

Abstract
Neuroinflammation and microglial activation are pathological markers of a number of central nervous system (CNS) diseases. Chronic activation of microglia induces the release of excessive amounts of reactive oxygen species (ROS) and pro-inflammatory cytokines. Additionally, chronic microglial activation has been implicated in several neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease. Thymoquinone (TQ) has been identified as one of the major active components of the natural product Nigella sativa seed oil. TQ has been shown to exhibit anti-inflammatory, anti-oxidative, and neuroprotective effects. In this study, lipopolysaccharide (LPS) and interferon gamma (IFNγ) activated BV-2 microglial cells were treated with TQ (12.5 μM for 24 h). We performed quantitative proteomic analysis using Orbitrap/Q-Exactive Proteomic LC-MS/ MS (Liquid chromatography-mass spectrometry) to globally assess changes in protein expression between the treatment groups. Furthermore, we evaluated the ability of TQ to suppress the inflammatory response using ELISArray™ for Inflammatory Cytokines. We also assessed TQ's effect on the gene expression of NFκB signaling targets by profiling 84 key genes via real-time reverse transcription (RT2) PCR array. Our results indicated that TQ treatment of LPS/IFNγ-activated microglial cells significantly increased the expression of 4 antioxidant, neuroprotective proteins: glutaredoxin-3 (21 fold; p < 0.001), biliverdin reductase A (15 fold; p < 0.0001), 3-mercaptopyruvate sulfurtransferase (11 fold; p < 0.01), and mitochondrial lon protease (> 8 fold; p < 0.001) compared to the untreated, activated cells. Furthermore, TQ treatment significantly (P < 0.0001) reduced the expression of inflammatory cytokines, IL-2 = 38%, IL-4 = 19%, IL-6 = 83%, IL-10 = 237%, and IL-17a = 29%, in the activated microglia compared to the untreated, activated which expression levels were significantly elevated compared to the control microglia: IL-2 = 127%, IL-4 = 151%, IL-6 = 670%, IL-10 = 133%, IL-17a = 127%. Upon assessing the gene expression of NFκB signaling targets, this study also demonstrated that TQ treatment of activated microglia resulted in > 7 fold down-regulation of several NFκB signaling targets genes, including interleukin 6 (IL6), complement factor B (CFB), chemokine (C–C motif) ligand 3 (CXCL3), chemokine (C–C) motif ligand 5 (CCL5) compared to the untreated, activated microglia. This modulation in gene expression counteracts the > 10-fold upregulation of these same genes observed in the activated microglia compared to the controls. Our results show that TQ treatment of LPS/IFNγ-activated BV-2 microglial cells induce a significant increase in expression of neuroprotective proteins, a significant decrease in expression inflammatory cytokines, and a decrease in the expression of signaling target genes of the NFκB pathway. Our findings are the first to show that TQ treatment increased the expression of these neuroprotective proteins (biliverdin reductase-A, 3-mercaptopyruvate sulfurtransferase, glutaredoxin-3, and mitochondrial lon protease) in the activated BV-2 microglial cells. Additionally, our results indicate that TQ treatment decreased the activation of the NFκB signaling pathway, which plays a key role in neuroinflammation. In conclusion, our results demonstrate that TQ treatment reduces the inflammatory response and modulates the expression of specific proteins and genes and hence potentially reduce neuroinflammation and neurodegeneration driven by microglial activation.
Keywords: Thymoquinone, Microglia, Neuroinflammation, NFκB, Neuroprotection
1. Introduction
Microglia serve as the brain's resident macrophages providing innate immunity for the CNS (González-Scarano and Baltuch, 1999; Schwab and Schluesener, 2004; Kim and de Vellis, 2005; Block et al., 2007). In a normal, healthy brain they remain in the “resting” state as sentinels of the CNS, constantly surveilling their microenvironment to remain ready for immediate activation (Kreutzberg, 1996; Davalos et al., 2005; Nimmerjahn et al., 2005). When triggered by an immunological challenge such as local injury or invading pathogens, activated microglia produces a phagocytic response in addition to increased expression of inflammatory cytokines as a central part of the brain's defense mechanism to ensure healthy neuronal function (Lynch, 2009; Solito and Sastre, 2012). This innate immune response helps to restore CNS homeostasis during pathological conditions via removal of unwanted cellular debris and pathogens and secreting neurotrophic agents in support of surrounding neurons (Lefkowitz and Lefkowitz, 2008; Ransohoff and Perry, 2009). However, excessive, or prolonged microglial activation results in chronic inflammatory response can lead to the overproduction of pro-inflammatory cytokines and reactive oxygen/nitrogen species (ROS/RNS). Chronic neuroinflammation and excessive oxidative stress have more recently been recognized as important pathological events in neurodegenerative disease (Akiyama et al., 2000; Bamberger et al., 2003; Sheng et al., 2003; Streit et al., 2004; Rojo et al., 2008).
Over the last few decades, there has been rapidly growing interest in naturally occurring phytochemical compounds with antioxidant, anti-inflammatory, as well as a neuroprotective potential. TQ, an abundant bioactive component in the oil extracted from the seeds of the Nigella sativa plant (Padhye et al., 2008; Ahmad et al., 2013; Khazdair, 2015) may be amongst the most promising recently studied phytochemical compounds. The oil and TQ have also shown potent anti-inflammatory effects on several inflammation-based models including experimental encephalomyelitis, peritonitis, asthma, and arthritis through suppression of pro-inflammatory mediators (Mahgoub, 2003; Salem, 2005; Umar et al., 2012; Fahmy et al., 2014; Keyhanmanesh et al., 2014). Additionally, studies show that TQ possess anti-oxidant (Ismail et al., 2010), neuroprotective (Kanter, 2008; Radad et al., 2009; Radad et al., 2009), anticancer (Yi et al., 2008; Banerjee et al., 2010; Al-Malki and Sayed, 2014) and beneficial immunomodulatory properties (Salem, 2005; Gholamnezhad et al., 2015).
Given what is now known about the common role of neuroinflammation in the development and progression of an array of neurodegenerative diseases and because TQ has shown to possess anti-inflammatory and neuroprotective pharmacological properties, we examined the TQ's anti-inflammatory effects as well as its effect on the NFκB signaling targets in the BV-2 microglia activated by the presence of LPS/IFNγ. Moreover, we performed quantitative proteomic analysis using Orbitrap/Q-Exactive Proteomic LC-MS/MS (Liquid chromatography-mass spectrometry) to globally assess changes in protein expression between the treatment groups.
2. Materials & methods
2.1. Materials
High glucose Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 4 mM GlutaMAX™, penicillin-streptomycin (10,000 U/ ml), interferon gamma recombinant mouse protein (IFNγ), and trypsin/ EDTA (0.25%) with phenol red were purchased from Thermo Fisher Scientific. Heat-inactivated fetal bovine serum (FBS) was purchased from Atlanta Biologicals. TQ (99% purity), lipopolysaccharides from Escherichia coli (LPS), and the reagents and Microcon-30 kDa centrifugal filter units used in the sample preparation for the LC/MS/MS proteome analyses were purchased from Sigma-Aldrich. The Mouse Inflammatory Cytokines Multi-Analyte ELISArray™ Kit (MEM-004A) as well as the supplies and reagents for the NFκB PCR Array assay, including RT2 Profiler PCR Array (PAMM 225Z), RNeasy Mini Kit, RNase-Free DNase Set, QIAshredders, RT2 First Strand Kit, and RT2 SYBR Green Mastermix were purchased from Qiagen.
2.2. Cell culture
The BV-2 cell line is an immortalized murine microglial cell line supplied by the lab of Elisabeth Blasi at the University of Perugia (Blasi et al., 1990). The BV-2 cells were cultured in high glucose DMEM-GlutaMAX™ media containing phenol red, 10% heat-inactivated FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. The cells were maintained at 37 °C in a 5% CO2 humidified atmosphere with the media changed every 2–3 days. For experiments, BV-2 cells were seeded at 5 × 105 cells/ml overnight. Next day, LPS/IFNγ was added to the culture media at a working concentration of 500 ng/ml LPS + 0.5 ng/ml IFNγ, respectively. The TQ stock was freshly prepared by initially dissolving in DMSO then diluting further with experimental media to the appropriate concentration so that the concentration of DMSO did not exceed 0.025%, which was used for the control (0 μM TQ). The concentration of TQ used in all the experiments was 12.5 μM, which was established by cell viability in our previous study (Cobourne-Duval, Taka, et al. 2016).
2.3. Quantitative proteomic analysis
The BV-2 cells were cultured and treated as previously described in T-75 flasks with or without TQ and LPS/IFNγ. The cells were then harvested after 24hs cells using 0.25% trypsin-EDTA and washed twice with cold PBS. Lysis buffer was added to the cell pellet, and the sample preparation protocol was followed. Thirty microliters of cell lysate were mixed with 200 μl of 8 M urea in 0.1 M Tris/HCl, pH 8.5 (UA) in the filter unit and centrifuged at 14,000 ×g for 15 min. Another 200 μl UA was added to the filter unit, the centrifugation was repeated, and the flow-through from the collection tube was discarded. One hundred microliters (100 μl) of 0.05 M iodoacetamide in UA (IAA) solution was added followed by centrifugation at 14,000 ×g for 10 min. 100 μl of UA was added to the filter unit, and the previous centrifugation was repeated. 100 μl of 0.05 M NH4HCO3 in water (ABC) was added to the filter unit and centrifuged at 14,000 ×g for 10 min. The addition of ABC to the filter unit followed by centrifugation was repeated twice. Next, 40 μl ABC with trypsin (enzyme to protein ratio 1:100) was added. The filter units were incubated at 37 °C for 18 h and transferred to new collection tubes. Fifty microliters (50 μl) 0.5 M NaCl was added to the filter units and centrifuged at 14,000 ×g for 10 min. The filtrate was acidified with CF3COOH and desalted. The samples were then sent to the Translational Science Laboratory at Florida State University for Orbitrap/Q-Exactive Proteomic LC-MS/MS (Liquid chromatography-mass spectrometry) for complex mixture analysis. The results were further analyzed by the ‘Scaffold version 4.6’ software.
