

Abstract
We report a series of tubulin targeting agents, some of which demonstrate potent antiproliferative activities. These analogs were designed to optimize the antiproliferative activity of 1 by varying the heteroatom substituent at the 4′-position, the basicity of the 4-position amino moiety, and conformational restriction. The potential metabolites of the active compounds were also synthesized. Some compounds demonstrated single digit nanomolar IC50 values for antiproliferative effects in MDA-MB-435 melanoma cells. Particularly, the S-methyl analog 3 was more potent than 1 in MDA-MB-435 cells (IC50 = 4.6 nM). Incubation of 3 with human liver microsomes showed that the primary metabolite of the S-methyl moiety of 3 was the methyl sulfinyl group, as in analog 5. This metabolite was equipotent with the lead compound 1 in MDA-MB-435 cells (IC50 = 7.9 nM). Molecular modeling and electrostatic surface area were determined to explain the activities of the analogs. Most of the potent compounds overcome multiple mechanisms of drug resistance and compound 3 emerged as the lead compound for further SAR and preclinical development.
Keywords: cyclopenta[d]pyrimidine, microtubule targeting agent, metabolism
Graphical abstract
1. Introduction
Tubulin binding agents (TBAs) (Figure 1) are a structurally diverse group of compounds and many have utility in the treatment of cancer.1 Five well established classes of TBAs are classified on the basis of their interactions within the αβ heterodimers of microtubules.2 Taxoid and laulimalide site agents stabilize microtubules, while vinca, maytansine and colchicine site agents inhibit microtubule formation and cause loss of cellular microtubules. However, triazolopyrimidines, which bind to vinca site, promote microtubule stability.3 Drugs, including paclitaxel, cabazitaxel, docetaxel, ixabepilone and several other chemotypes bind to β-tubulin at a site on the interior of the microtubule known as the taxoid site.4 Taxoids are useful in the treatment of breast, lung, head and neck, ovarian, and prostate cancers.5 Laulimalide and peloruside A also stabilize microtubules, but they bind to a non-overlapping site on β tubulin on the exterior of the microtubule.6 The vinca domain agents bind to β-tubulin at the αβ interface between different tubulin dimers. Vincristine, vinblastine, vinorelbine and eribulin are successful anticancer agents. Maytansine binds to a site on β tubulin, which is distinct from the vinca site. Maytansine is used as the payload of an antibody drug conjugate Kadcyla®, a drug used to treat advanced HER2 positive breast cancer.7 A diverse collection of small molecules along with colchicine and the combretastatins bind to the colchicine site on β-tubulin at its interface with α-tubulin within a single tubulin heterodimer.8
Figure 1.

Structures of microtubule targeting agents.
The expression of P-glycoproteins (Pgp), a drug efflux pump, and βIII-tubulin, one of many β-tubulin isotypes, are clinically established mechanisms of drug resistance to microtubule targeting drugs.9 Most colchicine site binding agents are not Pgp substrates and also possess the ability to circumvent βIII-tubulin mediated drug resistance.10 Although colchicine itself is not used as an anticancer agent, several colchicine site agents have been evaluated in clinical trials, including 2-methoxyestradiol, combretastatin A-4 (CA-4), the phosphorylated prodrug combretastatin A-4 phosphate, the combretastatin A-1 phosphate prodrug, BNC105P (sodium 6-methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl)benzofuran-7-yl phosphate), verubulin (MPC-6827), ABT-751 (N-[2-[(4-hydroxyphenyl)amino]-3-pyridinyl]-4-methoxybenzenesulfonamide) and plinabulin (NPT-2358).11 However, to date no colchicine site agent has advanced to Phase III studies, attesting to the necessity to develop viable colchicine site agents for evaluation as potential cancer chemotherapeutic drugs.
2. Rationale
We previously reported compound 1 (Table 1) as a potent colchicine site antiproliferative agent (IC50 = 7.0 nM in MDA-MB-435 cells antiproliferative assay).12 In the structure-activity relationship13 studies of cyclopenta[d]pyrimidine based antitubulin agents, we demonstrated the importance of the para-methoxyphenyl moiety for biological activity. However, as illustrated by marketed drugs anisindione, adapalene and olodaterol, the major metabolic pathway of the para-methoxyphenyl moiety is O-demethylation.14,15 The O-demethylated metabolite of 1, compound 2 was synthesized and found to be much less potent (IC50 = 405 nM MDA-MB-435 cells) than 1. The potential metabolic instability of 1 and poor activity of its probable metabolite 2 propelled us to pursue cytotoxic compounds with the cyclopenta[d]pyrimidine scaffold and different substituents at the 4′-anilino site.
