

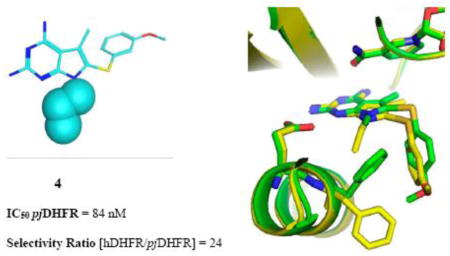
Abstract
To combine the potency of trimetrexate (TMQ) or piritrexim (PTX) with the species selectivity of trimethoprim (TMP), target based design was carried out with the X-ray crystal structure of human dihydrofolate reductase (hDHFR) and the homology model of Pneumocystis jirovecii DHFR (pjDHFR). Using variation of amino acids such as Met33/Phe31 (in pjDHFR/hDHFR) that affect the binding of inhibitors due to their distinct positive or negative steric effect at the active binding site of the inhibitor, we designed a series of substituted-pyrrolo[2,3-d]pyrimidines. The best analogs displayed better potency (IC50) than PTX and high selectivity for pjDHFR versus hDHFR, with 4 exhibiting a selectivity for pjDHFR of 24-fold.
Keywords: DHFR inhibitors, pjDHFR, hDHFR, Pneumocystis pneumonia, opportunistic infections, pyrrolo[2, 3-d]pyrimidines
Graphical Abstract
1. Introduction
Pneumocystis jirovecii (pj) is a fungus that infects the lungs of a majority of humans around the world. However, the immune system in healthy individuals keeps the infection under control. In immunocompromised patients, pj infection causes Pneumocystis pneumonia (PCP).1, 2 PCP can be fatal for patients with HIV/AIDS (most common), patients undergoing chemotherapy for cancer, patients on immunosuppressive medications, patients undergoing organ or bone-marrow transplantation or those who are malnourished.3, 4 PCP presents itself when the patients’ CD4 count is below 200 cells/mm3.5 Although PCP prophylaxis and antiretroviral therapy (ART) have changed the face of the HIV/AIDS epidemic, the incidence of HIV cases persist due to non-adherence to the medication, toxicity to the medications, emergence of drug resistant HIV strains, late diagnosis of HIV and the rise in the number of cases in developing countries.6, 7 Thus PCP continues to be a significant public health concern. In the US, 9% of the hospitalized HIV/AIDS and 1% of organ transplant patients develop PCP infection.8 In these patients, the mortality rate is from 5–40% while being treated for PCP and approaches 100% if left untreated.8
Both the prophylaxis and treatment for PCP involves the combination of trimethoprim (TMP)-sulfamethoxazole (SMX) (co-trimoxazole).9, 10 TMP (Figure 1) is a selective, but weak inhibitor of dihydrofolate reductase (DHFR), the enzyme necessary for the reduction of dihydrofolate to tetrahydrofolate,11 while SMX is an inhibitor of the dihydropteroate synthase (DHPS), the enzyme necessary for the synthesis of folates in fungi.12 The low activity of TMP against DHFR is augmented by SMX, in the treatment regimen. The efficacy, low cost and activity against a variety of infections has propelled co-trimoxazole to be used indiscriminately. Due to the rampant use, mutations in the DHPS locus of P. jirovecii (the fungal species that causes PCP in humans) encoding DHPS have been documented as the cause of TMP/SMX resistant strains of PCP.12–14 Various studies have also reported mutations discovered in pjDHFR after treatment or prophylaxis using DHFR inhibitors.15–19 Treatment failure and discontinuation of co-trimoxazole occurs in several cases due to such resistant strains or toxicity/allergy caused by SMX.20–24 When treatment fails with TMP/SMX, the second-line treatment in mild to moderate PCP is TMP-dapsone or clindamycin-primaquine, which also leads to low efficacy and often lethal side-effects.9, 25–27 Piritrexim (PTX) and trimetrexate (TMQ) are potent, but non-selective inhibitors of pjDHFR, which cause dose-limiting toxicities and have been discontinued.9, 28, 29 For patients that do not respond to first line treatment as well the inevitable appearance of resistance, new drugs for the treatment of PCP are critically needed.
Figure 1.

DHFR inhibitors for treatment for PCP
One of the most efficient strategies to treat PCP infection is to target P. jirovecii DHFR (pjDHFR).30 DHFR catalyzes the reduction of 7,8-dihydrofolate to the 5,6,7,8-tetrahydrofolate. Inhibition of DHFR interferes with folate cofactor requiring transformations including thymidylate and purine biosynthesis and resuls in inhibition of DNA synthesis.11 This inhibition causes a disruption in DNA, RNA and protein synthesis of the organism and eventually leads to death of the fungus. Pneumocystis infection is host specific. Pneumocystis carinii, however, is a distinct species that infects rats, different from P. jirovecii, responsible for human infections. The amino acid sequence of the DHFR of Pneumocystis carinii (pcDHFR) differs by 38% when compared to the DHFR of Pneumocystis jirovecii (pjDHFR).31 Hence, drug’s activity against the surrogate pcDHFR in-vitro may not translate into activity in the treatment of PCP infection in humans caused by P. jirovecii.
We have recently isolated pjDHFR31 and used it to evaluate clinically used agents such as TMP, PTX and novel DHFR inhibitors.32 These studies demonstrated that the inhibition of human(h)DHFR compared with pjDHFR allows the calculation of a selectivity ratio (IC50 hDHFR/IC50 pjDHFR) that provides a measure of the selective inhibition of the agent for pjDHFR over hDHFR. Compounds, such as PTX and TMQ, though highly potent, show poor selectivity for pjDHFR over hDHFR and are much too toxic in vivo; this lack of selectivity is responsible for their discontinuation for the treatment of infections caused by P. jirovecii. The selectivity of TMP however, for pjDHFR over hDHFR is 266-fold and contributes to its clinical success in PCP treatment. Besides the selectivity for pjDHFR another aspect that is highly desirable in an agent is potency for pjDHFR. TMP has a low potency as an inhibitor of pjDHFR and must be used with SMX for clinical efficacy. Our long-term goal is to provide analogs with excellent potency along with high selectivity for pjDHFR. Such agents could be used alone as well as with sulfonamides and other drugs for PCP infections in humans.
Rational design of pjDHFR inhibitors is hampered due to a lack of crystal structure information for pjDHFR. However, homology models can be used with refinement to model pjDHFR in the absence of crystal structures.32 Thus along with known hDHFR X-ray crystal structures,33 pjDHFR homology models can be used to design and predict potent and selective pjDHFR inhibitors. Another significant impediment in the drug discovery of inhibitors of pjDHFR is the inability to grow the organism outside the human lung and hence to develop a tissue culture for in vitro studies or an animal model for in vivo evaluation of the synthesized compounds. Due to this drawback, isolation and use of pjDHFR enzyme is currently the only direct indicator that a compound could be effective (or ineffective) in the treatment of PCP infection in humans.
3. Synthesis
Synthesis of 1–18 utilized a modification of the literature method.34 To a solution of hydroxyacetone 19 and malononitrile in ethanol, triethylamine was added and stirred overnight under argon to afford 20 (Scheme 1). The cyclisation of 20 without purification was carried out with guanidine and sodium methoxide at reflux to obtain 21 (10–35%). To a solution of iodine and the appropriate thiophenol (2:1 ethanol: water), 21 was added and maintained at reflux to afford 1 and 7–12. The pyrrole nitrogen on 1 and 7–12 was methylated using sodium hydride and methyl iodide in DMF to afford 2 and 13–18. For the N7-alkylated series, 1 was alkylated using appropriate alkyl halides to afford 3–6 (Scheme 2). Synthesis and characterization of compounds 1, 7, 8, and 10 has been presented previously by Gangjee et al.34
Scheme 1.

a) malononitrile, TEA, EtOH, rt, 12h; b) NaOMe, guanidine HCl, EtOH, reflux, 24h; c) thiophenol, I2, 2:EtOH:H2O, reflux, 24h; d) CH3I, NaH, DMF, rt, 0.5– 2h
Scheme 2.

a) R-Br, NaH, DMF, rt, 0.5– 2h
2. Design and docking studies
We published the X-ray crystal structure of hDHFR and pcDHFR with several pyrido[2,3-d]pyrimidines.33 In addition, using the published crystal structures for pcDHFR,33 a homology model of pjDHFR32 was refined to include the cofactor, nicotinamide adenine dinucleotide phosphate (NADPH). This refined pjDHFR homology model was utilized to evaluate the docking of the proposed compounds 1–18. The isolation of pjDHFR along with the development of the homology model for pjDHFR provided insight regarding the amino-acid sequence differences between active site of pjDHFR and pcDHFR, as well as that of hDHFR. The superimposition of the active site of pcDHFR and pjDHFR (Figure 2) displays the amino acid differences present in the active sites of the two enzymes. The active sites of pcDHFR has Glu32, Ile33, Ile65, and Phe69, which can affect the binding of the ligands designed. The active site of pjDHFR at the same positions in the active site has Asp32, Met33, Leu65, and Ser69. These amino acids differ in their size and electrostatics and thus would significantly influence the binding of the designed compounds considerably. These amino acid differences highlight the futility of designing and evaluating activity against the surrogate pcDHFR as inhibitors of pjDHFR.
