

Abstract
Aims
More than 50% of patients with heart failure have preserved ejection fraction characterized by diastolic dysfunction. The prevalance of diastolic dysfunction is higher in females and associates with multiple comorbidities such as hypertension (HT), obesity, hypercholesterolemia (HC), and diabetes mellitus (DM). Although its pathophysiology remains incompletely understood, it has been proposed that these comorbidities induce systemic inflammation, coronary microvascular dysfunction, and oxidative stress, leading to myocardial fibrosis, myocyte stiffening and, ultimately, diastolic dysfunction. Here, we tested this hypothesis in a swine model chronically exposed to three common comorbidities.
Methods and results
DM (induced by streptozotocin), HC (produced by high fat diet), and HT (resulting from renal artery embolization), were produced in 10 female swine, which were followed for 6 months. Eight female healthy swine on normal pig-chow served as controls. The DM + HC + HT group showed hyperglycemia, HC, hypertriglyceridemia, renal dysfunction and HT, which were associated with systemic inflammation. Myocardial superoxide production was markedly increased, due to increased NOX activity and eNOS uncoupling, and associated with reduced NO production, and impaired coronary small artery endothelium-dependent vasodilation. These abnormalities were accompanied by increased myocardial collagen content, reduced capillary/fiber ratio, and elevated passive cardiomyocyte stiffness, resulting in an increased left ventricular end-diastolic stiffness (measured by pressure–volume catheter) and a trend towards a reduced E/A ratio (measured by cardiac MRI), while ejection fraction was maintained.
Conclusions
The combination of three common comorbidities leads to systemic inflammation, myocardial oxidative stress, and coronary microvascular dysfunction, which associate with myocardial stiffening and LV diastolic dysfunction with preserved ejection fraction.
Keywords: Translational studies, Coronary circulation, Oxidant stress, Endothelium/nitric oxide, Heart failure
1. Introduction
More than 50% of patients with heart failure present with heart failure with preserved ejection fraction (HFpEF), characterized by diastolic dysfunction.1 Hospitalized patients with HFpEF have high mortality and rehospitalization rates, and there is currently no effective treatment available for these patients.2–4 Common metabolic and cardiovascular risk factors appear to be critical in the onset of diastolic dysfunction and its progression towards HFpEF, as the incidence of HFpEF increases with rising prevalence of obesity, hypertension (HT), chronic kidney disease, female sex, and type 2 diabetes mellitus (DM).5–7 Furthermore, studies in HFpEF patients have shown alterations in myocardial structure, function, and cell signaling that are unique to this form of heart failure.8–11 However, the pathophysiology of HFpEF is still not fully understood, particularly at the myocardial tissue level. Findings from these earlier studies have led to the proposition of a novel paradigm, in which multiple comorbidities, including obesity, HT, hypercholesterolemia (HC), and DM induce a systemic pro-inflammatory state that leads to coronary microvascular dysfunction and oxidative stress. In turn, these disease mechanisms result in myocardial stiffening and ultimately left ventricular diastolic dysfunction.12 Nevertheless, direct experimental evidence for this unifying hypothesis is still lacking. We set out to investigate the chain of events as proposed in this novel paradigm, using a large animal model chronically exposed to three common comorbidities that associate with diastolic dysfunction, i.e. hyperglycemia, HC, and HT. Since the prevalence of this disease is predicted to increase in our aging Western societies,13 such unique large animal model that mimics the complex disease mechanisms of diastolic dysfunction would offer a much needed platform for testing novel drug and lifestyle therapies.
2. Methods
2.1 Animals
Experiments were performed in accordance with the ‘Guiding Principles in the Care and Use of Laboratory Animals’ as approved by the Council of the American Physiological Society, and with approval of the Animal Care Committee at Erasmus University Medical Center, Rotterdam. Fourteen female (21.7 ± 0.3 kg at 2–3 months of age) Yorkshire x Landrace swine were included to study the effects of DM, HC, and HT (DM + HC + HT), while 12 healthy female swine of similar age were studied as controls (Control). Finally, six fresh control hearts from slaughterhouse female swine of similar body weight as the control animals (∼100 kg at sacrifice) were included to additionally study vascular function characteristics.
2.2 Induction of risk factors
The induction of risk factors in the DM + HC + HT group is described in detail in the Supplementary material online, Supplementary Methods. Briefly, DM was produced by injection of streptozotocin (50 mg/kg/day i.v. for 3 days, Bio-connect B. V., Huissen, The Netherlands). 9- to 11 days later, animals were sedated with intramuscular Zoletil (tiletamine/zolazepam; 5 mg/kg), Rompun (xylazine; 2.25 mg/kg) and atropine (1 mg), and artificially ventilated (O2 and N2 [1:2]), to which 1–2% (vol/vol) isoflurane was added. HT was produced by micro-embolization of the global right kidney as well as the lower pole of the left kidney using 75 mg of polyethylene microspheres (38–42 μm diameter, Cospheric, Santa Barbara, CA, USA) per kidney. One week after HT induction, a high fat diet (see Supplementary material online, Table S1), supplemented with 10 g NaCl/day was gradually introduced to produce HC.
2.3 Hemodynamic assessment
At 6 months follow-up, extensive in vivo hemodynamic assesment was performed with the animals under anesthesia (pentobarbital, 20 mg/kg i.v.) and in the awake state. All procedures are described in detail in the Supplementary material online, Supplementary Methods. Briefly, LV function was assessed using MRI (Discovery MR450, GE Medical System, Milwaukee, Wisconsin, USA), including end-diastolic volume (EDV), end-systolic volume (ESV), E/A ratio, stroke volume (SV), and ejection fraction (EF), and using a pressure–volume catheter (CD Leycom, The Netherlands), including EDV, ESV, SV, EF, end-diastolic (EDPVR), and end-systolic (ESPVR) pressure–volume relationships. Eight animals (4 DM + HC + HT, and 4 Control) were instrumented between 5- and 6-month follow-up with a Transonic flow probe around the ascending aorta and fluid filled catheters in the left atrium and aorta for hemodynamic measurements at rest and during exercise and the evaluation of kidney function.
2.4 Coronary small artery function in vitro
In order to assess coronary vascular endothelial function, coronary small arteries (∼300 µm diameter) were isolated from the epicardial surface of the LV apex and studied in vitro using a Mulvany wire myograph as presented in the Supplementary material online, Supplementary Methods. In short, the concentration–response curves (CRC) were measured for the endothelium-dependent vasodilator bradykinin (BK, 10−10 to 10−6 mol/l, Sigma–Aldrich, Zwijndrecht, The Netherlands) and the exogenous NO-donor, S-nitroso-N-acetylpenicillamine (SNAP, 10−10 to 10−5 mol/l, Sigma–Aldrich, Zwijndrecht, The Netherlands) following preconstriction with 10−6 mol/l thromboxane-A2 analogue U46619 (Sigma–Aldrich, Zwijndrecht, The Netherlands).
