

Summary
Mitochondrial fusion, fission and mitophagy form an essential axis of mitochondrial quality control. However, quality control may not be the only task carried out by mitochondrial dynamics. Recent studies link mitochondrial dynamics to the balance between energy demand and nutrient supply, suggesting changes in mitochondrial architecture as a mechanism for bioenergetic adaptation to metabolic demands. By favoring either connected or fragmented architectures, mitochondrial dynamics regulates bioenergetic efficiency and energy expenditure. Placing bioenergetic adaptation and quality control as competing tasks of mitochondrial dynamics may provide a new mechanism, linking excess nutrient environment to progressive mitochondrial dysfunction, common to age-related diseases.
E-TOC
Mitochondria have a very active social life style involving frequent fusion and fission events. Mitochondria that lose their ability to properly respire become excluded from the networking population and will be consumed by the cellular equivalent of a lion, the autophagosome. This forms a pathway of quality control. However, recent studies suggest that arrest of mitochondrial fusion at the cellular level, also termed “fragmentation”, is playing a role in the adaptation to excess nutrient environment. Recognizing that excess nutrient environment places mitochondria in a biological conflict of interest may help understanding the link between metabolic and aging associated conditions.
INTRODUCTION
As our relationship with mitochondria evolves, we remain fascinated with the impact of this organelle in two seemingly unrelated conditions: aging and metabolic diseases. While aging involves insufficiency of mitochondrial quality control and turnover mechanisms (such as autophagy), diabetes and obesity are influenced by the ability of the organism to deal with excess nutrient environment. The observation that both conditions are impacted by the duration of exposure to excess nutrient environment raises the question: Are the tasks of handling nutrients in excess and maintaining quality control ever in conflict? In this review, we discuss evidence to support a hypothesis that adaptation to excess nutrient environment interferes with quality control functions and, as a result, affects mitochondrial function in a magnitude that reflects the duration to which the organism was exposed to excess nutrient environment.
In response to changes in energy demand and supply the organism adapts by adjusting both its capacity and efficiency of ATP production. Bioenergetic efficiency is defined as the ATP produced in the mitochondria per molecule of nutrient (Figure 1) and mitochondrial ATP synthesis capacity is defined as ATP synthesized per unit of time.
Figure 1. Regulation of cellular bioenergetic efficiency under conditions of nutrient excess.
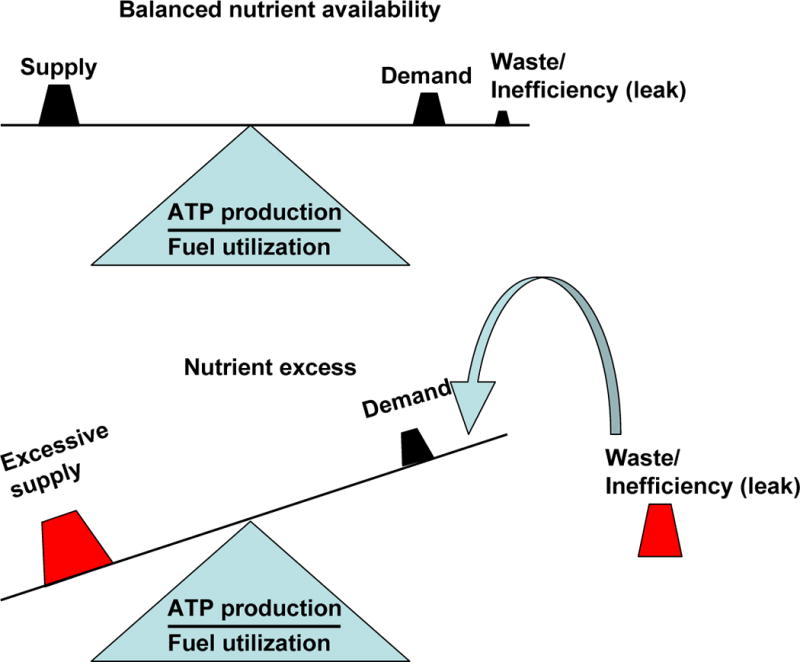
In the balanced state fuel/nutrient “supply” is sufficient to sustain energy (ATP) “demand”. Under this condition “waste” or inefficiency in the form of heat is minor. Nutrient excess, characterized by “excessive supply” in the absence of a parallel increase in “demand” represent a situation were the energy required to satisfy ATP demand is lower than the available energy. This is compensated for by adding an energy sink that does not involve ATP synthesis. This component is inefficiency/waste in the form of heat. The major mechanism for inefficiency/waste in the form of heat is mitochondrial proton “leak”. This mechanism can slow down nutrient accumulation and prevent the development of reductive stress (accumulation of NADH), and ROS production.
As an adaptation to excess nutrients, the organism recruits mechanisms to utilize nutrients first by storage and then by waste (heat generation). While spending time at the gym may be the appropriate way to waste energy and keep healthy, reducing energy efficiency may enable energy waste in tissues other than muscle and in individuals that are incompatible with the gym (such as the authors).
Studies in the field of mitochondrial dynamics have identified an intriguing link between energy demand/supply balance and mitochondrial architecture. Cells exposed to rich nutrient environment tend to keep their mitochondria in a separated (fragmented) state and mitochondria in cells under starvation tend to remain for a longer duration in the connected state (Molina et al., 2009; Gomes et al., 2011). Thus, it appears that bioenergetic adaptation that involves changes to bioenergetic efficiency and ATP synthesis capacity also implies remodeling of mitochondrial architecture.
However, bioenergetic adaptation is not the only mitochondrial task that involves changes to mitochondrial architecture. A vital task that engages the fusion and fission machinery is the Mitochondrial Life Cycle (Twig et al., 2008a). The Mitochondrial Life Cycle represents continuous changes to mitochondrial architecture through fusion and fission events. These brief transitions between connected and separated mitochondria enable the reorganization of mitochondrial components and the elimination of damaged material, thereby maintaining a healthy mitochondrial population. One can appreciate that the life cycle of mitochondria would be compromised if mitochondrial fusion or fission were disabled to allow for bioenergetic adaptation. Therefore, under certain nutrient environments, bioenergetic adaptation and quality control may represent conflicting tasks.
That mitochondrial quality control has evolved within the same mechanism that controls for bioenergetic efficiency is not surprising, given the understanding that low nutrient environment (caloric restriction, not starvation) may support increased longevity.
Adaptation of bioenergetic efficiency and ATP synthesis capacity to nutrient availability differs among tissues and is intimately linked to their specific physiology. Thus, we will focus on three paradigmatic tissues that show different bioenergetic efficiencies and mechanisms of adaptation to nutrient availability:
Brown adipose tissue: an example of low bioenergetic efficiency, as nutrient oxidation in the mitochondria mostly results in heat production upon its hormonal stimulation (reviewed in Cannon and Needergard, 2004).
Muscle: muscle cells harbor higher bioenergetic efficiency than beta cells (Affourtit and Brand, 2006) and activated brown fat, as nutrient oxidation driven by mitochondria is required to provide a constant and efficient supply of ATP to support contraction (Chappell and Perry, 1954). Thus, oxidative muscle is a good example of high mitochondrial ATP synthesis capacity and high bioenergetic efficiency.
Beta cells: their principal function is to secrete insulin according to the extracellular concentration of nutrients. The amount of nutrients is “sensed” by beta cells through their intracellular metabolism, which involves nutrient oxidation mediated by mitochondria (Ashcroft et al., 1984; reviewed in Deeney et al., 2000). Therefore, bioenergetic efficiency is expected to be highly regulated to allow proper insulin secretion.
Although the mechanisms for these tissue-specific differences are understood to a certain extent, less is known about the contribution of mitochondrial dynamics to tissue and diet-dependent bioenergetic efficiency and mitochondrial ATP synthesis capacity. Mitochondrial dynamics is a concept that comprises mitochondrial architecture resulting from movement, tethering, fusion and fission events. Multiple evidences demonstrate that mitochondrial dynamics are important for cell viability, senescence, mitochondria health, bioenergetic function, quality control and intracellular signaling (reviewed in Liesa et al., 2009; reviewed in Twig et al., 2008b). On the other hand, we are now beginning to understand how nutrients and the cellular metabolic state are regulating mitochondrial dynamics in different tissues and vice-versa; particularly in the beta-cell, brown adipose tissue and muscle (Molina et al., 2009; Quiros et al., 2012; Sebastian et al., 2012). Along with this, the relevance of mitochondrial dynamics in the specific physiology of different tissues has only been revealed recently, mostly thanks to different mouse models harboring tissue-specific deletions of core components regulating mitochondrial dynamics (Chen et al., 2007; Chen et al., 2011; Chen et al., 2010; Ishihara et al., 2009; Sebastian et al., 2012; Wakabayashi et al., 2009; Zhang et al., 2011).
In this context, the aim of this review is to summarize the knowledge about nutrients and the metabolic state regulation of mitochondrial bioenergetic function and efficiency in health and disease. We will discuss how this might be explained by differences in mitochondrial dynamics. In the last section, we will provide a model by which prolonged changes in dynamics through altered nutrient availability might alter mitochondrial quality control, and thus contribute to mitochondrial dysfunction associated with metabolic or other age-related diseases.
1. REGULATION OF CELLULAR BIOENERGETICS BY NUTRIENTS
a) How can bioenergetic efficiency affect cellular functionality and viability?
Not every tissue responds or adapts its function in a similar manner to starvation or to food abundance. For instance, nutrient sensors (i.e. beta cell), storage organs (i.e. adipose tissue) and “nutrient-consuming” organs (muscle and brain) respond to changes in nutrient availability through very different mechanisms. On the other hand, one could expect similar outcomes from these diverse mechanisms in tissues that rely on mitochondrial ATP synthesis in order to satisfy ATP demand. In the case of limited nutrient availability, one could expect that mechanisms enhancing bioenergetic efficiency (ATP produced per molecule of nutrient) may better serve the needs of a cell under these conditions. Enhanced efficiency over increased ATP synthesis capacity would be preferred, as increased capacity would deplete the limited nutrients in a shorter period of time. However, recent evidence demonstrated that starvation increases mitochondrial ATP synthesis capacity, by promoting mitochondrial ATP synthase dimerization (Gomes et al., 2011). Thus, it is likely that increased ATP synthesis capacity should be parallel to increased efficiency during starvation in vitro, in order to avoid a fast nutrient depletion (see section 1b).
The link between bioenergetic efficiency/ATP synthesis capacity and cell functionality or viability is less intuitive in the case of nutrient overload or exposure to high levels of high caloric nutrients (high fat or lipotoxicity). The two mechanisms by which decreased bioenergetic efficiency (nutrients supplying heat generation) may benefit cellular function are: by reduction of reactive oxygen species (ROS) production and by the enhanced removal of excess nutrients and their potentially cytotoxic metabolites (Figure 1).