Figure 2.

Superimposition of active sites of pcDHFR and pjDHFR. The blue ribbon and amino acid residues represent the active site of pcDHFR (PDB: 4QJZ, 1.61 Å).33 The pink ribbon and amino acid residues represent the homology model of pjDHFR active site. The ligand N6-methyl-N6-(naphthalen-2-yl)pyrido[2,3-d]pyrimidine-2,4,6-triamine (magenta) was co-crystallized with hDHFR.33
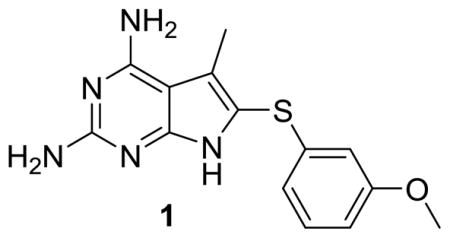
Gangjee et al.35 reportd 6-substituted pyrrolo[2,3-d]pyrimidines as inhibitors of pcDHFR. Reevaluation of compound 1 (Table 1) from this previous study35 in pjDHFR and hDHFR enzymes, indicated a moderate inhibitory potency for pjDHFR and marginal selectivity for pjDHFR over hDHFR. The reevaluation of 1 in isolated pjDHFR provided a lead analog for optimization of both potency and selectivity. We recognized that 1 was overall not as selective or potent as TMP; however, it was an improvement in its selectivity over PTX for pjDHFR and a good starting structure for improvement in both potency and selectivity.
Table 1.
Inhibition Concentrations (IC50) against pjDHFR and hDHFR and Selectivity Ratios
| |||
|---|---|---|---|
| # | pjDHFR (nM) | hDHFR (nM) | Selectivity Ratio [hDHFR/pjDHFR] |
| 1 | 213 | 970 | 5 |
| TMP | 92 | 24500 | 266 |
| PTX | 41 | 2 | 0.05 |
In order to determine the amino acid differences in the active site of pjDHFR and hDHFR, the pjDHFR homology model sequence was superimposed on the hDHFR X-ray crystal published with a pyrido[2,3-d]pyrimidine ligand (Figure 3).32, 33 The active site of hDHFR is partially composed of Glu30, Phe31, Asn64, and Val115. Analogous to these, the active site of pjDHFR is partially composed of the corresponding amino acids Asp32, Met33, Ser69, and Ile123. The side chains of these amino acids are different in shape, size and electronic properties, which allows the design of inhibitors with selectivity and potency for pjDHFR over hDHFR.
Figure 3.

Superimposition of active sites of hDHFR and pjDHFR. The amino acid residues shown are the residues that are different in the active site of the two species. The grey ribbon and amino acid residues co-crystallized with the ligand, N6-methyl-N6-(3,4,5-trifluorophenyl)pyrido[2,3-d]pyrimidine-2,4,6-triamine (magenta), represent the active site of hDHFR (PDB: 4QJC, 1.62 Å).33 The pink ribbon and amino acid residues represent the homology model of pjDHFR active site.
Following the evaluation of 1 in pjDHFR and hDHFR, we conducted docking studies of 1 in the pjDHFR homology model and in the hDHFR crystal structure (PDB: 4QJC, 1.62 Å)33 using the molecular modeling program LeadIt 2.1.636 and the parameters specified in the Experimental Section. Multiple low energy conformations were obtained on docking 1 in the active site of hDHFR and pjDHFR. As a representative example, Figure 4a shows the best docked conformation of 1 in the pjDHFR homology model. It displays a bi-dentate ionic bond between protonated N1 and 2-NH2 of 1 with Asp32. This interaction is most commonly observed in ligands in DHFR crystal structures.37 The 4-NH2 moiety of 1 forms hydrogen bonds with the backbones of Ile10 and the pyrrolo[2,3-d]pyrimidine scaffold is stabilized by pi-stacking interaction with Phe36. The 3′-methoxyphenyl moiety of 1 is oriented in the pocket formed by Leu 25 (not displayed), Met33, Ser64 and Leu65. The 3′-methoxyphenyl oxygen forms a hydrogen bonding interaction with Ser64 in the pocket. This docked pose generated a docking score of −34 kJ/mol. Figure 4b displays the best docked conformation of 1 in hDHFR crystal structure (PDB: 4QJC, 1.61 Å)33. It also exhibits a bi-dentate ionic interaction of the protonated N1 and 2-NH2 with Glu30. The 4-NH2 displayed a hydrogen bonding interaction of with the backbone of Val8 and Val15. The 3′-methoxyphenyl moiety is oriented in the pocket formed by Leu22 (not displayed), Phe31 and Ser59. The scaffold is similarly stabilized by pi-stacking interactions with Phe34. This docked pose generated a docking score of −29 kJ/mol in the hDHFR crystal structure. The docking score comparison between pjDHFR and hDHFR shows a difference of approximately 4 kJ/mol, suggesting selectivity of compound 1 for pjDHFR over hDHFR. This gain in selectivity could be a consequence of the steric clash of the side chain phenyl ring of 1 with Phe31 (in hDHFR) which is absent with Met33 (in pjDHFR). The N7-H of 1 presents itself towards a hydrophobic pocket formed by Phe31 in hDHFR and Met33 in pjDHFR (Figure 4a). We reason that this amino acid variation of Phe31 (in hDHFR) and Met33 (in pjDHFR) in the active sites can be further exploited to obtain selectivity for pjDHFR. Met33 is comparatively more flexible than Phe31 and hence can better accommodate larger inhibitors compared to Phe31. Thus appropriate substitutions on the N7 of the pyrrolo[2,3-d]pyrimidine scaffold of 1 could target this amino acid difference. The predicted distances of the N7 in 1 is approximately 4.89 Å from Met33 in the pjDHFR docked pose and approximately 3.57 Å from Phe31 in the hDHFR docked pose. Thus, a methyl substitution on N7 of 1 could create favorable hydrophobic interactions with Met33 in the pjDHFR active site and an unfavorable steric clash with the Phe31 in the hDHFR active site. To further validate our hypothesis, the N7-methyl analog of 1, 2 was also docked, synthesized, and evaluated.
Figure 4.

Docked pose of 1 (cyan) in (a) homology model of pjDHFR and (b) crystal structure of hDHFR (PDB: 4QJC, 1.62 Å)33.
The introduction of the N7-methyl moiety affords two significant changes in the molecule. First, it increases the hydrophobic interactions in both pjDHFR and hDHFR active sites. The second change is the decrease in the number of low energy conformations possible for 2 within 1 kcal/mol, compared to 1. This is a direct consequence of the further restricted rotation of the 6-aryl moiety due to the presence of the 5,7-dimethyl groups. The number of conformations possible for 1 and 2 were calculated using Sybyl38 and were found to be 122 and 72, respectively. Thus, conformational restriction induced by the N7-methyl group could afford the bioactive conformation or, at least, easier access to the bioactive conformation of 2 in pjDHFR. These two attributes resulting from the addition of the N7-methyl group could be responsible for an increase in potency of 2 over 1. The docking studies of 2 in the pjDHFR homology model and the hDHFR crystal structure (PDB: 4QJC, 1.62 Å);33 displays the interactions as expected (Figure 5a and 5b). The N7-methyl group is indeed oriented towards the hydrophobic pocket in both pjDHFR and hDHFR active sites. The docking scores of 2 in the pjDHFR homology model and the hDHFR crystal structure were −36 kJ/mol and −25 kJ/mol respectively. The difference in the docked scores between pjDHFR and hDHFR, of 11 kJ/mol also predicts an increased selectivity for pjDHFR.
Figure 5.

