

Abstract
Major depressive disorder (MDD) is one of the most common and debilitating neuropsychiatric illnesses. Accumulating evidence suggests a potential role of the immune system in the pathophysiology of MDD. The complement system represents one of the major effector mechanisms of the innate immune system, and plays a critical role in inflammation. However, the role of complement components in MDD is not well understood. Here, we found significant increase in component 3 (C3) expression in the prefrontal cortex (PFC) of depressed suicide subjects. We tested the role of altered C3 expression in mouse model of depression and found that increased C3 expression in PFC as a result of chronic stress causes depressive-like behavior. Conversely, mice lacking C3 were resilient to stress-induced depressive-like behavior. Moreover, selective overexpression of C3 in PFC was sufficient to cause depressive-like behavior in mice. We found that C3a (activated product of C3) receptor, C3aR+ monocytes were infiltrated into PFC following chronic stress. However, C3aR knockout mice displayed significantly reduced monocyte recruitment into PFC and reduced levels of the proinflammatory cytokine IL-1β in PFC after chronic stress. In addition, C3aR knockout mice did not exhibit chronic stress-induced behavior despair. Similarly, chronic stress-induced increases in C3aR+ monocytes and IL-1β in PFC, and depressive-like behavior were attenuated by myeloid cell depletion. These postmortem and preclinical studies identify C3aR signaling as a key factor in MDD pathophysiology.
Keywords: complement, C3, depression, animals, behavior
1. Introduction
Recent studies from both clinical as well as preclinical studies suggest that immune system plays an important role in the pathophysiology of Major Depressive Disorder (MDD)1,2. The role of inflammation in MDD is further supported by studies showing increased levels of pro-inflammatory cytokines in depressed subjects3,4. In particular, increased levels of IL-1β mRNA and protein levels have been found in the prefrontal cortex (PFC) of depressed and suicide subjects5. The PFC is most sensitive to the detrimental effects of stress exposure, exhibiting both structural as well as functional changes upon chronic stress, and has been especially implicated in depression6–10. Chronic stress has been shown to increase microglia activation in PFC, but not in hippocampus11. The association between immune activation and depressive behavior is further strengthened by findings from preclinical studies that show induction of depressive-like behaviors in rodents following cytokine administration, and increased proinflammatory signaling in rodent models of depressive-like behavior12–15. However, the endogenous primers for this activation are not well understood.
The innate immune system responds by recognition of conserved motifs in pathogens as well as a number of other indicators of cell stress such as cytokines leading to the activation of adaptive immunity16. The complement system is a vital component of innate immunity and is one of its major effector mechanisms17. Complement consists of a number of inactive components that are activated in a cascade-like manner by three major pathways: the classical pathway, lectin pathway, and alternate pathway18,19. All pathways converge upon generation of C3 convertase, a molecule that cleaves complement component C3 into C3a and C3b. C1q is the recognition molecule of the classical pathway, and its binding to antigens or antibodies can activate the associated serine proteases C1r and C1s, leading to cleavage of C2 and C4, which generates the C3 convertase, C4b2b; C4b2b cleaves C3 and activates downstream cascade components. The lectin pathway is initiated by the binding of mannose binding lectin (MBL) to mannose residues on the cell surface. This activates the MBL-associated proteases mannose binding lectin serine protease 1 (MASP1) and MASP2, which then cleave C4 to generate the C3 convertase, C4b2b. The alternative pathway is continuously activated by a “tickover” mechanism (via spontaneous C3 hydrolysis), and functions primarily as an amplification loop of C3b after initiation by the classical pathway and the lectin pathway. C3b opsonizes pathogens to facilitate phagocytosis. C3a, on the other hand, mediates immunomodulatory signals via its seven-transmembrane domain receptor, C3aR20.
In the CNS, complement has additional physiological activity outside of immune function. C3 is expressed in neurons21, glia22 and endothelial cells23. C3 signaling through C3aR has been shown to regulate synaptic function and dendritic morphology22. Complement components enhance proinflammatory TLR-mediated signaling leading to increased production of IL-1β24. Despite the intense interest into inflammation in depression, little is known about the role of complement system in depression. In the present study, we examined the role of C3 in depression using postmortem PFC samples from depressed suicide subjects and rodent models of depression. We demonstrate that selective C3 overexpression in the mPFC is sufficient to produce depressive-like behavior. C3 as well as C3aR KO mice were resilient to chronic stress. We found that C3a (activated product of C3) receptor, C3aR+ monocytes were infiltrated into PFC following chronic stress. However, C3aR knockout mice displayed significantly reduced monocyte recruitment into PFC and reduced levels of the proinflammatory cytokine IL-1β in PFC after chronic stress. Together, we identify a novel role of C3 in depressive behavior.
2. Materials and Methods
2.1. Animals
Adult (8 week old) male C57BL/6J mice were purchased from The Jackson Laboratory. Two month old C3−/− (129S4-C3tm1Crr/J), C3aR−/− (129S4-C3ar1tm1Cge/J) and their age-matched wild type (WT) controls were purchased from The Jackson Laboratory and housed and maintained in our animal housing facility at Augusta University. Mice were housed in groups of 4 mice in standard polypropylene cages in 12-h light-dark cycle in compliance with the US National Institute of Health guidelines and approved by Augusta University animal welfare guidelines. Mice were assigned to experimental groups based on their genotype. Selection of animal for lentiviral treatment was performed randomly and in a blinded manner.
2.2. Stereotaxic Injection of Lentivirus
C3 Lentiviral (LV) Activation Particles and Control LV Activation Particles were purchased from Santa Cruz Biotech, CA, USA. C3 LV Activation Particles contain the following SAM activation elements: a deactivated Cas9 (dCas9) nuclease (D10A and N863A) fused to the transactivation domain VP64, an MS2-p65-HSF1 fusion protein, and a target-specific 20 nt guide RNA. Lentiviral particles frozen stock contains a concentration of 1.0 × 106 infectious units of virus in Dulbecco’s Modified Eagle’s Medium with 25 mM HEPES pH 7.3. Lentiviral particles were injected sterotaxically into mouse PFC25,26. In brief, mice were received Buprenorphine (0.05 mg/kg, s.c.) for 15 min and then anesthetized with 1.5–2.5% isoflourane mixed with oxygen in an induction chamber. Mice were maintained under deep anesthesia and mounted in a stereotaxic apparatus. Around ~1 mm size burr hole were made to perform a very small craniotomy as per the recommended coordinates (x .5 mm (lateral), y 1.0 mm (anteriorposterior A–P), with respect to bregma at 0), z 1.0 mm (dorso−ventral D–V with brain surface at 0). A 30−gauge needle attached to a micro syringe containing lentvirus were inserted and lowered into the prefrontal cortical region of the brain. The microsyringe was left in place 5 min after each injection, and a total volume of 1.0 μl (1 × 106 infectious particles per milliliter) of lentivirus was administered at a rate of 0.2 μl/min at each site (Stoelting Co). Mice were allowed to recover from anesthesia, transferred to a clean cage with free access to chow diet and water. After the 2 wk of injection, the injection site was confirmed by imaging analysis for the fluorescence of lentiviral particles.
2.3. Chronic unpredictable stress (CUS) procedure
Two weeks prior to the beginning of the CUS paradigm, all animals were individually housed while all other aspects of housing remained as standard housing conditions. Mice underwent CUS for 21 days during the light phase of the 24 h period. CUS experiments were performed according to our previously published procedure26. Details on the duration and types of stressors used are provided in Table S1. The controls received the same temporal and spatial sequence of habituation and test days that the stressed animals experienced.