The flow of electron-mediated proton translocation in the respiratory chain can be compared to a flow of water in a garden hose (see section 1b for more detailed bioenergetics description). NADH, resulting from nutrient oxidation, feeds the hose inlet with water, while ATP synthase controls the hose final outlet. The pressure that the flow of water generates to the hose is the mitochondrial membrane potential (Δψm). The flow of water and pressure in the hose is determined by the rate of ATP synthesis. The minimum and maximum values of pressure that the hose can hold are determined by the material and integrity of the hose, not by the flow of water or the inlets and outlets (i.e. the range of Δψm in mV is determined by thermodynamics and the integrity of the organelle). ATP synthesis is determined by ATP demand, meaning that the hose outlet is controlled by ATP demand. If the hose could hold unlimited pressures, we would not have to be concerned with any parameter beyond ATP demand. However, this is not the case. The hose, as it turns out, has some cuts through which water can escape, when pressure builds up. A pressure valve that can divert excess water through a safe conduit can reduce the pressure in the hose, and prevent water leakage through the ‘cuts’ in the hose (increasing or maintaining the flow of water). In our analogy, the escape of water through the cuts represents the escape of electrons to produce ROS. The pressure valve represents induced uncoupling (increasing the flow) and basal proton leak (maintaining the flow). The balance between ATP demand and nutrient supply determines both the rate of ATP synthesis as well as the level of ROS produced by mitochondria.
Different tissues employ different mechanisms in their response to nutrient overload. The selection of specific compensatory mechanisms allows each tissue to maintain its unique primary function, while minimizing side effects related to ROS production. In certain cell types, compensatory mechanisms are placed upstream of the mitochondria and preventing its exposure to high levels of fuel. However, in beta cells, brown adipose tissue and muscle, mounting evidence suggest that conditions of nutrient excess that increase fuel availability to the mitochondria may modulate bioenergetic efficiency and mitochondrial ATP synthesis capacity (Koves et al., 2008; Bonnard et al., 2008; Rothwell and Stock, 1979; Wikstrom et al., 2007).
b) Understanding mechanisms of bioenergetic efficiency and changes in ATP synthesis capacity by respiration studies
Mitochondria from any tissue can provide energy in the form of ATP as a result of nutrient oxidation (Chance and Williams, 1955; Mitchell, 1961). Oxidation of nutrients will provide electrons to the mitochondrial electron transport chain (constituted by 4 complexes) in the form of NADH and FADH2. The sequential transport of electrons from complex I or II to III and IV extrudes protons from the matrix to the inter-membrane space, generating an electrochemical gradient (ΔμH+) resulting in a difference in charge (Δψ) and in proton concentration (ΔpH). The Δψm, which constitutes the mitochondrial membrane potential, is the main contributor to ΔμH+ (reviewed in Nicholls and Ferguson, 2002). In functional mitochondria, maximal and minimal Δψm values are around 225 and 90 mV respectively (absolute values in mV can vary depending on the study) (reviewed in Nicholls and Ferguson, 2002). This range in mV is dictated by the thermodynamic stability of functional mitochondria and represents the balance between proton extrusion and re-entry. In this regard, proton re-entry through complex V/ATP synthase provides energy that will be used to synthesize ATP from ADP or coupled respiration. The state at which isolated mitochondria are synthesizing ATP at maximal rates is named state 3 (Chance and Williams, 1955) and it occurs at intermediate Δψm values (~140 mV), as there is a high rate of both proton extrusion and re-entry (reviewed in Nicholls and Ferguson, 2002).
If proton re-entry takes place through other mechanisms, nutrient oxidation will result in increased proportion of heat generation versus ATP production or uncoupled respiration (reviewed in Nicholls and Ferguson, 2002). It is important to distinguish between two different types of respiratory states resulting from uncoupling. These two respiratory states show major functional differences and might occur under different physiological states:
-
1)
Respiration controlled by proton leak: it is typically measured during state 4 in isolated mitochondria (experimental state reached by ADP exhaustion) or by inhibition of the ATP synthase in intact cells or in isolated mitochondria. It is also referred to as respiration controlled by basal (not regulated) proton conductance (Parker et al., 2009).
-
2)
Respiration in the presence of uncouplers: this type of uncoupled respiration can be mediated by the addition of chemical compounds (i.e. FCCP) or by the activity of uncoupling proteins/molecules located in the inner mitochondrial membrane. The activity of these uncouplers decreases Δψm values by increasing proton reentry and respiration. It is also referred to as inducible (activated or inhibited) proton conductance (Parker et al., 2009).
The main difference between these two types of uncoupled respiration is that mitochondrial respiration under state 4 reaches the highest Δψm values (~175–225 mV) and maintains low oxygen consumption rates. This maximal Δψm value results from a combination of decreased rates of ATP synthase-mediated proton re-entry and the concomitant decrease in nutrient oxidation. The combination of these effects maintains Δψm values within the range dictated by thermodynamic stability of functional mitochondria. This state is associated with high ROS generation, as a consequence of the increase in Δψm.
In marked contrast, mitochondria treated with uncouplers (such as FCCP) have decreased Δψ value, which causes an increase in respiration rates to values higher or close to state 3. The concomitant increase in respiration maintains Δψm values within the range of thermodynamic stability (~90–120 mV). In this case, absolute values of calories from nutrients used for heat generation will be higher in uncoupler-induced respiration compared to state 4 (proton leak controlled respiration). Thus, this type of respiration decreases bioenergetic efficiency, as it drives nutrient oxidation towards heat generation. Furthermore, it is associated with lower ROS production, as Δψm values are reduced. The description of these basic differences between these two types of uncoupled respiration is relevant to understanding the physiological consequences of nutrient-mediated changes in respiration rates, Δψm and mitochondrial dynamics described in Section 2.
c) Nutrient availability control on mitochondrial respiration
Mitochondrial respiration is controlled by three different processes: 1) ATP turnover; determined by cellular ATP consumption and matrix ADP levels. 2) Substrate utilization; determined by fuel availability inside the mitochondrial matrix and its oxidation to generate NADH, FADH2. 3) Proton leak, determined by basal permeability of the inner membrane to protons. Understanding the contribution of each of these processes is essential to predict under which physiological and mitochondrial respiratory states, nutrient availability will be determining mitochondrial respiration and Δψm. For instance, in isolated mitochondria under state 3 (maximal ATP synthesis rates), both nutrient utilization and ATP turnover exert a similar control over respiration (measured as the flux control coefficient over respiration, with a value of approximately 0.4–0.5 for each process with a total value of 1) and thus over Δψm (Hafner et al., 1990). This finding supports the common idea that ATP demand determines mitochondrial respiration and dominates over the other processes (substrate/nutrient utilization and proton leak).
However, in an intact cell, the metabolic processes providing NADH/FADH2 to the mitochondrial matrix (glycolysis, fatty acid oxidation and TCA cycle) can control respiration with a flux control coefficient over respiration between 0.15–0.3 under basal conditions (reviewed in Nicholls and Ferguson, 2002; Hafner et al., 1990). Therefore, although ATP turnover has a major influence controlling respiration and membrane potential (coefficient values 0.4–0.5) under conditions of high ATP demand, nutrient utilization and its availability can still have a very significant control over respiration and the exact mitochondrial Δψ values in intact cells (within the range dictated by thermodynamics). Furthermore, it is expected that in intact cells or isolated mitochondria treated with uncouplers (or activation of endogenous uncouplers), nutrient utilization/availability will have even a greater control over respiration, as ATP turnover will not control mitochondrial respiration under these conditions. Overall, the fact that the flux control coefficient of each process over respiration can vary, allows the possibility that under certain physiological scenarios and specific cell types, nutrient availability/utilization might dominate in the determination of the exact mitochondrial Δψ values (always within the range dictated by thermodynamics; around 90–225 mV).
Of particular relevance for this review, in some cell types (i.e. nutrient sensors such as the beta cell), nutrient utilization has a higher flux control coefficient and greater control over mitochondrial respiration and membrane potential than in other cell types (i.e. muscle cells). Consistent with this, recent evidence confirmed previous findings that mitochondrial hyperpolarization is proportional to the increase in extracellular nutrient concentration (glucose and pyruvate) in INS1 cells (Goehring et al., 2012).
Furthermore, mitochondrial proteins determining basal proton conductance can be activated by nutrients per se (i.e. UCP1, specific for brown adipose tissue, see next section), increasing mitochondrial respiration and uncoupling (Rial et al., 1983; Parker et al., 2009; Shabalina et al., 2008). The existence of this regulatory mechanism in brown adipocytes suggests that nutrients with high caloric content can activate thermogenesis and exert important control over respiration per se, or even increase their own oxidation. This mechanism could promote “nutrient wasting” in the form of heat generation under conditions of increased nutrient supply (Figure 1). In addition, it suggests that similar regulatory pathways decreasing bioenergetic efficiency could exist in other tissues, but likely through other mediators and/or regulators. These regulatory pathways are expected to be relevant in nutrient sensors, which harbor high nutrient permeability. These mechanisms could involve and/or require changes in mitochondrial dynamics, as discussed in Section 2.
In this regard, obesity and diabetes research resulted in our appreciation of this mitochondrial “nutrient wasting” in the form of heat. This appreciation comes from the idea that inducing thermogenesis through increased mitochondrial nutrient oxidation in certain tissues (such as muscle, brown adipose tissue or beige adipocytes) could potentially compensate the deregulated energetic balance associated with nutrient excess (Levine et al., 1999; Schutz et al., 1984; Wu et al., 2012). Increased thermogenesis would help to prevent the storage of nutrients in excess in the form of triacylglycerides (Figure 1). Consequently, understanding how this mitochondrial “nutrient wasting” process is regulated in all cell types and in a tissue-specific manner may prove useful for the treatment of conditions associated with excess nutrients.