Docked pose of 2 (cyan) in (a) homology model of pjDHFR and (b) crystal structure of hDHFR (PDB: 4QJC, 1.62 Å)33
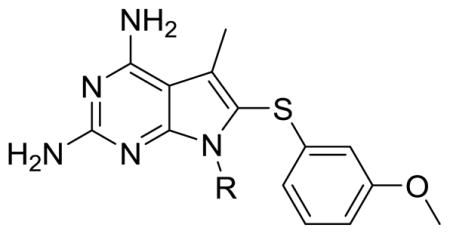
Evaluation of 2 in enzyme assays displayed increased potency towards pjDHFR and a 8-fold selectivity in inhibition of pjDHFR over hDHFR (Table 2). Compared to the activity of 1, the increase in potency towards pjDHFR can be attributed, in part, to the hydrophobic binding of the N7-methyl group in the pocket formed by Met33 and Leu25 (not displayed) and the easier access to the bound conformation, whereas the selectivity increase could be due to a probable steric clash between the N7-methyl group of 2 with Phe31 in hDHFR thus making the binding of 2 less favorable in hDHFR than 1. These evaluations (in vitro IC50) validate our homology model and docking methods. Owing to a large size of the pocket, the N7-methyl did not create a substantial increase in potency and/or selectivity, as expected. Hence it was of interest to synthesize and evaluate longer chain N7-substituetnts of 2, Series 1 (Table 2, compounds 3–6). These longer alkyl chains at the N7 exhibited an increased potency for pjDHFR, but 4 afforded a selectivity of 24-fold for pjDHFR over hDHFR, which was the highest observed for the pyrrolo[2,3-d]pyrimidine series. To structurally explain these results, we performed docking of 4 in the pjDHFR homology model (Figure 6a) and the hDHFR crystal structure (Figure 6b). In the homology model of pjDHFR, the terminus of the propyl chain was at a distance of 3.54 Å from Met33, in pjDHFR. This pose showed an excellent docking score of −36 kJ/mol, and the compound displayed an inhibitory potency IC50 of 74 nM for pjDHFR. The modelling of 4 in hDHFR showed the docked pose as depicted in Figure 6b and the docked score obtained was −24 kJ/mol. The low docking score suggested a less than appropriate fit of 4 in active site of hDHFR. The low score observed, also, reinforces the possibility of a steric clash of the propyl moiety with Phe31 (as observed in Figure 6c), which explains a decreased potency of 4 in the hDHFR. On homologation to a N7-n-butyl and branching to i-propyl, the activity and selectivity against pjDHFR does not increase significantly, indicating that the propyl chain is optimal at N7-position for this series.
Table 2.
Inhibition Concentrations (IC50) against pjDHFR and hDHFR and Selectivity Ratios
| ||||
|---|---|---|---|---|
| # | R | pjDHFR (nM) | hDHFR (nM) | Selectivity Ratio [hDHFR/pjDHFR] |
| 1 | H | 213 | 970 | 5 |
| 2 | CH3 | 160 | 1200 | 8 |
| 3 | CH2CH3 | 35 | 511 | 15 |
| 4 | CH2CH2CH3 | 84 | 2046 | 24 |
| 5 | CH(CH3)2 | 74 | 579 | 8 |
| 6 | CH2CH2CH2CH3 | 73 | 1130 | 15 |
Figure 6.


(a) Docked pose of 4 (cyan) in the homology model of pjDHFR; (b) docked pose of 4 (cyan) in the crystal structure of hDHFR (PDB: 4QJC, 1.62 Å)33 and (c) space-filled representation of Phe31 residue and N7-propyl group in the docked pose of 4 (cyan) in the crystal structure of hDHFR (PDB: 4QJC, 1.62 Å)33 to illustrate the high probability of steric clash.
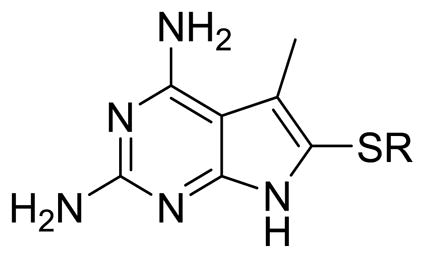
Our efforts at targeting the hydrophobic pocket containing Met33 (in pjDHFR) and Phe31 (in hDHFR) led to 4. It was of interest to study the effect of other amino acid differences within the active site where the side chain aryl group binds. The amino acids at a distance of 4.5 Å around the ligand were studied (Figure 6). The pocket in pjDHFR is composed of Met33, Ser64 and Leu65 and Ile123 and Asp21, Phe31, Ser59 and Val115 in hDHFR. Thus, the active sites have different electronics, shape and size which could affect the binding properties of the pocket. To achieve potency and selectivity by targeting these differing residues, the side-chain aryl substituents with electron withdrawing, electron donating and sterically bulky groups, as replacements for the 3′-methoxyphenyl group were attempted (Table 3, Compounds 7–12). Evaluation of 7–12 led to potent and selective compounds 9 and 12 (Table 3). The 2-napthyl and 4-trifluromethoxyphenyl substitutions showed a 2-fold increase in potency and a 2-fold increase in selectivity, compared to 1. The gain in potencies of 9 and 12 in pjDHFR, compared to 1, could be due to productive shape complementarity of the side chain aryl group and the pocket formed by Met33, Ser64 and Leu65. The gain in selectivity of 9 and 12 for pjDHFR over hDHFR, compared to 2, could be due to the steric clash of the bulkier side chain aryl moiety with Phe31 (in hDHFR), which is absent with Met33 (in pjDHFR). The high probability of a steric hindrance between the bulky side chain aryl group in these compounds with Phe31 in hDHFR is evident in the docking studies of 9 in the crystal structure of hDHFR (PDB: 4QJC, 1.62 Å)33 in Figure 7. The Phe31 in the hDHFR active site limits the movement of the side chain and forces a steric hindrance which decreases the potency of the larger side chain aryl groups, for hDHFR.
Table 3.
Inhibition Concentrations (IC50) against pjDHFR and hDHFR and Selectivity Ratios
| ||||
|---|---|---|---|---|
| # | R | pjDHFR (nM) | hDHFR (nM) | Selectivity Ratio [hDHFR/pjDHFR] |
| 1 | 3′-OCH3Ph | 213 | 970 | 5 |
| 7 | 2′-OCH3Ph | 177 | 624 | 4 |
| 8 | 4′-OCH3Ph | 252 | 1410 | 6 |
| 9 | 2-Naph | 101 | 2100 | 12 |
| 10 | 1-Naph | 167 | 1216 | 7 |
| 11 | 3′,4′-diFPh | 240 | 2318 | 10 |
| 12 | 4′-OCF3Ph | 81 | 811 | 10 |
Figure 7.

Docked pose of 9 (cyan) in the crystal structure of hDHFR (PDB ID: 4QJC)33. The Phe 31 residue is shown in a space fill view to illustrate the high probability of steric clash with the side chain aryl group of 9.
Since N7-methylation of 1 afforded an increase in potency and selectivity, we methylated the N7-position of 7–12 to afford 13–18 (Table 4). The N7-methlyation with varied side chain aryl group did not afford an increase in potency or selectivity and 4 remained the most selective compound in this series.
Table 4.
Inhibition Concentrations (IC50) against pjDHFR and hDHFR and Selectivity Ratios
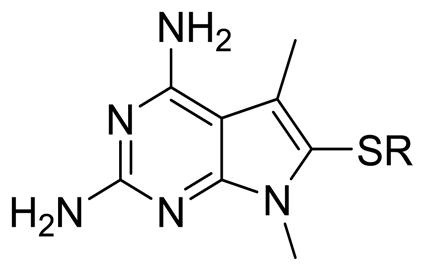
| ||||
|---|---|---|---|---|
| # | R | pjDHFR (nM) | hDHFR (nM) | Selectivity Ratio [hDHFR/pjDHFR] |
| 2 | 3′-OCH3Ph | 160 | 1200 | 8 |
| 13 | 2′-OCH3Ph | 210 | 1400 | 7 |
| 14 | 4′-OCH3Ph | 219 | 1372 | 6 |
| 15 | 2-Naph | 130 | 970 | 7 |
| 16 | 1-Naph | 177 | 1104 | 6 |
| 17 | 3′,4′-diFPh | 247 | 1917 | 8 |
| 18 | 4′-OCF3Ph | 110 | 1101 | 10 |
4. X-ray Crystal Structures (PDB Accession Numbers hDHFR-3 (5HT4); hDHFR-14 (5HT5) for compounds 3 and 14)
Structural data were measured for the ternary complexes of NADPH and native human DHFR with inhibitors 3 (Table 2) and 14 (Table 4), respectively, to validate the binding interactions of these inhibitors in the active site of hDHFR (Figure 8). Compound 3 was selected for its high selectivity and potency in pjDHFR. These data reveal that the presence of the N7-ethyl group of 3 causes the conformation of Phe31 to differ from that observed in the hDHFR complex with 14; Phe31 adopts alternate positions with partial occupancy. It is also interesting to note that the small shift in the binding orientation of inhibitors 3 and 14 allows the 3′-methoxy and the 4′-methoxy of 3 and 14 respectively to occupy similar positions in the binding site.
Figure 8.

