

Abstract
Background
Retinitis pigmentosa (RP) is the most common cause of inherited retinal degeneration and can occur in non-syndromic and syndromic forms. Syndromic RP is accompanied by other symptoms such as intellectual disability, hearing loss, or congenital abnormalities. Both forms are known to exhibit complex genetic interactions that can modulate the penetrance and expressivity of the phenotype.
Materials and methods
In an individual with atypical RP, hearing loss, ataxia and cerebellar atrophy whole exome sequencing was performed. The candidate pathogenic variants were tested by developing an in vivo zebrafish model and assaying for retinal and cerebellar integrity.
Results
Exome sequencing revealed a complex heterozygous protein-truncating mutation in RP1L1, p.[(Lys111Glnfs*27; Q2373*)], and a heterozygous nonsense mutation in C2orf71, p.(Ser512*). Mutations in both genes have previously been implicated in autosomal recessive non-syndromic RP, raising the possibility of a digenic model in this family. Functional testing in a zebrafish model for two key phenotypes of the affected person showed that the combinatorial suppression of rp1l1 and c2orf71l induced discrete pathology in terms of reduction of eye size with concomitant loss of rhodopsin in the photoreceptors, and disorganization of the cerebellum.
Conclusions
We propose that the combination of heterozygous loss-of-function mutations in these genes drives syndromic retinal dystrophy, likely through the genetic interaction of at least two loci. Haploinsufficiency at each of these loci is insufficient to induce overt pathology.
Keywords: Retinitis pigmentosa, digenic inheritance, hearing loss, zebrafish, ciliopathy, cerebellum
INTRODUCTION
Retinitis pigmentosa (RP) is an important cause of visual impairment with a prevalence of 1:4,000.1 The disorder is typically driven by degeneration of the rod photoreceptors, followed by cone cell death in a more advanced stage of the disease. The age of onset and the degree of progression are highly variable. Some forms are congenital, but most forms of RP start with night blindness in young adulthood, followed by mid-peripheral visual field loss and tunnel vision. In many instances, electrophysiological studies have revealed pathology that precedes gross structural defects in the photoreceptor and a concomitant asymptomatic latency period. Finally, also central vision can decrease, which may result in complete blindness. During funduscopy, pigment deposits and attenuated retinal vessels can be seen, although this can be absent at early stages of the disease. The diagnosis can be established by electroretinography (ERG).2 Persons with RP can have additional features, consisting of hearing impairment, dysmorphisms, intellectual disability or congenital abnormalities, collectively called syndromic RP. Syndromic RP is a genetic heterogeneous disease; at present, >80 genes are known to be involved, with most of them contributing mutations under an autosomal recessive inheritance paradigm (https://sph.uth.edu/Retnet/, November 2014). Exome sequencing is a powerful tool to analyze all known genes involved in syndromic RP and to identify new candidate genes.3 Here we present an individual with syndromic retinal dystrophy in whom exome sequencing revealed heterozygous null variants in RP1L1 (MIM 608581; NM_178857.5) and C2orf71 (MIM 613425; NM_001029883.2), two genes implicated in recessive forms of RP, for which haploinsufficiency at either locus does not impair vision. Functional testing revealed likely additive and multiplicative effects in different functional systems for these two transcripts, suggesting that the combinatorial effect of haploinsufficiency at the two loci might be the candidate driver mechanism.
MATERIALS AND METHODS
Clinical investigations
Clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. The study was approved by the Danish Research Ethical Committee (reference numbers KF 01-234/02 and KF 01-108/03). Written informed consent was obtained from the proband and her relatives. The female proband (age 35) was first audiometrically tested at age 1.5 years and treated with hearing aids. Later, she was found to have profound hearing loss, and hearing aids were not used after the age of five. Eye examinations took place at regular intervals, including funduscopy, visual field analysis, and electroretinography. Neurological examinations included a cerebral MRI-scan at 23 years of age.