2.5 Tissue analysis
All analyses are described in detail in the Supplementary material online, Supplementary Methods. Briefly, snap frozen samples of the subendocardium of the LV anterior free wall were analysed for mRNA expression levels of various genes (see Supplementary material online, Supplementary Methods Table S2), involved in different phases of development of diastolic dysfunction, reactive oxygen species (ROS), and NO production for NO-ROS balance, eNOS expression, and phosphorylation in order to asess eNOS uncoupling and activity. Myocardial levels of cyclic guanosine monophosphate (cGMP), and activity of phosphodiesterase 5 (PDE5) and protein kinase G (PKG) were measured using ELISA kits, to assess alterations in the downstream signalling pathway of NO. Calcium-force relations of single cardiomyocytes were performed for cardiomyocyte stiffness measurements. In addition, titin isoform- (N2BA and N2B) expression and phosphorylation were measured as previously described.14 Furthermore, histological analyses of myocardial collagen deposition, capillary density, and myocyte size were performed for myocardial structure characterization. Finally, the upper pole of the left kidney was used for analysis of tubulo-interstitial (TI) damage. Scored variables were the amount of inflammatory infiltrate between tubuli, interstitial fibrosis, tubular atrophy, and dilatation. A total TI damage score was calculated by summing the scores for the four variables. Fat deposition in the liver was also analysed for liver steatosis.
2.6 Data analysis
Data are presented as mean ± SEM. Comparison of variables between the two groups was performed by unpaired Student’s t-test (StatView 5.0 SAS Institute Inc.). Vasodilator responses to BK and SNAP were expressed as percentage of the preconstriction to U46619. Vasoconstrictor responses to U46619 were normalized to 10−1 mol/l KCl. Statistical analysis of CRCs, changes in mean aortic pressure over time and the measurements of Fpas were performed using two-way ANOVA and the analysis of the LAP measurements during exercise using regression analysis. P < 0.05 was considered statistically significant.
3. Results
3.1 Model characteristics
At 6-month follow-up, DM + HC + HT animals had lower body weights (79±3 kg) than their age-matched controls (102±4 kg, P < 0.05). No significant group differences were detected in LV-, left atrial-, or right ventricular weights, when normalized to body weight (see Supplementary material online, Table S3).
A significant decrease in insulin as well as significant increases in glucose, total cholesterol, LDL-, and HDL-cholesterol values, and the LDL/HDL ratio and to a lesser extent in triglycerides (P = 0.07), were observed in DM + HC + HT compared to controls (Table 1). Metabolic dysregulation was also accompanied by increased mRNA-expression of pyruvate dehydrogenase lipoamide kinase isozyme 4 (PDK4, a regulator of glucose metabolism and a marker of diastolic dysfunction15), in DM + HC + HT as compared to controls (1.91±0.24 vs. 1.00±0.22 AU, P < 0.05, see Supplementary material online, Table S4), which correlated with plasma glucose levels (P < 0.05). TNF-α plasma levels were significantly higher than those of healthy controls, consistent with a chronic inflammatory status in these animals, which correlated with the levels of glucose (see Supplementary material online, Figure S1A, P < 0.05), but not with plasma lipids (see Supplementary material online, Figure S1B). Plasma levels of ASAT were similar between groups (42±11 in DM + HC + HT vs. 40±2 U/l in Control), but ALAT was significantly lower in DM + HC + HT as compared to controls (24±4 vs. 51±2 U/l, P < 0.05), despite a significant increase in fat deposition in the liver of DM + HC + HT animals (0.86 ± 0.31% vs. 0.03 ± 0.01%; P < 0.05). These data are consistent with findings in other pig models of metabolic dysfunction and might be related to the high fructose and high sucrose content of the diet.16,17
Table 1.
Arterial blood characteristics in DM + HC + HT swine group vs. control, obtained at fasting state under anesthesia
| Parameter | Control (N = 8) | DM + HC + HT (N = 10) |
|---|---|---|
| Metabolic function | ||
| Glucose (mmol/l) | 6.1 ± 0.7 | 22.7 ± 0.9* |
| Insulin (ng/l) | 39 ± 14 | 12 ± 1* |
| Cholesterol (mmol/l) | 2.2 ± 0.1 | 16.8 ± 3.4* |
| LDL-cholesterol (mmol/l) | 1.1 ± 0.1 | 14.0 ± 3.2* |
| HDL-cholesterol (mmol/l) | 1.1 ± 0.1 | 5.1 ± 0.7* |
| LDL/HDL-Cholesterol | 1.1 ± 0.1 | 2.7 ± 0.4* |
| Triglycerides (mmol/l) | 0.35 ± 0.05 | 1.16 ± 0.36# |
| Renal function | ||
| Urea (mmol/l) | 4.2 ± 0.5 | 3.8 ± 0.4 |
| Creatinine (µmol/l) | 130 ± 6 | 129 ± 11 |
| Cystatin C (mg/l) | 0.42 ± 0.01 | 0.51 ± 0.03* |
| Aldosterone (pg/ml) | 1.4 ± 0.1 | 10.2 ± 4.1# |
| GFRa (ml/min) | 202 ± 7 | 123 ± 12* |
| Inflammation | ||
| TNF-α (pg/ml) | 74 ± 24 | 231 ± 64* |
| IL-6 (pg/ml) | 21 ± 8 | 67 ± 32 |
GFR was measured in chronically instrumented swine in the awake state.
LDL, low-density lipoprotein; HDL, high-density lipoprotein; GFR, glomerular filtration rate; TNF-α, tumor necrosis factor alpha; IL-6, interleukin-6.
N = 4 DM + HC + HT and 4 Controls.
P < 0.05,
P = 0.07, DM + HC + HT vs. Control.
Mean aortic pressure, measured under general anesthesia, rose from 62±3 mmHg immediately prior to injection to 87±4 mmHg following infusion of the polyethylene beads in the kidneys (P < 0.05). The increase in aortic pressure was well maintained over time, as indicated by the biweekly measurements in the awake state, with animals standing quietly in their cage (Figure 1A).
Figure 1.