2.4. Behavioral Tests
Behavioral testing was conducted 24 h following the final period of CUS exposure during the light phase of the 24 h period. Behavior tests were performed in the following sequential order in every animal: (1) open field test, (2) tail suspension test, (3) forced swim test, and (4) sucrose preference test with a period of rest of at least 24 h between each test. Immediately following the sucrose preference test, mice were sacrificed and brains were rapidly removed and processed for biological measures.
2.4.1. Sucrose preference test
Mice were habituated to 1% sucrose solution for 3 days at the start of the experiment, in which two bottles of 1% sucrose solution were placed in each cage. After adaptation, mice were deprived of fluid for 16 h. Mice were then presented with two identical bottles of 1% sucrose and water for 24 h. The position of the bottles containing water or sucrose solution was switched halfway through the test. At the end of the 24-h test, sucrose and water consumption (ml) were measured, and the preference for sucrose was calculated as a percentage of consumed sucrose solution of the total amount of liquid drunk.
2.4.2. Forced swim test
Mice were subjected to one 5-min session of swimming in water. On the test day, mice were placed in a clear cylinder with water (24 °C ± 1 °C, 45-cm depth and 20 cm diameter). The sessions were recorded from the side, and the immobility time was scored by a blind observer. Immobility was calculated as the least amount of movement of experimental mice to stay afloat.
2.4.3. Tail Suspension test
Each mouse was suspended with a tape 0.75 cm away from their tails and elevated 70 cm above the floor. Behavior was recorded for 6 min. Observer blinded to the subject’s treatment group scored video recordings for time spent struggling or immobile.
2.4.4. Open field test
Mouse locomotor activity in an open field was measured over a 10-min period. Time spent (sec.) in margin and center, total distance (cm) and mean speed (cm/sec.) were measured during the session. Ethovision XT 10 (Noldus Information Technologies Inc, USA) software and open field chamber (40 × 40 × 40 cm), and a video camera was fixed over the chamber by an adjacent rod, an activity monitor, a programmer and a printer were used for analysis. Sessions total for all parameters were taken. Observations were recorded for 10 min.
2.5. Monocyte/macrophage depletion
A Clodrosome Macrophage Depletion Kit, containing control liposomes (Encapsome) and clodronate liposomes (Clodrosome) (Encapsula NanoSciences, Brentwoood, TN), was used to deplete endogenous monocytes/macrophages. Intraperitoneal administration of 200 μl of Encapsome or clodronate liposomes (5 mg/ml) was performed three times at 7 days apart during CUS paradigm for 21 days. Following CUS, myeloid cell (CD11b+ F4/80+) depletion was confirmed by flow cytometry.
2.6. Western blotting
PFC samples collected immediately following decapitation under anesthesia were lysed in ice-cold lysis buffer containing protease inhibitor cocktail (1860932, ThermoFisher scientific, Waltham, MA) and protein concentration was determined by bicinchoninic acid (BCA) assay (MilliporeSigma St. Louis, MO). Protein was electrophoretically separated on a SDS PAGE gel and transferred to a nitrocellulose membrane. Blots were incubated in the appropriate primary antibody specific for C3 (sc-28294, Santa Cruz Biotech, Dallas, Texas), C3aR (sc-133172; Santa Cruz Biotech),VCAM-1 (114441-1-AP; Proteintech, Rosemont, IL), ICAM-1 (10831-1-AP; Proteintech), β-tubulin (2146, Cell Signaling, Danvers, MA) or β-actin (4967, Cell Signaling); and developed with the ECL Plus Western Blotting Detection System (GE Healthcare). Optical densities of the bands were analyzed using ImageJ software (NIH). For analysis, protein levels were normalized to β-tubulin or β-actin levels then expressed as a fold change of that in control animals.
2.7. Flow cytometry
Mice were anesthetized with isoflurane and perfused with ice-cold PBS. The brain was removed and the PFC separated and homogenized in cold PBS. Freshly harvested PFC tissue was sieved through a 100-μM cell strainer (BD Biosciences, San Diego, CA), followed by centrifugation (1000 rpm, 10 min) to prepare single-cell suspensions. Cells were incubated with Abs against cell surface markers, CD11b, F4/80, CD45 (all from eBioscience, San Diego, CA), Iba-1 (019-19741, Wako Chemicals USA, Inc. Richmond, VA) and C3aR (sc-133172; Santa Cruz Biotech). Following a PBS wash, cells were fixed and permeabilized using a Fixation/Permeabilization Concentrate (Affymetrix eBioscience). Cells were analyzed using a four-color flow cytometer (FACSCalibur) and CellQuest software (both from BD Biosciences, San Jose, CA)27. Isotype-matched controls were analyzed to set the appropriate gates for each sample. For each marker, samples were analyzed in duplicate. To minimize false-positive events, the number of double-positive events detected with the isotype controls was subtracted from the number of double-positive cells stained with the corresponding Abs (not isotype control). Cells expressing a specific marker were reported as a percentage of the number of gated events. Mean channel fluorescence intensity (MFI) derived from a fluorescence graph was used to study the level of cell surface C3aR expression.
2.8. Quantitative reverse transcriptase PCR (qRT-PCR)
For qRT-PCR analyses, the mouse PFC tissues were collected immediately following decapitation under anesthesia. RNA from mouse as well as human PFC samples were purified using a commercially available kit (SV RNA Isolation, Promega, Madison, WI, USA), qRT-PCR was performed on a MasterCycler (Eppendorf) using a SuperScript III Platinum SYBR Green One-Step qRT-PCR kit (Invitrogen, Carlsbad, CA, USA). Gene-specific primers were synthesized by Integrated DNA Technologies. Ct values of genes of interest were normalized to that of housekeeping genes (β-actin, ribosomal protein S3 (RPS3) or 18S). Primers used were mC3-FP, 5′-AGCAGGTCATCAAGTCAGGC-3′; mC3-RP, 5′-GATGTAGCTGGTGTTGGGCT-3′; mIL1β -FP, 5′-CCAAGCAACGACAAAATACC-3′, mIL1β-RP, 5′-GTTGAAGACAAACCGTTTTTCC-3′ mβ-actin-FP, 5′-GTTGTCGACGACGAGCG-3′; mβ-actin-RP, 5′-AATGAACCGAAGCACACCATA-3′; mRPS3-FP, 5′-GCACAGAGCCTCGCCTT-3′; mRPS3-RP, 5′-ATCAGAGAGTTGACCGCAGTT-3′; hC3-FP, 5′-GCTGCTCCTGCTACTAACCCA-3′; hC3-RP, 5′-AAAGGCAGTTCCCTCCACTTT-3′; h18S-FP, 5′-GGCCCTGTAATTGGAATGAGTC-3′; h18S-RP, 5′-CCAAGATCCAACTACGAGCTT-3′;
2.9. Immunofluorescence staining
Mice were anesthetized with isoflurane and perfused transcardially with PBS followed by 4% paraformaldehyde (PFA). Brains were removed and postfixed in PFA solution overnight and stored in 30% sucrose in PBS until the tissues sank. These were embedded in optimal cutting temperature (OCT) medium, frozen in isopentane (− 55 °C), and stored at − 80 °C until use. Serial sections (10 μm) were cut using a cryostat microtome (Leica CM 3050 S; Leica Microsystems, Chantilly, Virginia) and stored at − 20 °C. Sections were thawed, washed twice in PBS for 5 min and were incubated with blocking solution (10% normal donkey serum in PBS) with constant shaking for 30 min followed by overnight incubation with a cocktail of primary antibodies in 5% normal donkey serum in PBS containing 0.3% triton X-100. We used an antibody directed against the ionized calcium-binding adapter molecule-1 (Iba-1;1:100; 019-19741; Wako) which recognizes both microglia and macrophages28. To stain perivascular macrophages29, a myeloid cell type that resides in the perivascular space, we used an antibody to mannose receptor CD206 (1:100; ab64693; abcam). The above antibodies were used in combination with antibody to C3 (1:100; sc-28294, Santa Cruz Biotech, or 2934R; Bioss antibodies; Woburn, Massachusetts). After rinsing, sections were incubated for 2 h with Cy3 or Cy2-conjugated secondary antibodies (1:200; Jackson ImmunoResearch, PA) in 5% normal donkey serum in PBS containing 0.3% triton X-100. Sections were thoroughly rinsed, mounted, and cover-slipped in anti-fading aqueous medium. Confocal images were acquired using a Zeiss 510 LSM microscope.