2.4. ELISArray™ for inflammatory cytokines
Cells were cultured and treated as previously described in T-75 flasks. Approximately 650 μL the supernatant was transferred to labeled microcentrifuge tubes and centrifuged at 1000 ×g for 10 min to remove any particulate material, and the samples were prepared for analysis. The ELISA kit reagents including the wash buffer, assay buffer, and sample dilution buffer were prepared according to the kit's directions. Fifty microliters of assay buffer followed by 50 μL of each sample, Antigen Standard Cocktail (for the positive control), or assay buffer (for the negative control) were added to their corresponding wells of the ELISArray™ plate. The plate was then set for a 2-hr incubation at room temperature and processed according to the kit's instructions using the provided detection antibodies, kit reagents, and buffers. Within 30 min after adding Stop Solution to the ELISArray™ plate, absorbance at 450 nm and 570 nm (for wavelength correction) was measured.
2.5. RT2 profiler PCR array – NFκB signaling targets
Profiling the expression of 84 key genes responsive to NFκB signal transduction was performed using the NFκB Signaling Targets RT2 Profiler PCR Array to yield results that allow analysis of activation/ inhibition of NFκB signaling. The functional grouping of the genes involved in NFκB-related cellular processes in this array includes a selection of genes for cytokines & chemokines, inflammation, apoptosis, anti-apoptosis, immune response, type I interferon-responsive genes, development & differentiation, stress response, NFκB pathway, and transcription factors. Briefly, the BV-2 cells were seeded (5 × 105 cells/ ml) in T-75 flasks (20 mL/flask), treated as previously described, harvested using cell scrapers then collected via centrifugation in RNase-free polypropylene tubes. The supernatant was completely removed from the cell pellet via aspiration, and the RNA was extracted and purified according to the manufacturer's instructions using Qiagen's RNeasy Mini Kit with the assistance of QIAshredders to homogenize the cell pellets and the RNase-Free DNase Set to ensure a complete DNA removal. RNA quantity and purity were determined spectrophotometrically (Nanodrop) before converting the purified extracted RNA into first-strand cDNA using the Qiagen RT2 First Strand Kit according to the manufacturer's instructions. The prepared cDNA was then mixed with an appropriate amount RT2 SYBR Green Mastermix and RNase-free water in a 5 ml tube as directed and this mixture was aliquoted into the wells of the RT2 Profiler PCR Array. The RT2 Profiler PCR Array was tightly sealed, centrifuged for 1 min at 1000 ×g at room temperature (15–25 °C), and run on the PCR cycling program.
2.6. Statistical analysis
All data were expressed as a mean ± standard error from at least 3 independent experiments. Statistical significance of the difference between values for compared groups is considered at *P ≤.05, **P ≤.01, ***P ≤.001, and ****P ≤.0001.
The quantitative proteomic mass spectrometry data was analyzed using ‘Scaffold version 4.6’ software to identify, validate, organize, and perform quantitative analysis. Only those proteins with identities validated on the X! Tandem and SEQUEST search engines and identification confidence > 95% were evaluated. Quantitative analysis assessing the differential abundance between the experimental groups was performed using the t-test. Data generated from ELISArray™ for Inammatory Cytokines was statistically analyzed using GraphPad Prism 6 (version 6.07; Graph Pad Software Inc. San Diego, CA, USA by one-way ANOVA with Tukey's post hoc multiple comparisons test). The RT2 Profiler PCR Array data was analyzed via Qiagen's PCR Array Data Analysis Web Portal at www.SABiosciences.com/pcrarraydataanalysis.php, which calculates fold change/regulation using ΔΔCT (threshold cycle) method using the 2^ (−ΔΔCT) formula. The p-values are calculated based on a Student's t-test of the replicate 2^ (− ΔCT) values for each gene in the control group and experimental groups.
3. Results
Comparative quantitative proteomic analysis of LPS/IFNγ-activated BV-2 cells with and without TQ treatment revealed 35 differentially expressed proteins (> 95% identification confidence). Amongst these differentially expressed proteins, TQ treatment (12.5 μM for 24 h) of the LPS/IFNγ-activated microglia compared to the untreated, activated microglia resulted in the increased expression of 4 neuroprotective proteins: glutaredoxin-3 (21 fold), biliverdin reductase A (15 fold), 3-mercaptopyruvate sulfotransferase (11 fold), and mitochondrial lon protease (> 8 fold) (Table 1).
Table 1.
The Gene Ontology annotation showing molecular function, biological process, and expression levels of 4 differentially expressed key proteins identified and quantified by Orbitrap/Q Exactive LC-MS/MS technique. Mean fold-change corresponds to the upregulation of protein expression when comparing TQ + LPS/IFNγ vs LPS/INFγ treatment groups. P-values (t-test) are listed in the table; (n = 3).
| Protein
|
Mean fold change (TQ + LPS/IFNγ vs. LPS/IFNγ) |
p-Value (T-test) | GO Annotation
|
||||
|---|---|---|---|---|---|---|---|
| UniProtKB accession number |
Protein name | Protein ID | Molecular weight | Molecular function | Biological process | ||
| Q9CQM9 | Glutaredoxin-3 | GLRX3_MOUSE | 38 kDa | 21 | 0.00014 | Protein disulfide oxidoreductase activity | Oxidation-reduction process |
| Iron-sulfur cluster binding | Cell redox homeostasis | ||||||
| Electron carrier activity | Response to stress | ||||||
| Q9CY64 | Biliverdin reductase A | BIEA_MOUSE | 34 kDa | 15 | < 0.00010 | Biliverdin reductase activity | Heme catabolic process oxidation-reduction process |
| Q99J99 | 3-mercaptopyruvate sulfurtransferase | THTM_MOUSE | 33 kDa | 11 | 0.0022 | 3-Mercaptopyruvate sulfurtransferase activity | Hydrogen sulfide biosynthetic process |
| Immune system process | |||||||
| Response to toxic substance | |||||||
| Q8CGK3 | Lon protease homolog, mitochondrial | LONM_MOUSE | 106 kDa | 8.1 | 0.00047 | ATP & ADP binding | Aging |
| ATPase activity | Cellular response to oxidative stress | ||||||
| ATP-dependent peptidase activity | Response to hypoxia | ||||||
The LPS/IFNγ-activated BV-2 cells showed significantly higher protein expression of several inflammatory cytokines compared to the controls: IL-2 = 127%, IL-4 = 151%, IL-6 = 670%, IL-10 = 133%, and IL-17a = 127%. The protein expression of the same inflammatory cytokines in the TQ treated, LPS/IFNγ-activated cells were reduced significantly (P < .0001) compared to the protein expression levels activated cells without TQ treatment: IL-2 = 38%, IL-4 = 19%, IL-6 = 83%, IL-10 = 23%, and IL-17a = 29% (Fig. 1).
Fig. 1.
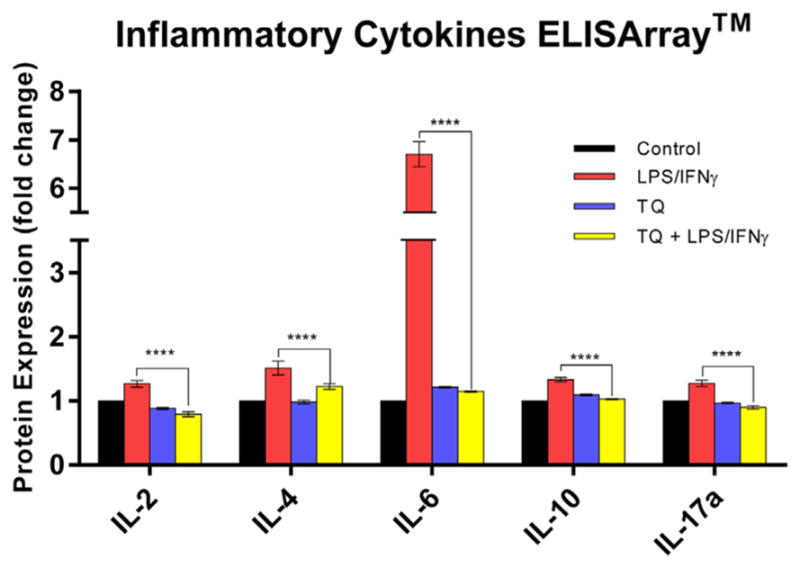
Multi-Analyte ELISArray™ for Inflammatory Cytokines & Chemokines protein expression fold change amongst the control, LPS/INFγ, TQ, and TQ + LPS/IFNγ groups. Data represent protein expression as the mean ± S.E.M (n = 3). Statistical significance of LPS/INFγ vs. TQ + LPS/IFNγ was evaluated by one-way ANOVA followed by Tukey's post hoc multiple comparisons test, p**** ≤0.0001.
TQ treatment alone significantly upregulated in NAD(P)H: quinone oxidoreductase 1 (NQO1) gene expression 10.62-fold (p < .05). Activation of BV-2 microglial cells with LPS/IFNγ lead to significant upregulation of several key genes related to the NFκB signaling targets compared to the control group. Interleukin 6 (IL6), complement factor B (CFB), complement component 3 (C3), chemokine (C–C motif) ligand 3 (CXCL3), and chemokine (C–C) motif ligand 5 (CCL5), are amongst the genes upregulated > 10-fold in the activated microglia compared to the controls. TQ treatment of the LPS/IFNγ-activated microglia caused a significant down-regulation of these same genes > 7 fold, excluding C3 which was a 3-fold down-regulation (Fig. 2 and Table 2).
Fig. 2.
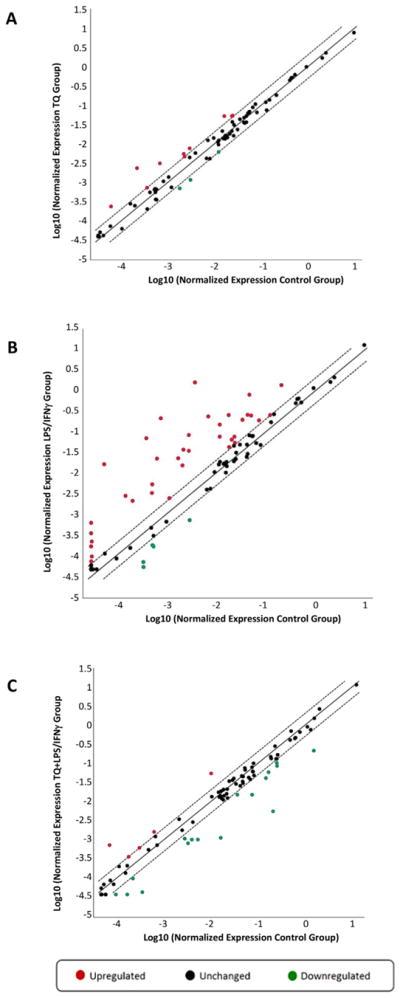
Scatter Plot of the Normalized Expression of NFκB Signaling Targets PCR Array in the Control, TQ, LPS/IFNγ, and TQ + LPS/IFNγ Experimental Groups. (A) TQ vs. Control, (B) LPS/IFNγ vs. Control, (C) TQ + LPS/IFNγ vs. LPS/IFNγ.