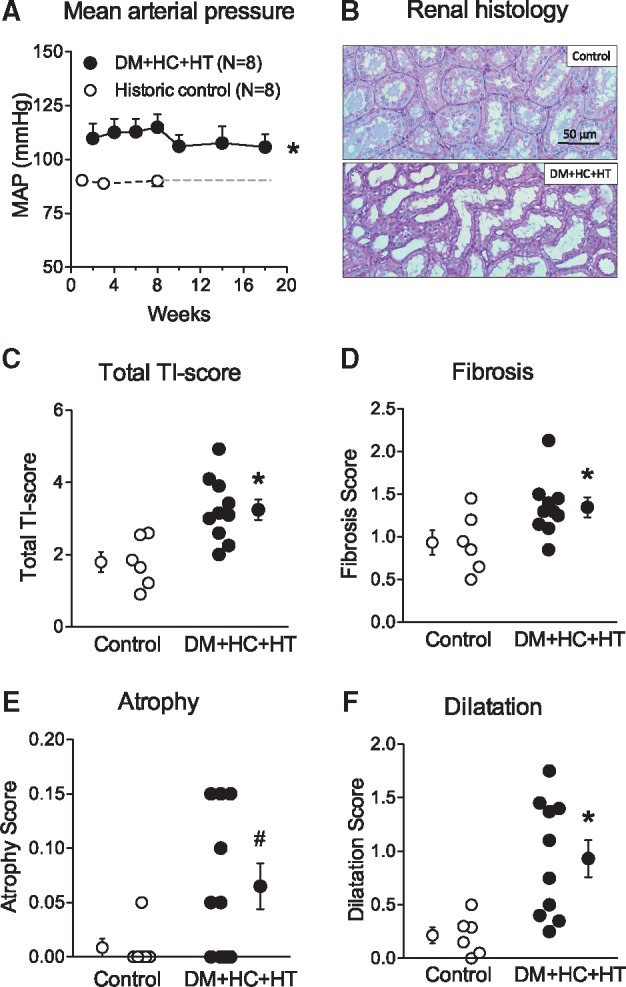
Mean arterial pressure (MAP), measured in resting awake state, was increased over time in DM + HC + HT as compared to healthy control swine from our laboratory (unpublished data) performed in the same time period (A). Representative PAS stained sections of the top part of the left kidney of control and DM + HC + HT swine at a magnification of 200×. Scale bar = 50 μm (B). Increased tubulo-interstitial damage in kidney sections of DM + HC + HT swine compared to control healthy swine (C). Total tubulo-interstitial damage score was calculated by summing the scores for peritubular inflammatory infiltrate (not shown), interstitial fibrosis (D), atrophy (E) and dilatation (F) (Control N = 8, DM + HC + HT N = 11). *P < 0.05, #P = 0.06.
Kidneys of the DM + HC + HT animals were smaller than kidneys of controls; however, when corrected for body weight, these differences were no longer apparent (see Supplementary material online, Table S3). Representative histology is shown for control and DM + HC + HT kidney cortex (Figure 1B). Total tubulo-interstitial (TI) damage score was higher in the DM + HC + HT group (P < 0.001, Figure 1C). The DM + HC + HT group showed no increase in peritubular infiltrate (data not shown), but showed more interstitial fibrosis (Figure 1D), a trend towards atrophy (Figure 1E, P = 0.06) and increased dilatation (Figure 1F) of the tubuli compared to controls. Consequently, GFR, measured by inulin clearance, was significantly reduced in DM + HC + HT as compared to healthy controls, indicative of kidney dysfunction (P < 0.05, Table 1). Although no differences in plasma creatinine or urea values were observed between the groups, cystatin C levels were significantly higher in the DM + HC + HT group (P < 0.05). Cystatin C strongly correlated with the levels of TNF-α (P = 0.005, see Supplementary material online, Figure S1C). There was a trend towards an increase in plasma aldosterone (10.4±4.1 vs. 1.4±0.01 pg/ml, P = 0.06).
3.2 Coronary small artery function
Small arteries from DM + HC + HT showed similar preconstriction to 10−6 M U46619 as control vessels, i.e. 65±6% vs. 70±16% of the response to 10−1 mol/l KCl in the BK experiments, and 62±8% vs. 64±8% in the SNAP experiments (both P = NS). The vasorelaxation to the endothelium-dependent vasodilator BK was significantly blunted in DM + HC + HT (Figure 2A), whereas the vasorelaxation to the endothelium-independent vasodilator SNAP was maintained (Figure 2B), indicative of endothelial dysfunction. Interestingly, in isolated small arteries obtained from five additional DM + HC + HT swine, pretreatment with the ROS scavenger N-2-mercaptopropionyl glycine (MPG) restored the vasodilator response to bradykinin (see Supplementary material online, Figure S2).
Figure 2.

Concentration response curves to bradykinin (BK, A) and the NO-donor S-nitroso-N-acetylpenicillamine (SNAP, B) in small arteries isolated from DM + HC + HT and healthy control hearts. *P < 0.05 DM + HC + HT vs. Control by 2-way ANOVA.
3.3 Myocardial ROS measurements
Myocardial production of the NO metabolites NO2− and NO3− was significantly lower in the DM + HC + HT compared to control suggesting reduced NO production (Figure 3A). However, neither myocardial cGMP levels (DM + HC + HT: 5.74 ± 2.14 pmol/mg protein, control swine: 8.12 ± 2.16 pmol/mg protein, P = 0.4), nor PDE5 activity (1.03 ± 0.49 vs. 1.40 ± 1.04 AU/µg protein, P = 0.6), were significantly altered, resulting in preserved PKG activity (0.49 ± 0.01 vs. 0.50 ± 0.02 AU/µg protein, P = 0.7). Although total eNOS expression was increased in DM + HC + HT as compared to control (Figure 3B), this increase was principally due to an increase in eNOS monomer, as the monomer/dimer ratio was markedly higher than in controls (Figure 3C). In addition, eNOS phosphorylation of residue Ser1177 was higher (possibly reflecting phosphorylation of the eNOS monomer18), suggesting that not only eNOS expression but also eNOS activity was increased (Figure 3D). The increase in monomer/dimer ratio in DM + HC + HT reflects uncoupling of eNOS. Accordingly, basal superoxide production was 3-fold higher in DM + HC + HT (Figure 3E) with the increased superoxide values correlating with inflammation (see Supplementary material online, Figure S1D) and the decrease in cardiac NO production (P = 0.008, see Supplementary material online, Figure S1E). This increase was suppressed by both L-NAME and VAS2870 (both P < 0.05), indicating that both NOS and NADPH oxidase contributed to superoxide production (Figure 3E). NADPH resulted in exaggerated—and VAS2870-inhibitable, but not L-NAME-inhibitable—superoxide production in DM + HC + HT as compared to controls (Figure 3F), confirming NADPH oxidase as a major source of superoxide in DM + HC + HT animals in addition to the uncoupled NOS-dependent superoxide production. NOX2 and 4 expression in the myocardium of DM + HC + HT did not differ from controls (see Supplementary material online, Table S4), suggesting that enzyme activity rather than transcriptional activation was higher in these animals. In contrast, gene expression of SOD-1 and catalase were higher in the DM + HC + HT as compared to controls (SOD-1 1.30 ± 0.09 vs. 1.00 ± 0.08, P < 0.05; catalase 2.44 ± 0.21 vs. 1.00 ± 0.22 AU, P < 0.05), likely representing a feedback mechanism to compensate for the increased oxidative stress. In accordance with the expression data, myocardial catalase activity was also increased in DM + HC + HT swine as compared to the healthy controls (22.3 ± 1.4 vs. 12.1 ± 0.3 nmol/min/mg protein, P = 0.0001). Both catalase (P = 0.0007) and SOD-1 (P = 0.02) expression correlated with superoxide production (see Supplementary material online, Figure S1F and G).