Cells that should be particularly susceptible to nutrient supply/demand imbalance are those allowing nutrient permeability regardless of their energy demand in form of ATP (in other words, regardless of their ATP turnover). Such cells are the nutrient sensors, regulators and storage organs: the beta cells, the hepatocytes and the adipocytes. In the case of white adipocytes, this high nutrient permeability allows storage of nutrients in the form of triacylglicerides. However, in the nutrient sensors (e.g. beta cell) nutrient oxidation and ATP/ADP ratio serve as a sensing mechanism and a signal generator for insulin secretion. This ability of the beta cell to control and modulate their mitochondrial bioenergetics according to nutrient supply is essential to maintain its functionality (nutrient stimulated insulin secretion) and viability (beta cell detoxification by increasing nutrient oxidation towards heat generation). These different susceptibilities are likely to rely, to a certain extent, on the distinct tissue-specific mitochondrial bioenergetic properties and their regulatory mechanisms. These are likely to include modulation of mitochondrial dynamics and morphology. The evidence linking changes in mitochondrial dynamics with nutrient wasting (basal and activated proton conductance), metabolic state and cell type specific processes will be discussed in Section 2.
d) Effects of nutrient excess on mitochondrial bioenergetics in brown adipose tissue, muscle and the beta-cell
Brown adipose tissue (BAT)
Mitochondria from brown adipose tissue harbor uncoupling protein 1 (UCP1), which generates heat through the dissipation of the mitochondrial membrane potential and thus increasing respiratory rates (Aquila et al., 1985; Heaton et al., 1978; Nicholls, 1974; Nicholls et al., 1978). UCP1 is used as a specific marker to detect brown adipocytes within other tissues. The brown adipocyte represents a model in which a large shift in bioenergetic efficiency can be acutely induced through hormonal stimulation. Activation of non-shivering thermogenesis in human brown adipocytes by cold is achieved by the increase in fatty acid availability to the mitochondria and their oxidation, which is the result of norepinephrine (NE)- induced lipolysis (reviewed in Cannon and Nedergaard, 2004; Ouellet et al., 2012). In the case of rodents, high fat diet (a form of nutrient excess) increases BAT mass. This is mainly thanks to the increase in brown fat proliferation/differentiation, which results in the increase in UCP1 expression and the expansion of mitochondrial mass per cell in rodent models (Himms-Hagen et al., 1981; Rothwell and Stock, 1979). Whether an increase in the activity of this diet-induced expanded BAT in rodents contributes to what was defined as diet-induced thermogenesis is controversial (reviewed in Kozak, 2010).
Mitochondrial expansion induced by high fat diet in rodent brown fat shows that when ATP demand is not the main drive for oxygen consumption (i.e. conditions characterized by increased uncoupling such as in the activated brown fat), nutrient excess and increased fuel availability to the mitochondria does not impair bioenergetic function. This lack of toxicity could be explained by the association between mitochondrial membrane potential and escape of electrons from the electron transport chain to generate ROS (Brand et al., 2004). Coupled respiration normally occurs at higher values of membrane potential when compared to uncoupled respiration to generate heat (through UCP1 activation or other uncouplers). This means that uncoupled mitochondria will potentially generate less ROS, when compared to coupled mitochondria under conditions of nutrient excess. Following the metaphor of the hose, mitochondria from brown fat would have a second valve, constituted by UCP1, which would allow increasing water flow, while avoiding high pressure and any damage to the hose. The lack of this second valve with high capacity in muscle mitochondria might explain why diets similar to the ones inducing mitochondrial expansion in brown fat cause mitochondrial oxidative damage and dysfunction (decreased citrate synthase activity and decreased expression of complex IV subunits) (Bonnard et al., 2008) (see next section). Thus, nutrient excess in the form of high fat diet can expand mitochondrial capacity in some tissues, whereas mitochondria from other tissues might be damaged by the same diet.
The case of the muscle
Current data suggest potential mechanisms by which nutrient supply/demand imbalance may affect muscle mitochondrial function. Nutrient excess in the form of long term high fat diet results in the accumulation of toxic levels of intermediates of fatty acid metabolism. Some of these intermediates were shown to be a result of incomplete mitochondrial fatty acid oxidation and to contribute to impaired insulin signaling and to decreased glucose oxidation (Koves et al., 2008). Furthermore, this accumulation could potentially contribute to the failure of mitochondrial electron transport chain function reported in skeletal muscle from type 2 diabetic patients (Kelley et al., 2002). Other studies show that increased ROS generation, caused by nutrient excess through long term feeding of a high sucrose and fat diet, likely causes self-inflicted oxidative damage to the mitochondria and their dysfunction, the latter taking place after the onset of insulin resistance (Bonnard et al., 2008). Thus, excessive ROS production would be a major contributor to insulin resistance. These mechanisms would suggest that decreased mitochondrial function is not a regulated process but rather caused by damaging effects caused by nutrient excess.
Other studies suggest that decreased mitochondrial electron transport chain (ETC) function reported in diabetic muscle might be a compensatory and a regulated mechanism that inefficiently counteracts insulin resistance. These studies characterized two mouse models of a “primary” reduction in ETC complexes activity, which are muscle-specific knock-outs of the apoptosis inducing factor (AIF) and the transcription factor A mitochondrial (TFAM) respectively (Pospisilik et al., 2007; Wredenberg et al., 2006). These knock-out mice showed improved insulin sensitivity (Wredenberg et al., 2006; Pospisilik et al., 2007) and protection from high fat diet-induced obesity (Pospisilik et al., 2007). These findings suggest that the observed decrease in mitochondrial bioenergetic function in type 2 diabetics could be preventing mitochondrial-mediated toxicity associated with nutrient excess. This would favor the hypothesis that inherited or induced transcriptional down-regulation of mitochondrial transcripts (Mootha et al., 2003; Patti et al., 2003, Petersen et al., 2004) is a protective mechanism which counteracts insulin resistance, rather than a pathogenic mechanism contributing to insulin resistance. A potential explanation for the beneficial effect of reduced ETC activity is that reduction in the mass of coupled mitochondria in the muscle exposed to nutrient excess and low ATP demand may serve as a mechanism to avoid ROS-mediated insulin resistance.
Another mechanism that could cope with toxicity associated with nutrient excess is muscle uncoupled respiration. Increasing proton conductance can decrease mitochondrial ROS production and can enhance the removal of toxic intermediates by completing their oxidation (see previous section). However, nutrient overload-induced uncoupling and their relationship to ROS production in muscle are still controversial and the conclusions are different depending on the study, diets, mouse models and even the mitochondrial population analyzed (subsarcolemal vs. intermyofibrillar mitochondria) (Asami et al., 2008; Mollica et al., 2006; Almind et al., 2007; Fink et al., 2007; Nabben et al., 2011a; Nabben et al., 2011b).
These inconsistent findings might reflect the inability of oxidative muscle to promote a large shift in bioenergetic efficiency. A large increase in uncoupling capacity by nutrient excess, as in brown fat, could severely compromise ATP synthesis and thus oxidative muscle contractile function and calcium homeostasis. Furthermore, muscle is a “nutrient-consuming organ” and it has a steady supply of nutrients in vivo (i.e. glucose during fed state and glycogen during the initial phase of starvation, fatty acids and ketone bodies during intermediate starvation). Therefore, it makes physiological sense that high caloric nutrients, such as fatty acids, do not by and large increase uncoupling capacity in oxidative muscle (inducible proton conductance) as in brown fat. On the other hand, it is of relevance to study basal proton conductance or proton leak in muscle, as this tissue accounts for the major part of nutrient oxidation and thus for the overall organism metabolic efficiency. Thus, the study of mechanisms controlling basal proton conductance in muscle might reveal mechanisms coping with nutrient excess.
The Beta-cell
The beta-cell gauges glucose, free fatty acids and aminoacids availability in the bloodstream and secretes insulin accordingly (Deeney et al., 2000; Rutter, 2001). This gauging is performed through nutrient oxidation and mitochondrial respiration. Mechanistically, the main signal stimulating insulin secretion is increased cytosolic ATP/ADP ratio, through glucose oxidation and likely increased mitochondrial ATP synthesis. In addition, it has been shown that mitochondrial oxidation by-products (known as insulin secretagogues, which can include Malonyl-CoA synthesized from Acetyl-CoA produced in the mitochondria, ROS or GTP) stimulate and foster this glucose stimulated insulin secretion (Pi et al., 2007; Prentki et al., 1997; Kibbey et al., 2007; Rutter, 2001). Some amino-acids can stimulate insulin secretion by providing Acetyl-CoA to the Krebs cycle and increasing mitochondrial ATP synthesis (Floyd, Jr. et al., 1966; Poitout and Robertson, 2008). Along with this, there are also additive effects on insulin secretion by simultaneous presence of different nutrients. Fatty acids can modulate glucose stimulated insulin secretion, either through their beta oxidation, through the generation of monoglycerides and acyl-CoA or by direct interaction with plasma membrane receptors (Poitout and Robertson, 2008). Since the beta cells import and metabolize nutrients based on availability, and not on demand, mechanisms that handle excess nutrient availability are of particular value.
How do beta-cell mitochondria respond to nutrient excess? Long term exposure of beta-cell to high levels of glucose, lipids or their combination has deleterious effects on beta-cell mitochondrial function, physiology and viability. The observation that glucose synergizes with FFA in producing the toxic effects of nutrient excess, suggests that the two converge onto a common product (Poitout and Robertson, 2008; Prentki et al., 2002; Deeney et al., 2000). The usual suspect would be a situation of reductive stress characterized by increase in NADH, which in the absence of increased ATP demand, generates mitochondrial hyperpolarization and produces excess ROS (see section 1a, hose metaphor).
Perhaps, since the ability to adapt to excess supply has rarely if ever been selected for, beta cells are designed to be sensitive to ROS as a mechanism for nutrient sensing. As such, the beta cells have low antioxidant activity. ROS production mediated by high nutrients is utilized in the beta cell to couple nutrient oxidation to insulin secretion independently of changes in mitochondrial ATP synthesis (Pi et al., 2007). Therefore, insulin secretion could occur under conditions in which the ATP demand in the beta-cell is low. However, an abnormal situation of permanent nutrient excess or continuous exposure to fat (such as type 2 diabetes) would cause mitochondrial damage or decreased function by sustained overproduction of ROS combined with reduced antioxidant activity.
Given the importance of ATP production, ROS and mitochondrial derived coupling factors in insulin secretion, one would expect that respiration would be very efficiently coupled to ATP synthesis in beta-cells. However, the case is exactly the opposite. Beta-cell mitochondria show higher levels of endogenous proton leak than mitochondria from other tissues (e.g. muscle derived cells) (Affourtit and Brand, 2006). Although it might seem counterintuitive, uncoupled respiration allows limiting ROS-mediated toxicity caused by nutrient excess. This is consistent with the fact that beta-cells require other mechanisms to control ROS production, as they harbor low antioxidant activity. Thus, mitochondrial uncoupling is one of the few antioxidant mechanisms described so far that maintains proportionality between nutrient oxidation and insulin secretion through ROS production (but changing the “stoichiometry” nutrient-secretion). At the same time, this uncoupling ability should be tightly regulated in a relatively short period of time, as ATP/ADP ratio is a signal for insulin secretion, which requires efficient and coupled ATP synthesis.
We can conclude that mitochondria in the beta-cell have some bioenergetic properties that fall in between mitochondria from muscle and brown fat, which permit executing their specific physiological function related to nutrient sensing.
