

This editorial refers to ‘Enhancing fatty acid utilization ameliorates mitochondrial fragmentation and cardiac dysfunction via rebalancing optic atrophy 1 processing in the failing heart’ by Y. Guo et al., pp. 979–991.
The heart is able to catabolize different substrates in order to meet its high metabolic demand. Such flexibility is crucial to face variations in workload and/or substrate availability and preserve an adequate ejection of blood. Conversely, in heart failure (HF), the heart develops a metabolic inflexibility characterized by shifts in metabolism, leading to impaired metabolic signalling and contractile dysfunction.
Shifting from fatty acid to glucose oxidation
The main feature of metabolic cardiac remodelling in HF concerns the changes in substrate utilization that leads to diminished fatty acid oxidation (FAO), mirroring a fetal-like metabolic pattern (Figure 1, top panels). The mechanisms underlying FAO impairment involve mitochondrial dysfunction, a reduced availability of fundamental cofactors such as CoA or carnitine, a downregulation of β-oxidation enzymes and increased reliance on alternative substrates, including glucose and ketone bodies.1 Hence, glucose uptake and glycolysis are increased (Figure 1), at least in early stages,2 and even exceed the rates of complete glucose oxidation, whereas in advanced HF insulin resistance leads to a decline in glucose utilization.3 Whether such metabolic remodelling is an adaptive or a maladaptive response to cardiac damage is debated: acutely, this phenomenon might represent a beneficial mechanism since it allows the shift towards more efficient metabolic pathways4; conversely, chronic persistence of this metabolic shift assumes maladaptive connotations; indeed, in dilated cardiomyopathy, glucose uptake and glycolysis are increased and uncoupled from glucose oxidation; moreover, remodelling of glucose metabolism has been shown to precede pressure-overload-induced left ventricular hypertrophy.5
Figure 1.
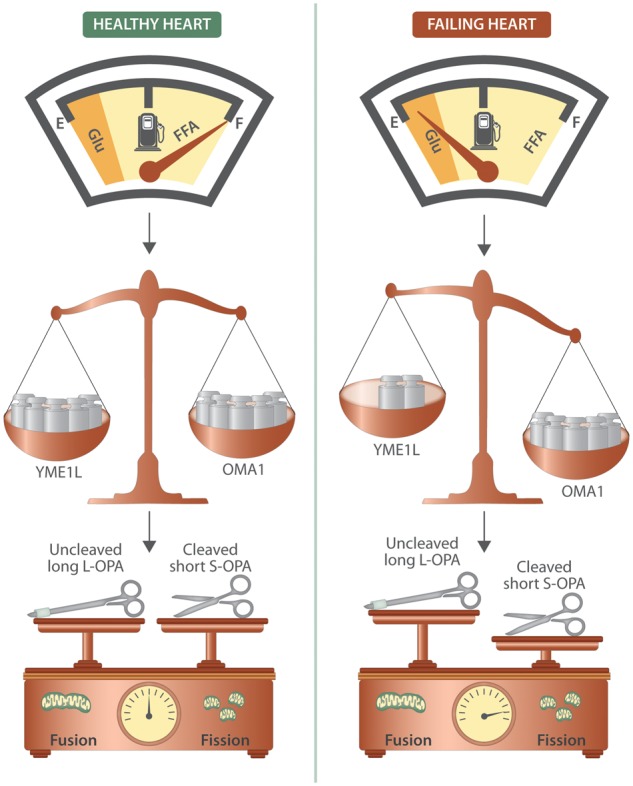
Myocardial energetics in HF: a matter of balance. During HF, the heart undergoes a metabolic remodelling which leads to reduced oxidation of free fatty acid and augmented utilization of glucose. A fine balance between the ATP-dependent metalloprotease (YME1L1) and the mitochondrial metalloendopeptidase (OMA1) is essential for regulating the cleavage of protein optic atrophy 1 (OPA1), which is a master regulator of mitochondrial dynamics. Indeed, OPA1, a dynamin-related protein associated with the inner mitochondrial membrane, is essential for mitochondrial fusion and cristae maintenance: the inner membrane-anchored long OPA1 (L-OPA1) undergoes proteolytic cleavage resulting in short OPA1 (S-OPA1); an imbalance in L-OPA/S-OPA ratio leads to increased mitochondrial fragmentation. Preclinical studies have demonstrated that enhancing FFA utilization in HF, thereby rebalancing the metabolic shift, ameliorates mitochondrial dynamics and cardiac dysfunction.