Figure 3.

NO production was decreased in the LV subendocardium of DM + HC + HT (N = 10) vs. Controls (N = 8) (A). However, myocardial eNOS expression was increased in DM + HC + HT (B), as was the monomer/dimer (MoDi) ratio (C), suggestive of eNOS uncoupling. Phosphorylation of eNOS (peNOS) was also significantly increased in DM + HC + HT (D) (N = 10) vs. Control (N = 8). Superoxide generation was increased in the LV subendocardium of DM + HC + HT vs. Controls and was suppressed by L-NAME and VAS2870 (E). Upon NADPH oxidase stimulation, the superoxide anion production was dramatically increased (F), which was inhibited by VAS2870 but not by L-NAME treatment. *P < 0.05 DM + HC + HT vs. Control; †P < 0.05 vs. corresponding basal.
3.4 Myocardial structure: collagen content, myocardial hypertrophy, and capillary density
The histological findings in the subendocardial layer of the LV anterior wall of animals from both groups are shown in Figure 4 and summarized in Figure 5. Total collagen deposition (collagen type I + III, Figure 4A and D) was assessed in the subendocardial and subepicardial layers of the LV. Collagen deposition was significantly increased in the subendocardium of DM + HC + HT as compared to control swine (Figure 5A), which correlated with the superoxide production (P = 0.0001, see Supplementary material online, Figure S1H). No difference was found between the subendocardium and the subepicardium in each group (data not shown). Moreover, molecular data suggest reduced matrix turnover in the subendocardium of DM + HC + HT animals, since expression of both MMP-2 and TIMP-2 was lower, while expression of MMP-9 and TIMP-1 was unaltered in DM + HC + HT animals as compared to controls (see Supplementary material online, Table S4). The MMP-2/TIMP-2 ratio was also significantly lower in the DM + HC + HT groups compared to control animals (0.72 ± 0.05 vs. 0.89 ± 0.03, P = 0.01), while the ratio MMP-9/TIMP-1 was similar (1.17 ± 0.21 vs. 1.73 ± 0.51, P = NS).
Figure 4.

Examples of histological staining for quantification of collagen deposition (Picrosirius Red A, D), myocyte size (Gomori B, E), and capillary density (Lectin C, F) in the LV subendocardium in Control (CON), and DM + HC + HT animals.
Figure 5.

Increased collagen deposition (A) but no myocyte hypertrophy (B) was recorded in LV subendocardium of the DM + HC + HT animals. Capillary density was similar between groups (C), however the capillary-to-fiber ratio was significantly reduced in DM + HC + HT (D). *P < 0.05 DM + HC + HT (N = 10) vs. Control (N = 8).
The measurement of the cardiomyocyte cross-sectional area (Figure 4B and E), revealed that cardiomyocytes from the DM + HC + HT were significantly smaller than those of control swine, which was observed in both subendocardium (413±42 μm2 vs. 598±23 μm2, P < 0.05; Figure 5B) and subepicardium (379±29 μm2 vs. 676±31 μm2, P < 0.05; data not shown in Figure 5B). Cardiomyocyte size was inversely correlated with the levels of cystatin C (P = 0.02), a potent risk factor for cardiovascular disease associated mortality and a strong marker for renal dysfunction. Interestingly, when normalized to body weight, the difference in myocyte area between Control and DM + HC + HT was no longer observed. In line with the observed reduction in cardiomyocyte size in DM + HC + HT, myocardial ATF4 expression was significantly higher in the DM + HC + HT as compared to controls (1.51 ± 0.12 vs. 1.00 ± 0.08 AU, P < 0.05), which may suggest increased ER stress in the cardiomyocytes as a result of the comorbidities. The gene expressions of ANF/NPPA, UBE2H, and ACTA1 and 2 in the myocardium were not different between groups (see Supplementary material online, Table S4), in line with the lack of myocyte hypertrophy in DM + HC + HT.
Capillary density (Figure 4C and F) was similar between the groups (Figure 5C) and also between the subendocardium and subepicardium in each group (data not shown). However, the capillary-to-fiber ratio was significantly lower in DM + HC + HT as compared to controls, suggestive of capillary rarefaction (Figure 5D). No differences were observed between the subendocardium and subepicardium of either group (not shown). Unpublished data from our laboratory obtained in weight-matched DM + HC + HT animals showed no difference in myocyte size vs. control animals, while showing a significant reduction in capillary density (1237 ± 81 in DM + HC + HT vs. 1548 ± 88 capillaries/mm2 in control, P = 0.03).
3.5 Single myocyte force measurements
Increased maximal force (Fmax, Figure 6A) as well as increased passive stiffness (Fpas, Figure 6B) of the cardiomyocytes isolated from the subendocardium of the LV anterior wall was observed in DM + HC + HT as compared to controls. Myocardial N2BA/N2B titin expression showed a shift towards the stiff N2B titin isoform (Figure 6C, P = 0.01), consistent with the increased passive stiffness of the cardiomyocytes, while no significant change in total phosphorylation of either isoform was observed (N2BA: 0.76 ± 0.06 AU in Control vs. 0.74 ± 0.05 AU in DM + HC + HT; N2B: 0.98 ± 0.11 AU in Control vs. 0.92 ± 0.07 AU in DM + HC + HT).
Figure 6.

Increased maximal (Fmax, A) and passive force (Fpas, B) were seen in DM + HC + HT as compared to Controls. Cardiomyocyte data were averaged for all measured cells and group averages are shown. Titin N2BA/N2B isoforms ratio was significantly decreased in DM + HC + HT animals (C). Typical examples of E and A waves in Control (D) and DM + HC + HT (E) animals and pressure–volume relationships (G) are presented. The early to late filling (E/A) ratio tended to be lower in the DM + HC + HT animals (F). End-diastolic elastance (slope of EDPVR, sEDPVR) was significantly increased (H) while a trend towards significance in the end-systolic elastance (slope of ESPVR, sESPVR) was recorded (I), *P < 0.05, #P = 0.10 DM + HC + HT vs. Control.