2. RELATIONSHIP BETWEEN BIOENERGETIC EFFICIENCY AND MITOCHONDRIAL DYNAMICS
In this section, we will summarize the changes observed in mitochondrial dynamics associated with conditions requiring a bioenergetic adaptation. This association raises different questions that are essential to answer in order to understand the relevance of this association:
-
1)
What comes first, changes in mitochondrial dynamics or changes in bioenergetic efficiency? Which factor serves the other? In this section, we will discuss evidence showing that changes in dynamics modulate bioenergetic efficiency and vice-versa. It is likely that the cell type and the metabolic state are major determinants in this relationship.
-
2)
If bioenergetic adaptation requires changes in mitochondrial dynamics, what are the consequences for mitochondrial quality control?
Regarding the first question, specific modulation of mitochondrial bioenergetics has been shown to cause profound changes to mitochondrial dynamics. These changes were to a large extent, interpreted in the context of quality control activation (Twig et al., 2008a; reviewed in Twig et al., 2008b). However, new evidence suggests that changes in mitochondrial structure mediated by nutrients and their metabolites may represent an adaptation to the changes in ATP demand and supply.
a) Summary of proteins regulating mitochondrial dynamics
Mitochondrial architecture is determined by motility, fusion and fission events. Mitochondrial fusion in mammals is mediated by Mitofusins (Mfn1 and Mfn2, located in the outer mitochondrial membrane) and Optic Atrophy gene 1 (Opa1, located in the inner membrane) (reviewed in Liesa et al., 2009). These three proteins require GTPase activity to mediate mitochondrial fusion. Proteolytic processing of Opa1 controls its fusion activity but also an Opa1 fusion-independent role, controlling cristae structure remodeling (reviewed in Liesa et al., 2009; Ishihara et al., 2006; Frezza et al., 2006). On the other hand, mitochondrial fission is mediated by Fission 1 protein (Fis1, located in the outer mitochondrial membrane), Mitochondrial fission factor (Mff, located in the outer mitochondrial membrane) and Dynamin related protein 1 (Drp1, is mostly cytosolic and translocates to the outer mitochondrial membrane during fission). Drp1 recruitment to the outer mitochondrial membrane and GTP hydrolysis are required for Drp1-mediated fission (reviewed in Liesa et al., 2009). Mff and Fis1 do not harbor GTPase activity and different studies show that they mediate fission by recruiting Drp1 (or other factors) to the mitochondria to a different extent. Of note, Drp1, Fis1 and Mff also control peroxisomal fission (Schrader, 2006; Waterham et al., 2007; Gandre-Babbe and Van der Bliek, 2008).
b) Mitochondrial fragmentation, proton leak and maximal respiratory capacity: effects of chemical uncouplers and nutrient excess
The addition of chemical uncouplers (i.e. FCCP or CCCP) causes complete mitochondrial network fragmentation, Drp1 recruitment to the outer membrane and OPA1 degradation (Duvezin-Caubet et al., 2006; Griparic et al., 2007; Ishihara et al., 2006; Song et al., 2007; Legros et al., 2002; Cereghetti et al., 2008). In addition, more recent studies show that depolarization by CCCP also triggers the proteasome-dependent degradation of additional mitochondrial fusion proteins (Mitofusin 1 and 2) and other outer membrane proteins. However, this proteasome-dependent degradation of Mfns requires the over-expression of the E3-ubiquitin-ligase Parkin (Tanaka et al., 2010; Ziviani et al., 2010; Chan et al., 2011). These studies demonstrated that mitochondrial fission is stimulated and fusion is inhibited in depolarized/uncoupled mitochondria through Drp1 recruitment and OPA1/Mfns degradation respectively. This suggests the possibility that fragmentation is advantageous for a system working at maximal respiratory capacity or for effective uncoupled respiration and depolarization.
Depolarization, decreased mitochondrial ATP synthesis or inhibition of fusion is not equivalent to mitochondrial dysfunction. Consistent with this, the use of uncouplers can mimic physiological conditions of nutrient excess and thus increase nutrient oxidation and electron transport chain activity, such as in the activated brown fat or in the beta-cell.
Consistent with this idea, studies exposing beta-cells to nutrient excess (Molina et al., 2009) or to conditions that uncouple mitochondria using a physiological stimulus, show increased respiration and robust fragmentation of the mitochondrial network (see Figure 2). Thus, it is likely that fragmentation is also associated with both maximal respiratory rates and increased proton leak.
Figure 2. Nutrient excess induces mitochondrial fragmentation in the beta-cell.
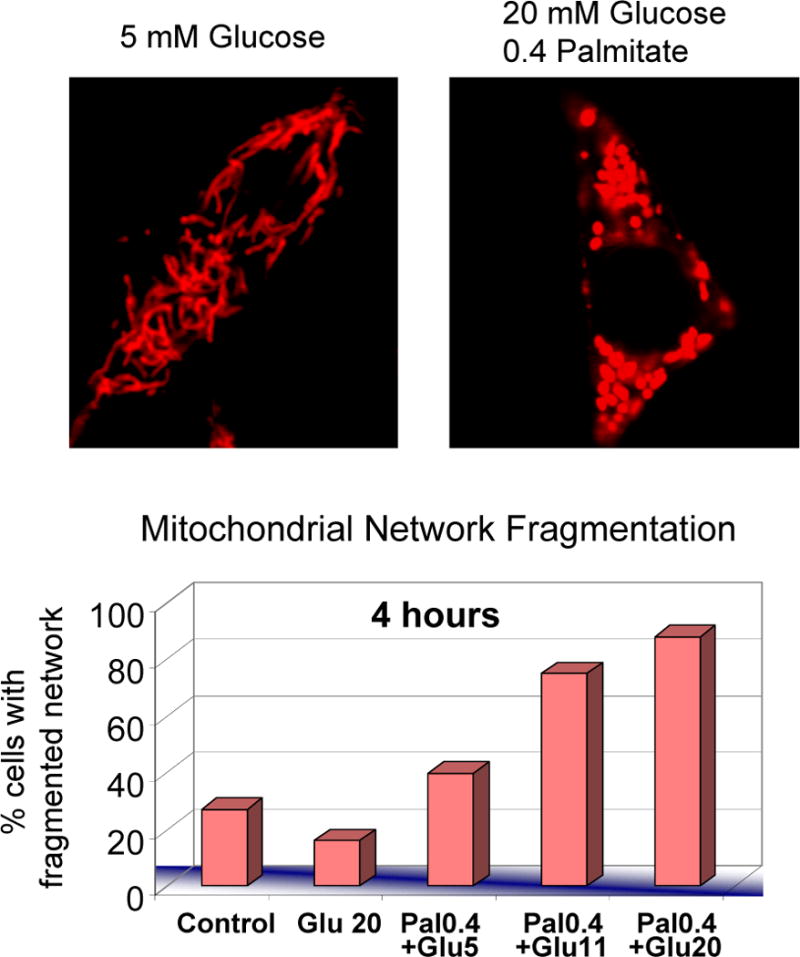
INS-1 cells treated for 4 hours with different concentrations of glucose and fatty acids (palmitate conjugated to BSA). The upper panel show representative images of INS-1 cells cultured with physiological glucose concentrations (5mM Glucose) and with high glucose and high fatty acids concentrations (20mM Glucose+ 0.4 mM Palmitate BSA at 4:1 ratio) for 4 hours. Mitochondria are shown in red and were stained with DsRed targeted to the mitochondria. Cells exposed to high levels of nutrients (20mM Glucose+ 0.4 mM Palmitate) show fragmentation and the formation of spherical mitochondria (ball-shape), whereas mitochondria with 5mM Glucose appear tubular. The bar graph shows the percentage of cells with fragmented mitochondria after 4 hours incubation with different concentrations of glucose and palmitate (in mM). Note the additive effect of glucose and fatty acids causing fragmentation. See Molina et al., 2009 for more details.
In this regard, there are some differences between the fragmentation observed under FCCP and the fragmentation observed under a rich nutrient environment or oxidative stress. Treatment with uncouplers results in fragmentation and the generation of doughnut shape mitochondria (Liu and Hajnoczky, 2011). Fragmentation in the beta cell is accompanied by increase in mitochondrial diameter to form ball shape instead of doughnut (bagel shape) mitochondria (Molina et al., 2009) (see Figure 2). The difference between the two conditions may hint to the potential different roles of the fragmentation and the increase in diameter. Fragmentation may support increased respiration and the increase in diameter may support increased leak. Indeed, these different morphologies can be explained by mitochondrial membrane potential values. FCCP causes massive mitochondrial depolarization (see section 1, it can reach 90 mV), whereas nutrient excess increases mitochondrial membrane potential (Goehring et al., 2012). Indeed, oligomycin, which markedly increases membrane potential (up to 220 mV, see section 1) was shown to cause fragmentation (Legros et al., 2002). Therefore, the increase in mitochondrial diameter with high nutrients could be a consequence of the increase in basal (endogenous) proton conductance associated with high membrane potential values. On the other hand, FCCP would artificially increase proton conductance by itself (induced) and would not activate the endogenous mechanisms to increase basal proton conductance (it is considered not inducible but it is always higher at high membrane potential values).
The common denominator between uncoupler-induced respiration and nutrient-induced proton leak/conductance is an increase in respiration and a decrease in ATP synthesis efficiency (less ATP per molecule of nutrient oxidized). The most conspicuous difference lies in the values of the mitochondrial membrane potential.
The mechanism by which fragmentation may benefit a condition of maximal respiration under uncoupler is not yet understood. Among other possibilities, fragmentation may represent a change in cristae structure that allows increased nutrient import. This would also be consistent with the dual role of OPA1 in mitochondrial fusion and cristae remodeling (Frezza et al., 2006). Thus, OPA1 processing/degradation could be one of the molecular mechanisms behind changes in cristae structure induced by uncouplers, facilitating nutrient import and/or inhibiting mitochondrial ATP synthase dimerization.
Since fragmentation is associated with increased leak, one may consider the possibility that mitochondrial fission proteins, such as Drp1, may facilitate it. At least in some systems there is evidence that Drp1-mediated fragmentation may promote leak through the permeability transition pore due to increased recruitment of Bax (Montessuit et al., 2010). In other systems, Drp1 recruitment to the outer mitochondrial membrane triggered cristae remodeling (Germain et al., 2005) and FCCP promoted Drp1 recruitment (Cereghetti et al., 2008). However, this does not mean that all forms of fragmentation facilitate leak. Nevertheless, it raises the potential role of fragmentation as a first step in the conversion of a cell into a high leak and high respiration state.
c) Mitochondrial elongation and bioenergetic function: changes in dynamics associated with situations requiring increased ATP synthesis capacity
The opposite condition to nutrient excess, starvation, causes an acute inhibition of mitochondrial fission, by inhibiting Drp1 recruitment to the mitochondria, and mitochondrial elongation due to unopposed fusion (Gomes et al., 2011; Rambold et al., 2011). These studies show that elongation prevented the removal of mitochondria by the starvation –induced autophagy. In addition, it causes an increase in mitochondrial cristae number, which is associated with the dimerization of the ATP synthase and thus higher ATP synthesis activity (Gomes et al., 2011). Therefore, starvation would elongate mitochondria in order to increase ATP synthesis capacity and thus sustain the ATP demand required during periods of limited nutrient availability. Furthermore, one could expect these mitochondria to be more coupled and to produce ATP more efficiently, as increased ATP synthesis capacity alone would deplete the limited amount of nutrients faster.