Table 2.
List of key genes related to NFκB signaling targets differentially expressed in LPS/IFNγ-activated and thymoquinone-treated BV-2 microglial cells. The fold regulation change of LPS/IFNγ vs. Control, TQ + LPS/IFNγ vs. LPS/IFNγ, and TQ vs. Control. P-values (t-test) are listed in the table; (n = 3).
| Gene symbol | Encoded protein | Genes upregulated in the LPS/IFNγ vs. control
|
Genes downregulated in the TQ + LPS/IFNγ vs. LPS/IFNγ
|
Function | ||||
|---|---|---|---|---|---|---|---|---|
| Direction | Fold regulation LPS-IFNγ/control | p-Value (T-test) | Direction | fold regulation TQ + LPS-IFNγ/LPS-IFNγ | p-value (T-test) | |||
| IL6 | Interleukin 6 | ↑ | 282.33 | < 0.001 | ↓ | 14.36 | < 0.001 | Acute & Chronic Inflammation; Innate Immune Response |
| CFB | Complement factor B | ↑ | 266.74 | < 0.001 | ↓ | 37.74 | < 0.001 | Acute Inflammation; Innate Immune Response |
| C3 | Complement component 3 | ↑ | 12.41 | < 0.001 | ↓ | 3.63 | < 0.001 | Acute Inflammation; Innate & Adaptive Immune Response |
| CCL5 | Chemokine (C–C motif) ligand 5 | ↑ | 402.43 | < 0.001 | ↓ | 7.22 | < 0.001 | Chemokine; Acute Inflammation; Immunoregulatory and Inflammatory process |
| CXCL3 | Chemokine (C-X-C motif) ligand 3 | ↑ | 11.17 | 0.011 | ↓ | 8.12 | 0.013 | Chemokine; Chemoattractant; Acute Inflammation |
| 1L12B | Interleukin 12B | ↑ | 5.36 | < 0.001 | ↓ | 4.47 | < 0.001 | Cytokine; Innate & Adaptive Immune Response; Apoptosis |
| IRF1 | Interferon regulatory factor 1 | ↑ | 6.36 | 0.007 | ↓ | 5.03 | 0.008 | Type I Interferon-Responsive Genes; Transcription Factor |
| Gene symbol | Encoded protein | Genes upregulated in the TQ-treated vs. control | Function | |||||
| Direction | Fold Regulation TQ/control | p-value (T-test) | ||||||
| NQO1 | NAD(P)H: Quinone Oxidoredutase | ↑ | 10.62 | 0.021 | Antioxidant & Detoxification | |||
4. Discussion
Neuroinflammation has been increasingly implicated in the onset and progression of multiple neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD) and Multiple Sclerosis (MS), despite their differing pathologies (Liu and Hong, 2003; Block and Hong, 2005; Gao and Hong, 2008; Chen et al., 2016; Kempuraj et al., 2016). Neuroinflammation is an innate, and initially, protective response mechanism in the brain, facilitated mostly by microglia and astrocytes producing a broad spectrum of inflammatory mediators (Azizi et al., 2015; Von Bernhardi et al., 2015). In neurodegenerative diseases, the role and consequences of inflammation may change dynamically over time (Gao and Hong, 2008). During acute neuroinflammation, the inflammatory mediators work together to restore the damaged glial cells and neuronal cells in the CNS (Kempuraj et al., 2016). However, chronic neuroinflammation commonly results in damaging and detrimental consequences for the CNS, rendering it more vulnerable to neurodegeneration (Kempuraj et al., 2016; Leszek et al., 2016).
The common thread between many neurodegenerative diseases is the fact that microglia, the CNS's resident macrophages, largely mediate the inflammatory immune responses (Amor et al., 2014; Von Bernhardi et al., 2015). Microglial activation is a key feature of neuroinflammation observed in nearly all CNS diseases (Kreutzberg, 1996; Hanisch and Kettenmann, 2007; Neumann et al., 2009). Activated microglia rapidly alter their transcriptional profile, leading to the secretion of a spectrum of inflammatory mediators (i.e., cytokines and chemokines) (Norden et al., 2015; Leszek et al., 2016). Activated microglia also contribute to the elevation of the levels reactive oxygen species (ROS). Excessive levels of inflammatory mediators and oxidative stress are both commonly observed in several neurodegenerative disorders (Block and Hong, 2005; Von Bernhardi et al., 2015; Kempuraj et al., 2016; Leszek et al., 2016). Furthermore, the inflammatory mediators released from microglia lead to the production of additional inflammatory mediators (Leszek et al., 2016). Therefore, exaggerated microglial activation can lead to prolonged neuroinflammation, triggering neurotoxic pathways and thereby lead to progressive neurodegeneration (Amor et al., 2014; Norden et al., 2015). It is hypothesized that damaged neurons signal microglia and induce reactive microgliosis; which further exacerbates neuronal damage via causing the release of excessive inflammatory and neurotoxic factors (Gao and Hong, 2008).
Inflammation and oxidative stress are distinctive biological processes that are closely intertwined and parallel in function in the brain, especially in neurodegenerative disorders. Oxidative stress can be generated from inflammatory responses as well as ROS released from activated microglia as defense agents against pathogens or their markers (Biswas, 2016). If the cell's antioxidant capacity is overwhelmed by excessive ROS, then the oxidative stress causes consequent damage to essential molecules and tissues. Moreover, inflammation can be enhanced by oxidative stress through the activation of NFκB, which is very sensitive to oxidative stress (Salzano et al., 2014). NFκB, commonly designated as a master regulator of inflammation, is a transcription factor that controls the expression of many genes involved in the inflammatory response (Leszek et al., 2016) including pro-inflammatory cytokines, chemokines, adhesion molecules, and inducible nitric oxide synthase (iNOS) (Tak and Firestein, 2001). NFκB is highly activated at sites of inflammation in an array of diseases and has been reported to be associated with chronic inflammation (Okamoto, 2006). Even moderate levels of ROS lead to the degradation of IκB, thereby releasing NFκB dimers from the cytoplasmic NFκB–IκB complex. This series of events allows them to translocate to the nucleus where they are free to bind to κB enhancer elements of target genes, inducing transcription of pro-inflammatory genes (Tak and Firestein, 2001, Von Bernhardi et al., 2015). This activation of NFκB pathway is evident in environments of elevated oxidative stress (Chen et al., 2009, Chongthammakun et al., 2009).
It has been hypothesized that in the diseased CNS the interactions between damaged neurons and dysregulated, over-activated microglia create a vicious self-propagating cycle causing uncontrolled, prolonged inflammation that drives the chronic progression of neurodegenerative diseases (Gao and Hong, 2008). Based on the current literature, we can extend that hypothesis to account for the fact that chronic inflammation also increases the levels of oxidative stress and thus increased activation of the NFκB pathway. This increased activation of the NFκB pathway leads to an upsurge in the release of additional pro-inflammatory cytokines, and thereby further drive and amplify neuroinflammation. This would support the chronic and progressive nature observed in neurodegenerative diseases. Modulation of the prolonged inflammatory response via disrupting this vicious cycle may be a disease-modifying therapeutic strategy for neurodegenerative diseases (Gao and Hong, 2008). An intervention that can interrupt this destructive cycle, when the inflammatory response is no longer beneflcial, should be effective in either halting or slowing down the progression of neurodegenerative diseases (Gao and Hong, 2008). Because the contributors to this vicious cycle can feed one another, a therapeutic with multiple actions to address these components may represent the most promising strategy to treat neurodegenerative diseases.
Numerous recent studies on thymoquinone (TQ) have revealed the array of therapeutic properties it possesses. Specifically, TQ has been shown to have anti-inflammatory and antioxidant effects in different disease states/models in vivo and in vitro. Studies have shown that TQ is a ROS scavenger (Mansour et al., 2002; Badary et al., 2003) capable of reducing the levels of several ROS and lipid/protein oxidation markers in liver and kidney tissue, microglia and macrophages (El-Mahmoudy et al., 2002; Attia et al., 2010; Mahmoud et al., 2014; Cobourne-Duval et al., 2016). Likewise, the anti-inflammatory properties of TQ treatment have been demonstrated in mast cells, microglia, kidney, and liver (El Gazzar et al., 2007; Al-Malki and Sayed, 2014; Mahmoud et al., 2014; Taka et al., 2015). Furthermore, studies have also indicated that TQ treatment provides neuroprotection against amyloid β-induced and α-synuclein-induced neurotoxicity (Khan et al., 2012, Alhebshi et al., 2013, Alhebshi et al., 2014), traumatic brain injury (Gülşen et al., 2016), environmental neurotoxins (Kanter, 2008; Radad et al., 2009; Radad et al., 2014), and transient forebrain ischemia (Al-Majed et al., 2006). Many studies have also indicated that TQ's protective effects involve the modulation of the NFκB pathway (Mohamed et al., 2005, El Gazzar et al., 2007, Sayed and Morcos, 2007, Sethi et al., 2008, Al-Malki and Sayed, 2014). In this study, we investigated whether the anti-inflammatory effects and modulation of the NFκB pathway were observed in the TQ-treated, LPS/IFNγ-activated BV-2 microglial cells.