Copy number variation and other genotyping studies
A 450k comparative genomic hybridization array (Agilent oligoarray, Santa Clara, CA, USA) analysis was performed, as well as targeted analysis of mitochondrial DNA variants at positions 1555, 3243 and 7445 implicated previously in forms of hearing loss (http://www.mitomap.org/bin/view.pl/MITOMAP/WebHome). In addition, Sanger sequencing of CEP290 (LCA, Meckel-Gruber and Joubert syndrome), OPA1 (optic atrophy), GJB2 (hearing loss), WFS1 (Wolfram syndrome or nonsyndromic deafness) and ZCD2 (Wolfram syndrome) was performed, as well as Multiplex Ligation-dependent Probe Amplification (MLPA) of OPA1 and WFS1 (http://www.ncbi.nlm.nih.gov/omim). Previously published mutations for LCA (version 6.0; 451 mutations in 11 genes, November 2007), Usher syndrome (version 6.0; 612 mutations in 9 genes, October 2009), autosomal recessive RP (version 5; 594 mutations in 19 genes, June 2010), and autosomal dominant RP (version2; 385 mutations in 16 genes, July 2010) were analyzed by Asper Biotech (Tartu, Estonia), using the Allele-specific Primer Extension (APEX) method (for references and further details, see: http://www.asperbio.com/asper-ophthalmics).
Whole exome sequencing
Whole exome sequencing of the affected individual, from whom written consent was obtained, was performed on a 5500XL sequencing platform from Life Technologies (Carlsbad, CA, USA). The exome was enriched according to the manufacturer’s protocol using the SureSelect Human All Exon v4 Kit (50 Mb; Santa Clara, CA, USA). LifeScope software v2.1 from Life Technologies (Carlsbad, CA, USA) was used to map color space reads on the hg19 reference genome assembly and call single nucleotide variants.
All variants with coverage above 5 reads were included. Variants were prioritized by excluding all variants present in fewer than 20% of reads, as well as all intergenic, intronic (other than canonical splice sites) variants and alterations leading to synonymous amino acid changes. Variants with a frequency >0.5% in dbSNP version 135 or the Nijmegen in-house database consisting of 1,152 exomes were also excluded. The remaining data were analyzed for homozygous variants (variant reads proportion >80%) and compound heterozygous variants. Furthermore, genes known to be implicated in retinal diseases, hearing impairment and intellectual disability were analyzed for the presence of causal variants.
The variants identified in the proband (ID: 062125) were registered in the respective Leiden Open-access Variation Databases: http://grenada.lumc.nl/LOVD2/eye/home.php?select_db=RP1L1 (for RP1L1) and http://grenada.lumc.nl/LOVD2/eye/home.php?select_db=C2orf71 (for C2orf71).
Modeling phenotypes in zebrafish embryos
Two morpholino oligonucleotides (MOs) were designed and ordered from Gene Tools (Philomath, OR) to suppress the expression of zebrafish homolog genes: a splicing blocker against rp1l1 (rp1l1 SB: 5′- AAGAAATAAAGTCCTGACCTTGCGC -3′, efficiency was examined by reverse transcription PCR (S1 Fig.), with primers rp1l1-E1F: 5′- GCTCGCAGTCTTCGAGTCTT -3′, rp1l1-E3R: 5′- TGATCTCGGTGGACCATTGG -3′, rp1l1-i1R: 5′- TTTCATCCTCCGAGCCACAC -3′.) and a translation blocker against C2orf71-like (c2orf71l TB: 5′- GGAGAGCAGCCCATTTCAGATAGAT -3′). CRISPR were designed to knockout rp1l1 (guide sequence: 5′- TGGACTCGATGCAGTCACGA -3′) and c2orf71l (guide sequence: 5′- AGTAGTGTTCTCCCCCGTGA -3′)4 (Knockout efficiency was examined by PCR (S2 Fig.), with primers rp1l1-CRtestF: 5′- GGCTTTTTCGACGCTGATCC -3′, rp1l1-CRtestR: 5′- TCTGGGCGTTGTGATGGTAC -3′, c2orf71l-CRtestF: 5′- GCTGCTTGATGTGCCTTCAC -3′, c2orf71-CRtestR: 5′- GTCAGATGGAGTGGTCTGCC -3′.). MOs or CRISPR gRNA plus Cas9 RNA were microinjected into the yolk of zebrafish embryos at 1–8 cell stage, and then convergent extension phenotype was examined at 10 somite stage (10 ss) as described before.5,6 At 5 days post fertilization (5 dpf), lateral images of zebrafish larvae were taking to measure eye size. Larvae were then either fixed in Dent’s for whole mount immunostaining with anti-acetylated-tubulin (Sigma T7451), or in 4% PFA for coronal cryosectioning, followed by histoimmunostaining with anti-rhodopsin (ZIRC, zpr-3).