3.6 Cardiac MRI and global LV function
MRI and PV loop variables as well as typical examples of E and A waves (Figure 6D and E) and pressure–volume loops (preload reduction, Figure 6G) are shown in Table 2 and Figure 6. LV EDV and SV, indexed to body weight, and ejection fraction were not different between the two groups, measured by either MRI or PV loop catheter (Table 2). Late gadolinium enhancement did not show any difference between the groups (data not shown), despite an increased collagen content in the LV of DM + HC + HT animals as measured histologically (Figure 5A). However, E/A ratio tended to be lower in the DM + HC + HT group, suggestive of early diastolic dysfunction (Figure 6F). These findings were complemented by the pressure–volume data, showing that DM + HC + HT animals had increased LV end-diastolic elastance, indicative of increased passive stiffness of the LV in vivo (Figure 6H, P < 0.05). The increase in LV end-diastolic elastance correlated with the increased collagen deposition in the subendocardium (see Supplementary material online, Figure S1I, P = 0.05). LV end-systolic elastance also tended to be higher in these animals, although this failed to reach statistical significance (Figure 6I, P = 0.10), which correlated well with the maximal force development of single cardiomyocytes (P = 0.006). Although no significant group differences were observed in LV end-diastolic pressure, dP/dtmax, LV dP/dtmin, or tau (Table 2), we did observe a shift in the relation between left atrial pressure and cardiac index (Figure 7) in chronically instrumented DM + HC + HT compared to control swine during exercise (P < 0.05). These data indicate that in swine in the awake state, at similar levels of cardiac index higher atrial filling pressures were observed in DM + HC + HT compared to control swine.
Table 2.
LV function in control and DM + HC + HT swine under anesthesia
| Parameter | Control | DM+HC+HT |
|---|---|---|
| Body weight (kg) | 102 ± 4 | 79 ± 3* |
| Pressure-volume catheter | N=8 | N=10 |
| Heart rate (bpm) | 85 ± 3 | 91 ± 5 |
| LV EDV (ml) | 174 ± 22 | 113 ± 11* |
| LV EDVi (ml/kg) | 1.5 ± 0.2 | 1.5 ± 0.2 |
| SV (ml) | 75 ± 3 | 55 ± 4* |
| SVi (ml/kg) | 0.75 ± 0.03 | 0.74 ± 0.08 |
| Ejection fraction (%) | 47 ± 5 | 50 ± 3 |
| Millar catheter | N=8 | N=10 |
| Heart rate (bpm) | 88 ± 4 | 85 ± 3 |
| dP/dt max (mmHg/s) | 1470 ± 138 | 1604 ± 214 |
| dP/dt min (mmHg/s) | −2510 ± 322 | −2687 ± 266 |
| Tau (ms) | 49 ± 3 | 48 ± 3 |
| LV EDP (mmHg) | 9 ± 2 | 9 ± 2 |
| MRI | N=6 | N=7 |
| Heart rate (bpm) | 89 ± 7 | 71 ± 3* |
| LV EDV (ml) | 189 ± 12 | 162 ± 11 |
| LV EDVi (ml/kg) | 1.8 ± 0.1 | 2.1 ± 0.1 |
| SV (ml) | 98 ± 9 | 73 ± 7* |
| SVi (ml/kg) | 0.93 ± 0.07 | 0.94 ± 0.06 |
| Ejection fraction (%) | 51 ± 3 | 45 ± 4 |
LV, left ventricle; SV, stroke volume; EDV, end-diastolic volume; EDVi, end-diastolic volume indexed for body weight; ESV, end-systolic volume; ESVi, end-systolic volume indexed for body weight; dP/dt min and dP/dt max, minimum and maximum rate of pressure change in the left ventricle; LV EDP, left ventriclular end diastolic pressure; Tau, time constant of isovolumic relaxation.
P < 0.05 compared to Control.
Figure 7.

Relation between left atrial pressure (LAP) and cardiac index (CI) at rest and during treadmill exercise in chronically instrumented Control (N = 4; 97 ± 6 kg, white circles) and DM + HC + HT (N = 4; 94 ± 7 kg, black circles) swine. *P < 0.05 DM + HC + HT vs. Control.
4. Discussion
In the present study, we report that in the absence of major geometrical alterations in the heart, the co-existence of three common comorbidities leads to LV diastolic dysfunction as evidenced by increased passive LV stiffness and a trend towards reduced LV diastolic early-to-late filling velocities, while EF was still preserved. The cascade of events leading to diastolic dysfunction is summarized in Figure 8,19 in which the events that were observed in the present study are highlighted in bold. These include the presence of chronic systemic inflammation and decreased endothelium-dependent vasodilation in the coronary arteries as assessed in vitro. We also observed reduced capillary-to-fiber ratio, elevated superoxide production due to NOX activation, eNOS uncoupling, and reduced myocardial NO production, all likely to be important mechanisms for the observed increases in LV collagen content and cardiomyocyte passive force contributing to LV stiffening and thereby reduced diastolic function.12
Figure 8.

In a large animal, chronic exposure to multiple common comorbidities results in systemic inflammation, endothelium-dependent coronary artery dysfunction, capillary rarefaction, oxidative stress, and perturbed nitric oxide production, which are associated with increased myocardial fibrosis and passive cardiomyocyte stiffness, resulting in LV diastolic dysfunction. Adapted with permission from Ter Maaten et al.19 The findings of the present study that are in agreement with the hypothesis are presented in bold.