(a) Comparison of the crystal structures of human DHFR as a ternary complex with 3 (yellow) and 14 (green) showing the electron density for the complex with hDHFR-3 (2Fo-Fc, 1σ, blue, 3σ, green) and (b) Comparison of the binding pocket for hDHFR-3 (yellow) and 14 (green). Note that Phe31 occupies two alternative conformations in these two structures. This change is in response to the larger N7-ethyl substituent of the inhibitor 3 as compared to N7-methyl substituent of 14.
The overall structures of hDHFR in complex with 3 and 14 are similar to those reported for other hDHFR inhibitor complexes.19, 32, 33 As observed in Figure 8, for 3 and 14, the 3′-methoxy and the 4′-methoxy substituents occupy the same binding pocket. In 3, the amino group of the side chain of Asn64 is within hydrogen bonding distance to the 3′-methoxy oxygen (2.9 Å) and the 4′-methoxy oxygen is within 3.4 Å of the Asn64 amino moiety in 14. The interactions of the N7-methyl substituent of 3 and N7-ethyl substituent of 14 results in Phe31 having two alternate conformations. Analysis of the intermolecular interactions involving the C5-methyl substituent of 3 and 14 shows hydrophobic contacts (4.3 and 4.6 Å, respectively) with the C5 of Val115. The 4-NH2 group of the inhibitors 3 and 14 form a hydrogen bond with the backbone carbonyl of Val115 (3.0 and 3.3 Å, respectively). Docking studies of 3 and 14 in the hDHFR crystal structure (PDB ID: 4QJC)33 afforded poses which mimic the conformation obtained from the crystal structures of hDHFR as a ternary complex with 3 and 14 (Figure 8). This further validates our docking protocols.
5. Summary
The X-ray crystal structures of 3 and 14 in hDHFR validate our hypothesis that bulk at the N7-position of the pyrrolo[2,3-d]pyrimidine scaffold results in a steric clash with Phe31. We have successfully designed, synthesized and evaluated novel series of analogs to explore active site amino acid residue differences in hDHFR and pjDHFR enzymes in our attempt to afford selective inhibitors of pjDHFR over hDHFR. This effort led to several compounds (3–6 and 12) exhibiting potency greater than TMP (92 nM) and selectivity greater than PTX (0.05-fold). The docking studies and crystal structures reveal the importance of targeting the differences in amino acid residues in the active site of pjDHFR and hDHFR. These predictions from the docking studies and the X-ray crystal studies were corroborated by the biological evaluation results. Compound 4 afforded the best selectivity for pjDHFR over hDHFR (24 fold) with a potency of 84 nM for pjDHFR. This suggested that the optimum bulk at the N7-position of the pyrrolo[3,2-d]pyrimidine scaffold, that can be tolerated for increased binding to pjDHFR active site and causing steric clash with hDHFR active site, is equivalent to a propyl group. Compound 4 maintained the potency equivalent to PTX, but exhibited a 48-fold increase in selectivity for pjDHFR over hDHFR. Utilizing the information provided in this study has allowed the design and synthesis of potentially more potent and selective compounds that are currently underway and will be the subject of future publications.
6. Experimental Section
6.1 Synthesis
All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo (0.2 mm Hg) in a CHEM-DRY drying apparatus over P2O5 at 70 °C. Melting points were determined on a MEL-TEMP II melting point apparatus with FLUKE 51 K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for proton (1H NMR) was recorded on a Bruker 400/500 MHz NMR spectrometer. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as an internal standard: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad singlet. The relative integrals of peak areas agreed with those expected for the assigned structures. High-resolution mass spectra (HRMS) were recorded on a MICROMASS AUTOSPEC (EBE Geometry) double focusing mass spectrometer (Electron Impact – EI) or Waters Q-TOF (quadrupole/time-of-flight tandem instrument) mass spectrometer (Electro-Spray Ionization – ESI). Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Element compositions are within 0.4% of the calculated values. Fractional moles of water or organic solvents frequently found in some analytical samples of antifolates could not be prevented in spite of 24–48 h of drying in vacuo and were confirmed where possible by their presence in the 1H NMR spectra. Thin-layer chromatography (TLC) was performed on WHATMAN UV254 silica gel plates with a fluorescent indicator, and the spots were visualized under 254 and/or 365 nm illumination. Proportions of solvents used for TLC are by volume. Column chromatography was performed on a 230–400 mesh silica gel purchased from Fisher Scientific. All solvents and chemicals were purchased from Sigma-Aldrich Chemical Co. or Fisher Scientific.
Procedure for the Synthesis of Compounds 1, 7–12
2-Amino-4-methyl-furan-3-carbonitrile (20)
To a solution of acetol (10 g, 135 mmol) in methanol (200mL) at room temperature was added malononitrile (8.9 g, 135 mmol) and triethylamine (13.7 g, 135 mmol). The resulting mixture was stirred at room temperature overnight. The reaction mixture was then stripped of solvent in vacuo. The residue was washed with hexane-ethyl acetate (5:1) (250mL × 5). The resulting hexane-ethyl acetate solution of the product was collected. After the evaporation of solvent under reduced pressure, 13 g (79%) of the crude product was obtained as an orange powder and was used directly in the next reaction without analysis.
2,4-Diamino-5-methyl-pyrrolo[2,3-d]pyrimidine (21)
To a solution of guanidine free base (from 82 mmol of NaOMe) in anhydrous ethanol (150 mL) was added aminonitrile 20 (10.0 g, 82 mmol). The mixture was refluxed for 24 h, cooled, and filtered. The filtrate was evaporated in vacuo, and the residue was chromatographed on silica gel with 10% MeOH/CHCl3 as the eluent. Fractions containing the product were combined and evaporated to give 24 (7.3g, 55%) as a light brown solid; TLC Rf 0.63 (MeOH/ CHCl3/NH4OH, 1:5:0.5); mp, 166–168 °C. 1H NMR (400 Hz) (Me2SO-d6) δ 2.23 (s, 3 H, 5-CH3), 5.25–5.78 (br, 2 H, 2-NH2, exch.), 6.19 (s, 2 H, 4-NH2, exch.), 6.42 (s, 1 H, 6-H), 10.43 (s, 1 H, 7-H, exch.).
5-methyl-6-(naphthalen-2-ylthio)thieno[2,3-d]pyrimidine-2,4-diamine (9)
Compound 9 (0.32 g, 21%) was obtained from 21 (0.8 g, 4.4 mmol), 2-thionapthalene (1.42 g, 8.8 mmol), and iodine (2.25 g, 8.8 mmol); TLC Rf 0.50 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 191.8–193.5°C. 1H NMR (400 Hz) (Me2SO-d6) δ 2.09 (s, 3H, 5-CH3), 5.66 (s, 2 H, 2-NH2, exch.), 6.32 (s, 2 H, 4-NH2, exch.), 7.17 (dd, 1H, C6H4, J=1.8Hz, J=8.7Hz), 7.45 (m, 3H, C6H4), 7.76 (d, 1H, J=8.3Hz), 7.82 (s, 1H, C6H4), 7.84 (m, 1H, C6H4), 11.05 (s, 1 H, 7-H, exch.). Anal. Calcd. for C17H15N5S: C, 63.53; H, 4.70; N, 21.79; S, 9.98. Found: C, 62.22; H, 4.86; N, 20.99; S, 9.60.
6-((3,4-difluorophenyl)thio)-5-methylthieno[2,3-d]pyrimidine-2,4-diamine (11)
Compound 11 (0.35 g, 24.3%) was obtained from 21 (0.8 g, 4.4 mmol), 3,4-difluorothiophenol (1.30 g, 8.8 mmol), and iodine (2.25 g, 8.8 mmol); TLC Rf 0.50 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 291.7–295.8 °C. 1H NMR (500 Hz) (Me2SO-d6) δ 2.33 (s, 3H, 5-CH3), 5.65 (s, 2 H, 2-NH2, exch.), 6.28 (s, 2 H, 4-NH2, exch.), 6.81 (d, 1 H, C6H4, J=8.7Hz), 7.06 (d, 1 H, C6H4, J=19.2Hz), 7.37 (dd, 1 H, C6H4, J=8.6Hz, J=19.2Hz), 10.99 (s, 1 H, 7-H, exch.). Anal. Calcd. for C13H11F2N4S 0.34 CH3OH: C, 50.87; H, 4.05; F, 11.76; N, 21.68; S, 9.93. Found: C, 50.97; H, 3.88; F, 11.55; N, 21.65; S, 10.06.