In a similar manner, mitochondrial elongation occurs during G1/S phase of the cell cycle, which is characterized by a large increase in ATP demand to support biogenic processes. Consequently, mitochondrial elongation during G1/S phase could permit high ATP synthesis rates that can sustain cell duplication (Mitra et al., 2009). These observations are consistent with respirometry studies, where it was demonstrated that cells at G1 phase have increased levels of coupled respiration and membrane potential (Schieke et al., 2008).
Consistent with the notion that mitochondrial elongation promotes increased mitochondrial ATP synthesis capacity is the association of elongation with cell senescence (Lee et al., 2007; Yoon et al., 2006). Senescence involves a decreased capacity of proliferation, homeostatic imbalance and thus decreased capacity of mitochondrial biogenesis. Under this condition increased ATP synthesis capacity and/or bioenergetic efficiency serves as an adaptation to reduced mitochondrial biogenesis. Mitochondrial fusion provides additional benefit, as it allows for sustaining functional mitochondria with higher number of mutated mitochondrial DNA copies per senescent cell by complementation. Indeed, senescent cells show increased inherent proton leak that might be caused by damage to the inner mitochondrial membrane (Hutter et al., 2004). This leak is compensated by increasing absolute values of basal respiration (compared to non-senescent cells) and thus maintaining the fraction of respiration coupled to ATP synthesis (Hutter et al., 2004). It will be interesting to test whether senescent cells can maintain the same degree of coupling if mitochondrial fragmentation is induced or elongation is prevented.
The senescent cell represents a situation of decreased bioenergetic capacity and decreased work load, while the starved cell has both capacity and workload increased. These different needs may explain the difference in the molecular mechanism under each condition: senescent cells show reduced Fis1 and Drp1 expression and slightly increased Mfn protein levels, while starved cells show no changes in total proteins levels, only in Drp1 recruitment to the mitochondria (Mai et al., 2010; Lee et al., 2007; Yoon et al., 2006; Gomes et al., 2011).
Other acute stresses, such as apoptosis activation (early stages) and oxidative stress (hydrogen peroxide treatment), have been shown to induce mitochondrial elongation. These changes were shown to facilitate ATP synthesis (Jendrach et al., 2008; Tondera et al., 2009).
The examples reviewed here illustrate that respiration under uncoupling as found in nutrient excess (or treatment with uncouplers) is associated with fragmentation and inhibition of fusion, whereas the opposite situation, starvation, is associated with inhibition of fission and increased ATP synthesis (and potentially with more coupled mitochondria under starvation). This comparison strengthens the hypothesis that mitochondrial dynamics plays an active role in changes in mitochondrial bioenergetic efficiency and capacity (see summary in Figure 3).
Figure 3. The balance of energy supply/demand is associated with corresponding changes to mitochondrial architecture and to bioenergetic efficiency.
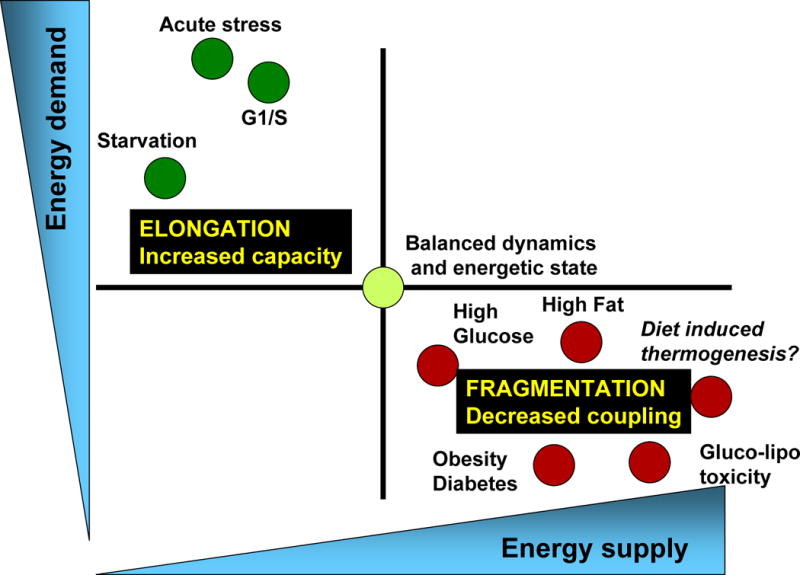
Physiological processes associated with increased energy demand and decreased energy supply, such as acute stress, starvation, G1/S phase, are characterized by mitochondrial elongation and by respiration coupled to ATP synthesis. On the other hand, physiological processes associated with decreased energy demand and increased supply (high levels of nutrients, obesity and diabetes…) are associated with mitochondrial fragmentation and decreased coupling (associated with heat generation or decreased mitochondrial function).
d) The link between bioenergetic efficiency and mitochondrial architecture: the pancreatic beta-cell
As discussed in the previous chapter, the beta-cell exquisitely adapts nutrient oxidation to nutrient availability, thereby coupling the latter to insulin secretion. This makes the beta-cell an attractive model for the study of the relationship between mitochondrial dynamics and cellular bioenergetic efficiency.
Beta cell mitochondria respond to nutrient excess by profound changes to mitochondrial architecture and dynamics. Exposure of beta-cells (INS-1) to high fat alone or in combination with glucose leads to mitochondrial fragmentation, which is detected after 4 and 24 hours of adding high glucose/fat (Molina et al., 2009) (Figure 2). Remarkably, the two nutrients show an additive effect in terms of inducing fragmentation. This suggests that the two are likely to activate the same fragmentation mechanism.
Thus, mitochondrial fragmentation in the beta cell is an early event that could be directly associated with increased nutrient oxidation. Mechanistically, the observed nutrient-induced fragmentation is mediated by inhibition of mitochondrial fusion (as shown by decreased sharing in mitochondrial matrix protein content; see Figure 4). Similar studies revealed a marked decrease in mitochondrial fusion in primary mouse islets exposed to high glucose/fatty acids for 48 hours (Molina et al., 2009).
Figure 4. Mitochondrial fragmentation induced by nutrient excess in the beta cell is caused by decreased mitochondrial fusion.
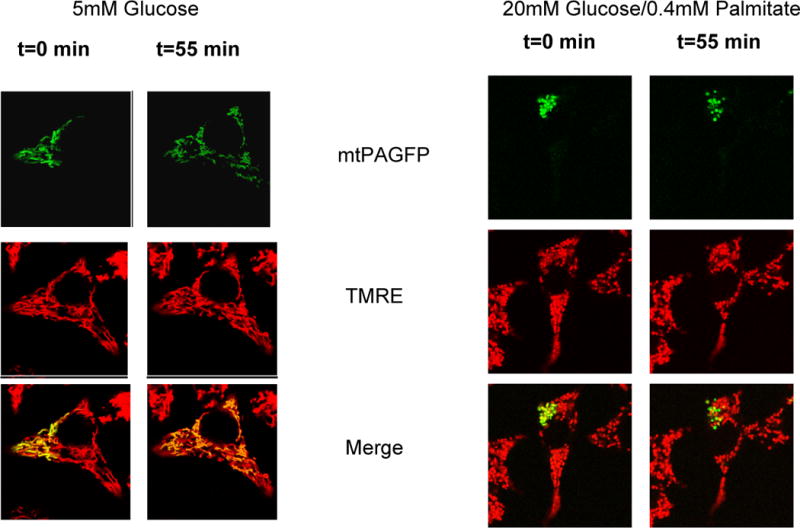
Mitochondrial fusion activity was quantified using mitochondrial matrix-targeted photoactivatable GFP (mtPAGFP, green). A portion of the mitochondrial population within a cell is labeled by laser photo-conversion and the sharing of the photo-converted molecules across the mitochondrial population through fusion events is monitored. Over 55 minutes the majority of mitochondria acquire photoconverted PA-GFP molecules. As a result of the dilution of the signal across the cell the intensity is diminished. TMRE (red) labeling was used to visualize the entire mitochondrial population. Right panels: INS-1 cells expressing mtPAGFP exposed to nutrient overload (20 mM Glucose + 0.4 mM Palmitate-BSA) for 4 hours show a dramatic reduction in fusion rates, as shown by the lack of mtPAGFP sharing with other mitochondria. Note that due to the lack of fusion the mtPAGFP intensity in the labeled mitochondria remains unchanged. Left Panels: INS-1 control cells (5 mM Glucose) present almost all mitochondria labeled with mtPAGFP 55 min after photoactivation. The images are adapted from Molina et al., 2009 with permission.
Is nutrient-induced fragmentation unique to the beta-cell? A model in which this question can be addressed is the brown adipocyte. The brown adipocyte allows for hormonal mediated induction of a leak in less than five minutes. This system is an example of a sharp increase in nutrient availability and an uncoupling mediated leak, moving from efficient respiration to the most inefficient respiration.
Activated brown fat preferably oxidizes fatty acids produced by lipolysis, which would be a similar situation to high fat exposure in the beta cell. Brown adipocytes go through complete mitochondrial network fragmentation upon induction of uncoupled respiration, supporting the observed correlation between the two (our unpublished data). Consequently, determining the importance of mitochondrial fragmentation to brown fat activation can be a strong proof that fragmentation is required to stimulate/enhance uncoupled respiration.
Increase in uncoupled respiration in the beta cell may serve as a mechanism to remove excess nutrient and set bioenergetic efficiency to balance beta cell nutrient supply and demand (see Figure 1). High glucose and particularly high fatty acids have multiple toxic effects in the beta cell, not only related to excessive ROS production (Las et al., 2011; Poitout and Robertson, 2008). The increase in uncoupled respiration could be a mechanism to decrease bioenergetic efficiency in the beta cell, and thus getting rid of the excess nutrients within the beta cell, by oxidizing them to generate heat. In this regard, Barbara Corkey and Marc Prentki suggested that increased nutrient oxidation and metabolic cycling in response to nutrient excess were mechanisms acting to permit beta cell detoxification. Consistent with this, increased uncoupled respiration and heat generation would be a mechanism to permit beta cell detoxification from the excess of nutrients through their oxidation without overproduction of ROS. Interestingly, fatty acid excess is more toxic for beta cells than high glucose (Poitout and Robertson, 2008). This may explain why fragmentation is higher in the presence of fatty acids excess than in the presence of high glucose in the beta cell. Fatty acids might require extra detoxification capacity within the beta cell because of their higher caloric content and their potential cytotoxic intermediates as a result of their incomplete oxidation (as in muscle) (Koves et al., 2008).