4.1. Thymoquinone increased the expression of key proteins with neuroprotective effects identified and quantified by Orbitrap/Q Exactive LC-MS/MS proteomics
Investigations on the effects of TQ treatment on LPS/IFNγ-activated BV-2 microglial cells were performed using an Orbitrap/Q Exactive LC-MS/MS proteomics. Four proteins were upregulated in the TQ treated, LPS/IFNγ-activated microglia compared to the untreated, activated microglia: biliverdin reductase A (BVR-A), glutaredoxin- 3 (Grx-3), 3-mercaptopyruvate sulfotransferase (3-MST), and mitochondrial lon protease (LONM). All four proteins possess antioxidant and neuroprotective properties as demonstrated in several recent studies (Panahian et al., 1999; Liu et al., 2006; Shibuya et al., 2009; Kimura et al., 2010; Kim et al., 2011; Zhang et al., 2013; Pham et al., 2015; Zhang et al., 2017). Prolonged oxidative stress and chronic neuroinflammation play significant roles in the pathogenesis and progression of neurodegenerative disorders (Emerit et al., 2004; Frank-Cannon et al., 2009). In response to such conditions, the brain responds via the expression of antioxidant and anti-inflammatory proteins as well as the upregulation of genes involved in the cell stress response (Calabrese et al., 2009). The induction of intrinsic antioxidant/anti-inflammatory pathways to decrease pro-oxidant/pro-inflammatory agents at the site of prolonged oxidative stress and chronic inflammation may be effective in slowing/halting the progression of neurodegenerative disorders such as AD and PD.
In this study, TQ treatment of LPS/IFNγ-activated microglial cells induced an increased expression of biliverdin reductase A (BVR-A) 15-fold compared to the untreated stimulated cells. Biliverdin reductase A is a pleiotropic enzyme that plays a pivotal role in the antioxidant defense against free radicals and cell homeostasis (Barone et al., 2011). BVR-A reduces its substrate biliverdin alpha (BV-α) into the powerful antioxidant and anti-nitrosative molecule, bilirubin (BR)-IX-αγ(Baranano et al., 2002, Barone et al., 2014). Studies have shown that bilirubin (BR) has strong antioxidant potential, specifically against peroxyl radicals (Stocker et al., 1987), and acts as a physiologic anti-oxidant neuroprotectant, protecting brain cultures from H2O2 neurotoxicity (Doré et al., 1999). Bilirubin (BR) is yielded as the final product of heme catabolism, which starts with heme oxygenase (HO) cleaving the heme ring to form biliverdin (BV), which is then reduced to BR (Maines and Panahian, 2001). HO is co-expressed with BVR-A in rat brain cells under normal conditions (Mancuso, 2004) but also pronounced and localized at neurofibrillary tangles, senile plaque neurites in AD brain (Smith et al., 1994). The upregulation of HO/BVR-A system is observed as a neuroprotective, antioxidant response that reduces intracellular levels of pro-oxidant heme and increases levels of the ROS and NO scavenger, bilirubin, to counteract the increased oxidative stress associated with the onset and progression of neurodegenerative disorders like AD (Butterfield et al., 2001; Butterfield and Lauderback, 2002; Mancuso, 2004; Poon et al., 2004; Calabrese et al., 2006; Barone et al., 2014).
TQ treatment of LPS/IFNγ-activated microglial cells also increased the protein expression of glutaredoxin-3 (Grx-3) 25-fold compared to the untreated, activated microglial cells. Grx-3 is a small redox enzyme that utilizes the reducing power of glutathione (GSH) as a cofactor to decrease oxidative stress via catalyzing disulfide reductions in the presence of NADPH and glutathione reductase (GR) in the glutathione system (Fernandes and Holmgren, 2004). Glutaredoxin efficiently reduces glutathionylated proteins to protein thiols and helps maintain redox status of proteins during oxidative stress (Pujol-Carrion and de la Torre-Ruiz, 2010; Sabens Liedhegner et al., 2012; Pham et al., 2015). Decreased levels of Grx-3 render cells susceptible to cellular oxidative stress (Zhang et al., 2017), whereas overexpression of nuclear-targeted Grx-3 is sufficient to suppress cells' sensitivity to oxidant treatments and reduce reactive oxygen species production (Kenchappa et al., 2004; Pham et al., 2015). Mitochondrial dysfunction, in addition to excessive oxidative stress, plays a significant role in an array of neurodegenerative diseases (Chen, 2011; Cozzolino and Carrì, 2012; Federico et al., 2012; Ferreira et al., 2013; Smeyne and Smeyne, 2013; Yan et al., 2013; Camilleri and Vassallo, 2014; Saharan and Mandal, 2014; Wang et al., 2014; Gu et al., 2015). Grx, likewise, helps maintain mitochondrial integrity by preventing the loss of mitochondrial membrane potential (MMP) caused by oxidative insult (Saeed et al., 2008).
3-mercaptopyruvate sulfotransferase (3-MST) is another neuroprotective protein that was upregulated in the TQ treated versus untreated LPS/IFNγ-activated BV-2 microglial cells. 3-MST protein expression was 11-fold higher in the TQ treated compared to the untreated activated cells. 3-MST acts as an antioxidant and, in combination with cysteine aminotransferase, are important producers of hydrogen sulfide (H2S) in the brain, retina, and vascular endothelial cells (Shibuya et al., 2009; The consortium, U, 2017). Neuro2a cells (mouse brain neuroblastoma) expressing 3MST and CAT showed significant resistance to oxidative stress (Kimura et al., 2010). H2S is peroxynitrite (ONOO−) scavenger and acts as a neuroprotectant, as well as an import0ant synaptic modulator, a signaling molecule, and smooth muscle contractor (Whiteman et al., 2004; The consortium, U, 2017). H2S-releasing compounds have been demonstrated in several studies to possess considerable anti-inflammatory, neuroprotective effects and may be candidates for treating neurodegenerative disorders that have a prominent neuroinflammatory component such as Alzheimer disease and Parkinson's disease (Kimura and Kimura, 2004; Hu et al., 2010; Liu and Bian, 2010; Kida et al., 2011). Studies have also demonstrated that treating BV-2 microglial cells with sodium hydrosulfide (NaHS; an H2S donor compound) attenuates Aβ-induced cell toxicity and suppressed the release of nitric oxide and the upregulation of inducible nitric oxide synthase (Liu and Bian, 2010). Moreover, systemic administration of NaHS, as well as inhaled H2S has been demonstrated to be highly effective protecting neurons in the SN and striatum in the 6-OHDA and MPTP-induced PD models, respectively (Hu et al., 2010; Kida et al., 2011). Furthermore, H2S-releasing compounds reduce the LPS-induced release of the pro-inflammatory mediators such as IL-6, IL-1β, TNFα, and NO from activated microglia and macrophages through NFκB–dependent pathways (Hu et al., 2010, Lee et al., 2010, Whiteman et al., 2010).
TQ treatment of activated microglial cells also increased the protein expression of mitochondrial lon protease (LONM), which is an ATP-dependent serine protease that selectively degrades misfolded, un-assembled or oxidatively damaged polypeptides of the mitochondrial matrix (The consortium, U, 2017). LONM serves in a key role concerning neurodegenerative disorders that involve misfolded proteins in their pathologies, such as AD and PD. Mitochondrial polypeptides are constantly exposed to reactive oxygen species (ROS) generated by “electron leakage” from the respiratory chain (Ngo et al., 2013). In healthy conditions, oxidized mitochondrial proteins are quickly removed via proteolytic degradation to prevent them from aggregating or cross-linking and resulting in cellular toxicity. LONM is a key enzyme involved in the elimination of oxidized proteins within the mitochondrial matrix which is crucial to maintaining cellular homeostasis (Ngo et al., 2013; Bota and Davies, 2016). It is also a key cytoprotective enzyme involved in aging and cellular response to oxidative stress/ hypoxia and the regulation of mitochondrial gene expression under such stress conditions (Ngo and Davies, 2009; Ngo et al., 2013; Bota and Davies, 2016; The consortium, U, 2017). The loss of LONM responsiveness may contribute to the increased levels of protein damage, and mitochondrial dysfunction observed in aging and age-related diseases (Ngo and Davies, 2009; Ngo et al., 2013). TQ treatment increased LONM expression > 8 fold compared to the untreated, activated BV-2 microglial cells.
4.2. Thymoquinone attenuated the expression of inflammatory cytokines in activated microglia
Increasingly more studies have shown that neuroinflammation and the flux of inflammatory mediators play significant roles in cognitive impairment via cytokine-mediated interactions between glial cells and neurons (Azizi et al., 2015). The over-expression of inflammatory cytokines in the brain may increase its susceptibility to the onset of neurodegenerative disease. Furthermore, it has been demonstrated that the early stages of the AD are associated with the upregulation of pro-inflammatory cytokines, which can initiate plaque production and enhance nerve cell degeneration (Azizi and Mirshafiey, 2012).
We evaluated the effects of TQ treatment in LPS/IFNγ-activated microglia on the inflammatory cytokine proflle using ELISArray™. Previously our lab has demonstrated the anti-inflammatory properties of TQ in BV-2 microglia solely activated with LPS (Taka et al., 2015). However, in this study we co-activated the BV-2 microglial cells with LPS and INFγ because the two pro-inflammatory agents work synergistically to induce maximal transcriptional responses, enhancing the release of NO in microglia upon activation (Paludan, 2000, Pawate et al., 2004, Yoo et al., 2008). LPS induces NO production through the stimulation of NFκB activation involving the MAP kinase pathway (Bhat et al., 2002, Pawate et al., 2004, Shen et al., 2005) while IFNγ induces NO production involves extracellular signal-regulated kinases cascade, mediated through phosphate kinase C (Shen et al., 2005). Excessive NO can react with superoxide and yield the highly cytotoxic RNS, peroxynitrite (ONOO−) which has been implicated in several CNS disorders, including AD (Heales et al., 1999; Torreilles et al., 1999). Inducible nitric oxide synthase (iNOS) and NADPH oxidase are the major sources of NO and superoxide ion production, respectively, in the activated microglia (Wilkinson and Landreth, 2006; Yoo et al., 2008). Moreover, superoxide ion plays a critical role in the cytokine-mediated inflammatory response in microglia (Choi et al., 2005). Stimulating the BV-2 cells with both LPS and IFNγ allowed us to take all these factors into account, given the fact that increased levels of both inflammation and oxidative stress are observed in several neurodegenerative disorders.