Table 1.
Structures of lead compound 1 and the designed compounds 2–14.
| Compd | structure | Compd | structure | Compd | structure |
|---|---|---|---|---|---|
| 1 |
|
6 |
|
11 |
|
| 2 |

|
7 |
|
12 |

|
| 3 |
|
8 |
|
13 |
|
| 4 |
|
9 |
|
14 |
|
| 5 |

|
10 |
|
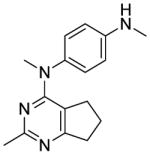
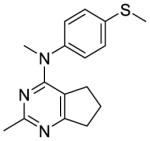
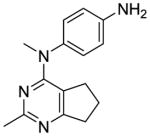
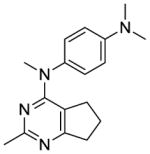
The distance and the nature of heteroatom substitution affects hydrogen bond (HB) strength.16 The HB acceptor (HBA) ability of sulfur is slightly weaker than that of oxygen.17 Nitrogen has a comparable HBA ability as oxygen.18,19 Thus, it was of interest to isosterically replace the oxygen atom of the 4′-OCH3 of 1 with a sulfur moiety as in 3 or an amino moiety as in 4. The primary metabolite of drugs containing the methylthiophenyl moiety (e.g., marketed drug thioridazine) is the methylsulfinylphenyl (e.g., marketed drug mesoridazine).20 To identify the metabolic pathway of compound 3, an in vitro metabolism study of 3 in human liver microsomes was performed.21 The principal metabolite of the methylthio moiety in 3 was the methylsulfinyl, and one of the minor metabolites was the methylsulfonyl moiety. Thus, the methylsulfinyl analog 5 and the methylsulfonyl analog 6 were also synthesized to determine their antiproliferative activities. The major metabolic pathway of the dimethylaminophenyl moiety (e.g., marketed drug mifepristone) is N-demethylation.22 Similarly, the 4′-dimethylamino compound 4 is expected to be metabolized to the monomethylamino 7, and the amino 8 via N-demethylation. Hence, compounds 7 and 8 were also synthesized to evaluate the antiproliferative activity of the potential metabolites of 4.
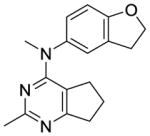
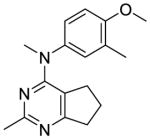
Compounds 9 and 10 were designed to restrict the conformation of the 4′-methoxy moiety. In addition, replacement of the aromatic phenyl ring with a saturated cyclohexyl ring, as in compound 11, was also synthesized to determine the importance of the aromatic phenyl moiety to activity.
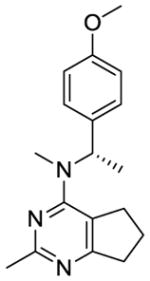
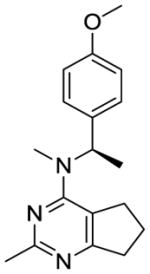
We were also interested in the homologation of the para-methoxy phenyl moiety. This was accomplished by adding a methylene moiety between the phenyl ring and the 4-position amine, as in 12. This homologation decreases the 4′-methoxy H-bonding distance to water molecule HOH728 in the x-ray crystal structure of colchicine at the colchicine site (PDB:4O2B23) and could increase potency. Substitution of a methyl group on the methylene of 12 affords a chiral center and sterically restricts the conformational preferences of the C4-N bond (bond a, Table 1), which could affect biological activity. Conformational search about bond a was carried out using Sybyl-X 2.1.1 with 1° rotation.24 There were 79 low energy conformations generated for compound 12 within 1 kcal/mol. For compounds 13 and 14, the number of low energy conformations generated were 46 and 34, respectively, indicating that low energy conformations of 13 and 14 were indeed decreased compared to 12.