The difference between fatty acids and glucose in mediating fragmentation can be explained by the following hypotheses.
Respiration with fatty acids as substrates is associated with increased mitochondrial proton leak and concomitantly with lower values of membrane potential, whereas glucose oxidation (feeding pyruvate to the mitochondria) occurs with relatively higher membrane potential and thus lower proton leak. Therefore, one could hypothesize that fatty acids are more efficient at inducing fragmentation because their oxidation and other additional effects mediated by fatty acids per se are associated with a higher proton leak.
Fatty acid oxidation has been associated with higher ROS production by the electron transport chain. This is in part due to an additional site for superoxide formation (ETF-Qo, an exclusive site for electron entry into the ETC through fatty acid beta oxidation) (Seifert et al., 2010). Fragmentation and uncoupling may therefore be a protective mechanism that prevents oxidative damage. This would suggest that ROS, and not the fatty acids or their beta oxidation, are the main activators of fragmentation and uncoupling. In this regard, mitochondrial superoxide has been shown to activate uncoupled respiration (Echtay et al., 2002). Therefore, one would expect antioxidants to decrease fragmentation and proton leak induced by high fatty acids.
An alternative mechanism would be that the fatty acids per se could be causing fragmentation by directly interfering with the fusion and fission machinery. Indeed, fatty acids were shown to activate uncoupled respiration in brown fat through UCP1 (Nicholls and Locke, 1984; Williamson, 1970). The mechanism for this activation would be more likely related to their chemical structure, rather than to fatty acid metabolism or an intrinsic protonophoric activity (Shabalina et al., 2008). In this context, a proportion of this activation related to fatty acid chemical structure was UCP1-independent (Shabalina et al., 2008). Therefore, one could expect that certain fatty acids could be simultaneously signaling mitochondria fragmentation and consequently uncoupled respiration in a UCP1-independent manner, in addition to being the fuels oxidized by mitochondria. Consistent with this, phospholipase activity in the mitochondria is required for mitochondrial fusion mediated by mitofusins (Choi et al., 2006). This study shows a direct connection between acidic lipids generated in the mitochondria by phospholipase activity and fusion (Choi et al., 2006). Thus, fatty acid excess or acidic lipid moieties could be modulating fusion by interfering in these phospholipase-dependent processes or others currently unknown (Huang et al., 2011). However, this pathway has not been described in beta cells or brown adipocytes so far.
Ultimately, reductive stress and increased ROS generation are associated with mitochondrial fragmentation. In some cases, this fragmentation could relieve from reductive stress and ROS generation by decreasing mitochondrial membrane potential through cristae remodeling and OPA1 processing (i.e. nutrient excess). At the same time, mitochondrial fragmentation could be recruited by mechanisms or physiological processes depolarizing the mitochondria, to amplify or enhance the capacity of these processes lowering the mitochondrial membrane potential.
3. THE PRIMARY ROLE OF MITOCHONDRIAL DYNAMICS IN BIOENERGETIC EFFICIENCY: LESSONS FROM GENETIC MODELS
Thus far we described the association of mitochondrial network fragmentation and elongation with bioenergetic efficiency. Examination of genetic models in which alteration of mitochondrial dynamics proteins is the primary change may allow us to better understand the cause and effect relationship between the two.
a) Effects of specific changes in mitochondrial dynamics on mitochondrial bioenergetic efficiency
Early studies showed that shifting the balance towards fusion protected from cell death and shifting it towards fission increased susceptibility to apoptosis (Frank et al., 2001; Lee et al., 2004). Consistent with this observation, apoptosis has been associated with complete mitochondrial fragmentation (Frank et al., 2001). Furthermore, smaller and fragmented mitochondria were found in skeletal muscle from type 2 diabetic and obese subjects, conditions that are associated with decreased electron transport chain activity and decreased Mfn2 expression (Bach et al., 2003; Kelley et al., 2002). Together, these findings led to the initial impression that mitochondrial fragmentation impairs mitochondrial respiratory function and is deleterious to cell viability.
However, these generalizations were found to be inaccurate. For example, inhibition of mitochondrial fission through Drp1 modulation impairs mitochondrial function. HeLa cells with reduced Drp1 expression showed decreased complex IV activity and a decrease in both state 3 respiration (maximal ATP synthesis rates) and state 4 respiration (proton leak or uncoupling) (Benard et al., 2007). Drp1 knockdown induced a decrease in mtDNA copy number in cell culture (Parone et al., 2008) and complete Drp1 abrogation in mice and humans caused lethality with brain developmental defects and severe neurodegeneration (Ishihara et al., 2009; Wakabayashi et al., 2009; Waterham et al., 2007). Therefore, Drp1-mediated fission is important to maintain proper quality control (Twig et al., 2008a), electron transport chain function, mtDNA integrity and cell viability. Drp1 also mediates peroxisomal fission and some of the physiological changes induced by Drp1 modulation can be attributed to effects on peroxisome function (Schrader, 2006; Waterham et al., 2007).
Mitochondrial fusion and fission occur sequentially in a repeating cycle (see Figure 5). The direct implication of this realization is that inhibition of either fusion or fission arrest the cycle. Indeed, similar bioenergetic defects are observed in cells in which fusion is inhibited. As an example, skeletal muscle harboring simultaneous deletions in Mfn1 and Mfn2 expression (Mfn double KO) show decreased number of mitochondrial DNA copies, increased mutation/deletion load and decreased mitochondrial respiration (Chen et al., 2010). On the other hand, an ineffective compensatory increase in mitochondrial mass and complex II activity has been observed in Mfn double KO muscles (Chen et al., 2010). The bioenergetic defect and the accompanying expansion of mitochondrial mass resemble the histopathology of patients harboring mutations in mitochondrial DNA causing MERRF (myoclonic epilepsy with red ragged fibers). Thus, absence of fusion alters mtDNA homeostasis and electron transport chain function in a similar manner to the inhibition of fission. The mechanisms by which lack of fusion or fission would decrease mitochondrial DNA levels are not clear. While mitochondrial fusion is the main mechanism proposed to allow complementation of functional components in mitochondria harboring mutated mtDNA copies, lack of complementation per se cannot explain why decreased fusion should decrease mitochondrial DNA levels (along with a compensatory increase in mass and transcription of nuclear-encoded mitochondrial components).
Figure 5. The life cycle of mitochondria and its regulation by nutrient availability.
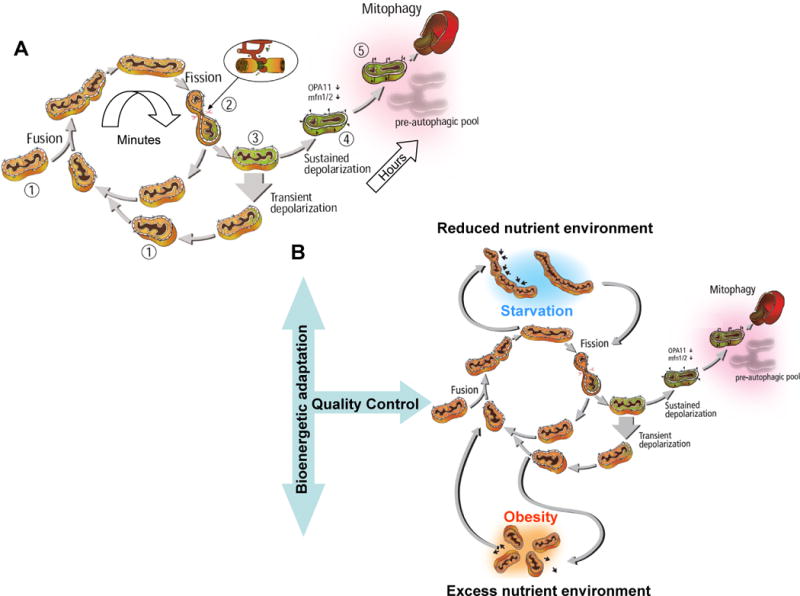
A) The life cycle of the mitochondria. The cycle is characterized by fusion and fission events. Fusion generates a network in which components of the two mitochondria are mixed and reorganized (1). Fission that follows within minutes splits fused mitochondria into two daughter mitochondria with disparate membrane potential (2). The daughter with the higher membrane potential is the first to return to the cycle of fusion/fission, while the daughter with more depolarized membrane potential will remain solitary until its membrane potential recovers (3). If membrane potential remains depolarized, this mitochondrion will loose its ability to fuse and become part of the pre-autophagic pool characterized by solitary, depolarized mitochondria (4). With a delay of 1–3 hours, these mitochondria are eliminated by autophagy (5).
B) Changes to nutrient availability and energy demand can divert mitochondria from the life cycle and extend their stay in the post fusion state (elongation) or the post fission state (fragmentation).
Elongation of mitochondria is a result of increased fusion or decreased fission activity (top section). This is typical for states of increased energy efficiency (starvation, acute stress, senescence). Shortening of mitochondria is a result of decreased fusion activity or increased fission activity (bottom section). This is typical for states of reduced bioenergetic efficiency (increased respiratory leak). Since bioenergetic adaptation high energy supply requires the arrest of the mitochondria life cycle, extended exposure to excess nutrient environment is expected to impact quality control, a condition that will contribute to reduced longevity.
b) Mfn2 deletion in skeletal muscle exacerbates the effects of nutrient excess on bioenergetic efficiency
Some of the first observations that associated fragmentation with excess nutrients were the decreased size of mitochondria in muscle from type 2 diabetic and obese humans or mouse models (Bach et al., 2003; Kelley et al., 2002). An accompanying decrease in Mfn2 expression provided a potential mechanism to the reduction in mitochondrial size (Bach et al., 2003; Bach et al., 2005). Consistent with this, specific deletion of Mfn2 in the muscle is associated with decreased mitochondrial ATP synthesis efficiency in permeabilized muscle fibers and with lower protein levels of different ETC subunits (Sebastian et al., 2012). This lower efficiency is explained by a mild decrease in ADP-stimulated respiration and by a mild increase in the leak, accompanied by an increase in ROS production (Sebastian et al., 2012). Therefore, as in the beta cell, fragmentation through inhibition of mitochondrial fusion is associated with increased proton leak in muscle. In addition, this specific deletion of Mfn2 in the muscle and mild repression in other tissues is sufficient to impair insulin signaling and exacerbates the deleterious effects of nutrient excess (high fat diet) (Sebastian et al., 2012). These results suggest that Mfn2 expression in the muscle is required for a proper bioenergetic adaptation to nutrient excess in the form of high fat diet. Thus, Mfn2 deletion in skeletal muscle causes a pro-diabetic effect that involves increased ROS generation and oxidative damage.