6-((4-trifluromethoxyphenyl)thio)-5-methylthieno[2,3-d]pyrimidine-2,4-diamine (12)
Compound 12 (0.38 g, 28%) was obtained from 21 (0.8 g, 4.4 mmol), 4-trifluromethoxythiophenol (1.26 g, 8.8 mmol), and iodine (2.25 g, 8.8 mmol); TLC Rf 0.50 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 291.7–295.8 °C. 1H NMR (500 Hz) (Me2SO-d6) δ 2.33 (s, 3 H, 5-CH3), 5.66 (s, 2 H, 2-NH2, exch.), 6.28 (s, 2 H, 4-NH2, exch.), 7.08 (d, 2 H, C6H4, J=8.9 Hz), 7.30 (d, 2 H, C6H4, J= 8.1 Hz), 10.98 (s, 1 H, 7-H, exch.). Anal. Calcd. for C14H12F3N5OS: C, 47.32; H, 3.40; F, 16.04; N, 19.71; O, 4.50; S, 9.02. Found: C, 47.13; H, 3.50; F, 15.99; N, 19.57; S, 8.91
General Procedure for the Synthesis of Compounds 2–6, 13–18
Mixture of 6-substituted pyrrolo[2,3-d]pyrimidine and sodium hydride was added to a three neck RBF. The RBF was made anhydrous using argon gas balloon. To this mixture, anhydrous DMF (10 mL) was added and stirred for 30 minutes with vigorous stirring. Subsequently, appropriate alkyl halide was injected in the reaction mixture, and the resulting reaction mixture was stirred and monitored by TLC until reaction was completed. The DMF was evaporated under vacuum and silica plug was prepared. The final compound was purified by flash chromatography using methanol-chloroform gradient elution.
6-((3-methoxyphenyl)thio)-5,7-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (2)
Reaction of 1 (0.150 g, 0.32 mmol), sodium hydride (0.012g, 0.5 mmol) and iodomethane (31 mmL, 0.5 mmol) using the general procedure described above gave 2 (0.120 g, 76.44%) as white solid; TLC Rf 0.58 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 277.4–279.4 °C; 1H NMR (400 Hz) (Me2SO-d6) δ 2.38 (s, 3 H, 5-CH3), 3.37 (s, 3 H, 7-CH3), 3.69 (s, 3 H, 3-OCH3), 5.81 (s, 1.68 H, 2-NH2, exch.), 6.37 (s, 1.58 H, 4-NH2, exch.), 6.49 (dd, 2 H, C6H4, J=1.76, 10.55 Hz), 6.73 (dd, 1 H, C6H4, J=2.35, 8.20 Hz), 7.19 (t, 1 H, C6H4, J=7.96, 7.96 Hz). HRMS (ESI) calculated for C15H17N5OS [M+H]+, 316.12266. Found: 316.12198. Found: 316.12198. HPLC analysis: retention time, 22.79 min; peak area, 96.08%; eluent A, H2O: eluent B, ACN; gradient elution (100% H2O to 10% H2O) over 60 min with flow rate of 0.5 mL/min and detection at 245 nm; column temperature, rt
7-ethyl-6-((3-methoxyphenyl)thio)-5-methyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (3)
Reaction of 1 (0.150 g, 0.50 mmol), sodium hydride (0.012g, 0.5 mmol) and bromoethane (53 mmL, 0.5 mmol) using the general procedure described above gave 3 (0.095 g, 60.5%) as white solid; TLC Rf 0.58 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 136.9–139.4 °C; 1H NMR (400 Hz) (Me2SO-d6) δ 1.04 (t, 3 H, J = 7.0 Hz, -CH3), 2.37 (s, 3 H, 5-CH3), 3.68 (s, 3 H, 3-OCH3), 3.92 (q, 2 H, J = 7.0 Hz, -CH2-), 5.80 (s, 2 H, 2-NH2, exch.), 6.35 (s, 2 H, 4-NH2, exch.), 6.49 (d, 2 H, J = 7.2 Hz, C6H4), 6.72 (dd, 1 H, C6H4, J = 2.1 Hz, J = 7.2 Hz), 7.19 (t, 1 H, C6H4, J = 8.2 Hz). Anal. Calcd. for C16H19N5OS: C, 58.34; H, 5.81; N, 21.26; O, 4.86; S, 9.73. Found: C, 58.0.; H, 5.97; N, 21.05; S, 9.52
6-((3-methoxyphenyl)thio)-5-methyl-7-propyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (4)
Reaction of 1 (0.120 g, 0.50 mmol), sodium hydride (0.012g, 0.5 mmol) and 1-bromopropane (62 mmL, 0.5 mmol) using the general procedure described above gave 4 (0.050 g, 37%) as white solid; TLC Rf 0.60 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 266.4–268.2 °C; 1H NMR (400 Hz) (Me2SO-d6) δ 0.73 (t, 3H, J=7.4Hz, -CH3), 1.51 (qd, 2H, J=7.2Hz, J=14.5Hz, -CH2-), 2.37 (s, 3 H, 5-CH3), 3.35 (s, 3 H, 3-OCH3), 3.83 (t, 2H, J=14.5Hz, -CH2-), 3.92 (q, 2 H, J = 7.0 Hz, -CH2-), 5.79 (s, 2 H, 2-NH2, exch.), 6.35 (s, 2 H, 4-NH2, exch.), 6.49 (d, 2 H, J = 7.2 Hz, C6H4), 6.72 (dd, 1 H, C6H4, J = 2.1 Hz, J =7.2 Hz), 7.19 (t, 1 H, C6H4, J = 8.2 Hz).). Anal. Calcd. for C17H21N5OS: C, 59.45; H, 6.16; N, 20.39; O, 4.66; S, 9.34. Found: C, 58.72; H, 6.23; N, 19.97; S, 8.98.
7-isopropyl-6-((3-methoxyphenyl)thio)-5-methyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (5)
Reaction of 1 (0.090 g, 0.30 mmol), sodium hydride (0.009g, 0.36 mmol) and 2-romopropane (38 mmL, 0.36 mmol) using the general procedure described above gave 5 (0.060 g, 59%) as white solid; TLC Rf 0.60 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 157.4–160.1 °C; 1H NMR (400 Hz) (Me2SO-d6) δ 1.40 (d, 6H, J=6.7 Hz, -CH3), 2.38 (s, 3 H, 5-CH3), 3.68 (s, 3 H, 3-OCH3), 4.13(m, 1 H, -CH-), 5.80 (s, 2 H, 2-NH2, exch.), 6.49 (s, 2 H, 4-NH2, exch.), 6.49 (d, 2 H, J = 7.2 Hz, C6H4), 6.72 (dd, 1 H, C6H4, J = 2.1 Hz, J = 7.2 Hz), 7.19 (t, 1 H, C6H4, J = 8.2 Hz). Anal. Calcd. for C17H21N5OS: C, 59.45; H, 6.16; N, 20.39; O, 4.66; S, 9.34. Found: C, 59.12; H, 6.08; N, 19.65; S, 8.87
7-butyl-6-((3-methoxyphenyl)thio)-5-methyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (6)
Reaction of 1 (0.100 g, 0.33 mmol), sodium hydride (0.010g, 0.4 mmol) and 1-bromobutane (55 mmL, 0.5 mmol) using the general procedure described above gave 6 (0.065 g, 55%) as white solid; TLC Rf 0.62 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 282.4–284.2 °C; 1H NMR (400 Hz) (Me2SO-d6) δ 0.76 (t, 3H, J=7.3Hz, -CH3), 1.14 (qd, 2H, J=7.2Hz, J=14.6Hz, -CH2-), 1.45 (td, 2 H, J=7.5Hz, J=14.7Hz, -CH2-), 2.38 (s, 3 H, 5-CH3), 3.68 (s, 3 H, 3-OCH3), 3.87 (t, 2 H, J=14.5Hz, -CH2-),3.92 (q, 2 H, J = 7.0 Hz, -CH2-), 5.80 (s, 2 H, 2-NH2, exch.), 6.49 (s, 2 H, 4-NH2, exch.), 6.49 (d, 2 H, J = 7.2 Hz, C6H4), 6.72 (dd, 1 H, C6H4, J = 2.1 Hz, J = 7.2 Hz), 7.19 (t, 1 H, C6H4, J = 8.2 Hz). Anal. Calcd. for C18H23N5OS 0.03 CHCl3: C, 59.98; H, 6.43; N, 19.40; O, 4.48; S, 8.88. Found: C, 59.94; H, 6.25; N, 19.38; S, 8.78.
6-((2-methoxyphenyl)thio)-5,7-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (13)
Reaction of 7 (0.150 g, 0.5 mmol), sodium hydride (0.012g, 0.5 mmol) and iodomethane (31 mmL, 0.5 mmol using the general procedure described above gave 13 (0.1 g, 64%) as a white solid; TLC Rf 0.58 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 208.6–209.6 °C; 1H NMR (400 Hz) (Me2SO-d6) δ 2.34 (s, 3 H, 5-CH3), 3.37 (s, 3 H, 7-CH3), 3.89 (s, 3 H, 2-OCH3), 5.76 (s, 2 H, 2-NH2, exch.), 6.35 (s, 2 H, 4-NH2, exch.), 6.81 (m, 2 H, C6H4), 7.01 (d, 1 H, J=6.7 Hz, C6H4), 7.11 (d, 1 H, J=7.8 Hz, C6H4). HRMS (ESI) calculated for C15H17N5OS [M+H]+, 316.12266. Found: 316.12402. HPLC analysis: retention time, 21.99 min; peak area, 97.37 %; eluent A, H2O: eluent B, ACN; gradient elution (100% H2O to 10% H2O) over 60 min with flow rate of 0.5 mL/min and detection at 245 nm; column temperature, rt.