RESULTS
Clinical features of proband with syndromic retinal dystrophy
We ascertained a 35-year-old woman born to healthy unrelated parents of Scandinavian ancestry. She was born without perinatal complications as the first child after an uneventful pregnancy in week 42. The family history was negative regarding hearing and vision problems. At the age of 1.5 years she presented with a bilateral hearing loss of 80–90 dB and was initially treated with hearing aids. Later, she was found to have profound hearing loss, and hearing aids were not used after the age of five. She communicates with Swedish sign language. Absent otoacoustic emissions indicated a cochlear involvement in the deafness.
At the age of one strabismus was diagnosed, and at 4 nystagmus was documented by an ophthalmologist. When she was 12, she had visual problems in scotopic conditions and decreased visual acuity, and she was clinically suspected to have Usher syndrome. At age 18, funduscopy revealed an atypical retinitis pigmentosa (RP) with bull’s eye-like macular abnormalities and pale optic discs (Figs 1A,B). Bone spicules were absent. The ERG showed progressive rod and cone dystrophy. At the age of 21 she had a bilateral visual acuity of 0.02, excentric fixation, nystagmus, light adaptation problems, and night blindness. At age 23, atrophy of the retinal pigment epithelium (RPE) was noted, reminiscent of RPE changes in Leber congenital amaurosis. She did not show facial dysmorphic features.
Fig. 1. Clinical and genetic characterization of a family with syndromic retinal dystrophy.
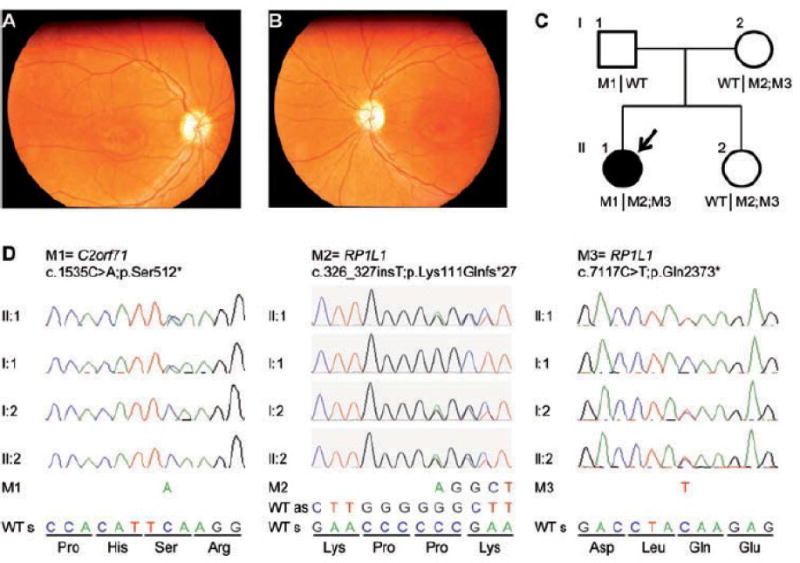
(A, B) Fundus photographs (A = right eye, B = left eye) taken at 18 years of age show normal retinal vessels, maculae with small wall-shaped degenerative changes and pale optic discs in both eyes. (C) Pedigree of the family. The arrow indicates the person in whom exome sequencing was performed. (D) Validation by Sanger sequencing of the identified RP1L1 c.[326_327insT;7117C>T] and C2orf71 c.1535C>A mutations in the affected individual, the unaffected sibling and her parents. Segregation analyses by Sanger sequencing point towards a digenic inheritance. As = antisense, M = mutation, s = sense, WT = wild-type.
Neurological examination showed unilateral (left side) vestibular areflexia, tremor and low normal intelligence. She has anxiety, a mild degree of depression, displays some obsessive behavior and has attended a psychiatrist several times. She works as an assistant in a stable with horses. A cerebral MRI-scan at 23 years of age demonstrated vermis atrophy and a smaller cerebellum. There was a normal distribution of white and grey matter. Pons, brainstem, visual cortex, inner ear structures, optic nerve and acoustic nerve were normal. She has one healthy younger sister, aged 31.
Heterozygous null mutations in C2orf71 and RP1L1 in syndromic retinal dystrophy
CNV analysis did not reveal significant deletions or duplications. Similarly, we found no mutations in the genes analyzed by Sanger sequencing, or in mitochondrial variants at positions 1555, 3243 and 7445. Finally, the APEX analysis of genes implicated previously in LCA, USH, adRP and arRP (known in 2010) also revealed no causal variants. In the exome sequencing results of the affected individual, the mean coverage was 91x per target with a median coverage of 71x. In total, we identified 49,333 variants. After applying quality filtering and excluding common and synonymous variants, 252 variants remained (S1 Table). Under the assumption of an autosomal recessive inheritance pattern, two variants were identified in RP1L1 (c.326_327insT, p.(Lys111Glnfs*27) and c.7117C>T, p.(Gln2373*)) (Table 1, S3 Fig.).