2.10. Il-1β protein levels by enzyme linked immunosorbent assay (ELISA)
Mouse PFC samples collected immediately following decapitation under anesthesia were lysed in ice-cold lysis buffer containing protease inhibitor cocktail and protein concentration was determined by BCA assay. Il-1β protein levels in mouse PFC samples were measured by ELISA according to the manufacturer’s instructions (#MLB00C; R&D Systems). The values are expressed as pg/mg protein.
2.11. Postmortem samples
Human postmortem PFC tissue samples (Brodmann’s area 10) from suicide completers (N=15) and psychiatrically normal controls (N=15) were used in the current study. The samples were obtained from the Quebec Suicide Brain Bank (QSBB; Douglas Institute; www.douglas.qc.ca/suicide). The study was granted by the Douglas Hospital Institutional Review Board and, written informed consent was obtained from each participating family (Declaration of Helsinki, 1964).
2.10. Statistical Analysis
For mouse studies, data were analyzed using two-tailed Student’s t-tests (for two-group comparisons) or Analysis of Variance (ANOVA; for multiple-group comparisons). p < 0.05 was considered significant. Post hoc analyses were carried out using Bonferroni’s test. For the postmortem analysis, Analysis of Covariance (ANCOVA) was used to examine differences between depressed-suicide subjects and the controls on C3 expression. Group differences in C3 expression were examined while age, post mortem interval (PMI), pH, RNA integrity, lifetime substance use, substance use at death, and psychiatric medication use were evaluated as possible covariates for inclusion in the model. It was decided apriori that candidate covariates with at least small correlations with C3 expression would be included as a covariate in the ANCOVA. The effect size difference between depressed-suicide and control subjects was estimated as partial eta-square (η2p). All analyses were completed using SPSS Statistics 20 software (IBM).
3. Results
3.1. C3 expression is increased in the prefrontal cortex of depressed subjects
There were no significant differences between postmortem samples obtained from the depressed-suicide sample and controls in any of the evaluated covariates (Table S2 and Table S3). Only lifetime substance use (phi=0.333), substance use at death (phi=0.208), and psychiatric medication use (phi =0.283) were retained as covariates following the initial evaluation. To determine whether C3 expression is altered in depressed subjects, we conducted quantitative PCR analysis of postmortem PFC samples collected from a cohort of suicide victims diagnosed with depression and age-matched individuals without psychiatric diagnoses26. We found a significant increase in C3 mRNA levels in postmortem PFC samples obtained from depressed suicide subjects as compared to control subjects (Fig 1; P<0.001). A key limitation in the studies using postmortem samples is the presence and history of substance use and/or medications at the time of death. Out of 15 depressed suicide subjects, 11 had the presence of substance use and 7 had medication at the time of death. To determine if the observed increase in C3 mRNA levels in the PFC of depressed suicide subjects are related to these potential covariates, we performed ANCOVA. None of the three covariates—substance use [F(1,25) = 1.709, p = 0.203, η2p = 0.064], substance use at death [F(1,25) = 0.109, p = 0.744, η2p = 0.004], and medication use [F(1,25) = 2.126, p = 0.157, η2p = 0.078] were significantly associated with C3 mRNA in the ANCOVA. Depressed-suicide status was a significant predictor of C3 mRNA [F(1, 25) = 40.27, p < 0.001, η2p = 0.617, Observed Power = 1.00]. Additional analysis compared the effect of antidepressant treatment on C3 mRNA levels, but no significant effect was observed.
Figure 1. Increase in C3 mRNA levels in the prefrontal cortex of depressed suicide subjects.
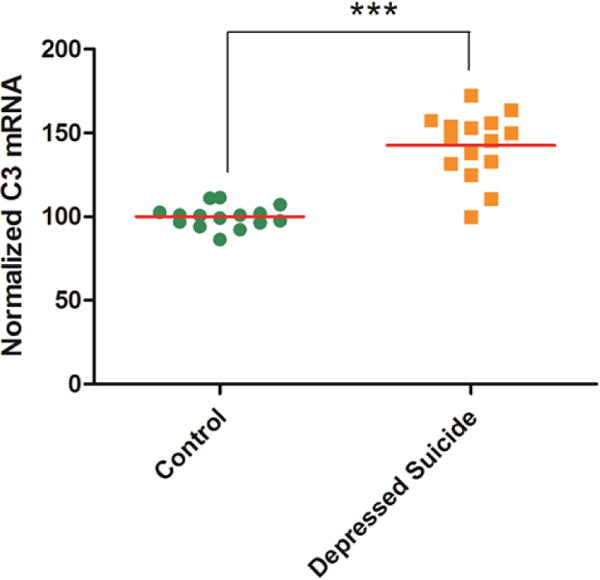
C3 mRNA levels in the prefrontal cortex of depressed suicide (n=15) and control (n=15) subjects were measured by RT-PCR. mRNA levels were normalized to housekeeping gene 18S. ***p<0.001.
3.2. C3 knockout mice are resilient to CUS-induced depressive-like behavior
Because C3 is a converging point of all three complement activation pathways and is highly expressed in the PFC of depressed suicide subjects, we further examined the function of C3 in depressive-like behavior using rodent model of depression. We used a mouse chronic unpredictable stress (CUS) paradigm that is a well validated rodent model of depressive-like behavior26. In this paradigm, mice were exposed to a serious of mild unpredictable stressors for 21 days. At the end of the stress paradigm, the animals were tested for tail suspension test (TST), forced swim test (FST), open field activity (OF) and sucrose preference test (SPT). The CUS-exposed mice gained significantly less weight over 21 days (Fig 2A; p<0.05). There was a significant increase in C3 mRNA (Fig 2B; p<0.05and protein (Fig 2C; p<0.05) levels in PFC of CUS-exposed mice, compared to non-stressed (NS) mice. Immunofluorescence analysis of PFC sections from CUS-exposed mice showed that C3 is colocalized with Iba-1, a marker for myeloid cells (monocyte/macrophage lineage) and CD206, a marker for perivascular macrophages, a myeloid cell type that resides in the perivascular space (Fig 2D).
Figure 2. C3 knockout mice are resilient to CUS-induced depressive-like behavior.
