Google Scholar]
- 42.Ahmed CMI, Burkhart MA, Mujtaba MG, Subramaniam PS, Johnson HM. The role of IFNγ nuclear localization sequence in intracellular function. J Cell Sci. 2003;116:3089–3098. doi: 10.1242/jcs.00528. [DOI] [PubMed] [Google Scholar]
- 43.Ahmed CMI, Johnson HM. IFN-γ and its receptor subunit IFNGR1 are recruited to the IFN-γ-activated sequence element at the promoter site of IFN-γ-activated genes: evidence of transactivational activity in IFNGR1. J Immunol. 2006;177:315–321. doi: 10.4049/jimmunol.177.1.315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.