Table 1.
Variants of interest in the affected individual II:1.
| Chromosome | chr2 | chr8 | chr8 |
|---|---|---|---|
| Position | 29295593 | 10480385 | 10464491 |
| Gene name | C2orf71 | RP1L1 | RP1L1 |
| Reference nucleotide | G | G | |
| Number of reference reads | 89 | 13 | 86 |
| Variant nucleotide | T | T | A |
| Number of variant reads | 64 | 7 | 62 |
| Percentage of variant reads | 42% | 35% | 42% |
| Mutation type | Nonsense | Frameshift | Nonsense |
| Refseq | NM_001029883 | NM_178857 | NM_178857 |
| Mutation DNA | c.1535C>A | c.326_327insT | c.7117C>T |
| Mutation protein | p.(Ser512*) | p.(Lys111Glnfs*27) | p.(Gln2373*) |
| Sanger verification | Yes | Yes | Yes |
Segregation analysis showed that they were both present on the maternal allele (Figs. 1C–D). By analyzing for the presence of variants in genes implicated in intellectual disability, hearing impairment and inherited retinal disease, a heterozygous stop mutation in C2orf71 (c.1535C>A, p.(Ser512*)), a gene previously shown to be involved in autosomal recessive RP. The analysis of the raw exome data and additional Sanger sequencing of the distal parts of exons 1 and 2 of C2orf71, which were not covered by whole exome sequencing, did not reveal a second mutation. The list of the intellectual disability, hearing impairment and inherited retinal disease genes and their coverage data are presented in S2–4 Tables. The median overlap of the targets of these genes were 95%, 96%, 97%, with a median coverage of 74x, 71x, 73x, respectively.
Additive effect of morpholino knockdown of rp1l1 and c2orf71l on convergent extension and eye size in zebrafish
The presence of RP1L1 p.[(Lys111Glnfs*27; Gln2373*)] and C2orf71, p.(Ser512*), both of which are by definition pathogenic in the context of autosomal recessive RP, raised the possibility of digenic inheritance of syndromic retinal dystrophy in this individual. To test this hypothesis in vivo, we sought to model the individual’s phenotypes in zebrafish embryos. We first performed reciprocal BLAST searches, through which we identified a sole ortholog of each: rp1l1 and c2orf71l (51% similarity with 36% identity, and 60% similarity with 40% identity, respectively). Next, we designed morpholino oligonucleotides (MO) for both genes: a splice blocker for rp1l1, and a translational blocker for c2orf71l due to the lack of useable designs for a splice blocker. As a first test, we evaluated the impact of suppression of each of the two genes on early gastrulation; both genes have been shown to be relevant to ciliary function,7–9 perturbation of which induces convergent extension (CE) defects in mid-somitic embryos.10,11 Consistent with this expectation, suppression of each of rp1l1 and c2orf71l induced CE defects; embryos with an expansion of the “gap angle” (>45°) were considered affected and were plotted as such (Figs. 2A-B; triplicated). Lack of available reagents, limited expression,7,8,12,13 and relative large gene size precluded the generation of rescue constructs to test for MO specificity. As such, we sought an alternative method, which was employed to induce small deletions in these two genes by CRISPR/Cas9. Masked evaluation of embryos showed that CRISPR mutants phenocopied MOs. Relevant to the human genotype, 20% (rp1l1) and 43% (c2orf71l) of the single MOs showed CE defects, concomitant suppression induced CE in 64% of embryos, a significant difference (p<0.05) that appears to reflect an additive effect of the two MOs.
Fig. 2. Genetic interaction between rp1l1 and c2orf71l in zebrafish.