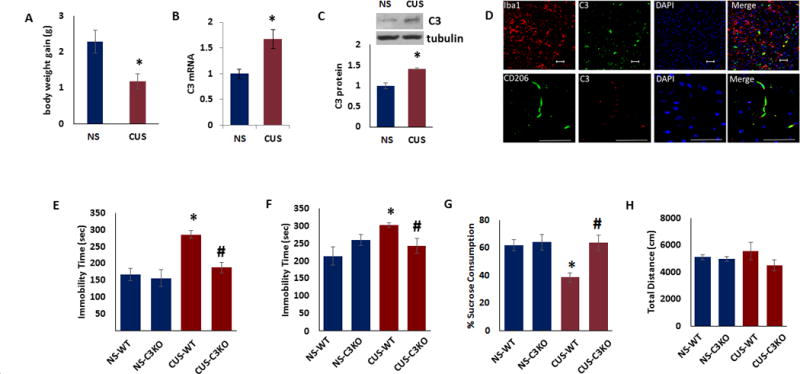
(a) Body weight gain for male mice exposed to CUS (n=6) or no stress (NS) (n=6) over 21 days. (b) C3 mRNA in the prefrontal cortex of male mice exposed to CUS (n=6) or no stress (NS) (n=6). The Ct values were normalized to RPS3. (c) Top. Representative immunoblot data showing C3 and β-tubulin expression in the PFC of male mice exposed to CUS (n=3) or no stress (NS) (n =5). Bottom. C3 protein levels normalized to β-tubulin. (d) Iba1+ myeloid cells and perivascular macrophages express C3 after CUS. Representative images showing Iba1, and CD206 (perivascular macrophages) staining with C3. (Scale bar: 50 μm.) (e–h) Adult male C3 knockout and wildtype (WT) mice were exposed to CUS and depressive-like behavior was determined (N=6–7). (e) Tail suspension test, (f) forced swim test, (g) preference for sucrose in sucrose preference test and (h) distance traveled in the open field test. Data are expressed as mean ± s.e.m. *P < 0.05 vs NS (a,b,c) or NS-WT (e–g); #P < 0.05 vs CUS-WT (e–g); Student’s t test (a,b,c); two-way ANOVA (e–h).
To further examine the role of C3 in depressive-like behavior, we used C3 KO mice. Our baseline behavioral analysis (before exposure to CUS) showed no significant difference between C3 KO and WT mice in TST, FST, OF and SPT (data not shown). Next, we examined whether deletion of C3 influences the response to CUS. As expected CUS exposed WT animals showed significant increases in immobility time in TST (Fig 2E) and FST (Fig 2F). However, the immobility time in C3 KO mice exposed to CUS were similar to WT mice in both TST (Fig 2E; two-way ANOVA, stress, F(1,24) = 15.06, p < 0.001; genotype, F(1,24) = 7.84, p < 0.01; stress × genotype, F(1,24) = 5.12, p < 0.05; post hoc, NS-WT vs CUS-WT, p < 0.001, CUS-WT vs CUS-C3 KO, p < 0.001) and FST (Fig 2F; two-way ANOVA, stress × genotype, F(1,24) = 6.50, p < 0.05; post hoc, NS-WT vs CUS-WT, p < 0.05, CUS-WT vs CUS-C3 KO, p < 0.05). In sucrose preference test, CUS resulted in a significant reduction in sucrose consumption in WT mice, indicative of stress-induced anhedonia (Fig 2G). However, C3 KO mice exposed to CUS consumed sucrose volumes that were similar to the levels in WT group (Fig 2G; two-way ANOVA, stress, F(1,24) = 5.61, p < 0.05; genotype, F(1,24) = 7.28, p < 0.05; stress × genotype, F(1,24) = 4.50, p < 0.05; post hoc, NS-WT vs CUS-WT, p < 0.01, CUS-WT vs CUS-C3 KO, p < 0.01). We did not find any significant effect on locomotor activity in the open field test (Fig 2H). These results demonstrate that C3 KO mice show normal behavior in the absence of stress, but are resistant to CUS-induced depressive-like behavior.
3.3. Overexpression of C3 in PFC causes depressive-like behavior in mice
To determine whether restricting C3 overexpression to a pathologically relevant brain region would suffice to create the behavioral deficits, we infused mice with control or C3 lentiviral activation particles in the medial PFC (mPFC) by a lentiviral approach (Fig. 3A). Following two weeks to allow for expression of viral constructs, mice were tested in measures of depressive-like behavior. We found significant increase in C3 mRNA expression in PFC following C3 administration (Fig 3B; p<0.05). In behavior tests, mice infused with C3 demonstrated significant increases in immobility time in TST (Fig 3C; p<0.05) and FST (Fig 3D; p<0.05). C3 overexpression resulted in a significant reduction in sucrose consumption compared with control mice (Fig 3E; p<0.05). No significant difference in locomotor activity was observed between the two groups (Fig 3F; p<0.05) suggesting that change in immobility is not due to a generalized decrease in ambulation.
Figure 3. Overexpression of C3 in PFC causes depressive-like behavior in mice.
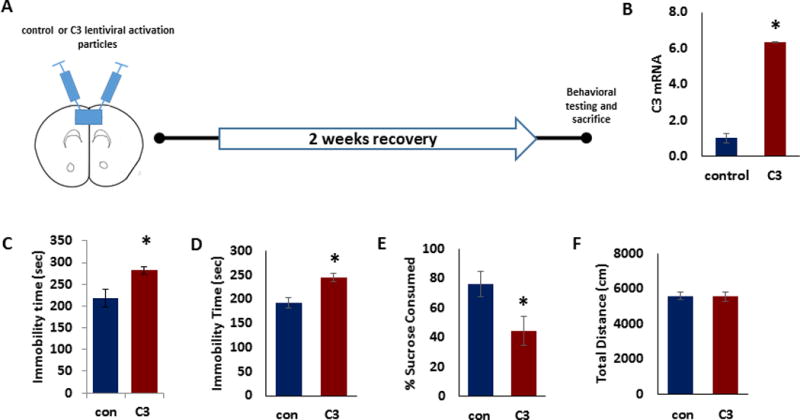
(a) Schematic representation of stereotaxic injection of control (con) or C3 lentiviral activation particles into mouse PFC followed by behavior tests. (b) C3 mRNA in the PFC of mice injected with control or C3 activation particles (n=6). The Ct values were normalized to RPS3. (c) Tail suspension test, (d) forced swim test, (e) preference for sucrose in sucrose preference test and (f) distance traveled in the open field test for the mice injected with control or C3 activation particles (n=6–8). Data are expressed as mean ± s.e.m. *P < 0.05 vs control; Student’s t test.
3.4. C3aR knockout mice are resilient to CUS-induced despair behavior
It is known that C3a (activated product of C3) induces immunomodulatory signals via its receptor, C3aR20. We found a significant increase in C3aR protein levels in PFC of CUS-exposed mice, compared to non-stressed mice (Fig 4A; p<0.05). To further determine the role of C3aR in depressive-like behavior, we used C3aR KO mice. We found significant increases in immobility time in tail suspension and forced swim tests in WT mice following CUS as compared to non-stressed (NS) mice (Fig 4B–C). However, the immobility time in C3aR KO mice exposed to CUS were similar to WT mice in both TST (Fig 4B; two-way ANOVA, stress × genotype, F(1,20) = 6.18, p < 0.05; post hoc, NS-WT vs CUS-WT, p < 0.05, NS-WT vs CUS-C3aR KO, p > 0.05) and FST (Fig 4C; two-way ANOVA, stress, F(1,21) = 5.49, p < 0.05; genotype, F(1,21) = 4.55, p < 0.05; stress × genotype, F(1,21) = 10.03, p < 0.01; post hoc, NS-WT vs CUS-WT, p < 0.05, NS-WT vs CUS-C3aR KO, p > 0.05). In sucrose preference test, CUS failed to induce anhedonia-like behavior in both WT and C3aR KO mice (Fig 4D). In the open field test, CUS exposed WT as well as C3aR KO mice showed hyperactivity as determined by the distance travelled in the chamber (Fig 4E; two-way ANOVA, stress, F(1,25) = 23.12, p < 0.001; post hoc, NS-WT vs CUS-WT, p < 0.001, NS-C3aR KO vs CUS-C3aR KO, p < 0.05). These results demonstrate that C3aR KO mice are resistant to CUS-induced despair behavior.