Compounds 1–14 were docked in the x-ray crystal structure of the colchicine site in tubulin (PDB: 4O2B, 2.3 Å)23 using the parameters specified in the experimental section. Multiple low energy conformations were obtained on docking. As representative examples, Fig. 2A shows the docked conformation of 3 (magenta), superimposed on the co-crystallized ligand, colchicine (pink), and lead compound 1 (green). The cyclopenta[d]pyrimidine scaffold of 3 forms hydrophobic interactions with Alaα180, Valα181, Asnβ258, Metβ259 and Lysβ352. The N4-methyl in 1 is oriented towards a hydrophobic pocket lined by residues Alaβ250, Lysβ254 and Leuβ255, but, in 3, the N-methyl group adopts a different conformation and interacts with Valβ315 and Thrβ353. The N-phenyl group of 3 superimposes on the A ring of colchicine and interacts with Leuβ248, Alaβ250, Leuβ255 and Alaβ316. The 4′-methoxy group in 1 interacts with Valβ238, Cysβ241, and Ileβ378, and in 3 it interacts with Cysβ241 and Leuβ242. In addition, the oxygen atom of the 4′-methoxy group lies within H-bonding distance (3.0 Å for 1 and 2.16 Å for 3) with a water molecule (HOH728) present in the crystal structure in the vicinity of Cysβ241. The best docked poses of 1 and 3 had scores of −7.91 kcal/mol and −8.07 kcal/mol, respectively, indicating that the conformation of 3 was similar to that of 1. Fig. 2B shows the docked pose of 5 (orange), superimposed on the crystallized ligand colchicine (pink) and lead compound 1 (green). The mode of binding of 5, the principal metabolite of 3, in tubulin was similar to that of 1, except that for 5 the 4′-sulfoxide group undergoes H-bonding with HOH728. Compound 5 had a docked score of −8.19 kcal/mol, similar to those of 1 and 3. All the remaining compounds, except 2 and 6, docked similarly in the colchicine site of tubulin and had scores that ranged between −7.66 and −8.35 kcal/mol, similar to that of lead compound 1. Compounds 2 and 6 failed to dock in a pose similar to that shown for 1, suggesting that these analogs may show reduced binding or no binding at the colchicine site and were included as potential negative controls.
Figure 2.

A. Superimposition of the docked poses of 1 (green), 3 (magenta) and colchicine (pink) in the colchicine site of tubulin (PDB ID: 4O2B). A dash line represented the border of α-tubulin and β-tubulin. B. Superimposition of the docked poses of 1 (green), 5 (orange) and colchicine (pink) in the colchicine site of tubulin (PDB ID: 4O2B).
3. Chemistry
Cyclization of commercially available 15 (Scheme 1) with acetamidine hydrochloride and potassium tert-butoxide in DMF afforded the cyclopenta[d]pyrimidine 16 in 79% yield.25 Chlorination of 16 with excess POCl3 gave 4-chlorocyclopenta[d]pyrimidine 17 in 73% yield.
Scheme 1.

Synthesis of intermediate 4-chloro-2-methylcyclopenta[d]pyrimidine, 17.
Reagents and conditions: (a) Acetamidine hydrochloride, KOt-Bu, DMF, 120 °C, 79%; (b) POCl3, 100 °C, 4 h, 73%.
Monomethylation of 18, 22 and 24 (Scheme 2) with CH3I and NaH in THF provided the N-methyl anilines 19, 23 and 25, respectively, in 65–75% yields. Oxidation of the N-methyl-4-methylthioaniline 19 with meta-chloroperoxybenzoic acid (mCPBA) (1.1 eq.) in acetonitrile (ACN) at 0 °C afforded the sulfoxide congener 20 in 67% yield.26 Further oxidation of the sulfoxide with an additional 1.1 equivalent of mCPBA under similar oxidation condition afforded the sulfone 21 in 59% yield.
Scheme 2.

Synthesis of anilines and methylthio derivatives.
Reagents and conditions: (a) NaH, THF, CH3I, 0 °C, 65–75%; (b) mCPBA (1.1 eq.), ACN, 0 °C, 1 h, 67%; (c) mCPBA (1.1 eq.), ACN, 0 °C, 1 h, 59%.
Formyl protection of the amines in 26a-b and 33a–c (Scheme 3) with formic acid and acetic anhydride afforded the N-formyl anilines 27a-b and 34a-c. Reduction of the formyl group in 27a-b and 34a-c with LiAlH4 in THF afforded the corresponding N-methyl anilines in 53–72% yields. Protection of the amino group in trans-4-hydroxycyclohexylamine 29 with CBzCl and TEA in ACN afforded 30 in 90% yield. N- and O- methylation of 30 with NaH and CH3I afforded 31 in 68% yield. Reductive deprotection of 31 with Pd catalyzed hydrogenation provided 32 in 92% yield.