Paulus and Tschöpe12 proposed that a systemic inflammatory state produced by cardiovascular comorbidities is common in many HF patients, particularly those with HFpEF.20,21 The inflammatory state promotes coronary microvascular endothelial dysfunction, which is characterized by generation of reactive oxygen species and reduced NO bioavailability, and leads to a cascade of signaling events that ultimately promotes cardiac fibrosis and myocyte stiffness. Indeed, it is now well established that multiple common comorbidities, including obesity, DM, and HT, are strongly associated with HFpEF.5–7,22–24 This cascade of events could be faithfully recapitulated in the present study, although the causality between the various steps remains to be tested. Firstly, a pro-inflammatory state was documented by markedly elevated levels of circulating TNF-α in diseased animals. TNF-α values correlated with the levels of glucose and cystatin C, confirming that DM and kidney dysfunction (as evidenced by high tubulo-interstitial damage score, increased cystatin C levels, and impaired GFR, despite normal urea and creatinine levels), are important contributors to increased systemic inflammation. Secondly, coronary small artery endothelial dysfunction was also evident as relaxation responses to bradykinin were diminished, while smooth muscle cell function was preserved as indicated by the preservation of vascular responses to the exogenous NO donor SNAP and the maintained cGMP-PDE5-PKG signaling, although the latter was measured in the myocardium and might also, partly, reflect cardiomyocyte signaling. Thirdly, and perhaps most importantly, ROS production was markedly increased and correlated with the decreased cardiac NO production. Specifically, the basal superoxide production was increased 3-fold in DM + HC + HT as compared to control swine. This increase was significantly suppressed by L-NAME as well as by VAS2870, indicating that both NOS and NADPH oxidase were sources of superoxide production. Upon stimulation of NOX, myocardial superoxide production was markedly increased in DM + HC + HT as compared to controls, confirming NOX as the major source of superoxide. This was probably due to the increase in NOX-activity, as myocardial gene expression was not significantly different between groups. Furthermore, although increased eNOS expression and phosphorylation levels were observed in DM + HC + HT animals, the monomer/dimer ratio was increased almost 3-fold, indicating that eNOS-uncoupling accounted for the aggravated NOS-dependent superoxide production. The increased myocardial superoxide levels in conjunction with low NO levels suggest direct quenching of NO by superoxide as a major factor contributing to the reduced NO bioavailability in the present animal model, both basal and agonist induced. This is in agreement with data from Paolocci et al. in isolated rat hearts.25 In our model, the increase in superoxide production correlated with the levels of TNF-α, confirming that inflammation is one of the important mechanisms leading to the increase in oxidative stress in the diseased animals. Interestingly, catalase activity, as well as the expression of catalase and SOD-1 were also significantly increased in the diseased animals, representing a possible compensatory mechanism for increased oxidative stress as both expression levels correlated with superoxide production.
The observed functional alterations were associated with structural vascular impairments in the myocardium of DM + HC + HT animals. We observed capillary rarefaction as evidenced by lower capillary-to-fiber ratio in the diseased hearts. Capillary rarefaction has been asociated with metabolic syndrome in animal models26 and has only been sparsely investigated in previous studies of diastolic dysfunction, but our findings are in accordance with a study by Mohammed et al. who reported a reduced capillary density in the myocardium of HFpEF patients.27 Capillary rarefaction may have important clinical implications, particularly when occurring simultaneously with the observed coronary arterial endothelial dysfunction, as it will impair myocardial oxygenation especially during physical exercise.28 These microvascular alterations, at the level of both small arteries and capillaries, may contribute to reduced exercise tolerance in HFpEF patients.29 The observed vascular changes were accompanied by an increase in total collagen, which together with the increased passive stiffness of single cardiac myocytes (likely due to the shift towards the stiffer titin N2B isoform), translated into higher LV stiffness30–32 as demonstrated by increased LV end-diastolic elastance and the trend towards a decreased E/A ratio, both clear indicators of diastolic dysfunction. Indeed, the increase in collagen correlated with the increased LV end-diastolic elastance, indicating collagen deposition in the myocardium as an important determinant of increased stiffness. The activation of pro-fibrotic pathways in the myocardium and the resulting increase in collagen deposition is likely due to the increased oxidative stress and inflammation modulating the TGF-beta/SMAD3 signalling pathway.33 Furthermore, Westermann et al.34 have shown in patients with HFpEF that cardiac inflammation results in accumulation of extracellular matrix contributing to the development of diastolic dysfunction. Disturbed turnover of extracellular matrix, mediated through dysregulation of MMPs and their tissue inhibitors (TIMPS) is likely to play a role in this process. Our data suggest a reduced, rather than increased, matrix turnover in the myocardium of the animals with comorbidities at this stage of the disease. In addition, data obtained in a dog model of subacute heart failure indicate that persistent neurohumoral activation, which is also likely to be present in our porcine model, may result in high-energy phosphate depletion and enhancement of AMP deaminase activity contributing to myocardial stiffening irrespective of changes in extracellular matrix.35 The structural abnormalities observed in the present study translated into a shift in the relation between left atrial pressure and cardiac index in exercising swine, indicating that also during exercise higher filling pressures were required to achieve a similar level of cardiac index, consistent with a stiffer heart. Interestingly, the increased maximal force of single cardiomyocytes correlated with the observed trend towards increased LV end-systolic elastance. It could therefore be speculated that the increase in contractility acted as a compensatory mechanism to maintain LV function particularly in the face of the elevated blood pressure in the absence of LV hypertrophy.36
4.1 Aspects of the experimental model
The animal model presented here is complementary to a recently reported porcine model of diastolic dysfunction.37 Schwarzl et al. induced HT and HC using DOCA-salt and western diet containing high amounts of salt, fat, and cholesterol. An important difference between the two studies, besides the longer follow-up in our study (6 months vs. 12 weeks), was the induction of diabetes and kidney dysfunction in our model. The type of diabetes induced in our study is not consistent with either DM type I or with early DM type II, but rather with a late stage DM type II with impaired insulin production. In addition, the animals with comorbidities had a lower body weight, despite increased lipid plasma levels which may be due to growth retardation, which has also been shown in children with chronic kidney disease.38 Although Schwarzl et al.37 reported several similar functional findings, including increased LV end-diastolic stiffness, increased superoxide production, and eNOS uncoupling, some findings are distinctly different. Diabetes, which is a common comorbidity in HFpEF patients, likely contributed to the endothelial dysfunction and activation of NOX, which are also observed in HFpEF patients.18 Moreover, whereas concentric LV hypertrophy occurred in response to the pressure-overload in the study of Schwarzl et al. cardiomyocyte area was reduced in our model, indicating that in the present model, myocyte hypertrophy is not a factor contributing to myocardial stiffening. The lack of myocyte hypertrophy, is in agreement with the maintained PKG activity levels in our animals and in accordance with the concept that severe hyperglycemia induces muscle cell atrophy both in skeletal39,40 and cardiac muscle.41,42 Furthermore, myocyte size inversely correlated in our animals with the levels of cystatin C suggesting a role of the chronic kidney dysfunction in this process. Schwarzl et al. only evaluated renal function by creatinine and urea, but not inulin clearance and cystatin C. Our study shows that marked reductions in glomerular filtration rate are not detected by urea and creatinine in hypertensive swine. Increased LV fibrosis was documented in the present study and correlated with the increased diastolic elastance, but was not observed in the study by Schwarzl et al., most likely because hyperglycemia in addition to inflammation is an important trigger for extracellular matrix remodeling in HFpEF.43,44 Diabetes is known to lead to vascular deficiency, mediated by miR-320 according to a recent study45 as documented here as capillary rarefaction. In the study by Schwarzl et al.37 reduced phosphorylation of titin, the giant molecular ‘spring’ that is considered as one of the most important factors responsible for cardiomyocyte passive stiffness, was observed and a shift towards its stiffer isoform N2B was reported, supporting the concept that both collagen and titin contribute to myocardial stiffness in HFpEF patients.30 In the present model, while the shift in titin isoform towards the stiff N2B isoform was also documented, a change in total phosphorylation was not observed. Altogether, data from both models suggest that the presence of different comorbidities may initiate partially different pathologic pathways, that may provide potential targets for further study and mechanistic interventions. Based on the data from the present animal model, chronic treatment with antioxidants aiming at increasing NO bioavailability and alleviating oxidative stress, lowering inflammation, as well as sGC stimulators might result in reduction of collagen deposition, cardiomyocyte stiffening, and delay of onset of diastolic dysfunction.