Recent attempts to coherently unify all metabolic alterations observed in HF have led to consider failing heart as an engine flooded with fuel: according to this model, the failing heart would be in a state of dyssnchrony for the imbalance between higher energetic demand and substrate availability vs. lower oxidative capacity and cofactor(s) availability such as carnitine and CoA. These phenomena eventually lead to accumulation of intermediates, which impair cardiac function, or substrates, including non-oxidized FA, which are diverged towards lipotoxic signalling pathways.1
Lipotoxicity or lipoprotection? The answer is in your mitochondria
The fundamental role played by cardiac metabolic remodelling in the development and progression of HF has led to the concept that supporting myocardial energetics through interventions on diet and macronutrient availability provides an important adjuvant therapy for HF. However, the potential success of such a strategy depends on whether the reversion of cardiac metabolism towards preferential FAO through increased fat intake would exert detrimental or beneficial effect. The paradigm of lipotoxicity is based on cardiac imbalance between reduced capacity of the cardiomyocyte to oxidize FA and normal/increased FA delivery. This phenomenon finally leads to subsequent accumulation of either intermediates (e.g. diacylglycerol and ceramide) that impair cardiac function or substrates (e.g. non-oxidized FA), which are diverged towards signalling or lipotoxic pathways.1,6 However, emerging evidence deriving from both genetic and dietary strategies indicates that normalization of FAO deficit would exert a cardioprotective, rather than detrimental, effect. Indeed, the deletion of acetyl-CoA carboxylase 2 preserves FAO and contractile function after pressure overload by decreasing rates of production of malonyl-CoA (a potent inhibitor of β-oxidation).7 In addition, feeding rodents high-fat diets might preserve or even improve cardiac function in HF. In a rat model of post-ischemic HF, feeding high fat chow improves myocardial function at rest and during physiological stress compared to normal chow but may negatively affect the contractile reserve in sham conditions.8 Equally important, in the Dahl salt-sensitive rat, a model that develops hypertension-induced HF with salt feeding, high saturated fat chow prevented LV remodelling and increased survival rate when compared to a standard chow.9 Myocardial function can be also preserved by manipulating the exposure of the failing heart to different FA species: e.g. supplementation with oleate (the primary FA in the Mediterranean diet) or n-3 polyinsaturated FA (fish oil), rather than palmitate or stearate, improves cardiac contractility of failing hearts.1,6
Alterations in mitochondrial dynamics, respiratory capacity and ATP synthesis are considered key factors leading to the chronic cardiac energy deficit observed in HF.10 The delicate regulation of mitochondrial dynamics is a crucial step that allows the cell to face stressing conditions and to meet changes in energetic demands.11 Hence, imbalanced mitochondrial fusion/fission can lead to HF.12,13 In the current issue of Cardiovascular Research, Guo and colleagues demonstrate that enhancing FA utilization, via dietary modifications and via cardiac overexpression of CD36 (the predominant form of FA translocase in the heart) significantly ameliorates mitochondrial fragmentation and cardiac dysfunction in HF via rebalancing mitochondrial dynamics14 – finely orchestrated by YME1L, OMA1 and OPA1 (Figure 1, middle and bottom panels).
Dietary management of HF patients: the tricky obesity paradox
Specific metabolic interventions in HF patients are lacking since no agreement has been reached concerning the beneficial or detrimental implications of cardiac metabolic remodelling. One crucial issue is the management of weight in patients with HF. Indeed, according to the obesity paradox, there is discordance between the increased risk of developing HF in obese patients and the better short-intermediate term prognosis exhibited by overweight and obese patients with established HF compared to their lean counterparts. A meta-analysis investigating the influence of body mass index (BMI) in HF revealed the highest risk in lowest BMI categories, the lowest in overweight and obese class I patients while increasing degree of obesity failed to achieve a statistically significant effect on CV mortality and hospitalization.15 Mechanisms potentially involved in the obesity paradox include: greater fat stores availability, which could provide resistance to the catabolic progression of the underlying disease to the stage of cachexia, an independent risk factor for mortality in HF patients; secretion of anti-inflammatory cytokines; increased strength and muscular mass; a higher cardiorespiratory fitness.