Scheme 3.

Synthesis of anilines, derivatives and related intermediates.
Reagents and conditions: (a) HCOOH, Ac2O, DCM, rt, 4–6 h; (b) THF, LiAlH4, 60 °C, 53–72%; (c) CBzCl, ACN, TEA, 0 °C, 90%; (d) NaH, THF, CH3I, 0 °C, 68%; (e) MeOH, Pd/C, H2 (55 psi), 2 h, 92%.
Nucleophilic displacement of the 4-chlorocyclopenta[d]pyrimidine 17 (Scheme 4) with the appropriate secondary amines in compounds 19–21, 23, 25, 28a–b, 32 and 35a–c afforded compounds 3–7 and 9–14 (Table 1) in 61–93% yields.
Scheme 4.
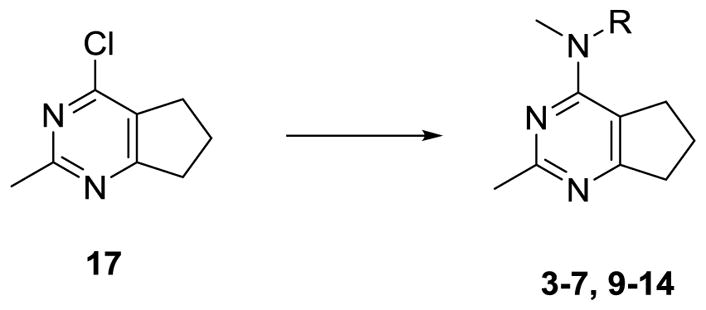
General synthesis of target compounds 3–7, 9–14.
Reagents and conditions: compounds 19–21, 23, 25, 28a–b, 32 and 35a–c, dioxane, HCl (2 N in dioxane, 1 drop), μW, 120–160 °C, 3–6 h, 61–93%.
HBr induced O-demethylation27 of 1 (Scheme 5) afforded 2 in 72% yield.
Scheme 5.
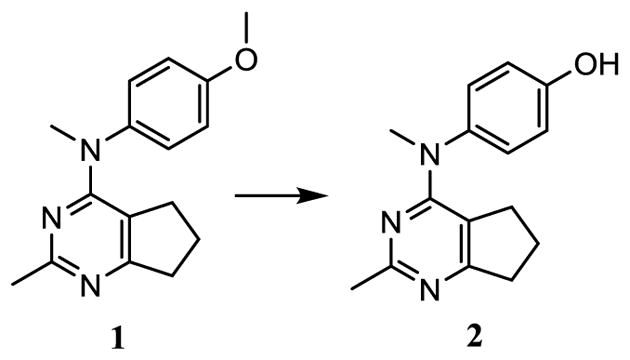
Synthesis of target compound 2.
Reagents and conditions: HBr (48% aq.), 80 °C, 3–6 h, 72%.
Nucleophilic displacement of 4-chlorocyclopenta[d]pyrimidine 17 (Scheme 6) with N-methyl-4-nitroaniline afforded 36 in 79% yield. Pd catalyzed hydrogenation of 36 gave 8 in 69% yield.
Scheme 6.
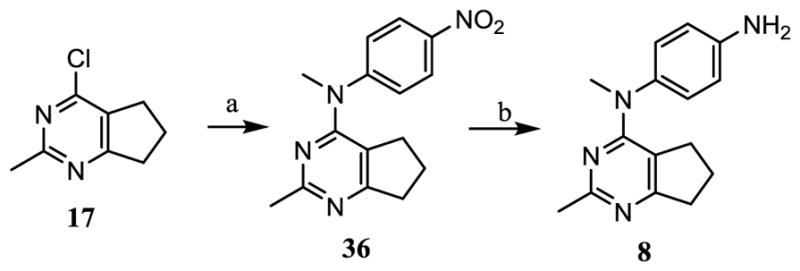
Synthesis of target compound 8.
Reagents and conditions: (a) N-methyl-4-nitroaniline, dioxane, HCl (2 N in dioxane, 1 drop), μW, 130 °C, 3 h, 79%; (b) MeOH, Pd/C, H2 (55 psi), rt, 69%;
4. Biological Evaluation and SAR
Compounds 2–14 were tested for their antiproliferative effects against the drug sensitive MDA-MB-435 cancer cells in culture using a sulforhodamine B (SRB) assay28,29 with the antitubulin agents CA-4 and compound 1 as positive controls. The microtubule disrupting effects of these compounds were evaluated in a cell-based phenotypic screen29 and the EC50 for microtubule depolymerizing effects determined.