4.2 Conclusions
As summarized in Figure 8, the present study demonstrates that in a large animal, chronic exposure to multiple common comorbidities results in systemic inflammation, endothelium-dependent coronary microvascular dysfunction, capillary rarefaction, oxidative stress, and perturbed nitric oxide production, which are associated with increased myocardial fibrosis and passive cardiomyocyte stiffness in the absence of myocyte hypertrophy, resulting in LV diastolic dysfunction. Future studies are needed to address the exact mechanisms connecting the different steps in the process; however, this large animal model provides an excellent translational tool for improving our understanding of the early pathophysiology of heart failure with preserved ejection fraction and for testing novel therapeutic interventions, including drugs, exercise, and diet interventions, for the treatment of the patients with this type of heart failure.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
The authors thank Wies Lommen and Max Goebel (VUMC, Amsterdam), Adele Dijk (UMC, Utrecht), Ilona Krabbendam, Lau Blonden, Ruben van Drie (ErasmusMC, Rotterdam, The Netherlands), and Marion von Frieling-Salewsky (University of Muenster, Germany) for their expert technical support.
Conflict of interest: none declared.
Funding
This study was supported by grants from the European Commission FP7-Health-2010 grant MEDIA-261409, the Netherlands CardioVascular Research Initiative: an initiative with support of the Dutch Heart Foundation [CVON2011-11 (ARENA), CVON2012-08 (PHAEDRA), CVON2014-11 (RECONNECT)] and The Academy of Finland 251272, Finnish Diabetes Research Foundation, and Finnish Foundation for Cardiovascular Research.
References
- 1. Borlaug BA, Paulus WJ.. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 2011;32:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CS, Sato N, Shah AN, Gheorghiade M.. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol 2014;63:1123–1133. [DOI] [PubMed] [Google Scholar]
- 3. Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JG, Cody RJ, Chioncel O, Collins SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O’Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR, Georgiopoulou VV, Gheorghiade M.. Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail 2014;2:97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwarzl M, Ojeda F, Zeller T, Seiffert M, Becher PM, Munzel T, Wild PS, Blettner M, Lackner KJ, Pfeiffer N, Beutel ME, Blankenberg S, Westermann D.. Risk factors for heart failure are associated with alterations of the LV end-diastolic pressure-volume relationship in non-heart failure individuals: data from a large-scale, population-based cohort. Eur Heart J 2016;37:1807–1814. [DOI] [PubMed] [Google Scholar]
- 5. Lam CS, Carson PE, Anand IS, Rector TS, Kuskowski M, Komajda M, McKelvie RS, McMurray JJ, Zile MR, Massie BM, Kitzman DW.. Sex differences in clinical characteristics and outcomes in elderly patients with heart failure and preserved ejection fraction: the Irbesartan in Heart Failure with Preserved Ejection Fraction (I-PRESERVE) trial. Circ Heart Fail 2012;5:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taylor AL. Heart failure in women. Curr Heart Fail Rep 2015;12:187–195. [DOI] [PubMed] [Google Scholar]
- 7. Ather S, Chan W, Bozkurt B, Aguilar D, Ramasubbu K, Zachariah AA, Wehrens XH, Deswal A.. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol 2012;59:998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zile MR, Baicu CF, Gaasch WH.. Diastolic heart failure – abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med 2004;350:1953–1959. [DOI] [PubMed] [Google Scholar]
- 9. Falcao-Pires I, Gavina C, Hamdani N, Van Der Velden J, Stienen GJM, Niessens HWM, Leite-Moreira AF, Paulus WJ.. Mechanisms underlying diastolic dysfunction induced by pressure overload differ between aortic valve stenosis and hypertension. Cardiovas Res 2012;93:S107–S107. [Google Scholar]
- 10. Czuriga D, Musters RJP, Borbely A, Falcao-Pires I, Bogaards SJP, Leite-Moreira AF, Papp Z, Van der Velden J, Stienen GJM, Paulus WJ.. High diastolic stiffness modifies sarcomeric structure in failing human cardiomyocytes. Eur Heart J 2011;32:904–905.20675661 [Google Scholar]
- 11. Czuriga D, Paulus WJ, Czuriga I, Edes I, Papp Z, Borbely A.. Cellular mechanisms for diastolic dysfunction in the human heart. CPB 2012;13:2532–2538. [PubMed] [Google Scholar]
- 12. Paulus WJ, Tschope C.. A novel paradigm for heart failure with preserved ejection fraction comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 13. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM.. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006;355:251–259. [DOI] [PubMed] [Google Scholar]
- 14. Unger A, Beckendorf L, Böhme P, Kley R, von Frieling-Salewsky M, Lochmüller H, Schröder R, Fürst DO, Vorgerd M, Linke WA.. Translocation of molecular chaperones to the titin springs is common in skeletal myopathy patients and affects sarcomere function. Acta Neuropathol Commun 2017;5:72.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Le Page LM, Rider OJ, Lewis AJ, Ball V, Clarke K, Johansson E, Carr CA, Heather LC, Tyler DJ.. Increasing pyruvate dehydrogenase flux as a treatment for diabetic cardiomyopathy: a combined 13C hyperpolarized magnetic resonance and echocardiography study. Diabetes 2015;64:2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang SL, Xia JH, Zhang YY, Fan JG, Wang H, Yuan J, Zhao ZZ, Pan Q, Mu YL, Xin LL, Chen YX, Li K.. Hyperinsulinemia shifted energy supply from glucose to ketone bodies in early nonalcoholic steatohepatitis from high-fat high-sucrose diet induced Bama minipigs. Sci Rep 2015;5:13980.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, Sturek M, Chalasani N.. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology 2009;50:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franssen CCS, Unger A, Korkmaz HI, De Keulenaer GW, TschöPe C, Leite-Moreira AF, Musters R, Niessen HWM, Linke WA, Paulus WJ, Hamdani N.. Myocardial Microvascular Inflammatory Endothelial activation in heart failure with preserved ejection fraction. JACC Heart Failure 2015;4:312–324. [DOI] [PubMed] [Google Scholar]