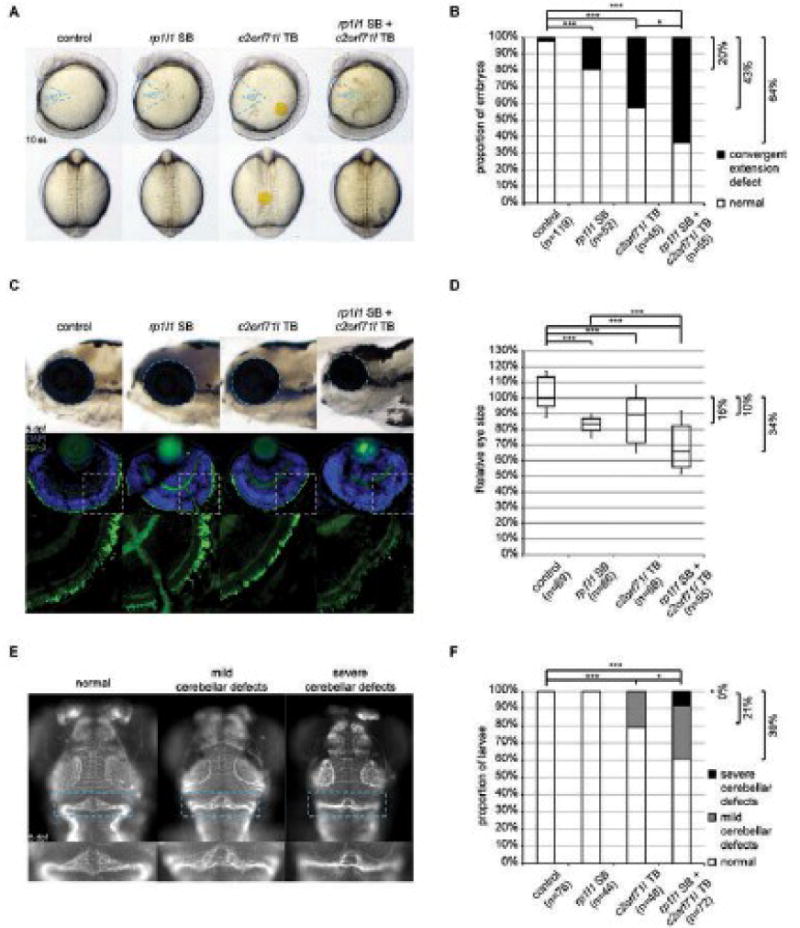
(A) Convergent extension defects were observed in 10 somites stage (ss) embryos injected with a splice-blocking (SB) morpholino oligonucleotide (MO) against rp1l1 and/or a translation blocking (TB) MO against c2orf71l. Lateral view is shown in upper panels, with body gap angles marked by blue dashed lines; dorsal view is shown in lower panels. (B) Proportion of embryos in (A) shows additive effect between rp1l1 and c2orf71l on convergent extension defects. Fisher exact test results were shown with * indicating p<0.05, and *** indicating p<0.001. (C) In the first row, the size of the eye (marked with blue dashed lines) in 5 days post fertilization (dpf) larvae was measured in lateral view. In the second row, cryosections of 5 dpf larva eyes were stained with anti-rhodopsin (zpr-3, green) and nuclear counterstain DAPI (blue). The green channel of delineated areas in second row was shown in the third row. (D) B-box plot of relative eye size (as marked in first row of (C)) was plotted, percent decrease of median compared to control was shown, suggesting additive effect between rp1l1 and c2orf71l on eye size phenotype. Two-tail t-test results were shown with *** indicating p<0.001. (E) Dorsal view (anterior end pointing up) of the heads of 5 dpf embryos stained with acetylated-tubulin antibody, showing mild and severe defects in cerebellum (marked with blue dashed lines) induced by injection of rp1l1 SB and/or c2orf71 TB. (F) Proportion of embryos with mild and severe cerebellum defects shown in (E) was plotted, suggesting synergistic effect between rp1l1 and c2orf71 on cerebellum defects. Fisher exact test results were shown with * indicating p<0.05, and *** indicating p<0.001.
Next, we sought to assess this possible interaction in the context of pathology directly relevant to the affected individual, by evaluating the eye (macro- and microscopically). Suppression of numerous RP-causing genes in zebrafish gives rise to microphthalmia in zebrafish embryos.14,15 Consistent with this, suppression of either rp1l1 or c2orf71l caused 10–20% reduction in eye size at five days post fertilization (5 dpf) (Figs. 2C-D). Reminiscent of the CE interaction, co-suppression of both genes gave rise to a 34% reduction of eye size. This is a significant reduction of eye size compared to the effects of suppressing the single genes (p<0.001), and also suggestive for an additive effect. Moreover, whereas each MO exhibited disorganization and likely partial loss of photoreceptors as visualized by rhodopsin staining in cryosectioned retinas, double morphants had no appreciable rhodopsin staining (Fig. 2C). Once again, these phenotypes could be reproduced in our CRISPR/Cas9 mutants (data not shown).