Figure 4. C3aR deficiency attenuates CUS-induced depressive-like behavior and increase in infiltration of monocytes in PFC.
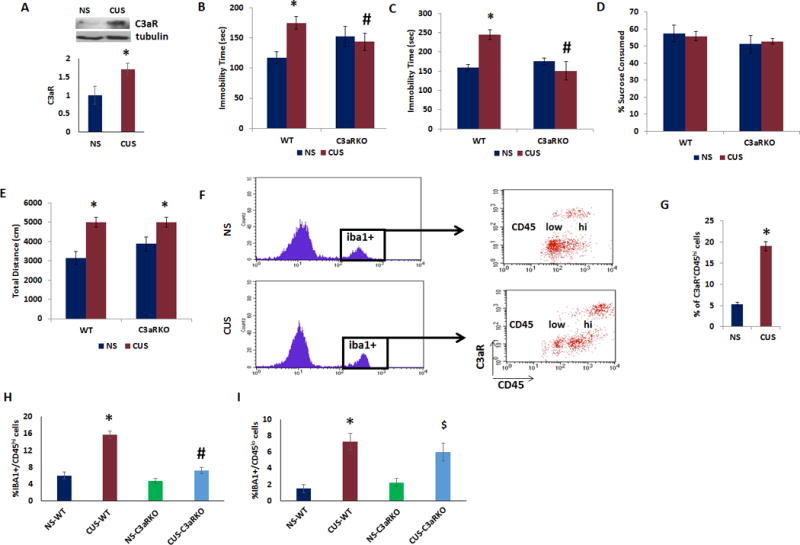
(a) Top. Representative immunoblot data showing C3aR and β-tubulin expression in the PFC of male mice exposed to CUS (n=3) or no stress (NS) (n=3). Bottom. C3aR protein levels normalized to β-tubulin. Adult male C3aR knockout and wildtype (WT) mice were exposed to CUS or no stress (NS) and depressive-like behavior was determined. (b) Tail suspension test, (c) forced swim test, and (d) preference for sucrose in sucrose preference test and (e) open field test. Data are expressed as mean ± s.e.m. *P < 0.05 vs NS-WT; #P < 0.05 vs CUS-WT; two-way ANOVA. N=8 per group. (f) Adult male mice were exposed to CUS or no stress (NS) and PFC tissues were collected for flow cytometry analysis. Iba1+/C3aR+ cells were analyzed using CD45 to differentiate infiltrating monocytes (CD45hi) from resident microglia (CD45lo). (g) % of Iba1+ cells expressing C3aR and CD45hi in PFC of mice exposed to CUS or no stress (NS). Data are expressed as mean ± s.e.m. *P < 0.05 vs NS. N=4 per group. (h–i) Adult male C3aR knockout and wildtype (WT) mice were exposed to CUS or no stress and PFC tissues were collected for flow cytometry analysis. Iba1+ cells were analyzed using CD45 to differentiate (h) infiltrating monocytes (CD45hi) from (i) resident microglia (CD45lo). *P < 0.05 vs NS-WT; #P < 0.05 vs CUS-WT; $P < 0.05 vs NS-C3aRKO; two-way ANOVA. N=4 per group.
3.5. C3aR deficiency reduces the number of chronic stress-induced infiltrating monocytes into PFC
We used flow cytometry analysis to identify the cell types expressing C3aR in PFC following CUS. Differences in the levels of CD45 expression was used to determine if ionized calcium binding adapter molecule 1 (Iba1)–positive cells are infiltrating monocytes from the peripheral circulation, which show high levels of CD45, or part of a resident microglial population that express CD45 at low levels. We found that CUS increased the proportion of Iba1+C3aR+ cells expressing CD45hi in PFC (Fig 4F,G).
If C3aR mediates the recruitment of monocytes into PFC of CUS-exposed mice, then in the absence of C3aR we would expect monocyte infiltration to be significantly reduced. Therefore, we examined monocyte migration into PFC of C3aR KO mice following CUS. We found that in CUS exposed C3aR KO mice, the number of Iba1+CD45hi cells (i.e., infiltrating monocytes) in PFC was significantly reduced compared with CUS exposed WT mice (Fig 4H; two-way ANOVA, stress, F(1,12) = 63.74, p < 0.001; genotype, F(1,12) = 40.38, p < 0.001; stress × genotype, F(1,12) = 22.33, p < 0.001; post hoc, NS-WT vs CUS-WT, p < 0.001, NS-C3aR KO vs CUS-C3aR KO, p > 0.05). Also, we found a significant increase in the number of resident microglial cells (Iba1+CD45lo) in PFC following CUS (Fig 4I; two-way ANOVA, stress, F(1,12) = 33.32, p < 0.001; post hoc, NS-WT vs CUS-WT, p < 0.001, NS-C3aR KO vs CUS-C3aR KO, p > 0.05). However, C3aR deficiency did not result in any significant change in the number of Iba1+CD45lo cells in PFC following CUS. These results indicate that C3aR+ monocytes infiltrate the PFC following CUS and demonstrate that CUS-induced infiltration of monocytes is C3aR-dependent.
3.6. C3aR deficiency reduces CUS-induced increase in VCAM-1, ICAM-1 and IL-1β levels
We examined the protein levels of two key monocyte cell adhesion molecules, VCAM-1 and ICAM-1 which have been well implicated in monocyte infiltration into brain following stress30. We found a significant increase in the short isoform of VCAM-1 (~50 KDa) in the PFC of WT mice following CUS [Fig 5A,B; two-way ANOVA, stress, F(1,10) = 45.07, p < 0.001; genotype, F(1,10) = 46.56, p < 0.001; stress × genotype, F(1,10) = 70.12, p < 0.001; post hoc, NS-WT vs CUS-WT, p < 0.001, NS-C3aR KO vs CUS-C3aR KO, p > 0.05]. However, the increase of VCAM-1 was attenuated in the C3aR KO mice exposed to CUS compared with WT mice subjected to CUS. Similarly, C3aR deficiency significantly attenuated CUS-induced increase in ICAM-1 protein levels in mouse PFC [Fig 5A,C; two-way ANOVA, stress, F(1,10) = 8.21, p < 0.05; genotype, F(1,10) = 27.28, p < 0.001; stress × genotype, F(1,10) = 39.67, p < 0.001; post hoc, NS-WT vs CUS-WT, p < 0.001, NS-C3aR KO vs CUS-C3aR KO, p > 0.05]. No change in the levels of long isoform of VCAM-1 (~100 KDa) was found in any of the treatment groups (Fig 5). To further understand the role of C3aR+ monocytes in CUS-induced inflammation, we investigated the expression of IL-1β, a key inflammatory mediator in chronic stress conditions in WT and C3aR KO mice at 3 weeks following CUS, when C3aR+ monocytes have infiltrated PFC. Significant increases in IL-1β mRNA [Fig 5D; two-way ANOVA, stress × genotype, F(1,11) = 12.24, p < 0.05; post hoc, NS-WT vs CUS-WT, p < 0.05, NS-C3aR KO vs CUS-C3aR KO, p > 0.05] and protein [Fig 5E; two-way ANOVA, stress, F(1,8) = 8.49, p < 0.01; genotype, F(1,8) = 22.61, p < 0.001; post hoc, NS-WT vs CUS-WT, p < 0.05, NS-C3aR KO vs CUS-C3aR KO, p > 0.05] levels were found in PFC of CUS-exposed WT mice. However, the induction of IL-1β was attenuated in the C3aR KO mice exposed to CUS compared with WT mice subjected to CUS (Fig. 5D–E).