Compound 2 (Table 2), the possible phenolic (4′-hydroxylphenyl) metabolite of 1, had low activity, and this finding emphasized the necessity to optimize 1 structurally. The S-methyl analog 3 was more potent in the MDA-MB-435 antiproliferative assay (IC50 = 4.6 nM) and as a microtubule depolymerization agent (EC50 = 13 nM) than 1. The 4′-dimethylamino analog 4 was as potent as the lead compound 1, for antiproliferative activity in MDA-MB-435 cells (IC50 = 8.2 nM).
Table 2.
IC50 values for inhibition of proliferation of MDA-MB-435 cells and EC50 values for microtubule depolymerizing activities in A-10 cells.
| Compd | EC50 in A-10 cells (nM) | IC50 ± SD (MDA-MB-435) Cancer cells (nM) | Compd | EC50 in A-10 cells (nM) | IC50 ± SD (MDA-MB-435) Cancer cells (nM) |
|---|---|---|---|---|---|
| 1 | 26 | 7.0 ± 0.7 | 9 | 70 | 12.1 ± 0.9 |
| 2 | 5,560 | 405 ± 75 | 10 | 56 | 18.4 ± 1.9 |
| 3 | 13 | 4.6 ± 0.5 | 11 | >10,000 | ND |
| 4 | 40 | 8.2 ± 0.6 | 12 | >10,000 | 450 ± 14 |
| 5 | 31 | 7.9 ± 0.5 | 13 | >10,000 | ND |
| 6 | >10,000 | ND | 14 | >10,000 | ND |
| 7 | 45 | 20 ± 1.9 | CA-4 | 13.0 | 3.4 ± 0.6 |
| 8 | 5,120 | 1223 ± 70 |
The major metabolite of 3, i.e., the sulfoxide analog 5, was equipotent with lead compound 1 in both the antiproliferative (IC50 = 7.9 nM) and microtubule depolymerization assays (EC50 = 31 nM). One of the minor metabolites of 3, the sulfonyl analog 6, was inactive. The 4′-methylamino compound 7 (MDA-MB-435, IC50 = 20 nM), one of the probable metabolites of 4, was 2.9-fold less potent than 1. 4′-Amino compound 8, the other probable metabolite of 4, had low activity.
The conformationally restricted dihydrobenzofuran analog 9 was 1.7-fold less potent than 1 in MDA-MB-435 cells (IC50 = 12.1 nM). Analog 10, in which the conformation of the 4′-methoxy moiety was partially restricted, was about 2.6-fold less potent than 1 (MDA-MB-435: IC50 = 18.4 nM). In 9, the methylene group resides at an angle with the phenyl ring that is dictated by the envelope shape of the dihydrofuran ring, thus providing restriction to rotation. In 10, the 3′-methyl group could create steric hindrance and partially restrains the free rotation of the 4′-methoxy moiety. The lower activities of 9 and 10 compared with 1 indicate that an ortho substitution to 4′-methoxy group on phenyl ring is not favorable to antiproliferative activity.
The docking of compounds 1–14 in the colchicine site suggested the compounds to be potent inhibitors of tubulin, except 2 and 6. To further rationalize the low activity of 2, 6, 7, and 8, the electrostatic surface of the colchicine site pocket (Fig. 3A), of colchicine (Fig. 3B) and of compounds 1–8 (Fig. 3C) were generated using MOE 2016.08.30 Fig 3A indicates that the surface of the binding pocket, where the methoxy groups of colchicine bind, to be electron deficient (blue surface). This suggests that compounds with electron rich groups (red surface) will bind more favorably and hence should afford potent inhibition. This is corroborated from the activities of 1, 3, 4 and 5, all of which, like colchicine, have a red surface in this area (Fig 3B and 3C). Compound 6 which has a 4′-sulfone group makes the aryl ring highly electron deficient, and, moreover, the sulfone group would probably lie in an area lined by the carbonyl backbones of Cysβ241, Valβ248 and Aspβ251 (Fig. 3A), and this results in an unfavorable repulsive interaction. This can explain, in part, the failure of 6 to dock in the colchicine site and consequently its low activity. The poor activity of 2 (compared to 1) and of 7 and 8 (compared to 4) can also be rationalized on the basis of the electrostatic surface of these compounds. The electron withdrawing effect of the 4′-OH group in 2 and the 4′-NH2 in 8 (encircled blue surfaces in Fig. 3C) causes an unfavorable interaction in an already electron deficient region of the binding site and provides an explanation for the low activity of 2 and 8. The 4′-NHMe group in 7 however shows both blue and red surfaces and shows a slightly reduced activity when compared to 4. Compounds 9 and 10 had electrostatic surfaces similar to that of compound 1 (not shown here). Electrostatic complementarity has been discussed as an approach to improve compound selectivity,31 and, based on the electrostatic surfaces observed for compounds 1–8, we propose that these surfaces can be invoked to explain the differences in the biological activities of these compounds. However, compounds 11–14 with the slightly different docking result as compound 1 (Supporting information Figure S2), the electrostatic surfaces generated were similar to that observed for compound 1 (Supporting information Figure S3), but could not explain the inactivity of these compounds.