Synergistic effect of morpholino knockdown of rp1l1 and c2orf71l on cerebellar development in zebrafish
Finally, we turned our attention to the cerebellar atrophy of our individual, a phenotype that is, to our knowledge, not associated typically with recessive mutations in either RP1L116 or C2orf71.8,13 Suppression of either gene and imaging of the cerebellum gave no or modest pathology at 5 dpf, with 0% (rp1l1) and 21% (c2orf71l) of embryos exhibiting defects in size and shape of the cerebellum; by contrast, concomitant suppression of both genes induced cerebellar pathology in 39% of embryos a significant exacerbation (p<0.05) that is not additive (Figs. 2E-F). CRISPR-mediated deletion of either gene also gave rise to cerebellar defects (data not shown).
DISCUSSION
In an individual with a syndromic form of RP we identified heterozygous variants in two genes encoding ciliary proteins, namely RP1L1, p.[(Lys111Glnfs*27; Gln2373*)], and C2orf71, p.(Ser512*). Mutations in genes encoding ciliary proteins are known to cause a wide spectrum of diseases, including RP, hearing impairment, intellectual disability and cerebellar abnormalities, making them obvious candidates for the phenotype in this person. RP1L1 is expressed primarily in cones and rods.17 The first 350 amino acids are conserved and share high similarity with RP1, a protein that can be mutated in autosomal dominant and recessive forms of RP.18–20 This conserved region contains two doublecortin domains. For RP1, it was shown that these domains are important for stabilization of the microtubule-based axoneme in the photoreceptor outer segment.12 RP1 and RP1L1 co-localize to the axoneme, but RP1L1 is also present in the connecting cilium.7 A subset of heterozygous mutations in RP1L1 are known to cause autosomal dominant occult macular dystrophy (MIM 613587), a disease characterized by progressive decrease of visual acuity down to 0.1, a normal fundus, but specific ERG- and optical coherence tomography (OCT) abnormalities.21 Recently, other homozygous and compound heterozygous mutations in this gene were identified to cause autosomal recessive RP.16 The C2orf71 gene is predominantly expressed in the retina, and the protein is localized in the primary cilia of transfected human retinal pigmented epithelial cells.8,13 Mutations in C2orf71 are known to cause autosomal recessive RP (MIM 613428).13
Although, we cannot fully exclude a mutation on the other allele of these genes (e.g. in a non-coding region) or a de novo variant in another gene leading to a dominant disorder, the most parsimonious interpretation of our data is a digenic model, not only because of the obvious deleterious nature of the discovered mutations in two ciliary genes but because of the synthetic cerebellar phenotype that could only be observed in the double morphant/mutant. Naturally, this postulate can only be substantiated by the discovery of additional families with similar mutational architecture.
Thus far, a limited number of examples of digenic inheritance have been reported.22 Double heterozygous mutations in PRPH2 and ROM cause RP.23 Usher syndrome can be the result of double heterozygous mutations in CDH23 and PCDH15.24 Another type of digenic inheritance is reported in Bardet-Biedl syndrome, a ciliopathy characterized by RP, learning disability, obesity, polydactyly, renal abnormalities, and hypogonadism in males.25 In this model, two mutant alleles in one gene and a heterozygous variant in another gene were postulated to cause the disease, essentially acting as a modifier of penetrance.26 However, not in all affected persons a third allele is necessary to give rise to the disease phenotype,27 and it was hypothesized that in some cases, the expression level of the mutated genes determines the penetrance or variable expressivity of the disease.28 Interestingly, a heterozygous C2orf71 null allele in combination with a homozygous nonsense mutation in CEP250 was associated with atypical Usher syndrome, characterized by early-onset sensorineural hearing loss and a relatively mild form of RP.9 In a double homozygote of these variants the retina was more severely affected suggesting an additive effect in these ciliary proteins.