Figure 5. C3aR deficiency attenuates CUS-induced increase in VCAM-1, ICAM-1 and IL-1β levels in PFC.

PFC tissues from C3aR knockout and WT mice exposed to CUS or no stress (NS) were analyzed for VCAM-1, ICAM-1 and IL-1β levels. (a) Representative immunoblot data showing VCAM-1, ICAM-1 and β-actin expression in the PFC. (b) VCAM-1 (short isoform) and (c) ICAM-1 protein levels normalized to β-actin. (d) IL-1β mRNA in the PFC. The Ct values were normalized to RPS3. (e) IL-1β protein levels were determined by ELISA. Data are expressed as mean ± s.e.m. *P < 0.05 vs NS-WT; #P < 0.05 CUS-WT; Two-way ANOVA. N=3–5 per group.
3.7. Monocyte/macrophage cell depletion attenuates CUS-induced depressive-like behavior
Trafficking of monocytes/macrophages into the CNS is a pathological feature of stress-induced behavioral changes in rodent models31. To test whether the infiltration of monocyte-macrophages into PFC contributes to the development of CUS-induced depressive-like behavior, we depleted mice of these myeloid cells by three administrations of liposome-encapsulated clodronate (Clodrosome) at 7 days apart during the CUS paradigm. Administration of Clodrosome significantly reduced the number of circulating CD11b+ F4/80+ myeloid cells (Fig. 6A) compared with Encapsome-treated (CON) mice. CUS significantly increased the number of Iba1+/C3aR+/CD45hi cells in PFC in Encapsome-treated mice. However, Clodrosome administration completely inhibited the CUS-induced increase in Iba1+/C3aR+/CD45hi cells in PFC (Fig 6B,C; two-way ANOVA, stress, F(1,13) = 130.3, p < 0.001; treatment, F(1,13) = 33.28, p < 0.001; stress × treatment, F(1,13) = 74.89, p < 0.001; post hoc, NS-CON vs CUS-CON, p < 0.001, NS-CON vs CUS-CL, p > 0.05). Clodrosome administration significantly attenuated CUS-induced despair behavior in TST (Fig 6D; two-way ANOVA, stress, F(1,20) = 6.46, p < 0.05; post hoc, NS-CON vs CUS-CON, p < 0.01, NS-CON vs CUS-CL, p > 0.05) and FST (Fig 6E; two-way ANOVA, stress, F(1,20) = 11.38, p < 0.01; treatment, F(1,20) = 31.09, p < 0.001; stress × treatment, F(1,20) = 25.69, p < 0.001; post hoc, NS-CON vs CUS-CON, p < 0.001, NS-CON vs CUS-CL, p > 0.05), and anhedonia-like behavior in SPT (Fig 6F; two-way ANOVA, stress, F(1,18) = 8.67, p < 0.01; treatment, F(1,18) = 4.82, p < 0.05; post hoc, NS-CON vs CUS-CON, p < 0.01, NS-CON vs CUS-CL, p > 0.05) as compared to CUS-exposed Encapsome-treated mice. No change in the locomotor activity was observed in the open field test between groups (Fig 6G). These results suggest that infiltrating monocyte-macrophages play a critical role in CUS-induced depressive-like behavior.
Figure 6. Monocyte/macrophage cell depletion attenuates CUS-induced depressive-like behavior.
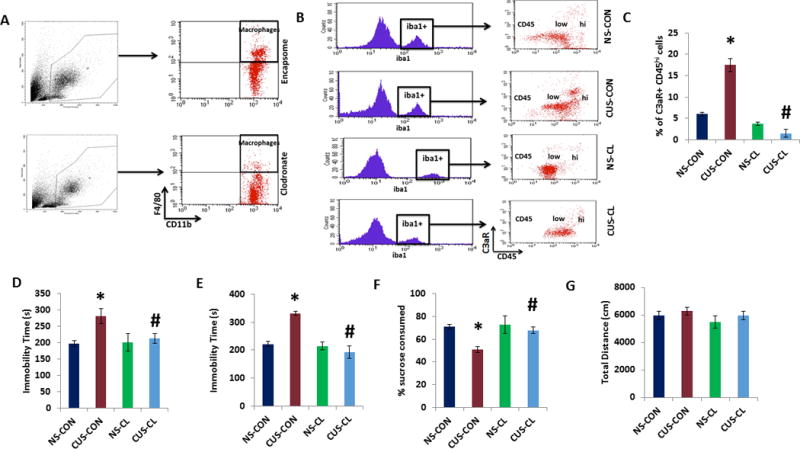
(a) Adult male mice were treated with three administrations of liposome-encapsulated clodronate (Clodrosome; CL) or Encapsome (control; CON) at 7 days apart during the CUS paradigm of 21 days. At the end of the CUS procedure, mice were tested for depressive-like behavior, and PFC samples were collected for flow cytometry analysis. (a) Myeloid cells (CD11b+F4/80+) cells quantified by flow cytometry. (b) Iba1+/C3aR+ cells were analyzed using CD45 to differentiate infiltrating monocytes (CD45hi) from resident microglia (CD45lo). (c) % of Iba1+ cells expressing C3aR and CD45hi in PFC. Data are expressed as mean ± s.e.m. *P < 0.05 vs CON; #P < 0.05 vs CON+CUS; two-way ANOVA. N=4 per group. (d) Tail suspension test, (e) forced swim test, (f) preference for sucrose measured in sucrose preference test, and (g) open field test. Data are expressed as mean ± s.e.m. *P < 0.05 vs NS-CON; #P < 0.05 vs CUS-CON; two-way ANOVA. N=8 per group.
4. Discussion
Our results demonstrate that increased C3 signaling induces depressive-like behavior in the TST and FST, and is sufficient to cause anhedonic behavior in the absence of chronic stress. Conversely, inhibition of C3 signaling using C3 or C3aR KO mice blocks the depressive-like behavior caused by CUS exposure, suggesting resilience to chronic stress. Moreover, C3aR deficiency was sufficient to attenuate stress-induced infiltration of monocytes and IL-1β mRNA levels in PFC. Together, these findings suggest that altered C3 signaling is a potential molecular mechanism underlying the pathology of MDD.
In the brain, C3 may have a role in non-immune neuronal functions23,32,33,34. More specifically, studies in C3 knockout mice suggest C3’s involvement in the induction of behavioral deficits and regulation of synapses. C3 KO mice demonstrate resiliency towards anxiety and protection against age-related cognitive declines in learning and special memory35,36, as well as an excessive number of synapses37,38 and increased synaptic efficacy in response to a high-frequency burst46. C3 signaling through C3aR has been shown to regulate synaptic function and dendritic morphology22. C3a, the cleaved form of C3, has been implicated in recruiting microglia to synaptically enriched regions39. Thus, increased function of the C3 signaling pathway could contribute to chronic stress-induced loss of synapses and impairments in synaptic connectivity in PFC40,41,42. Our data on the increased expression of C3 in postmortem PFC of depressed subjects support this possibility and indicate the possible role of C3 in the loss of synapses seen in depression43. The increase in C3 expression following chronic stress conditions and attenuation of stress-induced depressive behavior in C3 KO mice as well as in C3aR KO mice further substantiates the role of C3 signaling in depressive-behavior.