Figure 3.


A. Electrostatic surface of the colchicine site binding pocket in tubulin. B. Electrostatic surface of colchicine. C. Electrostatic surfaces of compounds 1–8. Red surface indicates electron rich surface, blue surface indicates electron deficient surface, and white surface represents hydrophobic surface. Residues Val238, Cys241, Leu248 and Asp251 belong to the β-chain of tubulin.
We also calculated the number of low energy conformations for 1–14 within 7 kcal/mol (Table 3). Compounds 1–10 had a very low number of such conformations (2–8). In contrast, compounds 11–14, owing to their increased flexibility, showed a 5 to 21-times higher number of conformations. Biological evaluation of these compounds (Table 2) showed that compounds 11, 13 and 14 were devoid of any microtubule depolymerization activity, and 12 had poor MDA-MB-435 inhibitory activity. The electrostatic surface area calculations were, like the docking studies, unable to explain the inactivity of compounds 11–14. We therefore conclude that the increased conformational flexibility at the C4 substitution in 11–14 is detrimental to cytotoxic effects of these compounds, despite the docking studies and electrostatic surface area calculations that suggest these compounds should have potent inhibitory activities.
Table 3.
Number of conformations generated by compounds 1–14 calculated using MOE2016.08.
| Compound No. | No. of conformations within 7 kcal/mol | Compound No. | No. of conformations within 7 kcal/mol |
|---|---|---|---|
| 1 | 4 | 8 | 2 |
| 2 | 2 | 9 | 4 |
| 3 | 4 | 10 | 4 |
| 4 | 5 | 11 | 42 |
| 5 | 4 | 12 | 34 |
| 6 | 4 | 13 | 36 |
| 7 | 8 | 14 | 34 |
The effects of 3, 4, 5, 9 and 10 on the polymerization of purified tubulin were evaluated (Table 4). This allows a study of the direct interaction of these compounds with their intracellular target. The ability of these compounds to bind to the colchicine site on tubulin was evaluated by measuring inhibition of [3H]colchicine binding to tubulin (Table 4).32–35 The data show that 3, 4, 5, 9 and 10 are equipotent with 1 and CA-4 as tubulin assembly inhibitors (Table 4). At a 5 μM concentration, these compounds inhibited [3H]colchicine binding from 83 to 93%; while at a 1 μM concentration, they inhibited colchicine binding from 59 to 79%. Since the tubulin assembly inhibitory values for these compounds were very similar, the improved activity of 3 in MDA-MB-435 cell lines (Table 2) compared to 1 can be attributed, in part, to improved passive diffusion of 3 (SlogP = 3.76) when compared to 1 (SlogP = 3.05). The 4′-SMe in 3 imparts more hydrophobicity to the molecule and, therefore, better cellular inhibitory activity. Similarly, the more potent activity of 3 as compared to 5 in the MDA-MB-435 cells (Table 2) can also be ascribed to enhanced passive diffusion of 3 when compared to 5 (SlogP = 2.78), even though 5 has a better inhibitory value for tubulin assembly than 3. These data show that compounds 3, 4, 5, 9 and 10 primarily bind to the colchicine site of tubulin and with potency similar to that of the standard CA-4.
Table 4.