On the other hand, Mfn2 and other mitochondrial dynamics components are regulated by pathways activated during conditions of increased energy demand (i.e exercise and cold exposure) and down-regulated in type 2 diabetic patients (nutrient excess), involving the transcriptional co-activators PGC-1α and PGC-1β (Cartoni et al., 2005; Liesa et al., 2008; Soriano et al., 2006; Mootha et al., 2003; Patti et al., 2003). This regulation also supports, to a certain extent, the link between increased energy demand and mitochondrial elongation described before (see illustration in Figure 3).
In conclusion, maintaining both fusion and fission events is the key parameter to the homeostasis of the bioenergetic function of the mitochondrial population within the cell. Specific defects in mitochondrial dynamics can generate cellular energetic states similar to conditions with altered nutrient supply/demand balance, as shown in muscle. In the specific case of long-term nutrient excess, it is possible that the extension in time of a short-term protective response to nutrient overload, such as fragmentation to reduce reductive stress, ROS and membrane potential, has deleterious effects on mitochondrial quality control in the long term (Mouli et al., 2009; Twig et al., 2008a). These effects on mitochondrial quality control mediated by altered morphology can potentially explain the abnormal mitochondrial bioenergetic function and cumulative damage associated with metabolic diseases or aging.
4. EFFECTS OF NUTRIENT AVAILABILITY ON MITOCHONDRIAL QUALITY CONTROL
Changes in mitochondrial dynamics affect quality control and can therefore influence bioenergetic capacity indirectly (Twig et al., 2008a). Moreover, recent evidence suggests that nutrients influence quality control function (Las et al., 2011; Singh et al., 2009). Therefore, for appropriate consideration of the relationship between mitochondrial dynamics and bioenergetics one has to consider how both interact with mitochondrial quality control mechanisms.
a) Mechanisms for mitochondrial quality control and its regulation by bioenergetics, mitophagy and mitochondrial dynamics
The use of confocal microscopy allowed the visualization of single mitochondria units and specifically to track them over time (Twig et al., 2008a; Twig et al., 2010; reviewed in Liesa et al., 2009). A very relevant observation to our discussion is the finding that single mitochondrial units were heterogeneous in terms of their bioenergetic activity (Wikstrom et al., 2007; Wikstrom et al., 2009). This was reflected by the difference in mitochondrial membrane potential between the different units. Furthermore, this heterogeneity was modulated by nutrient excess and other metabolic changes (Wikstrom et al., 2007), which regulate mitochondrial dynamics (Molina et al., 2009). These data demonstrate that mitochondrial fusion and fission does not completely equilibrate the bioenergetic properties of the entire mitochondrial population. This stands in contrast to the mitochondrial complementation theory which hypothesizes that mitochondrial fusion homogenizes the entire population, a conclusion drawn from the observation of shared matrix soluble components. Remarkably, however, decreasing mitochondrial fusion rate resulted in increased heterogeneity illustrating the contribution of mitochondrial dynamics to the maintenance of the mitochondrial bioenergetic function. The paradox could be settled by the understanding that fusion, fission and autophagy are all connected by one axis (Figure 5).
The quality control axis is centered on the fission event which may generate two bioenergetically different mitochondria, one with a higher membrane potential and one with lower membrane potential. The single daughter mitochondrion with lower membrane potential has two options: 1) Recover its membrane potential and regain the capacity to reconnect with the network 2) Remain in the solitary period, depolarized. If membrane potential is not restored during the solitary period, OPA1 will be degraded. Thus, the solitary mitochondria will not be able to re-engage with the network and will be degraded by mitophagy. One can conclude that fission is an important process isolating a potentially damaging organelle and that selective fusion governs the fate of the mitochondria to be autophagocytosed (Twig et al., 2008a). Within this context, long-term inhibition (days) of fission by Drp1 dominant negative over-expression can reduce the increase in respiration induced by uncouplers in intact cells (Twig et al., 2008a). These results should not be interpreted as evidence for the requirement of fragmentation to achieve maximal respiratory capacity (see section 3a). The effects on bioenergetics caused by long-term inhibition of fission can be explained by accumulation of irreversibly damaged mitochondria that cannot be segregated (Twig et al., 2008a). This finding is supported by changes of membrane fluidity in isolated mitochondria from cells with down-regulation of Drp1 (Benard et al., 2007), showing that the alteration is maintained when mitochondria are taken out of the cells and mitochondrial dynamics are absent.
While it is widely accepted that fission events produce uneven daughters that are selected by autophagy it may be appropriate to indicate that this was only shown in the beta cell and COS7 cells. Similarly, that mitochondrial autophagy is a housekeeping process that targets spontaneously depolarized mitochondria was thus far shown only in the beta cells.
Multiple studies have identified additional mechanisms for the inability of mitochondria in the solitary period to fuse and the signals that label them to be recognized and removed by the autophagic machinery. The U3-ubiquitin ligase Parkin (mutated in Parkinson’s disease), through PINK1 Serine-threonine kinase activity, is recruited to depolarized mitochondria to target them for mitophagy (Narendra et al., 2008; Vives-Bauza et al., 2010; Ziviani et al., 2010). In addition, Parkin ubiquitinates Mfn, promoting its degradation by the proteasome system and thus contributing to fusion inhibition of the solitary depolarized mitochondria (Chan et al., 2011; Tanaka et al., 2010; Ziviani et al., 2010). Therefore, we can define that these solitary and dysfunctional mitochondria, with no fusion capabilities, comprise the pre-autophagic pool of mitochondria.
A key component dictating the efficiency of mitochondrial quality control by fusion, fission and autophagy is the ability of a full cycle to be completed and the number of cycles per day (Mouli et al., 2009). A mathematical model that runs multiple iterations of the cycle predicts that the rate of fusion/fission cycles determines the capacity of the pathway to restore quality upon damage. In this context the effect of nutrient on the rate of fusion, fission and the formation of the mitochondrial pre-autophagic pool may be considered as important in its effect on autophagy (Las et al, 2011; Singh et al., 2009).
b) Evidence of mitochondrial quality control, mitophagy and autophagy modulation by nutrients and their relationship to the energetic state
Nutrient excess leads to the inhibition of fusion, resulting in fragmentation and an incomplete cycle of fusion, fission and autophagy (Molina et al., 2009; Las et al., 2011). In addition, it does not allow for mitochondrial complementation and thus increases subcellular mitochondrial heterogeneity (Wikstrom et al., 2007). Given this lack of selective removal, one could expect that mitochondrial mass would decrease, as the population will be mostly comprised of small and depolarized mitochondria (Figure 5). Therefore, to maintain mitochondrial health, it would only be required to stimulate mitochondrial biogenesis. However, nutrient excess can impair autophagic flux by inhibiting lysosomes, which are required to digest autophagosomes and thus damaged mitochondria (Las et al., 2011). As a consequence, dysfunctional mitochondria will accumulate and they will affect even mitochondria generated de novo (by unselective fusion and/or increased ROS production). These alterations can explain different reports demonstrating mitochondrial dysfunction in pathologies associated with an imbalance in nutrient supply/demand.
Turnover requires both fusion events and the segregation of damaged components by fission, which will not enter again into the network because fusion is bioenergetically selective (Figure 5). We suggest that the interaction between mitochondrial life-cycle, dynamics and bioenergetics evolved to adapt to changes in nutrient availability, which are physiologically comprised of feeding and fasting states. Any prolongation in the feeding or fasting state requires a bioenergetic adaptation that will shift the mitochondrial dynamics balance. A prolonged shift will have deleterious effects on mitochondrial health and quality control. In the case of the fasting state, the shift in dynamics required for bioenergetic adaptation will homogenize the mitochondrial population, preventing the segregation, the formation of the pre-autophagic pool and the removal of damaged components by mitophagy. In the fed state and/or nutrient excess (particularly high fat), fragmentation and high respiratory rates can lead to damage, in addition to mechanisms affecting the autophagic machinery downstream of the pre-autophagic pool of mitochondria. This would cause the accumulation of dysfunctional units and increase ROS generation. In this context, it is likely that caloric restriction (or proper fed/fasting cycles) would promote a bioenergetic adaptation and a change in mitochondrial dynamics permitting the most efficient mitochondrial quality control mechanisms. Thus, this interaction between bioenergetics adaptation, mitochondrial dynamics and quality control could explain some of the beneficial effects associated with caloric restriction.
Acknowledgments
We thank Professors Daniel Dagan, Susan K. Fried and Barbara E. Corkey for insightful comments. M.L. is an Evans Center Fellow and was the recipient of a post-doctoral fellowship from Fundación Ramón Areces. O.S. is funded by NIH R01HL071629–03 and R01DK074778 grants.