6-((4-methoxyphenyl)thio)-5,7-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (14)
Reaction of 8 (0.150 g, 0.32 mmol), sodium hydride (0.012g, 0.5 mmol) and iodomethane (31 mmL, 0.5 mmol) using the general procedure described above gave 14 (0.135 g, 86%) as white solid; TLC Rf 0.58 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 266.0–267.8 °C; 1H NMR (400 Hz) (Me2SO-d6) δ 2.38 (s, 3 H, 5-CH3), 3.37 (s, 3 H, 7-CH3), 3.69 (s, 3 H, 3-OCH3), 5.81 (s, 2 H, 2-NH2), 6.37 (s, 2 H, 4-NH2), 6.49 (dd, J=1.76, 10.55 Hz, 2 H, C6H4), 6.73 (dd, J=2.35, 8.20 Hz, 1 H, C6H4), 7.19 (t, J=7.96, 7.96 Hz, 1 H, C6H4). Anal. Calcd. for C15H17N5OS: C, 57.12; H, 5.43; N, 22.21; O, 5.07; S, 10.17. Found: C, 56.90; H, 5.48; N, 21.94; S, 10.01.
5,7-dimethyl-6-(naphthalen-2-ylthio)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (15)
Reaction of 9 (0.18 g, 0.56 mmol), sodium hydride (0.016g, 0.67 mmol) and iodomethane (40 mmL, 0.64 mmol) using the general procedure described above gave 15 (0.11 g, 59%) as white solid; TLC Rf 0.57 (MeOH/CHCl3/NH4OH, 1:5:0.5);); mp, 266.0–267.8 °C 1H NMR (400 Hz) (Me2SO-d6) δ 2.43 (s, 3 H, 5-CH3), 3.40 (s, 3H, 7-CH3), 5.82 (s, 2 H, 2-NH2, exch.), 6.39 (s, 2 H, 4-NH2, exch.), 7.13 (d, 1H, J=8.7Hz), 7.45 (dd, 3 H, J=6.7Hz, J=12.8Hz)), 7.78 (d, 1 H, J=7.8Hz), 7.85 (d, 2 H, J=8.6Hz). Anal. Calcd. for C18H17N5S: C, 64.45; H, 5.11; N, 20.88; S, 9.56. Found: C, 64.21; H, 5.25; N, 20.68; S, 9.29
5,7-dimethyl-6-(naphthalen-1-ylthio)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (16)
Reaction of 10 (0.20 g, 0.62 mmol), sodium hydride (0.017g, 0.75 mmol) and iodomethane (46 mmL, 0.72 mmol) using the general procedure described above gave 16 (0.12 g, 57%) as white solid; TLC Rf 0.57 (MeOH/CHCl3/NH4OH 1:5:0.5); mp, 232.6–235.6 °C 1H NMR (400 Hz) (Me2SO-d6) δ 2.51 (s, 3H, 5-CH3), 3.39 (s, 3H, 7-CH3), 5.89 (s, 2 H, 2-NH2, exch.), 6.49 (s, 2 H, 4-NH2, exch.), 6.64 (d, 1 H, C6H4, J=7.3 Hz), 7.34 (t, 1 H, J= 7.8 Hz), 7.63 (td, 2 H, J=6.9Hz, J=14.9Hz,), 7.72 (d, 1 H, J=8.1Hz), 7.96 (d, 1 H, J=8.0Hz), 8.28 (d, 1 H, J=8.3Hz). Anal. Calcd. for C18H17N5S: C, 64.45; H, 5.11; N, 20.88; S, 9.56. Found: C, 64.68; H, 4.91; N, 20.82; S, 9.59
6-((3,4-difluorophenyl)thio)-5,7-dimethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (17)
Reaction of 11 (0.120 g, 0.39 mmol), sodium hydride (0.012g, 0.5 mmol) and iodomethane (31 mmL, 0.5 mmol) using the general procedure described above gave 17 (0.08 g, 64%) as white solid; TLC Rf 0.57 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 266.0–267.8 °C 1H NMR (500 Hz) (Me2SO-d6) δ 2.38 (s, 3 H, 5-CH3), 3.34 (s, 3 H, 7-CH3), 5.83 (s, 2 H, 2-NH2, exch.), 6.38 (s, 2 H, 4-NH2, exch.), 6.75 (d, 1 H, C6H4, J=8.7Hz), 7.06 (ddd, 1 H, C6H4, J=2.3Hz, J=7.4Hz, J=10.8Hz), 7.37 (ddd, 1 H, C6H4, J=2.3Hz, J=7.4Hz, J=10.8Hz). Anal. Calcd. for C14H13F2N5S 0.04 CHCl3: C, 51.71; H, 4.03; F, 11.65; N, 21.47; S, 9.64. Found: C, 51.75; H, 4.01; F, 11.47; N, 21.30; S, 9.74
5,7-dimethyl-6-((4-(trifluoromethoxy)phenyl)thio)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (18)
Reaction of 12 (0.150 g, 0.42 mmol), sodium hydride (0.012g, 0.5 mmol) and iodomethane (32 mmL, 0.5 mmol) using the general procedure described above gave 18 (0.1 g, 48%) as white solid; TLC Rf 0.57 (MeOH/CHCl3/NH4OH, 1:5:0.5); mp, 282.0–283.8 °C 1H NMR (400 Hz) (Me2SO-d6) δ 2.39 (s, 3 H, 5-CH3), 3.38 (s, 3 H, 7-CH3), 5.88 (s, 2 H, 2-NH2, exch.), 6.47 (s, 2 H, 4-NH2, exch.), 7.06 (d, 2 H, C6H4, J=8.2 Hz), 7.29 (d, 2 H, C6H4, J= 8.3 Hz). Anal. Calcd. for C15H14F3N5OS: C, 48.78; H, 3.82; F, 15.43; N, 18.96; O, 4.33; S, 8.68. Found: C, 49.03; H, 4.01; F, 18.93; N, 18.93; S, 8.61
6.2 Molecular Modeling
Docking of compounds 1–18 was carried out using the published X-ray crystal structure of N6-methyl-N6-(3,4,5-trifluorophenyl)pyrido[3,2-d]pyrimidine-2,4,6-triamine in hDHFR(PDB: 4QJC, 1.62 Å)33 and in the homology model of pjDHFR32 using LeadIT 2.1.636. The docking in LeadIT was constrained to the active site of the protein. Polar hydrogen atoms of amino acids were not constrained, thereby permitting them free rotation. Base placement of fragments for docking was carried out using triangle docking. Default parameters were used for scoring and clash handling. The maximum number of solutions per iteration and maximum number of fragmentation were set to 200. Ten poses were obtained per molecule. The docked poses were exported to MOE 2016.08 for visualization.39 The validation of LeadIT as a suitable docking system for pjDHFR and hDHFR was carried out by re-docking the native ligands in the x-ray crystal structures of pcDHFR (PDB: 2FZI)40 and hDHFR (PDB: 4QJC). The ligands were sketched in MOE 2016.0839 and docking was carried out with LeadIT 2.1.6 as described above. The best docked pose of the ligands had RMSD of 0.7060 Å in pjDHFR and 0.8860 Å in hDHFR. Thus, LeadIT 2.1.6 was validated and chosen for the docking studies.
6.3 Pharmacological assay
The expression and purification of recombinant pj- and hDHFR was carried out as previously described.18 Standard DHFR assays were conducted at 37°C with continuous recording of change of absorbance at 340 nM. The assay contained 41 mM sodium phosphate buffer at pH 7.4, 8.9 mM 2-mercaptoethanol, 150 mM KCl, and saturating concentrations of NADPH (117 μM). Dihydro folic acid (DHFA) was used at an optimum concentration of 9 μM. The results reported previously by Namjoshi et al.31 were carried out at 18 M of DHFA.