Both C2orf71 and RP1L1 are expressed primarily in the retina and are involved in non-syndromic retinopathies. Therefore, it might be plausible that the RP could be ascribed to the mutations we identified. Consistent with this hypothesis, co-suppression of both genes lead to exacerbation of retinal disorganization and revealed an additive effect on the microphthalmia phenotype. The possible link between cerebellar hypotrophy and our two candidate genes, RP1L1 and C2orf71, might be a novel discovery. The primary cilium is necessary for cerebellar development,29 and cerebellar defects have been reported in persons with recessive mutations in ciliary genes.30 Loss of function of either gene in human or mouse is not known to drive cerebellar defects.7,8,13,16 Our data showed that in vivo suppression of either rp1l1 or c2orf71l gave rise to no or mild cerebellar defects; however, combined suppression of both genes has a multiplicative effect and leads to severe cerebellar defects in a zebrafish model. Therefore, within the limits of evidence a different species can provide, we hypothesize that epistasis between these two loci is a driver behind the observed pathology. Naturally, we are cautious at reaching such conclusions with data derived from a single individual; however, we encourage the evaluation of exomes from persons with complex syndromic presentations for mutations in ciliary genes that might not necessarily conform to an expected, simpler model of autosomal recessive inheritance with complete penetrance.
Supplementary Material
S1 Fig: knockdown efficiency of rp1l1 SB morpholino.
(A) A diagram of rp1l1 gene structure. The position of SB is marked with a blue line, and the position and direction of primers (E1F, I1R, and E3R) are marked with blue arrows. (B) PCR on cDNA from zebrafish embryos with two pairs of primers (E1F+I1R, and E1F+E3R). The primer set E1F+I1R picked up a band in zebrafish embryos injected with rp1l1 SB (marked with blue dashed lines) but not in control embryos, suggesting that blocking of splicing at exon 1 and intron 1 junction caused insertion of at least part of intron 1. PCR of beta-actin was used as cDNA quality control. (C) Sequencing of the band marked with blue dashed lines in (B), confirming that the mRNA of rp1l1 in SB-injected zebrafish embryos extends from exon 1 into intron 1, and consequently generates a premature stop codon (marked with blue lines).
S2 Fig. knockout efficiency of rp1l1 and c2orf71l CRISPR.
(A) PCR on gDNA from one control zebrafish embryo and four embryos (1–4) injected with rp1l1 gRNA and Cas9 RNA, showing results of T7 endonuclease I assay. (B) PCR on gDNA from one control zebrafish embryo and four embryos (1–4) injected with c2orf71l gRNA and Cas9 RNA, showing results of T7 endonuclease I assay.
S3 Fig. Identification of the variants in raw WES data.
Identification of the variants in the raw WES data (BAM-file) (A) RP1L1 c.326_327insT, (B) RP1L1 c.7117C>T, and (C) C2orf71 c.1535C>A.
S1 Table. Variants that remained after applying quality filtering and excluding common and synonymous variants
S2 Table. List of the retinal disease genes and coverage data.
S3 Table. List of the hearing impairment genes and coverage data.
S4 Table. List of the intellectual disability genes and coverage data.
Acknowledgments
We are grateful to the proband and her family for their cooperation, Marijke N. Zonneveld-Vrieling and Ellen Blokland for expert technical assistance, and Anneke I. den Hollander for her contribution to genotyping studies. We thank the Swedish National Expert Team for Diagnosing Deafblindness. N.K. is a Distinguished Brumley Professor.
Footnotes
DECLARATION OF INTEREST
The authors report no conflicts of interest.