The LPS/IFNγ-activated BV-2 microglial cells showed significantly increased expression of inflammatory cytokines, IL-2, –4, –6, –10, and 17a compared to the controls. TQ treatment markedly decreased the expression of these inflammatory cytokines in the activated microglial cells. Amongst these inflammatory cytokines, the protein expression of IL-6 was most drastically affected by LPS/IFNγ activation and TQ treatment. There was > 6-fold increased protein expression of IL-6 in the activated microglia compared to the control. TQ treatment of the activated microglia resulted in a > 5-fold decrease in IL-6 expression in the treated versus untreated, activated microglial cells.
IL-6 is one of the most potent pleiotropic pro-inflammatory cytokines vastly produced in the brain by activated microglia (Ye and Johnson, 1999, McGeer and McGeer, 2001). IL-6 is involved in mediating cellular communication in physiological as well as pathological states. It can induce an acute phase response triggered during the early course of an infection (Ye and Johnson, 1999). During inflammation, the IL-6 released by the activated microglia can stimulate other microglia to release a cascade of pro-inflammatory cytokines (Wang et al., 2015). The IL-6 produced in the brain plays a significant role in neuroinflammation. Studies indicate that if IL-6 is chronically over-expressed in the CNS, it creates a state that predisposes the development and contributes to the progression of several neurodegenerative disorders, including AD (Ye and Johnson, 1999; Azizi and Mirshafiey, 2012). Moreover, AD patients and animal models are found to have significantly higher levels of IL-6 in the plasma, cerebrospinal fluid, and brains, especially locally around amyloid plaques (Wang et al., 2015). A study by Hull and colleagues has demonstrated that although IL-6 levels around amyloid plaques were found to be elevated, neuritic pathology had not yet developed, suggesting IL-6 may be a cause, and not just a consequence, of neuritic degeneration (Hüll et al., 1996a). Hence, IL-6 is implicated in the early pathology of AD with acute or chronic inflammatory components (Hüll et al., 1996b).
4.3. Thymoquinone downregulated several key NFκB signaling target genes
Several studies have indicated that TQ imparts its therapeutic effects partly via modulating the NFκB activation pathway (Mohamed et al., 2005, El Gazzar et al., 2007, Sayed and Morcos, 2007, Sethi et al., 2008, Al-Malki and Sayed, 2014). In this study, we investigated whether TQ treatment would modulate the gene expression of NFκB signaling targets in LPS/IFNγ-activated microglia. TQ treatment alone caused upregulation of the NQO1 gene which encodes for the anti-oxidant and detoxifying enzyme NAD(P)H: quinone oxidoreductase 1 (Brown et al., 2015). NQO1 enzyme performs two-electron reduction of quinones to hydroquinones while preventing one electron reduction (Brown et al., 2015). This two-electron reduction ensures complete oxidation of the quinone substrate without the formation of semiquinones and species with reactive oxygen radicals that are damaging to cells. NQO1 has a preference for short-chain acceptor quinones (Sparla et al., 1996) and plays a role in ubiquinone and vitamin E quinone metabolism, which protect cellular membranes from peroxidative injury in their reduced state. Furthermore, reduced forms of ubiquinone and vitamin E quinone have been shown to possess anti-oxidant properties that are superior to their non-reduced forms (Kohar et al., 1995). Similarly, TQ antioxidant activity and radical-scavenging capacity have been found to be attributed to it reduced form (thymohydroquinone) (Staniek and Gille, 2010). Therefore, the enzymatic activity of NQO1 is crucial for the protective effects of TQ. A recent study by Velagapudi and colleagues have confirmed the involvement of NQO1 in TQ's ability to inhibit neuroinflammation (Velagapudi et al., 2017). Additionally, in the LPS/IFNγ-activated microglial cells, thymoquinone treatment caused the downregulation of specific NFκB signaling target genes that were upregulated compared to the controls. Each of these genes encodes for proteins that are involved in inflammation and the immune response regulated by NFκB. Our study demonstrated that IL-6 gene expression was elevated in the activated microglial cells compared to the controls and the elevated expression was decreased > 14 fold when the cells were treated with TQ.
Nuclear factor kappa B (NFκB) is the main transcription factor that regulates the encoding of the IL-6 gene and plays a significant role in the age-related increase in IL-6 gene expression observed in the brain (Libermann and Baltimore, 1990). It has been demonstrated that NFκB binding to the IL-6 gene promoter is increased in glia and brain of aged mice compared to juvenile and adult (Ye and Johnson, 2001). Furthermore, NFκB's DNA-binding activity is higher in the forebrain and hippocampus of the aged rat as well as in the hippocampal and cerebral cortical neurons of Alzheimer's patients (Godbout and Johnson, 2004). Both circumstances lead to increased expression of the inflammatory cytokine IL-6. The ability to effectively and efficiently defend against oxidative stress declines with age (Ye and Johnson, 2001) allowing for more reactive oxygen species (ROS) to be available to react with surrounding molecules in the cellular environment. The increased levels of ROS precipitate the phosphorylation and ubiquitination of IκBa, which liberates NFκB, and thereby allows the transcription factor to translocate from the cell cytoplasm to the nucleus, resulting in the activation of the NFκB pathway (Schreck et al., 1992). This series of connections suggest that oxidative stress in the aged brain initiates a cascade that yields increased NFκB DNA-binding activity and enhanced expression of cytokines, specifically IL-6 (Ye and Johnson, 2001). Hence, the age-associated increase in brain IL-6 is due to increased binding of NFκB to the IL-6 promoter, which is related to the increasing ROS levels in an aged system (Ye and Johnson, 2001, Godbout and Johnson, 2004). TQ's ability to significantly reduce IL-6 gene expression further confirms our ELISArray assay results which illustrated that TQ treatment of activated microglia reduced the levels of the IL-6 pro-inflammatory cytokine.
Increasing evidence suggests that inflammation and neurodegeneration in AD brains are also partially mediated by complement activation which is correlated with cognitive impairment (Shen et al., 2013). The levels of complement mRNAs and their protein products have been found to be significantly higher in the livers of AD patients compared to those in the livers of healthy individuals (Tuppo and Arias, 2005). The complement system consists of a tightly regulated network of proteins that make up an integral part of the innate immune system (Dunkelberger and Song, 2010). The C3 and CFB genes encode for complement component 3 and complement factor B proteins, respectively, of the complement cascade. Complement Component 3 (C3) is an abundant plasma protein that plays a central role in the activation of the complement system in all three pathways (classic, alternative, and lectin) and hence, contributes considerably to innate immunity and the modulation of the inflammatory response (Sarma and Ward, 2011). Likewise, complement factor B (CFB) is a critical component protein of the complement system involved in the activation of the alternative complement pathway. Complement factor B binds to C3b, a product of the spontaneous hydrolysis of C3 in the alternative activation cascade, to form C3bB which precipitates the formation of C3 convertase and, through a series of steps, ultimately leads to the killing and clearing of invading pathogens and damaged cells (Janeway et al., 2001; Sarma and Ward, 2011). The complement system mediates immune responses to inflammatory triggers to attract additional phagocytes, enhances the ability of phagocytic cells to clear microbes and damaged cells, and precipitates lysis of foreign microbes via the membrane attack complex (MAC) (Janeway, Travers et al. 2001, Sarma and Ward, 2011). However, inappropriate complement activation can also cause cell injury or death and has been recognized as an important pathogenic factor in many diseases including neurodegenerative diseases such as AD (Crehan et al., 2012; Orsini et al., 2014). The gene expression of CFB and C3 were upregulated 266 and 12-fold, respectively, in the LPS/ IFNγ-activated microglial cells compared to the controls. TQ treatment of the activated microglia decreased the expression of CFB and C3 genes 37 and 3-fold, respectfully, indicating that TQ treatment decreases activation of the alternative pathway of the complement cascade in activated microglia.
Alongside the complement cascade, chemokines also play active roles in the in the innate immune response. In this study, the gene expression of CCL5, which encodes for chemokine (C–C motif) ligand 5, and CXCL3 which encodes for and chemokine (C-X-C motif) ligand 3, were elevated 402 and 11-fold, respectively, in the activated microglial cells compared to the controls. TQ treatment of the activated microglia decreased gene expression 7 and 8-fold compared to the untreated, activated cells. The CCL5 and CXCL3 chemokines are involved in acute inflammation and immunoregulation of the inflammatory process. Both CCL5 and CXCL3 chemokines emulated act as a chemoattractant and play an active role in recruiting leukocytes into inflammatory sites. CCL5 as a prominent chemokine that mediates the chemotaxis of microglia toward beta-amyloid (Aβ) aggregates observed in Alzheimer's disease (Huang et al., 2009), and it is commonly observed in the microcirculatory system of AD-affected brains, upregulated as a response to a cytokine-mediated increase of ROS (Tripathy et al., 2010). The microglial clustering around neuritic plaques contribute the neuroinflammation, and progressive neurodegeneration and CCL5 down-regulation reduce chemotaxis of microglia toward Aβγaggregates (Huang et al., 2010). Furthermore, it has also been demonstrated that CCL5 is up-regulated in the substantia nigra of PD mouse models and the neutralization of CCL5 protects against nigrostriatal degeneration (Chandra et al., 2016). CXCL3 has also been found to be upregulated other disease states (Martín-Fuentes et al., 2009; See et al., 2014; Gui et al., 2016).
TQ treatment in control microglial cells significantly upregulated the expression of antioxidant enzyme NAD(P)H dehydrogenase quinone 1. Additionally, TQ treatment in the activated microglial cells significantly downregulated the same genes that were upregulated by LPS/ IFNγ activation thereby reducing NFκB pathway activation/signaling and attenuating the pro-inflammatory response.
5. Conclusion
Our findings are the first to show that TQ treatment in the activated BV-2 microglial cells increased the expression of antioxidant and neuroprotective proteins, biliverdin reductase-A, 3-mercaptopyruvate sulfurtransferase, glutaredoxin-3, and mitochondrial lon protease. TQ also reduced the expression of several inflammatory cytokines in the LPS/ IFNγ activated BV-2 microglial cells. Furthermore, our studies showed TQ modulated the expression of genes involved in the NFκB signaling pathway, which play a key role in neuroinflammation. By modulating the expression of NFκB pathway genes, TQ may regulate the production of pro-inflammatory cytokines, and therefore, it could explain the mechanism by which TQ exhibited an inhibitory effect on the expression IL-2, IL-4, IL-6, IL-10, and IL-17a. Thus, our findings demonstrate TQ's potential in reducing neuroinflammation and neurodegeneration driven by microglial activation.