6.4 Crystallization and X-ray Data Collection and Refinement
Expression and purification of wild type human dihydrofolate reductase (hDHFR) were carried out as previously described.41 Recombinant hDHFR was washed in a Centricon-10 with 100 mM K2HPO4 buffer pH 6.9 with 30% saturated ammonium sulfate and concentrated to 7.9 mg ml−1. The hDHFR samples were incubated for 1 h on ice with a tenfold excess of NADPH and compounds 3 and 14, respectively, prior to crystallization using the hanging-drop vapor diffusion method using siliconized glass cover slips and storage at 14°C. Protein droplets of the hDHFR complexes contained K2HPO4 pH 6.9 with 30% saturated ammonium sulfate equilibrated against a reservoir solution consisting of 100 mM K2HPO4 pH 6.9 with 60% saturated ammonium sulfate, 3% (v/v) ethanol. Crystals of hDHFR-3-NADPH and hDHFR-14-NADPH ternary complex were hexagonal and belonged to the space group H3. Data were collected at 100K to 1.46Å resolution for both crystals using the remote access robot on beamline 14.7 at the Stanford Synchrotron Radiation Laboratory.19, 42–44 The data were processed using HKL2000 program package.45 The diffraction statistics are shown in Table 5. Both crystal structures were solved by molecular replacement methods using the coordinates for hDHFR (1u72)46 in the program Molref.47 Inspection of the resulting difference electron density maps made using COOT48 running on an iMac workstation revealed density for the ternary complex of both crystals. The final cycles of refinement were carried out using the program Refmac5 in the CCP4 suite of programs.47 The Ramachandran conformational parameters from the last cycle of refinement generated by RAMPAGE49 showed that more than 96% of the residues refined have the most favored conformation and none are in the disallowed regions. Coordinates for these structures have been deposited with the Protein Data Bank.
Table 5.
Crystal Properties and Refinement Statistics of 3 and 14 bound to hDHFR
| Compound | (B2-282) 3 | (A6-283) 14 |
|---|---|---|
| PDB accession | 5HT4 | 5HT5 |
| Space Group | H3 | H3 |
| Lattice constants (Å)/° | ||
| a | 85.68 | 85.45 |
| b | 85.68 | 85.45 |
| c | 77.03 | 77.69 |
| α | 90.0 | 90.0 |
| β | 90.0 | 90.0 |
| γ | 120.0 | 120.0 |
| Beamline | SSRL 14-1 | SSRL 14-1 |
| Resolution Å | 1.46 (1.49) | 1.46 (1.49) |
| Wavelength Å | 0.979 | 0.979 |
| Rmerge %a,b | 0.05 (0.067) | 0.174 (0.136) |
| Completeness % | 92.6 (46.2) | 78.9 (70.6) |
| Observed Reflect | 121,242 | 49,944 |
| Unique Reflections | 37,327 | 36,629 |
| I/σ(I) | 35.0 (0.90) | 34.5 (2.4) |
| Multiplicitya | 2.0 (1.4) | 1.3 (1.2) |
| Reflections used | 25,428 | 12,777 |
| Resolution Å | 34.2 – 1.60 | 26.3 – 1.90 |
| R-factor | 0.24 | 0.19 |
| Rfree | 0.30 | 0.28 |
| Total protein atoms | 1677 | 1653 |
| Total water atoms | 78 | 75 |
| Average B-factor Å2 | 28.2 | 33.2 |
| Error in Luzzati plot | 0.27 | 0.24 |
| Rms deviation from ideal | ||
| Bond length Å | 0.021 | 0.021 |
| Bond angle | 2.38 | 2.23 |
| Ramachandran plot | ||
| Most favored % | 96.7 | 95.7 |
| Additional allowed % | 2.7 | 3.3 |
| Disallowed % | 0.5 | 1.1 |
The values in parentheses refer to data in the highest resolution shell.
Rsym = ΣhΣi|Ih,i − <Ih>| / ΣhΣi|Ih,i|, where <Ih> is the mean intensity of a set of equivalent reflections.
R-factor = Σ|Fobs − Fcalc| / ΣFobs, where Fobs and Fcalc are observed and calculated structure factor amplitudes.
Rfree-factor was calculated for R-factor for a random 5% subset of all reflections.
Supplementary Material
Acknowledgments
This work was supported, in part, by grants from the National Institute of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID), RO1AI098458, Adrian van Kaam Chair in Scholarly Excellence (AG) Duquesne University, and NSF for NMRs. We would like to acknowledge Dr. Sudhir Raghavan for his input in conducting the molecular modeling.
Abbreviations
- Pj
Pneumocystis jirovecii
- PCP
Pneumocystis pneumonia
- ART
antiretroviral therapy
- TMP
trimethoprim
- SMX
sulfamethoxazole
- DHFR
dihydrofolate reductase
- DHPS
dihydropteroate synthase
- PTX
Piritrexim
- TMQ
trimetrexate
- PC
Pneumocystis carinii
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kovacs JA. 349 - Pneumocystis Pneumonia A2 - Goldman, Lee. In: Schafer AI, editor. Goldman’s Cecil Medicine (Twenty-Fourth Edition) W.B. Saunders; Philadelphia: 2012. pp. 1997–2005. [Google Scholar]
- 2.Huang YS, Yang JJ, Lee NY, Chen GJ, Ko WC, Sun HY, Hung CC. Treatment of Pneumocystis jirovecii pneumonia in HIV-infected patients: a review. Expert Rev Anti Infect Ther. 2017;15:873–892. doi: 10.1080/14787210.2017.1364991. [DOI] [PubMed] [Google Scholar]
- 3.Sokulska M, Kicia M, Wesołowska M, Hendrich AB. Pneumocystis jirovecii—from a commensal to pathogen: clinical and diagnostic review. Parasitol Res. 2015;114:3577–3585. doi: 10.1007/s00436-015-4678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yiannakis EP, Boswell TC. Systematic review of outbreaks of Pneumocystis jirovecii pneumonia: evidence that P. jirovecii is a transmissible organism and the implications for healthcare infection control. The Journal of hospital infection. 2016;93:1–8. doi: 10.1016/j.jhin.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Goldman L, Schafer AI. Goldman-Cecil Medicine. Elsevier Health Sciences; 2015. [Google Scholar]
- 6.Bernheimer JM, Patten G, Makeleni T, Mantangana N, Dumile N, Goemaere E, Cox V. Paediatric HIV treatment failure: a silent epidemic. J Intl AIDS Soc. 2015;18:20090. doi: 10.7448/IAS.18.1.20090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armstrong-James D, Meintjes G, Brown GD. A neglected epidemic: fungal infections in HIV/AIDS. Trends in microbiology. 2014;22:120–7. doi: 10.1016/j.tim.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Harris JR, Balajee SA, Park BJ. Pneumocystis Jirovecii Pneumonia: Current Knowledge and Outstanding Public Health Issues. Curr Fungal Infect Rep. 2010;4:229–237. [Google Scholar]
- 9.Masur H, Brooks JT, Benson CA, Holmes KK, Pau AK, Kaplan JE. Prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: Updated Guidelines from the Centers for Disease Control and Prevention, National Institutes of Health, and HIV Medicine Association of the Infectious Diseases Society of America. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2014;58:1308–11. doi: 10.1093/cid/ciu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller RF, Huang L, Walzer PD. Pneumocystis Pneumonia Associated with Human Immunodeficiency Virus. Clin Chest Med. 2013;34:229–241. doi: 10.1016/j.ccm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Hawser S, Lociuro S, Islam K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem Pharmacol. 2006;71:941–8. doi: 10.1016/j.bcp.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 12.Yoon C, Subramanian A, Chi A, Crothers K, Meshnick SR, Taylor SM, Beard CB, Jarlsberg LG, Lawrence GG, Avery M, Swartzman A, Fong S, Roth B, Huang L. Dihydropteroate Synthase Mutations in Pneumocystis Pneumonia: Impact of Applying Different Definitions of Prophylaxis, Mortality Endpoints and Mutant in a Single Cohort. Med Mycol. 2013;51:568–575. doi: 10.3109/13693786.2013.770604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L, Crothers K, Atzori C, Benfield T, Miller R, Rabodonirina M, Helweg-Larsen J. Dihydropteroate Synthase Gene Mutations in Pneumocystis and Sulfa Resistance. Emerg Infect Dis. 2004;10:1721–1728. doi: 10.3201/eid1010.030994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponce CA, Chabe M, George C, Cardenas A, Duran L, Guerrero J, Bustamante R, Matos O, Huang L, Miller RF, Vargas SL. High Prevalence of Pneumocystis jirovecii Dihydropteroate Synthase Gene Mutations in Patients with a First Episode of Pneumocystis Pneumonia in Santiago, Chile, and Clinical Response to Trimethoprim-Sulfamethoxazole Therapy. Antimicrob Agents Chemother. 2017;61:e01290–16. doi: 10.1128/AAC.01290-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Queener SF, Cody V, Pace J, Torkelson P, Gangjee A. Trimethoprim resistance of dihydrofolate reductase variants from clinical isolates of Pneumocystis jirovecii. Antimicrob Agents Chemother. 2013;57:4990–8. doi: 10.1128/AAC.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nahimana A, Rabodonirina M, Bille J, Francioli P, Hauser PM. Mutations of Pneumocystis jirovecii dihydrofolate reductase associated with failure of prophylaxis. Antimicrob Agents Chemother. 2004;48:4301–5. doi: 10.1128/AAC.48.11.4301-4305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siripattanapipong S, Leelayoova S, Mungthin M, Worapong J, Tan-Ariya P. Study of DHPS and DHFR genes of Pneumocystis jirovecii in Thai HIV-infected patients. Med Mycol. 2008;46:389–392. doi: 10.1080/13693780701883722. [DOI] [PubMed] [Google Scholar]