Footnotes
There is no supplementary material
Reference List
- Affourtit C, Brand MD. Stronger control of ATP/ADP by proton leak in pancreatic beta-cells than skeletal muscle mitochondria. Biochem J. 2006;393:151–159. doi: 10.1042/BJ20051280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almind K, Manieri M, Sivitz WI, Cinti S, Kahn CR. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc Natl Acad Sci U S A. 2007;104:2366–2371. doi: 10.1073/pnas.0610416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquila H, Link TA, Klingenberg M. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. EMBO J. 1985;4:2369–2376. doi: 10.1002/j.1460-2075.1985.tb03941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asami DK, McDonald RB, Hagopian K, Horwitz BA, Warman D, Hsiao A, Warden C, Ramsey JJ. Effect of aging, caloric restriction, and uncoupling protein 3 (UCP3) on mitochondrial proton leak in mice. Exp Gerontol. 2008;43:1069–1076. doi: 10.1016/j.exger.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg-Henriksson H, Manco M, Calvani M, Castagneto M, Palacin M, Mingrone G, Zierath JR, Vidal H, Zorzano A. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes. 2005;54:2685–2693. doi: 10.2337/diabetes.54.9.2685. [DOI] [PubMed] [Google Scholar]
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- Cadenas S, Buckingham JA, Samec S, Seydoux J, Din N, Dulloo AG, Brand MD. UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Lett. 1999;462:257–260. doi: 10.1016/s0014-5793(99)01540-9. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Leger B, Hock MB, Praz M, Crettenand A, Pich S, Ziltener JL, Luthi F, Deriaz O, Zorzano A, Gobelet C, Kralli A, Russell AP. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol. 2005;567:349–358. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JB, Perry SV. The Respiratory and Adenosinetriphosphatase Activitiesof Skeletal-Muscle Mitochondria. Biochem J. 1954;55:586–595. doi: 10.1042/bj0550586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu Y, Dorn GW. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- Deeney JT, Prentki M, Corkey BE. Metabolic control of beta-cell function. Semin. Cell Dev Biol. 2000;11:267–275. doi: 10.1006/scdb.2000.0175. [DOI] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- Fink BD, Herlein JA, Almind K, Cinti S, Kahn CR, Sivitz WI. Mitochondrial proton leak in obesity-resistant and obesity-prone mice. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1773–R1780. doi: 10.1152/ajpregu.00478.2007. [DOI] [PubMed] [Google Scholar]
- Floyd JC, Jr, Fajans SS, Conn JW, Knopf RF, Rull J. Stimulation of insulin secretion by amino acids. J Clin Invest. 1966;45:1487–1502. doi: 10.1172/JCI105456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Gandre-Babbe S, Van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring I, Gerencser AA, Schmidt S, Brand MD, Mulder H, Nicholls DG. Plasma membrane potential oscillations in insulin secreting Ins-1 832/13 cells do not require glycolysis and are not initiated by fluctuations in mitochondrial bioenergetics. J Biol Chem. 2012;287:15706–15717. doi: 10.1074/jbc.M111.314567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di BG, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner RP, Brown GC, Brand MD. Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force using the “top-down” approach of metabolic control theory. Eur J Biochem. 1990;188:313–319. doi: 10.1111/j.1432-1033.1990.tb15405.x. [DOI] [PubMed] [Google Scholar]
- Heaton GM, Wagenvoord RJ, Kemp A, Jr, Nicholls DG. Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur J Biochem. 1978;82:515–521. doi: 10.1111/j.1432-1033.1978.tb12045.x. [DOI] [PubMed] [Google Scholar]
- Himms-Hagen J, Triandafillou J, Gwilliam C. Brown adipose tissue of cafeteria-fed rats. Am J Physiol. 1981;241:E116–E120. doi: 10.1152/ajpendo.1981.241.2.E116. [DOI] [PubMed] [Google Scholar]
- Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell. 2011;20:376–387. doi: 10.1016/j.devcel.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter E, Renner K, Pfister G, Stockl P, Jansen-Durr P, Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. 2004;380:919–928. doi: 10.1042/BJ20040095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jendrach M, Mai S, Pohl S, Voth M, Bereiter-Hahn J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion. 2008;8:293–304. doi: 10.1016/j.mito.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, Shulman GI. Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab. 2007;5:253–264. doi: 10.1016/j.cmet.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Kozak LP. Brown fat and the myth of diet-induced thermogenesis. Cell Metab. 2010;11:263–267. doi: 10.1016/j.cmet.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in beta-cells. J Biol Chem. 2011;286:42534–42544. doi: 10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, Youle RJ, Cho H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem. 2007;282:22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JA, Eberhardt NL, Jensen MD. Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science. 1999;283:212–214. doi: 10.1126/science.283.5399.212. [DOI] [PubMed] [Google Scholar]
- Liesa M, Borda-d’Agua B, Medina-Gomez G, Lelliott CJ, Paz JC, Rojo M, Palacin M, Vidal-Puig A, Zorzano A. Mitochondrial fusion is increased by the nuclear coactivator PGC-1beta. PLoS One. 2008;3:e3613. doi: 10.1371/journal.pone.0003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- Liu X, Hajnoczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011;18:1561–1572. doi: 10.1038/cdd.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci. 2010;123:917–926. doi: 10.1242/jcs.059246. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, Shirihai OS. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315. doi: 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica MP, Lionetti L, Crescenzo R, D’Andrea E, Ferraro M, Liverini G, Iossa S. Heterogeneous bioenergetic behaviour of subsarcolemmal and intermyofibrillar mitochondria in fed and fasted rats. Cell Mol Life Sci. 2006;63:358–366. doi: 10.1007/s00018-005-5443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P, Martinou JC. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mouli PK, Twig G, Shirihai OS. Frequency and selectivity of mitochondrial fusion are key to its quality maintenance function. Biophys J. 2009;96:3509–3518. doi: 10.1016/j.bpj.2008.12.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabben M, Hoeks J, Moonen-Kornips E, Van BD, Briede JJ, Hesselink MK, Glatz JF, Schrauwen P. Significance of uncoupling protein 3 in mitochondrial function upon mid- and long-term dietary high-fat exposure. FEBS Lett. 2011a;585:4010–4017. doi: 10.1016/j.febslet.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Nabben M, Shabalina IG, Moonen-Kornips E, van BD, Cannon B, Schrauwen P, Nedergaard J, Hoeks J. Uncoupled respiration, ROS production, acute lipotoxicity and oxidative damage in isolated skeletal muscle mitochondria from UCP3-ablated mice. Biochim Biophys Acta. 2011b;1807:1095–1105. doi: 10.1016/j.bbabio.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Hamster brown-adipose-tissue mitochondria. The control of respiration and the proton electrochemical potential gradient by possible physiological effectors of the proton conductance of the inner membrane. Eur J Biochem. 1974;49:573–583. doi: 10.1111/j.1432-1033.1974.tb03861.x. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Bernson VS, Heaton GM. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl. 1978;32:89–93. doi: 10.1007/978-3-0348-5559-4_9. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ. Bioenergetics 3. Academic Press; 2002. [Google Scholar]
- Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiol Rev. 1984;64:1–64. doi: 10.1152/physrev.1984.64.1.1. [DOI] [PubMed] [Google Scholar]
- Ouellet V, Labbe SM, Blondin DP, Phoenix S, Guerin B, Haman F, Turcotte EE, Richard D, Carpentier AC. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest. 2012;122:545–552. doi: 10.1172/JCI60433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker ND, Crichton PG, Vidal-Puig AJ, Brand MD. Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria. J Bioenerg Biomembr. 2009;41:335–342. doi: 10.1007/s10863-009-9232-8. [DOI] [PubMed] [Google Scholar]
- Parone PA, Da CS, Tondera D, Mattenberger Y, James DI, Maechler P, Barja F, Martinou JC. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One. 2008;3:e3257. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51(Suppl 3):S405–S413. doi: 10.2337/diabetes.51.2007.s405. [DOI] [PubMed] [Google Scholar]
- Prentki M, Tornheim K, Corkey BE. Signal transduction mechanisms in nutrient-induced insulin secretion. Diabetologia. 1997;40(Suppl 2):S32–S41. doi: 10.1007/s001250051395. [DOI] [PubMed] [Google Scholar]
- Quiros PM, Ramsay AJ, Sala D, Fernandez-Vizarra E, Rodriguez F, Peinado JR, Fernandez-Garcia MS, Vega JA, Enriquez JA, Zorzano A, Lopez-Otin C. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rial E, Poustie A, Nicholls DG. Brown-adipose-tissue mitochondria: the regulation of the 32000-Mr uncoupling protein by fatty acids and purine nucleotides. Eur J Biochem. 1983;137:197–203. doi: 10.1111/j.1432-1033.1983.tb07815.x. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Stock MJ. A role for brown adipose tissue in diet-induced thermogenesis. Nature. 1979;281:31–35. doi: 10.1038/281031a0. [DOI] [PubMed] [Google Scholar]
- Rutter GA. Nutrient-secretion coupling in the pancreatic islet beta-cell: recent advances. Mol Aspects Med. 2001;22:247–284. doi: 10.1016/s0098-2997(01)00013-9. [DOI] [PubMed] [Google Scholar]
- Schieke SM, McCoy JP, Jr, Finkel T. Coordination of mitochondrial bioenergetics with G1 phase cell cycle progression. Cell Cycle. 2008;7:1782–1787. doi: 10.4161/cc.7.12.6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M. Shared components of mitochondrial and peroxisomal division. Biochim Biophys Acta. 2006;1763:531–541. doi: 10.1016/j.bbamcr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Schutz Y, Bessard T, Jequier E. Diet-induced thermogenesis measured over a whole day in obese and nonobese women. Am J Clin Nutr. 1984;40:542–552. doi: 10.1093/ajcn/40.3.542. [DOI] [PubMed] [Google Scholar]
- Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert EL, Estey C, Xuan JY, Harper ME. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J Biol Chem. 2010;285:5748–5758. doi: 10.1074/jbc.M109.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina IG, Backlund EC, Bar-Tana J, Cannon B, Nedergaard J. Within brown-fat cells, UCP1-mediated fatty acid-induced uncoupling is independent of fatty acid metabolism. Biochim Biophys Acta. 2008;1777:642–650. doi: 10.1016/j.bbabio.2008.04.038. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA 1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Liesa M, Bach D, Chan DC, Palacin M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006;55:1783–1791. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- Sugden MC, Watts DI, Marshall CE, McCormack JG. Brown-adipose-tissue lipogenesis in starvation: effects of insulin and (−) hydroxycitrate. Biosci Rep. 1982;2:289–297. doi: 10.1007/BF01115114. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da CS, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008a;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008b;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Liu X, Liesa M, Wikstrom JD, Molina AJ, Las G, Yaniv G, Hajnoczky G, Shirihai OS. Biophysical properties of mitochondrial fusion events in pancreatic beta-cells and cardiac cells unravel potential control mechanisms of its selectivity. Am J Physiol Cell Physiol. 2010;299:C477–C487. doi: 10.1152/ajpcell.00427.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Welle S, Lilavivat U, Campbell RG. Thermic effect of feeding in man: increased plasma norepinephrine levels following glucose but not protein or fat consumption. Metabolism. 1981;30:953–958. doi: 10.1016/0026-0495(81)90092-5. [DOI] [PubMed] [Google Scholar]
- Wikstrom JD, Katzman SM, Mohamed H, Twig G, Graf SA, Heart E, Molina AJ, Corkey BE, de Vargas LM, Danial NN, Collins S, Shirihai OS. beta-Cell mitochondria exhibit membrane potential heterogeneity that can be altered by stimulatory or toxic fuel levels. Diabetes. 2007;56:2569–2578. doi: 10.2337/db06-0757. [DOI] [PubMed] [Google Scholar]
- Wikstrom JD, Twig G, Shirihai OS. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int J Biochem Cell Biol. 2009;41:1914–1927. doi: 10.1016/j.biocel.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Williamson JR. Control of energy metabolism in hamster brown adipose tissue. J Biol Chem. 1970;245:2043–2050. [PubMed] [Google Scholar]
- Wredenberg A, Freyer C, Sandstrom ME, Katz A, Wibom R, Westerblad H, Larsson NG. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun. 2006;350:202–207. doi: 10.1016/j.bbrc.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, Malka F, Jou MJ, Martinou JC, Yoon G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fisl. J Cell Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S, Clerc P, Polster BM, Aja SM, Pletnikov MV, Kensler TW, Shirihai OS, Iijima M, Hussain MA, Sesaki H. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell. 2011;22:2235–2245. doi: 10.1091/mbc.E10-12-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]