Acknowledgments
This research was supported by NIH-National Institute on Minority Health and Health Disparity Grants G12 MD007582 and P20 MD 006738.
List of abbreviations
- 3-MST
3 mercaptopyruvate sulfurtransferase
- AD
Alzheimer's disease
- ANOVA
analysis of variance
- ATP
adenosine triphosphate
- BVR-A
biliverdin reductase A
- C3
complement component 3
- CCL5
chemokine (C–C) motif ligand 5
- CFB
complement factor B
- CNS
central nervous system
- CXCL3
chemokine (C–C motif) ligand 3
- Grx
glutaredoxin
- GSH
glutathione
- IFNγ
interferon gamma
- IκB
inhibitor of kappa B
- IL
interleukin
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- LONM
lon protease, mitochondrial homolog
- LPS
lipopolysaccharide
- NFκB
nuclear factor kappa B
- NQO1
NAD(P)H:quinone oxidoreductase 1
- PD
Parkinson's disease
- ROS
reactive oxygen species
- TQ
thymoquinone
Footnotes
Conflict of interest
None declared.
References
- Ahmad A, Husain A, Mujeeb M, Khan SA, Najmi AK, Siddique NA, Damanhouri ZA, Anwar F. A review on the therapeutic potential of Nigella sativa: a miracle herb. Asian Pac J Trop Biomed. 2013;3(5):337–352. doi: 10.1016/S2221-1691(13)60075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mark R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhebshi AH, Gotoh M, Suzuki I. Thymoquinone protects cultured rat primary neurons against amyloid β-induced neurotoxicity. Biochem Biophys Res Commun. 2013;433(4):362–367. doi: 10.1016/j.bbrc.2012.11.139. [DOI] [PubMed] [Google Scholar]
- Alhebshi AH, Odawara A, Gotoh M, Suzuki I. Thymoquinone protects cultured hippocampal and human induced pluripotent stem cell-derived neurons against α-synuclein-induced synapse damage. Neurosci Lett. 2014;570:126–131. doi: 10.1016/j.neulet.2013.09.049. [DOI] [PubMed] [Google Scholar]
- Al-Majed AA, Al-Omar FA, Nagi MN. Neuroprotective effects of thymoquinone against transient forebrain ischemia in the rat hippocampus. Eur J Pharmacol. 2006;543(1–3):40–47. doi: 10.1016/j.ejphar.2006.05.046. [DOI] [PubMed] [Google Scholar]
- Al-Malki AL, Sayed AA. Thymoquinone attenuates cisplatin-induced hepatotoxicity via nuclear factor kappa-β. BMC Complement Altern Med. 2014;14:282. doi: 10.1186/1472-6882-14-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM. Inflammation in neurodegenerative diseases—an update. Immunology. 2014;142(2):151–166. doi: 10.1111/imm.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attia A, Ragheb A, Sylwestrowicz T, Shoker A. Attenuation of high cholesterol-induced oxidative stress in rabbit liver by thymoquinone. Eur J Gastroenterol Hepatol. 2010;22(7):826–834. doi: 10.1097/MEG.0b013e328336000d. [DOI] [PubMed] [Google Scholar]
- Azizi G, Mirshafiey A. The potential role of proinflammatory and anti-inflammatory cytokines in Alzheimer disease pathogenesis. Immunopharmacol Immunotoxicol. 2012;34(6):881–895. doi: 10.3109/08923973.2012.705292. [DOI] [PubMed] [Google Scholar]
- Azizi G, Navabi SS, Al-Shukaili A, Seyedzadeh MH, Yazdani R, Mirshafiey A. The role of inflammatory mediators in the pathogenesis of Alzheimer's disease. Sultan Qaboos Univ Med J. 2015;15(3):e305–e316. doi: 10.18295/squmj.2015.15.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badary OA, Taha RA, Gamal el-Din AM, Abdel-Wahab MH. Thymoquinone is a potent superoxide anion scavenger. Drug Chem Toxicol. 2003;26(2):87–98. doi: 10.1081/dct-120020404. [DOI] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23(7):2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Padhye S, Azmi A, Wang Z, Philip PA, Kucuk O, Sarkar FH, Mohammad RM. Review on molecular and therapeutic potential of thymoquinone in cancer. Nutr Cancer. 2010;62(7):938–946. doi: 10.1080/01635581.2010.509832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci U S A. 2002;99(25):16093–16098. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, Di Domenico F, Cenini G, Sultana R, Coccia R, Preziosi P, Perluigi M, Mancuso C, Butterfield DA. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer's disease and amnestic mild cognitive impairment. J Alzheimers Dis. 2011;25(4):623–633. doi: 10.3233/JAD-2011-110092. [DOI] [PubMed] [Google Scholar]
- Barone E, Di Domenico F, Mancuso C, Butterfield DA. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: it's time for reconciliation. Neurobiol Dis. 2014;62:144–159. doi: 10.1016/j.nbd.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat NR, Feinstein DL, Shen Q, Bhat AN. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells. Roles of nuclear factors, nuclear factor kappa B, cAMP response element-binding protein, CCAAT/ enhancer-binding protein-beta, and activating transcription factor-2. J Biol Chem. 2002;277(33):29584–29592. doi: 10.1074/jbc.M204994200. [DOI] [PubMed] [Google Scholar]
- Biswas SK. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxidative Med Cell Longev. 2016;2016:5698931. doi: 10.1155/2016/5698931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76(2):77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bota DA, Davies KJ. Mitochondrial Lon protease in human disease and aging: including an etiologic classification of Lon-related diseases and disorders. Free Radic Biol Med. 2016;100:188–198. doi: 10.1016/j.freeradbiomed.2016.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O, Tolstoy I, Pruitt KD, Maglott DR, Murphy TD. Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. 2015;43:D36–D42. doi: 10.1093/nar/gku1055. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress1,2. Free Radic Biol Med. 2002;32(11):1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: a central role for amyloid β-peptide. Trends Mol Med. 2001;7(12):548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Sultana R, Scapagnini G, Guagliano E, Sapienza M, Bella R, Kanski J, Pennisi G, Mancuso C, Stella AM, Butterfield DA. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer's disease. Antioxid Redox Signal. 2006;8(11–12):1975–1986. doi: 10.1089/ars.2006.8.1975. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Mancuso C, Barone E, Calafato S, Bates T, Rizzarelli E, Kostova AT. Vitagenes, dietary antioxidants and neuroprotection in neuro-degenerative diseases. Front Biosci (Landmark Ed) 2009;(14):376–397. doi: 10.2741/3250. [DOI] [PubMed] [Google Scholar]
- Camilleri A, Vassallo N. The centrality of mitochondria in the pathogenesis and treatment of Parkinson's disease. CNS Neurosci Ther. 2014;20(7):591–602. doi: 10.1111/cns.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra G, Rangasamy SB, Roy A, Kordower JH, Pahan K. Neutralization of RANTES and Eotaxin prevents the loss of dopaminergic neurons in a mouse model of Parkinson disease. J Biol Chem. 2016;291(29):15267–15281. doi: 10.1074/jbc.M116.714824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM. Mitochondrial dysfunction, metabolic deficits, and increased oxidative stress in Huntington's disease. Chang Gung Med J. 2011;34(2):135–152. [PubMed] [Google Scholar]
- Chen L, Liu L, Yin J, Luo Y, Huang S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int J Biochem Cell Biol. 2009;41(6):1284–1295. doi: 10.1016/j.biocel.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Chen WW, Zhang X, Huang WJ. Role of neuroinflammation in neurodegenerative diseases (review) Mol Med Rep. 2016;13(4):3391–3396. doi: 10.3892/mmr.2016.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Lee DY, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: the role of microglial NADPH oxidase. J Neurosci. 2005;25(16):4082–4090. doi: 10.1523/JNEUROSCI.4306-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chongthammakun V, Sanvarinda Y, Chongthammakun S. Reactive oxygen species production and MAPK activation are implicated in tetrahydrobiopterin-induced SH-SY5Y cell death. Neurosci Lett. 2009;449(3):178–182. doi: 10.1016/j.neulet.2008.10.106. [DOI] [PubMed] [Google Scholar]
- Cobourne-Duval MK, Taka E, Mendonca P, Bauer D, Soliman KF. The antioxidant effects of Thymoquinone in activated BV-2 murine microglial cells. Neurochem Res. 2016;41(12):3227–3238. doi: 10.1007/s11064-016-2047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M, Carrì MT. Mitochondrial dysfunction in ALS. Prog Neurobiol. 2012;97(2):54–66. doi: 10.1016/j.pneurobio.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Crehan H, Hardy J, Pocock J. Microglia, Alzheimer's disease, and complement. Int J Alzheimers Dis. 2012;2012:983640. doi: 10.1155/2012/983640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates the rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Doré S, Takahashi M, Ferris CD, Zakhary R, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A. 1999;96(5):2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- El Gazzar MA, El Mezayen R, Nicolls MR, Dreskin SC. Thymoquinone attenuates proinflammatory responses in lipopolysaccharide-activated mast cells by modulating NF-kappaB nuclear transactivation. Biochim Biophys Acta Gen Subj. 2007;1770(4):556–564. doi: 10.1016/j.bbagen.2007.01.002. [DOI] [PubMed] [Google Scholar]
- El-Mahmoudy A, Matsuyama H, Borgan MA, Shimizu Y, El-Sayed MG, Minamoto N, Takewaki T. Thymoquinone suppresses the expression of inducible nitric oxide synthase in rat macrophages. Int Immunopharmacol. 2002;2(11):1603–1611. doi: 10.1016/s1567-5769(02)00139-x. [DOI] [PubMed] [Google Scholar]
- Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58(1):39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Fahmy HM, Noor NA, Mohammed FF, Elsayed AA, Radwan NM. Nigella sativa as an anti-inflammatory and promising remyelinating agent in the cortex and hippocampus of experimental autoimmune encephalomyelitis-induced rats. The Journal of Basic & Applied Zoology. 2014;67(5):182–195. [Google Scholar]
- Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322(1–2):254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal. 2004;6(1):63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- Ferreira B, Mendes F, Osório N, Caseiro A, Gabriel A, Valado A. Glutathione in multiple sclerosis. Br J Biomed Sci. 2013;70(2):75–79. doi: 10.1080/09674845.2013.11669939. [DOI] [PubMed] [Google Scholar]
- Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29(8):357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholamnezhad Z, Rafatpanah H, Sadeghnia HR, Boskabady MH. Immunomodulatory and cytotoxic effects of Nigella sativa and thymoquinone on rat splenocytes. Food Chem Toxicol. 2015;86:72–80. doi: 10.1016/j.fct.2015.08.028. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Interleukin-6 in the aging brain. J Neuroimmunol. 2004;147(1–2):141–144. doi: 10.1016/j.jneuroim.2003.10.031. [DOI] [PubMed] [Google Scholar]
- González-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Gu F, Chauhan V, Chauhan A. Glutathione redox imbalance in brain disorders. Curr Opin Clin Nutr Metab Care. 2015;18(1):89–95. doi: 10.1097/MCO.0000000000000134. [DOI] [PubMed] [Google Scholar]
- Gui SL, Teng LC, Wang SQ, Liu S, Lin YL, Zhao XL, Liu L, Sui HY, Yang Y, Liang LC, Wang ML, Li XY, Cao Y, Li FY, Wang WQ. Overexpression of CXCL3 can enhance the oncogenic potential of prostate cancer. Int Urol Nephrol. 2016;48(5):701–709. doi: 10.1007/s11255-016-1222-2. [DOI] [PubMed] [Google Scholar]
- Gülşen İ, Ak H, Çölçimen N, Alp HH, Akyol ME, Demir İ, Atalay T, Balahroğlu R, Rağbetli MÇ. Neuroprotective effects of Thymoquinone on the Hippocampus in a rat model of traumatic brain injury. World Neurosurgery. 2016;86:243–249. doi: 10.1016/j.wneu.2015.09.052. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Heales SJ, Bolaños JP, Stewart VC, Brookes PS, Land JM, Clark JB. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta. 1999;1410(2):215–228. doi: 10.1016/s0005-2728(98)00168-6. [DOI] [PubMed] [Google Scholar]
- Hu LF, Lu M, Tiong CX, Dawe GS, Hu G, Bian JS. Neuroprotective effects of hydrogen sulfide on Parkinson's disease rat models. Aging Cell. 2010;9(2):135–146. doi: 10.1111/j.1474-9726.2009.00543.x. [DOI] [PubMed] [Google Scholar]
- Huang WC, Yen FC, Shiao YJ, Shie FS, Chan JL, Yang CN, Sung YJ, Huang FL, Tsay HJ. Enlargement of Abeta aggregates through chemokine-dependent microglial clustering. Neurosci Res. 2009;63(4):280–287. doi: 10.1016/j.neures.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Huang WC, Yen FC, Shie FS, Pan CM, Shiao YJ, Yang CN, Huang FL, Sung YJ, Tsay HJ. TGF-beta1 blockade of microglial chemotaxis toward Abeta aggregates involves SMAD signaling and down-regulation of CCL5. J Neuroinflammation. 2010;7:28. doi: 10.1186/1742-2094-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüll M, Berger M, Volk B, Bauer J. Occurrence of interleukin-6 in cortical plaques of Alzheimer's disease patients may precede transformation of diffuse into neuritic plaques. Ann N Y Acad Sci. 1996a;777:205–212. doi: 10.1111/j.1749-6632.1996.tb34420.x. [DOI] [PubMed] [Google Scholar]
- Hüll M, Strauss S, Berger M, Volk B, Bauer J. The participation of inter-leukin-6, a stress-inducible cytokine, in the pathogenesis of Alzheimer's disease. Behav Brain Res. 1996b;78(1):37–41. doi: 10.1016/0166-4328(95)00213-8. [DOI] [PubMed] [Google Scholar]
- Ismail M, Al-Naqeep G, Chan KW. Nigella sativa thymoquinone-rich fraction greatly improves plasma antioxidant capacity and expression of antioxidant genes in hypercholesterolemic rats. Free Radic Biol Med. 2010;48(5):664–672. doi: 10.1016/j.freeradbiomed.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Janeway CAJ, Travers P, Walport M, Schlomchik M. Immunobiology: The Immune System in Health and Disease. Taylor & Francis, Inc; New York, Garland Science: 2001. The Complement System and Innate Immunity. [Google Scholar]
- Kanter M. Nigella sativa and derived thymoquinone prevent hippocampal neurodegeneration after chronic toluene exposure in rats. Neurochem Res. 2008;33(3):579–588. doi: 10.1007/s11064-007-9481-z. [DOI] [PubMed] [Google Scholar]
- Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, Zaheer S, Iyer SS, Zaheer A. Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1) [PMC free article] [PubMed] [Google Scholar]
- Kenchappa RS, Diwakar L, Annepu J, Ravindranath V. Estrogen and neuroprotection: higher constitutive expression of glutaredoxin in female mice offers protection against MPTP-mediated neurodegeneration. FASEB J. 2004;18(10):1102–1104. doi: 10.1096/fj.03-1075fje. [DOI] [PubMed] [Google Scholar]
- Keyhanmanesh R, Pejman L, Omrani H, Mirzamohammadi Z, Shahbazfar AA. The effect of single dose of thymoquinone, the main constituents of Nigella sativa, in guinea pig model of asthma. Bioimpacts. 2014;4(2):75–81. doi: 10.5681/bi.2014.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Vaibhav K, Javed H, Khan MM, Tabassum R, Ahmed ME, Srivastava P, Khuwaja G, Islam F, Siddiqui MS, Safhi MM, Shafi MM. Attenuation of Aβ-induced neurotoxicity by thymoquinone via inhibition of mitochondrial dysfunction and oxidative stress. Mol Cell Biochem. 2012;369(1–2):55–65. doi: 10.1007/s11010-012-1368-x. [DOI] [PubMed] [Google Scholar]
- Khazdair MR. The protective effects of Nigella sativa and its constituents on induced neurotoxicity. J Toxicol. 2015;2015:841823. doi: 10.1155/2015/841823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida K, Yamada M, Tokuda K, Marutani E, Kakinohana M, Kaneki M, Ichinose F. Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson's disease. Antioxid Redox Signal. 2011;15(2):343–352. doi: 10.1089/ars.2010.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81(3):302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Kim SY, Kang HT, Choi HR, Park SC. Biliverdin reductase A in the prevention of cellular senescence against oxidative stress. Exp Mol Med. 2011;43(1):15–23. doi: 10.3858/emm.2011.43.1.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18(10):1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12(1):1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- Kohar I, Baca M, Suarna C, Stocker R, Southwell-Keely PT. Is alpha-tocopherol a reservoir for alpha-tocopheryl hydroquinone? Free Radic Biol Med. 1995;19(2):197–207. doi: 10.1016/0891-5849(95)00010-u. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lee M, Sparatore A, Del Soldato P, McGeer E, McGeer PL. Hydrogen sulfide-releasing NSAIDs attenuate neuroinflammation induced by microglial and astrocytic activation. Glia. 2010;58(1):103–113. doi: 10.1002/glia.20905. [DOI] [PubMed] [Google Scholar]
- Lefkowitz DL, Lefkowitz SS. Microglia and myeloperoxidase: a deadly partnership in neurodegenerative disease. Free Radic Biol Med. 2008;45(5):726–731. doi: 10.1016/j.freeradbiomed.2008.05.021. [DOI] [PubMed] [Google Scholar]
- Leszek J, Barreto GE, Gąsiorowski K, Koutsouraki E, Ávila-Rodrigues M, Aliev G. Inflammatory mechanisms and oxidative stress as key factors responsible for progression of neurodegeneration: role of brain innate immune system. CNS Neurol Disord Drug Targets. 2016;15(3):329–336. doi: 10.2174/1871527315666160202125914. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, Bian JS. Hydrogen sulfide protects amyloid-βγinduced cell toxicity in microglia. J Alzheimers Dis. 2010;22(4):1189–1200. doi: 10.3233/JAD-2010-101002. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304(1):1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu J, Tetzlaff W, Paty DW, Cynader MS. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic Biol Med. 2006;40(6):960–967. doi: 10.1016/j.freeradbiomed.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40(2):139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- Mahgoub AA. Thymoquinone protects against experimental colitis in rats. Toxicol Lett. 2003;143(2):133–143. doi: 10.1016/s0378-4274(03)00173-5. [DOI] [PubMed] [Google Scholar]
- Mahmoud AM, Ahmed OM, Galaly SR. Thymoquinone and curcumin attenuate gentamicin-induced renal oxidative stress, inflammation, and apoptosis in rats. EXCLI J. 2014;13:98–110. [PMC free article] [PubMed] [Google Scholar]
- Maines MD, Panahian N. The heme oxygenase system and cellular defense mechanisms. Do HO-1 and HO-2 have different functions? Adv Exp Med Biol. 2001;502:249–272. doi: 10.1007/978-1-4757-3401-0_17. [DOI] [PubMed] [Google Scholar]
- Mancuso C. Heme oxygenase and its products in the nervous system. Antioxid Redox Signal. 2004;6(5):878–887. doi: 10.1089/ars.2004.6.878. [DOI] [PubMed] [Google Scholar]
- Mansour MA, Nagi MN, El-Khatib AS, Al-Bekairi AM. Effects of thymoquinone on antioxidant enzyme activities, lipid peroxidation and DT-diaphorase in different tissues of mice: a possible mechanism of action. Cell Biochem Funct. 2002;20(2):143–151. doi: 10.1002/cbf.968. [DOI] [PubMed] [Google Scholar]
- Martín-Fuentes P, Civeira F, Solanas-Barca M, García-Otín AL, Jarauta E, Cenarro A. Overexpression of the CXCL3 gene in response to oxidized low-density lipoprotein is associated with the presence of tendon xanthomas in familial hypercholesterolemia. Biochem Cell Biol. 2009;87(3):493–498. doi: 10.1139/o09-006. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging. 2001;22(6):799–809. doi: 10.1016/s0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- Mohamed A, Afridi DM, Garani O, Tucci M. Thymoquinone inhibits the activation of NF-kappaB in the brain and spinal cord of experimental autoimmune encephalomyelitis. Biomed Sci Instrum. 2005;41:388–393. [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132(Pt 2):288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JK, Davies KJ. Mitochondrial Lon protease is a human stress protein. Free Radic Biol Med. 2009;46(8):1042–1048. doi: 10.1016/j.freeradbiomed.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JK, Pomatto LC, Davies KJ. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress, but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013;1:258–264. doi: 10.1016/j.redox.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96(Pt A):29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T. NF-kappaB and rheumatic diseases. Endocr Metab Immune Disord Drug Targets. 2006;6(4):359–372. doi: 10.2174/187153006779025685. [DOI] [PubMed] [Google Scholar]
- Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci. 2014;8:380. doi: 10.3389/fncel.2014.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhye S, Banerjee S, Ahmad A, Mohammad R, Sarkar FH. From here to eternity - the secret of pharaohs: therapeutic potential of black cumin seeds and beyond. Cancer Ther. 2008;6(b):495–510. [PMC free article] [PubMed] [Google Scholar]
- Paludan SR. Synergistic action of pro-inflammatory agents: cellular and molecular aspects. J Leukoc Biol. 2000;67(1):18–25. doi: 10.1002/jlb.67.1.18. [DOI] [PubMed] [Google Scholar]
- Panahian N, Huang T, Maines MD. Enhanced neuronal expression of the oxidoreductase–biliverdin reductase–after permanent focal cerebral ischemia. Brain Res. 1999;850(1–2):1–13. doi: 10.1016/s0006-8993(99)01726-6. [DOI] [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferon gamma. J Neurosci Res. 2004;77(4):540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Pham K, Pal R, Qu Y, Liu X, Yu H, Shiao SL, Wang X, O'Brian Smith E, Cui X, Rodney GG, Cheng N. Nuclear glutaredoxin 3 is critical for protection against oxidative stress-induced cell death. Free Radic Biol Med. 2015;85:197–206. doi: 10.1016/j.freeradbiomed.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon HF, Calabrese V, Scapagnini G, Butterfield DA. Free radicals: the key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontol A Biol Sci Med Sci. 2004;59(5):478–493. doi: 10.1093/gerona/59.5.m478. [DOI] [PubMed] [Google Scholar]
- Pujol-Carrion N, de la Torre-Ruiz MA. Glutaredoxins Grx4 and Grx3 of Saccharomyces cerevisiae play a role in actin dynamics through their Trx domains, which contributes to oxidative stress resistance. Appl Environ Microbiol. 2010;76(23):7826–7835. doi: 10.1128/AEM.01755-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radad K, Moldzio R, Taha M, Rausch WD. Thymoquinone protects dopaminergic neurons against MPP+ and rotenone. Phytother Res. 2009;23(5):696–700. doi: 10.1002/ptr.2708. [DOI] [PubMed] [Google Scholar]
- Radad K, Hassanein K, Al-Shraim M, Moldzio R, Rausch WD. Thymoquinone ameliorates lead-induced brain damage in Sprague Dawley rats. Exp Toxicol Pathol. 2014;66(1):13–17. doi: 10.1016/j.etp.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer's disease. Arch Med Res. 2008;39(1):1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Sabens Liedhegner EA, Gao XH, Mieyal JJ. Mechanisms of altered redox regulation in neurodegenerative diseases—focus on S—glutathionylation. Antioxid Redox Signal. 2012;16(6):543–566. doi: 10.1089/ars.2011.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed U, Durgadoss L, Valli RK, Joshi DC, Joshi PG, Ravindranath V. Knockdown of cytosolic glutaredoxin 1 leads to loss of mitochondrial membrane potential: implication in neurodegenerative diseases. PLoS One. 2008;3(6):e2459. doi: 10.1371/journal.pone.0002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharan S, Mandal PK. The emerging role of glutathione in Alzheimer's disease. J Alzheimers Dis. 2014;40(3):519–529. doi: 10.3233/JAD-132483. [DOI] [PubMed] [Google Scholar]
- Salem ML. Immunomodulatory and therapeutic properties of the Nigella sativa L. seed. Int Immunopharmacol. 2005;5(13–14):1749–1770. doi: 10.1016/j.intimp.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Salzano S, Checconi P, Hanschmann EM, Lillig CH, Bowler LD, Chan P, Vaudry D, Mengozzi M, Coppo L, Sacre S, Atkuri KR, Sahaf B, Herzenberg LA, Mullen L, Ghezzi P. Linkage of inflammation and oxidative stress via the release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A. 2014;111(33):12157–12162. doi: 10.1073/pnas.1401712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343(1):227–235. doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed AA, Morcos M. Thymoquinone decreases AGE-induced NF-kappaB activation in proximal tubular epithelial cells. Phytother Res. 2007;21(9):898–899. doi: 10.1002/ptr.2177. [DOI] [PubMed] [Google Scholar]
- Schreck R, Albermann K, Baeuerle P. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells. Radical Res Communicat. 1992;17(4):221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Schluesener HJ. Microglia rules insights into microglial-neuronal signaling. Cell Death Differ. 2004;11(12):1245–1246. doi: 10.1038/sj.cdd.4401487. [DOI] [PubMed] [Google Scholar]
- See AL, Chong PK, Lu SY, Lim YP. CXCL3 is a potential target for breast cancer metastasis. Curr Cancer Drug Targets. 2014;14(3):294–309. doi: 10.2174/1568009614666140305222328. [DOI] [PubMed] [Google Scholar]
- Sethi G, Ahn KS, Aggarwal BB. Targeting nuclear factor-kappa B activation pathway by thymoquinone: role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol Cancer Res. 2008;6(6):1059–1070. doi: 10.1158/1541-7786.MCR-07-2088. [DOI] [PubMed] [Google Scholar]
- Shen S, Yu S, Binek J, Chalimoniuk M, Zhang X, Lo SC, Hannink M, Wu J, Fritsche K, Donato R, Sun GY. Distinct signaling pathways for induction of type II NOS by IFN gamma and LPS in BV-2 microglial cells. Neurochem Int. 2005;47(4):298–307. doi: 10.1016/j.neuint.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Shen Y, Yang L, Li R. What does complement do in Alzheimer's disease? Old molecules with new insights. Transl Neurodegener. 2013;2(1):21. doi: 10.1186/2047-9158-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14(1):133–145. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11(4):703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- Smeyne M, Smeyne RJ. Glutathione metabolism and Parkinson's disease. Free Radic Biol Med. 2013;62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, Wiggert B, Petersen RB, Perry G. Heme oxygenase-1 is associated with the neuroff-brillary pathology of Alzheimer's disease. Am J Pathol. 1994;145(1):42–47. [PMC free article] [PubMed] [Google Scholar]
- Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparla F, Tedeschi G, Trost P. NAD(P)H:(Quinone-acceptor) oxidoreductase of tobacco leaves is a Flavin mononucleotide-containing Flavoenzyme. Plant Physiol. 1996;112(1):249–258. doi: 10.1104/pp.112.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staniek K, Gille L. Is thymoquinone an antioxidant? BMC Pharmacol. 2010;10(Suppl 1):A9. [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1(1):14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taka E, Mazzio EA, Goodman CB, Redmon N, Flores-Rozas H, Reams R, Darling-Reed S, Soliman KF. Anti-inflammatory effects of thymoquinone in activated BV-2 microglial cells. J Neuroimmunol. 2015;286:5–12. doi: 10.1016/j.jneuroim.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The consortium U. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torreilles F, Salman-Tabcheh S, Guérin M, Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev. 1999;30(2):153–163. doi: 10.1016/s0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- Tripathy D, Thirumangalakudi L, Grammas P. RANTES upregulation in the Alzheimer's disease brain: a possible neuroprotective role. Neurobiol Aging. 2010;31(1):8–16. doi: 10.1016/j.neurobiolaging.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuppo EE, Arias HR. The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol. 2005;37(2):289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Umar S, Zargan J, Umar K, Ahmad S, Katiyar CK, Khan HA. Modulation of the oxidative stress and inflammatory cytokine response by thymoquinone in collagen-induced arthritis in Wistar rats. Chem Biol Interact. 2012;197(1):40–46. doi: 10.1016/j.cbi.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Velagapudi R, Kumar A, Bhatia HS, El-Bakoush A, Lepiarz I, Fiebich BL, Olajide OA. Inhibition of neuroinflammation by thymoquinone requires activation of Nrf2/ARE signaling. Int Immunopharmacol. 2017;48:17–29. doi: 10.1016/j.intimp.2017.04.018. [DOI] [PubMed] [Google Scholar]
- Von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. 2015;7:124. doi: 10.3389/fnagi.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta. 2014;1842(8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med. 2015;3(10):136. doi: 10.3978/j.issn.2305-5839.2015.03.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, Moore PK. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite 'scavenger'? J Neurochem. 2004;90(3):765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal. 2010;12(10):1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson BL, Landreth GE. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer's disease. J Neuroinflammation. 2006;3:30. doi: 10.1186/1742-2094-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. 2013;62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from the brain of aged mice. J Neuroimmunol. 1999;93(1–2):139–148. doi: 10.1016/s0165-5728(98)00217-3. [DOI] [PubMed] [Google Scholar]
- Ye S-M, Johnson RW. Regulation of interleukin-6 gene expression in the brain of aged mice by nuclear factor κB. J Neuroimmunol. 2001;117(1–2):87–96. doi: 10.1016/s0165-5728(01)00316-2. [DOI] [PubMed] [Google Scholar]
- Yi T, Cho SG, Yi Z, Pang X, Rodriguez M, Wang Y, Sethi G, Aggarwal BB, Liu M. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol Cancer Ther. 2008;7(7):1789–1796. doi: 10.1158/1535-7163.MCT-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Choi JW, Shin CY, Jeon SJ, Park SJ, Cheong JH, Han SY, Ryu JR, Song MR, Ko KH. Activation of p38 MAPK induced peroxynitrite generation in LPS plus IFN-gamma-stimulated rat primary astrocytes via activation of iNOS and NADPH oxidase. Neurochem Int. 2008;52(6):1188–1197. doi: 10.1016/j.neuint.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH, Liu LS, Jiang ZS. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol Cell Biol. 2013;33(6):1104–1113. doi: 10.1128/MCB.01215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Dong Y, Yu Q, Kai Z, Zhang M, Jia C, Xiao C, Zhang B, Li M. Function of glutaredoxin 3 (Grx3) in oxidative stress response caused by iron homeostasis disorder in Candida albicans. Future Microbiol. 2017;12:1397–1412. doi: 10.2217/fmb-2017-0098. [DOI] [PubMed] [Google Scholar]