The PFC is a highly vulnerable region in response to stress, undergoing structural, neurochemical and functional alterations that determine the deficits in PFC-mediated behaviors44. The increase in Iba1+/CD45hi cells in the PFC after CUS indicates the recruitment of the infiltrating monocytes. Mice lacking C3aR exhibited complete prevention of the recruitment of monocytes into PFC, indicating that the reduced number of infiltrating monocytes is attributed mainly to the absence of C3aR+ monocyte infiltration. These findings suggest that complement component links the central and peripheral immune systems. Indeed, C3a has been shown to enhance recruitment of peripheral immune cells to the brain leading to increased neuroinflammation45. Moreover, C3 KO and C3aR KO mice demonstrated a reduced amount of infiltrating peripheral cells in the brain compared to wild-type mice46. The reduction was attributed to decreased brain endothelial expression of vascular adhesion factors such as VCAM-1 and ICAM-1, which have also been shown as necessary for increased monocyte recruitment during social defeat stress30,46. Our data on VCAM-1 and ICAM-1 further suggest the roles of these molecules in monocyte recruitment into PFC following chronic stress. It is known that the trafficking and recruitment of monocytes into brain depends on chemokine receptors. In a mouse model of repeated social defeat (RSD) stress, CCR2KO and CX3CR1KO mice did not exhibit increased brain macrophages following RSD47. Moreover, RSD increased CCL2 expression in the brain47. Also, CCR2 knockout mice showed reduced monocyte recruitment into brain and reduced levels of the proinflammatory cytokine IL-1β in hippocampus after status epilepticus48. Although C3aR signaling has been shown to induce IL-1β, the role of chemokines and their receptors in C3aR-mediated monocyte infiltration and depressive-like behavior following chronic stress are not known, which needs further investigation.
Equally as important as the link between complement and monocytes is the link between complement and microglia. Increased activation of microglia has been found in postmortem brain tissues from depressed and suicide subjects49,50, and increased microglial activity in PFC was found to be positively correlated with the severity of depression51. In rodents, chronic stress has been shown to increase microglial activation in PFC of mice susceptible to anhedonia52. Microglia are the only native cell type expressing C3aR in the brain53, and are major sources of CNS-derived IL-1β54. A number of studies indicate that the activation of innate immune mechanisms, in particular proinflammatory cytokines IL-1β, IL-6 and tumor necrosis factor alpha (TNF-α) plays an important role in the pathophysiology of depression55. A recent study has found significant increases in IL-1β, IL-6 and TNF-α in the PFC of adult depressed suicide victims56. C3 enhances proinflammatory signaling and increases production of IL-1β57. Our findings on increased C3 mRNA in the PFC of depressed suicide subjects further strengthen the role of innate immune system in depression. In addition, IL-1β inhibition by pharmacological or genetic approach has been shown to attenuate chronic stress-induced depressive behavior in rodents14,58,59. We identified Iba1+ myeloid cells and CD206+ perivascular macrophages as the major cellular sources of C3 after CUS. We found that C3aR deficiency is associated with a significant attenuation of CUS-induced increase in IL-1β in the PFC. Additionally, NLRP3 inflammasome activation mediates IL-1β-related inflammation in the PFC of depressive rats59 and is required for complement-induced IL-1β release57. It is known that chronic stress causes an increase in hippocampal astrocyte release of extracellular ATP, which can activate the inflammasome via the receptor P2X7R60. Notably, the release of ATP connects C3a and the inflammasome; C3a has been found to bind to C3aR on monocytes and induce release of ATP into the extracellular space upon LPS stimulation in vitro57. It is possible that this paradigm is replicated in the brain; however, the question of source would then come into play, as microglia as well as invading peripheral monocytes express C3aR. Our data showing inhibition in CUS-induced increase in both the number of C3aR+ monocytes in PFC and depressive-like behavior in Clodrosome-treated mice suggest that C3aR present in peripheral monocytes may play a critical role in chronic stress-induced depressive-like behavior. It is important to note that monocyte secretion of IL-1β and activation of IL-1R1 on the endothelial surface has been shown as necessary for the development of anxiety behaviors46. Therefore, the source of ATP, the identity of the cells expressing C3aR, and the identity of the effector cells warrant further studies in the context of chronic stress.
C3 is regulated in an NFκB-dependent manner, as the C3 promoter contains two putative κB binding sites22. Chronic stress has been shown to induce the activation of NF-κB signaling pathway, which subsequently exerts antineurogenic and anhedonic effects15. In addition, NF-kB signaling induces expression of the NLRP3 inflammasome61. C3 cleavage can either lead to formation of the downstream MAC, or allow C3a to bind to monocytes to cause ATP release, both of which activate the inflammasome62.
In conclusion, our data from human postmortem brain samples and animal studies demonstrate an important role for C3 in mediating depressive behaviors. The high number of covariates in a relatively small cohort of human samples is a limitation in our study. Additional studies using more number of subjects with/without antidepressant medication would be of great interest. It is also important to determine whether CUS induces monocyte infiltration to brain regions other than PFC such as hippocampus and amygdala which are also implicated in depressive-like behavior. Moreover, it is likely that C3 as well as C3aR KO mice show alterations in peripheral monocytes that could impact on immune-to-brain communication following chronic stress and warrants further investigation.
Supplementary Material
Highlights.
Complement component 3 (C3) mediates chronic stress-induced depressive-like behavior in mice.
C3a receptor (C3aR) knockout mice were resilient to stress-induced depressive-like behavior.
C3aR knockout mice displayed significantly reduced monocyte recruitment into PFC after stress.
Myeloid cell depletion attenuated stress-induced C3aR+ monocyte infiltration into PFC.
Acknowledgments
The authors would like to acknowledge Quebec Suicide Brain Bank for postmortem tissue samples. The authors acknowledge the grant support from US National Institute of Mental Health grant R01 MH 097060 (A.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors’ contributions
A.P contributed to study design, data collection, analysis and writing. A.C., T.F., C.D.P., T.D., and A.N. contributed to data collection and analysis. B.B performed flow cytometry analysis. A.O.A contributed to statistical analysis. G.T provided the human postmortem tissues.
Biomedical Financial Interests and Potential Conflicts of Interest: All authors report no relevant biomedical financial interests or potential conflicts of interest.