- 18.Cody V, Pace J, Queener SF, Adair OO, Gangjee A. Kinetic and Structural Analysis for Potent Antifolate Inhibition of Pneumocystis jirovecii, Pneumocystis carinii, and Human Dihydrofolate Reductases and Their Active-Site Variants. Antimicrob Agents Chemother. 2013;57:2669–2677. doi: 10.1128/AAC.00172-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cody V, Pace J, Makin J, Piraino J, Queener SF, Rosowsky A. Correlations of Inhibitor Kinetics for Pneumocystis jirovecii and Human Dihydrofolate Reductase with Structural Data for Human Active Site Mutant Enzyme Complexes. Biochemistry. 2009;48:1702–1711. doi: 10.1021/bi801960h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang JJ, Huang CH, Liu CE, Tang HJ, Yang CJ, Lee YC, Lee KY, Tsai MS, Lin SW, Chen YH, Lu PL, Hung CC. Multicenter study of trimethoprim/sulfamethoxazole-related hepatotoxicity: incidence and associated factors among HIV-infected patients treated for Pneumocystis jirovecii pneumonia. PloS one. 2014;9:e106141. doi: 10.1371/journal.pone.0106141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordin FM, Simon GL, Wofsy CB, Mills J. Adverse reactions to trimethoprim-sulfamethoxazole in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1984;100:495–499. doi: 10.7326/0003-4819-100-4-495. [DOI] [PubMed] [Google Scholar]
- 22.Masters PA, O’Bryan TA, Zurlo J, Miller DQ, Joshi N. Trimethoprim-sulfamethoxazole revisited. Arch Intern Med. 2003;163:402–410. doi: 10.1001/archinte.163.4.402. [DOI] [PubMed] [Google Scholar]
- 23.Floris-Moore MA, Amodio-Groton MI, Catalano MT. Adverse Reactions to Trimethoprim/Sulfamethoxazole in AIDS. Ann Pharmacother. 2003;37:1810–1813. doi: 10.1345/aph.1D179. [DOI] [PubMed] [Google Scholar]
- 24.Ho JM, Juurlink DN. Considerations when prescribing trimethoprim-sulfamethoxazole. CMAJ: Canadian Medical Association journal = journal de l’Association medicale canadienne. 2011;183:1851–8. doi: 10.1503/cmaj.111152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sangiolo D, Storer B, Nash R, Corey L, Davis C, Flowers M, Hackman RC, Boeckh M. Toxicity and Efficacy of Daily Dapsone as Pneumocystis jiroveci Prophylaxis after Hematopoietic Stem Cell Transplantation: A Case-Control Study. Biol Blood Marrow Transplant. 2005;11:521–529. doi: 10.1016/j.bbmt.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 26.White NJ. Cardiotoxicity of antimalarial drugs. Lancet Infect Dis. 2007;7:549–558. doi: 10.1016/S1473-3099(07)70187-1. [DOI] [PubMed] [Google Scholar]
- 27.Nickel P, Schurmann M, Albrecht H, Schindler R, Budde K, Westhoff T, Millward J, Suttorp N, Reinke P, Schurmann D. Clindamycin-primaquine for pneumocystis jiroveci pneumonia in renal transplant patients. Infection. 2014;42:981–9. doi: 10.1007/s15010-014-0660-y. [DOI] [PubMed] [Google Scholar]
- 28.Kovacs JA, Allegra CJ, Swan JC, Drake JC, Parrillo JE, Chabner BA, Masur H. Potent antipneumocystis and antitoxoplasma activities of piritrexim, a lipid-soluble antifolate. Antimicrob Agents Chemother. 1988;32:430–433. doi: 10.1128/aac.32.4.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Short CES, Gilleece YC, Fisher MJ, Churchill DR. Trimetrexate and folinic acid: a valuable salvage option for Pneumocystis jirovecii pneumonia. AIDS. 2009;23:1287–1290. doi: 10.1097/QAD.0b013e32832d0792. [DOI] [PubMed] [Google Scholar]
- 30.Gangjee A, Kurup S, Namjoshi O. Dihydrofolate reductase as a target for chemotherapy in parasites. Curr Pharm Des. 2007;13:609–39. doi: 10.2174/138161207780162827. [DOI] [PubMed] [Google Scholar]
- 31.Cody V, Chisum K, Pope C, Queener SF. Purification and characterization of human-derived Pneumocystis jirovecii dihydrofolate reductase expressed in Sf21 insect cells and in Escherichia coli. Protein Expr Purif. 2005;40:417–23. doi: 10.1016/j.pep.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 32.Gangjee A, Namjoshi OA, Raghavan S, Queener SF, Kisliuk RL, Cody V. Design, synthesis, and molecular modeling of novel pyrido[2,3-d]pyrimidine analogues as antifolates; application of Buchwald-Hartwig aminations of heterocycles. J Med Chem. 2013;56:4422–41. doi: 10.1021/jm400086g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cody V, Pace J, Namjoshi OA, Gangjee A. Structure-activity correlations for three pyrido[2,3-d]pyrimidine antifolates binding to human and Pneumocystis carinii dihydrofolate reductase. Acta Crystallogr F Struct Biol Commun. 2015;71:799–803. doi: 10.1107/S2053230X15008468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor EC, Patel HH, Jun JG. A One-Step Ring Transformation/Ring Annulation Approach to Pyrrolo[2,3-d]pyrimidines. A New Synthesis of the Potent Dihydrofolate Reductase Inhibitor TNP-351. J Org Chem. 1995;60:6684–6687. [Google Scholar]
- 35.Gangjee A, Lin X, Queener SF. Design, synthesis, and biological evaluation of 2,4-diamino-5-methyl-6-substituted-pyrrolo[2,3-d]pyrimidines as dihydrofolate reductase inhibitors. Journal of medicinal chemistry. 2004;47:3689–92. doi: 10.1021/jm0306327. [DOI] [PubMed] [Google Scholar]
- 36.LeadIT 2.1.6. Biosolve IT; St. Augustin, Germany: www.biosolveit.de. [Google Scholar]
- 37.Cody V, Schwalbe CH. Structural characteristics of antifolate dihydrofolate reductase enzyme interactions. Crystallography Reviews. 2006;12:301–333. [Google Scholar]
- 38.SYBYL-X 2.1.1. Tripos International; 1699 South Hanley Rd. St. Louis, Missouri, 63144; USA: [Google Scholar]
- 39.Molecular Operating Environment (MOE), 2016.08. Chemical Computing Group ULC; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2017. [Google Scholar]
- 40.Cody V, Pace J, Chisum K, Rosowsky A. New insights into DHFR interactions: analysis of Pneumocystis carinii and mouse DHFR complexes with NADPH and two highly potent 5-(omega-carboxy(alkyloxy) trimethoprim derivatives reveals conformational correlations with activity and novel parallel ring stacking interactions. Proteins. 2006;65:959–69. doi: 10.1002/prot.21131. [DOI] [PubMed] [Google Scholar]
- 41.Cody V, Pace J, Makin J, Piraino J, Queener SF, Rosowsky A. Correlations of inhibitor kinetics for Pneumocystis jirovecii and human dihydrofolate reductase with structural data for human active site mutant enzyme complexes. Biochemistry. 2009;48:1702–11. doi: 10.1021/bi801960h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. An automated system to mount cryo-cooled protein crystals on a synchrotron beam line, using compact sample cassettes and a small-scale robot. J Appl Crystallogr. 2002;35:720–726. doi: 10.1107/s0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.González AMP, McPhillips S, Song J, Sharp K, Taylor J, Adams P, Sauter N, Soltis S. Web-Ice: integrated data collection and analysis for macromolecular crystallography. J Appl Crystallogr. 2008;41:176–184. [Google Scholar]
- 44.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. Journal of synchrotron radiation. 2002;9:401–6. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.Cody V, Luft JR, Pangborn W. Understanding the role of Leu22 variants in methotrexate resistance: comparison of wild-type and Leu22Arg variant mouse and human dihydrofolate reductase ternary crystal complexes with methotrexate and NADPH. Acta Crystallogr D Biol Crystallogr. 2005;61:147–55. doi: 10.1107/S0907444904030422. [DOI] [PubMed] [Google Scholar]
- 47.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.