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002:1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abu-Safieh L, Alrashed M, Anazi S, et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013;23:236–247. doi: 10.1101/gr.144105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwang WY, Fu Y, Reyon D, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaghloul NA, Liu Y, Gerdes JM, et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc Natl Acad Sci U S A. 2010;107:10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaghloul NA, Katsanis N. Zebrafish assays of ciliopathies. Methods Cell Biol. 2011;105:257–272. doi: 10.1016/B978-0-12-381320-6.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamashita T, Liu J, Gao J, et al. Essential and synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis pigmentosa. J Neurosci. 2009;29:9748–9760. doi: 10.1523/JNEUROSCI.5854-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura DY, Baye LM, Perveen R, et al. Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am J Hum Genet. 2010;86:686–695. doi: 10.1016/j.ajhg.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khateb S, Zelinger L, Mizrahi-Meissonnier L, et al. A homozygous nonsense CEP250 mutation combined with a heterozygous nonsense C2orf71 mutation is associated with atypical Usher syndrome. J Med Genet. 2014;51:460–469. doi: 10.1136/jmedgenet-2014-102287. [DOI] [PubMed] [Google Scholar]
- 10.Ross AJ, May-Simera H, Eichers ER, et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 11.Ferrante MI, Romio L, Castro S, et al. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet. 2009;18:289–303. doi: 10.1093/hmg/ddn356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Zuo J, Pierce EA. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collin RW, Safieh C, Littink KW, et al. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;86:783–788. doi: 10.1016/j.ajhg.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Nakaya N, Chavali VR, et al. A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:400–409. doi: 10.1016/j.ajhg.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu X, Zeng Z, Gautier P, et al. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Invest Ophthalmol Vis Sci. 2011;52:2960–2966. doi: 10.1167/iovs.10-6800. [DOI] [PubMed] [Google Scholar]
- 16.Davidson AE, Sergouniotis PI, Mackay DS, et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat. 2013;34:506–514. doi: 10.1002/humu.22264. [DOI] [PubMed] [Google Scholar]
- 17.Conte I, Lestingi M, den Hollander A, et al. Identification and characterisation of the retinitis pigmentosa 1-like1 gene (RP1L1): a novel candidate for retinal degenerations. Eur J Hum Genet. 2003;11:155–162. doi: 10.1038/sj.ejhg.5200942. [DOI] [PubMed] [Google Scholar]
- 18.Sullivan LS, Heckenlively JR, Bowne SJ, et al. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 20.Khaliq S, Abid A, Ismail M, et al. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J Med Genet. 2005;42:436–438. doi: 10.1136/jmg.2004.024281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akahori M, Tsunoda K, Miyake Y, et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum Genet. 2010;87:424–429. doi: 10.1016/j.ajhg.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaffer AA. Digenic inheritance in medical genetics. J Med Genet. 2013;50:641–652. doi: 10.1136/jmedgenet-2013-101713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science (80−) 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 24.Zheng QY, Yan D, Ouyang XM, et al. Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum Mol Genet. 2005;14:103–111. doi: 10.1093/hmg/ddi010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437–446. [PMC free article] [PubMed] [Google Scholar]
- 26.Badano JL, Leitch CC, Ansley SJ, et al. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nat New Biol. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- 27.Abu-Safieh L, Al-Anazi S, Al-Abdi L, et al. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet. 2012;20:420–427. doi: 10.1038/ejhg.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estrada-Cuzcano A, Koenekoop RK, Senechal A, et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130:1425–1432. doi: 10.1001/archophthalmol.2012.2434. [DOI] [PubMed] [Google Scholar]
- 29.Spassky N, Han YG, Aguilar A, et al. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol. 2008;317:246–259. doi: 10.1016/j.ydbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardenas-Rodriguez M, Badano JL. Ciliary biology: understanding the cellular and genetic basis of human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:263–280. doi: 10.1002/ajmg.c.30227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Fig: knockdown efficiency of rp1l1 SB morpholino.
(A) A diagram of rp1l1 gene structure. The position of SB is marked with a blue line, and the position and direction of primers (E1F, I1R, and E3R) are marked with blue arrows. (B) PCR on cDNA from zebrafish embryos with two pairs of primers (E1F+I1R, and E1F+E3R). The primer set E1F+I1R picked up a band in zebrafish embryos injected with rp1l1 SB (marked with blue dashed lines) but not in control embryos, suggesting that blocking of splicing at exon 1 and intron 1 junction caused insertion of at least part of intron 1. PCR of beta-actin was used as cDNA quality control. (C) Sequencing of the band marked with blue dashed lines in (B), confirming that the mRNA of rp1l1 in SB-injected zebrafish embryos extends from exon 1 into intron 1, and consequently generates a premature stop codon (marked with blue lines).
S2 Fig. knockout efficiency of rp1l1 and c2orf71l CRISPR.
(A) PCR on gDNA from one control zebrafish embryo and four embryos (1–4) injected with rp1l1 gRNA and Cas9 RNA, showing results of T7 endonuclease I assay. (B) PCR on gDNA from one control zebrafish embryo and four embryos (1–4) injected with c2orf71l gRNA and Cas9 RNA, showing results of T7 endonuclease I assay.
S3 Fig. Identification of the variants in raw WES data.
Identification of the variants in the raw WES data (BAM-file) (A) RP1L1 c.326_327insT, (B) RP1L1 c.7117C>T, and (C) C2orf71 c.1535C>A.
S1 Table. Variants that remained after applying quality filtering and excluding common and synonymous variants
S2 Table. List of the retinal disease genes and coverage data.
S3 Table. List of the hearing impairment genes and coverage data.
S4 Table. List of the intellectual disability genes and coverage data.