Weight management of HF patients remains controversial.6 Although weight loss could prevent negative effects of high BMI on cardiac hemodynamics, the obesity paradox would suggest a different approach. Thus, intentional weight loss is currently recommended only in Class II and III obese patients,6 while in overweight and class 1 obese patients an approach focused on aerobic exercise training and maintenance of body weight is preferable.
Funding
Dr Gaetano Santulli is supported by the National Institutes of Health (R00DK107895).
Conflict of interest: none declared.
References
- 1. Wende AR, Brahma MK, McGinnis GR, Young ME.. Metabolic origins of heart failure. JACC Basic Transl Sci 2017;2:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R.. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004;44:662–667. [DOI] [PubMed] [Google Scholar]
- 3. Ciccarelli M, Chuprun JK, Rengo G, Gao E, Wei Z, Peroutka RJ, Gold JI, Gumpert A, Chen M, Otis NJ, Dorn GW 2nd, Trimarco B, Iaccarino G, Koch WJ.. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 2011;123:1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grossman AN, Opie LH, Beshansky JR, Ingwall JS, Rackley CE, Selker HP.. Glucose-insulin-potassium revived: current status in acute coronary syndromes and the energy-depleted heart. Circulation 2013;127:1040–1048. [DOI] [PubMed] [Google Scholar]
- 5. Doenst T, Pytel G, Schrepper A, Amorim P, Farber G, Shingu Y, Mohr FW, Schwarzer M.. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res 2010;86:461–470. [DOI] [PubMed] [Google Scholar]
- 6. Butler T. Dietary management of heart failure: room for improvement? Br J Nutr 2016;115:1202–1217. [DOI] [PubMed] [Google Scholar]
- 7. Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R.. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res 2012;111:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berthiaume JM, Bray MS, McElfresh TA, Chen X, Azam S, Young ME, Hoit BD, Chandler MP.. The myocardial contractile response to physiological stress improves with high saturated fat feeding in heart failure. Am J Physiol Heart Circ Physiol 2010;299:H410–H421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okere IC, Chess DJ, McElfresh TA, Johnson J, Rennison J, Ernsberger P, Hoit BD, Chandler MP, Stanley WC.. High-fat diet prevents cardiac hypertrophy and improves contractile function in the hypertensive dahl salt-sensitive rat. Clin Exp Pharmacol Physiol 2005;32:825–831. [DOI] [PubMed] [Google Scholar]
- 10. Gambardella J, Trimarco B, Iaccarino G, Santulli G.. New insights in cardiac calcium handling and excitation-contraction coupling. Adv Exp Med Biol 2017;doi: 10.1007/5584_2017_106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franco A, Sorriento D, Gambardella J, Pacelli R, Prevete N, Procaccini C, Matarese G, Trimarco B, Iaccarino G, Ciccarelli M.. GRK2 moderates the acute mitochondrial damage to ionizing radiation exposure by promoting mitochondrial fission/fusion. Cell Death Discov 2018;4:doi: 10.1038/s41420-018-0028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T.. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015;350:aad0116.. [DOI] [PubMed] [Google Scholar]
- 13. Sorriento D, Gambardella J, Fiordelisi A, Trimarco B, Ciccarelli M, Iaccarino G, Santulli G.. Mechanistic role of kinases in the regulation of mitochondrial fitness. Adv Exp Med Biol 2017;982:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo Y, Wang Z, Qin X, Xu J, Hou Z, Yang H, Mao X, Xing W, Li X, Zhang X, Gao F.. Enhancing fatty acid utilization ameliorates mitochondrial fragmentation and cardiac dysfunction via rebalancing OPA1 processing in the failing heart. Cardiovasc Res 2018;114:979–991. [DOI] [PubMed] [Google Scholar]
- 15. Sharma A, Lavie CJ, Borer JS, Vallakati A, Goel S, Lopez-Jimenez F, Arbab-Zadeh A, Mukherjee D, Lazar JM.. Meta-analysis of the relation of body mass index to all-cause and cardiovascular mortality and hospitalization in patients with chronic heart failure. Am J Cardiol 2015;115:1428–1434. [DOI] [PubMed] [Google Scholar]