References
- 1.Capuron L, Dantzer R. Cytokines and depression: the need for a new paradigm. Brain Behavior and Immunity. 2003;17:S119–24. doi: 10.1016/s0889-1591(02)00078-8. [DOI] [PubMed] [Google Scholar]
- 2.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nature Reviews in Neuroscience. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biological Psychiatry. 2009;65:732–41. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nature Reviews Immunology. 2016;16:22–34. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandey GN. Inflammatory and Innate Immune Markers of Neuroprogression in Depressed and Teenage Suicide Brain. Modern Trends in Pharmacopsychiatry. 2017;31:79–95. doi: 10.1159/000470809. [DOI] [PubMed] [Google Scholar]
- 6.Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. Journal of Neuroscience. 2006;26:7870–4. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmes A, Wellman CL. Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neuroscience Biobehavior Reviews. 2009;33(6):773–83. doi: 10.1016/j.neubiorev.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bondi CO, Rodriguez G, Gould GG, Frazer A, Morilak DA. Chronic unpredictable stress induces a cognitive deficit and anxiety-like behavior in rats that is prevented by chronic antidepressant drug treatment. Neuropsychopharmacology. 2008;33:320–31. doi: 10.1038/sj.npp.1301410. [DOI] [PubMed] [Google Scholar]
- 9.Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. Journal of Neuroscience. 2007;27:2781–7. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koenigs M, Grafman J. The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behavioral Brain Research. 2009;201:239–43. doi: 10.1016/j.bbr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tramullas M, Finger BC, Moloney RD, Golubeva AV, Moloney G, Dinan TG, et al. Toll-like receptor 4 regulates chronic stress-induced visceral pain in mice. Biological Psychiatry. 2014;76:340–8. doi: 10.1016/j.biopsych.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Dunn AJ, Swiergiel AH, de Beaurepaire R. Cytokines as mediators of depression: what can we learn from animal studies? Neuroscience Biobehavior Reviews. 2005;29:891–909. doi: 10.1016/j.neubiorev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 13.Felger JC, Alagbe O, Hu F, Mook D, Freeman AA, Sanchez MM, et al. Effects of interferon-alpha on rhesus monkeys: a nonhuman primate model of cytokine-induced depression. Biological Psychiatry. 2007;62:1324–33. doi: 10.1016/j.biopsych.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:751–6. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2669–74. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain A, Pasare C. Innate Control of Adaptive Immunity: Beyond the Three-Signal Paradigm. Journal of Immunology. 2017;198:3791–3800. doi: 10.4049/jimmunol.1602000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Research. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 18.Sompayrac l: How the immune system works. 5. New York: Wiley; 2006. [Google Scholar]
- 19.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system. I. Molecular mecnisms of activation and regulation. Frontiers in Immunology. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. Journal of Immunology. 2015;194:3542–8. doi: 10.4049/jimmunol.1403068. [DOI] [PubMed] [Google Scholar]
- 21.Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, Reiner O. Developmental activities of the complement pathway in migrating neurons. Nature Communications. 2017;8:15096. doi: 10.1038/ncomms15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 2015;85:101–15. doi: 10.1016/j.neuron.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klegeris A, Bissonnette CJ, Dorovini-Zis K, McGeer PL. Expression of complement messenger RNAs by human endothelial cells. Brain Research. 2000;871:1–6. doi: 10.1016/s0006-8993(00)02253-8. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, Wetsel RA, Miwa T, Song WC. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–36. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fagan K, Crider A, Ahmed AO, Pillai A. Complement C3 Expression Is Decreased in Autism Spectrum Disorder Subjects and Contributes to Behavioral Deficits in Rodents. Molecular Neuropsychiatry. 2017;3:19–27. doi: 10.1159/000465523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandya CD, Hoda N, Crider A, Peter D, Kutiyanawalla A, Kumar S, et al. Transglutaminase 2 overexpression induces depressive-like behavior and impaired TrkB signaling in mice. Molecular Psychiatry. 2017;22:745–753. doi: 10.1038/mp.2016.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braun M, Vaibhav K, Saad N, Fatima S, Brann DW, Vender JR, et al. Activation of Myeloid TLR4 Mediates T Lymphocyte Polarization after Traumatic Brain Injury. Journal of Immunology. 2017;198:3615–3626. doi: 10.4049/jimmunol.1601948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007 Jul;55(7):687–700. doi: 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- 29.Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia. 2005 Feb;49(3):375–84. doi: 10.1002/glia.20124. [DOI] [PubMed] [Google Scholar]
- 30.Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci. 2015 Jan;8:21. 447. doi: 10.3389/fnins.2014.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez K, Fornaguera-Trías J, Sheridan JF. Stress-Induced Microglia Activation and Monocyte Trafficking to the Brain Underlie the Development of Anxiety and Depression. Current Topics in Behavioral Neuroscience. 2017;31:155–172. doi: 10.1007/7854_2016_25. [DOI] [PubMed] [Google Scholar]
- 32.Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Molecular Immunology. 2011;48:1592–603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM. The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Medicine. 2010;12:179–92. doi: 10.1007/s12017-009-8085-y. [DOI] [PubMed] [Google Scholar]
- 34.Barnum SR. Complement biosynthesis in the central nervous system. Critical Reviews in Oral Biology Medicine. 1995;6:132–46. doi: 10.1177/10454411950060020301. [DOI] [PubMed] [Google Scholar]
- 35.Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. Journal of Neuroscience. 2015;35(38):13029–42. doi: 10.1523/JNEUROSCI.1698-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Alcazar M, Daborg J, Stokowska A, Wasling P, Bjorefeldt A, Kalm M, et al. Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Experimental neurology. 2014;253:154–64. doi: 10.1016/j.expneurol.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 37.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 38.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009 Sep;46(14):2753–66. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004 Aug;60(2):236–48. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- 41.Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, McEwen BS, Morrison JH. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cerebral Cortex. 2006;16:313–20. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- 42.Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. Journal of Neurobiology. 2001;49:245–53. doi: 10.1002/neu.1079. [DOI] [PubMed] [Google Scholar]
- 43.Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Progress in Neuropsychopharmacology and Biological Psychiatry. 2011;35:1774–9. doi: 10.1016/j.pnpbp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu F, Zou Q, Ding X, Shi D, Zhu X, Hu W, et al. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. Journal of Neuroinflammation. 2016;13:23. doi: 10.1186/s12974-016-0485-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Molecular Psychiatry. 2017 Apr 4; doi: 10.1038/mp.2017.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013 Aug;33(34):21. 13820–33. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varvel NH, Neher JJ, Bosch A, Wang W, Ransohoff RM, Miller RJ, Dingledine R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A. 2016 Sep;113(38):20. E5665–74. doi: 10.1073/pnas.1604263113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? Journal of Neuroinflammation. 2011;8:94. doi: 10.1186/1742-2094-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. Journal of Psychiatric Research. 2008;42:151–7. doi: 10.1016/j.jpsychires.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 51.Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 2015;72:268–75. doi: 10.1001/jamapsychiatry.2014.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Couch Y, Anthony DC, Dolgov O, Revischin A, Festoff B, Santos AI, et al. Microglial activation, increased TNF and SERT expression in the prefrontal cortex define stress-altered behaviour in mice susceptible to anhedonia. Brain, behavior, and immunity. 2013;29:136–46. doi: 10.1016/j.bbi.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 53.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annual review of neuroscience. 2012;35:369–89. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 54.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. Journal of Clinical Investigation. 2012;122:1164–71. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haapakoski R, Ebmeier KP, Alenius H, Kivimäki M. Innate and adaptive immunity in the development of depression: An update on current knowledge and technological advances. Prog Neuropsychopharmacol Biol Psychiatry. 2016 Apr;66:3. 63–72. doi: 10.1016/j.pnpbp.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pandey GN. Inflammatory and Innate Immune Markers of Neuroprogression in Depressed and Teenage Suicide Brain. Mod Trends Pharmacopsychiatry. 2017;31:79–95. doi: 10.1159/000470809. Epub 2017 Jul 24. [DOI] [PubMed] [Google Scholar]
- 57.Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013;122:3473–81. doi: 10.1182/blood-2013-05-502229. [DOI] [PubMed] [Google Scholar]
- 58.Koo JW, Duman RS. Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Current Opinion Investigative Drugs. 2009;10:664–71. [PMC free article] [PubMed] [Google Scholar]
- 59.Pan Y, Chen XY, Zhang QY, Kong LD. Microglial NLRP3 inflammasome activation mediates IL-1β-related inflammation in prefrontal cortex of depressive rats. Brain, behavior, and immunity. 2014;41:90–100. doi: 10.1016/j.bbi.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 60.Yue N, Huang H, Zhu X, Han Q, Wang Y, Li B, et al. Activation of P2X7 receptor and NLRP3 inflammasome assembly inhippocampal glial cells mediates chronic stress-induced depressive-likebehaviors. J Neuroinflammation. 2017;14:102. doi: 10.1186/s12974-017-0865-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. Journal of Immunology. 2009;183:787–91. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. Journal of Immunology. 2013;191:1006–10. doi: 10.4049/jimmunol.1300489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.