- 19. Ter Maaten JM, Damman K, Verhaar MC, Paulus WJ, Duncker DJ, Cheng C, van Heerebeek L, Hillege HL, Lam CS, Navis G, Voors AA.. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail 2016;18:588–598. [DOI] [PubMed] [Google Scholar]
- 20. Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, Niebauer J, Hooper J, Volk HD, Coats AJ, Anker SD.. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 2000;102:3060–3067. [DOI] [PubMed] [Google Scholar]
- 21. Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL.. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001;103:2055–2059. [DOI] [PubMed] [Google Scholar]
- 22. Alagiakrishnan K, Banach M, Jones LG, Datta S, Ahmed A, Aronow WS.. Update on diastolic heart failure or heart failure with preserved ejection fraction in the older adults. Ann Med 2013;45:37–50. [DOI] [PubMed] [Google Scholar]
- 23. Mentz RJ, Kelly JP, von Lueder TG, Voors AA, Lam CS, Cowie MR, Kjeldsen K, Jankowska EA, Atar D, Butler J, Fiuzat M, Zannad F, Pitt B, O’Connor CM.. Noncardiac comorbidities in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol 2014;64:2281–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, Gong Y, Liu PP.. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med 2006;355:260–269. [DOI] [PubMed] [Google Scholar]
- 25. Paolocci N, Biondi R, Bettini M, Lee C-I, Berlowitz CO, Rossi R, Xia Y, Ambrosio G, L»Abbate A, Kass DA, Zweier JL.. Oxygen radical-mediated reduction in basal and agonist-evoked NO release in isolated rat heart. J Mol Cell Cardiol 2001;33:671–679. [DOI] [PubMed] [Google Scholar]
- 26. Trask AJ, Katz PS, Kelly AP, Galantowicz ML, Cismowski MJ, West TA, Neeb ZP, Berwick ZC, Goodwill AG, Alloosh M, Tune JD, Sturek M, Lucchesi PA.. Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J Appl Physiol 2012;113:1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM.. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015;131:550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laughlin MH, Davis MJ, Secher NH, van Lieshout JJ, Arce-Esquivel AA, Simmons GH, Bender SB, Padilla J, Bache RJ, Merkus D, Duncker DJ.. Peripheral circulation. Compr Physiol 2012;2:321–447. [DOI] [PubMed] [Google Scholar]
- 29. Borlaug BA, Melenovsky V, Russell SD, Kessler K, Pacak K, Becker LC, Kass DA.. Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation 2006;114:2138–2147. [DOI] [PubMed] [Google Scholar]
- 30. Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM.. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 2015;131:1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yarbrough WM, Mukherjee R, Stroud RE, Rivers WT, Oelsen JM, Dixon JA, Eckhouse SR, Ikonomidis JS, Zile MR, Spinale FG.. Progressive induction of left ventricular pressure overload in a large animal model elicits myocardial remodeling and a unique matrix signature. J Thorac Cardiovasc Surg 2012;143:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marshall KD, Muller BN, Krenz M, Hanft LM, McDonald KS, Dellsperger KC, Emter CA.. Heart failure with preserved ejection fraction: chronic low-intensity interval exercise training preserves myocardial O2 balance and diastolic function. J Appl Physiol 2013;114:131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Esposito G, Cappetta D, Russo R, Rivellino A, Ciuffreda LP, Roviezzo F, Piegari E, Berrino L, Rossi F, De Angelis A, Urbanek K.. Sitagliptin reduces inflammation, fibrosis and preserves diastolic function in a rat model of heart failure with preserved ejection fraction. Br J Pharmacol 2017;174:4070–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C.. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 2011;4:44–52. [DOI] [PubMed] [Google Scholar]
- 35. Paolocci N, Tavazzi B, Biondi R, Gluzband YA, Amorini AM, Tocchetti CG, Hejazi M, Caturegli PM, Kajstura J, Lazzarino G, Kass DA.. Metalloproteinase inhibitor counters high-energy phosphate depletion and AMP deaminase activity enhancing ventricular diastolic compliance in subacute heart failure. J Pharmacol Exp Ther 2006;317:506–513. [DOI] [PubMed] [Google Scholar]
- 36. Kawaguchi M, Hay I, Fetics B, Kass DA.. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction: implications for systolic and diastolic reserve limitations. Circulation 2003;107:714–720. [DOI] [PubMed] [Google Scholar]
- 37. Schwarzl M, Hamdani N, Seiler S, Alogna A, Manninger M, Reilly S, Zirngast B, Kirsch A, Steendijk P, Verderber J, Zweiker D, Eller P, Hofler G, Schauer S, Eller K, Maechler H, Pieske BM, Linke WA, Casadei B, Post H.. A porcine model of hypertensive cardiomyopathy: implications for heart failure with preserved ejection fraction. Am J Physiol. Heart Circ Physiol 2015;309:H1407–H1418. [DOI] [PubMed] [Google Scholar]
- 38. Salas P, Pinto V, Rodriguez J, Zambrano MJ, Mericq V.. Growth retardation in children with kidney disease. Int J Endocrinol 2013;2013:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hulmi JJ, Silvennoinen M, Lehti M, Kivela R, Kainulainen H.. Altered REDD1, myostatin, and Akt/mTOR/FoxO/MAPK signaling in streptozotocin-induced diabetic muscle atrophy. Am J Physiol Endocrinol Metab 2012;302:E307–E315. [DOI] [PubMed] [Google Scholar]
- 40. Jakobsen J, Reske-Nielsen E.. Diffuse muscle fiber atrophy in newly diagnosed diabetes. Clin Neuropathol 1986;5:73–77. [PubMed] [Google Scholar]
- 41. Kawaguchi M, Asakura T, Saito F, Nemoto O, Maehara K, Miyake K, Sugai N, Maruyama Y.. Changes in diameter size and F-actin expression in the myocytes of patients with diabetes and streptozotocin-induced diabetes model rats. J Cardiol 1999;34:333–339. [PubMed] [Google Scholar]
- 42. Kawaguchi M, Techigawara M, Ishihata T, Asakura T, Saito F, Maehara K, Maruyama Y.. A comparison of ultrastructural changes on endomyocardial biopsy specimens obtained from patients with diabetes mellitus with and without hypertension. Heart Vessels 1997;12:267–274. [DOI] [PubMed] [Google Scholar]
- 43. Hutchinson KR, Lord CK, West TA, Stewart JA. Jr.,. Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS One 2013;8:e72080.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao J, Randive R, Stewart JA.. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J Diabetes 2014;5:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, Chang J, Peng T, Fan GC.. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol 2014;74:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.