Inhibition of tubulin assembly and colchicine binding by CA-4 and 3, 4, 5, 9 and 10 and their SlogP (log octanol/water partition coefficient calculated using MOE2016.08) values.
| Compd. | SlogP | Inhibition of tubulin assembly IC50 (μM) ± SD | Inhibition of colchicine binding % Inhibition ± SD | |
|---|---|---|---|---|
| 5 μM inhibitor | 1 μM inhibitor | |||
| 1 | 3.05 | 1.6 ± 0.1 | 92 ± 0.7 | 70 ± 2 |
| 3 | 3.76 | 1.3 ± 0.1 | 92 ± 3 | 74 ± 5 |
| 4 | 3.11 | 1.2 ± 0.01 | 85 ± 3 | 65 ± 4 |
| 5 | 2.78 | 1.1 ± 0.06 | 93 ± 3 | 79 ± 4 |
| 9 | 2.98 | 1.2 ± 0.09 | 85 ± 4 | 64 ± 5 |
| 10 | 3.36 | 1.1 ± 0.007 | 83 ± 4 | 59 ± 5 |
| CA-4 | - | 1.3 ± 0.1 | 98 ± 0.6 | 87 ± 0.2 |
The ability to circumvent Pgp-mediated drug resistance was evaluated using an SK-OV-3 isogenic cell line pair (Table 5). Paclitaxel, a known Pgp substrate, was 864-fold less potent in cells expressing the Pgp drug transporter (Resistance ratio, Rr = 864). As with 1 and CA-4, compounds 3, 4, 5, 7, 9 and 10 were only 1.3 to 2-fold less potent as Pgp-overexpressing cells as compared with the parental cells. These data indicate that 3, 4, 5, 7, 9 and 10 are poor substrates for Pgp and therefore would be better than some clinically used microtubule targeting drugs that are substrates for transport by Pgp.
Table 5.
Compound activity in parental and Pgp expressing cell line
| Compd. | IC50 (nM) ± SD | ||
|---|---|---|---|
| Parental SK-OV-3 | Pgp expressing SK-OV-3/MDR1-6/-6 | Rr | |
| 1 | 11.3 ± 0.2 | 16.4 ± 0.4 | 1.5 |
| 3 | 7.2 ± 0.6 | 9.3 ± 0.4 | 1.3 |
| 4 | 13.0 ± 1.0 | 22.6 ± 2.3 | 1.7 |
| 5 | 12.6 ± 0.3 | 19.9 ± 5 | 1.6 |
| 7 | 36.3 ± 3.3 | 49.7 ± 7.9 | 1.4 |
| 9 | 18.7 ± 1.2 | 29.8 ± 5.6 | 1.6 |
| 10 | 23.9 ± 2.5 | 47.5 ± 2.4 | 2.0 |
| CA-4 | 4.5 ± 0.2 | 6.6 ± 1.3 | 1.5 |
| Paclitaxel | 3.0 ± 0.06 | 2600 ± 270 | 864 |
The abilities of the compounds to overcome Pgp-mediated drug resistance was evaluated in an isogenic cell line pair using the SRB assay28. Cellular protein was measured using the SRB assay.
Another clinically relevant drug resistance mechanism of tubulin-targeting agents is the overexpressing of the βIII subtype of tubulin. The ability of compounds 3, 4, 5, 7, 9 and 10 to overcome βIII-tubulin mediated drug resistance was examined using an isogenic HeLa cell line pair (Table 6). All these compounds had similar resistance ratio (Rr) values that ranged from 0.6 to 1.5, similar to the value obtained with CA-4 (Rr = 1.2). These data suggested that these compounds overcome βIII-tubulin mediated drug resistance compared with paclitaxel with a Rr value of 4.7.
Table 6.
Effects of compounds in parental and overexpressing βIII cells.
| Compd. | IC50 (nM) ± SD | ||
|---|---|---|---|
| Parental HeLa | Wild type βIII expressing HeLa | Rr | |
| 1 | 12.0 ± 1.2 | 9.6 ± 0.9 | 0.8 |
| 3 | 7.0 ± 0.5 | 5.3 ± 0.8 | 0.8 |
| 4 | 11.3 ± 0.7 | 9.8 ± 0.9 | 0.9 |
| 5 | 6.8 ± 0.4 | 10.0 ± 1.3 | 1.5 |
| 7 | 31.0 ± 1.6 | 19.1 ± 3.9 | 0.6 |
| 9 | 16.0 ± 1.2 | 15.1 ± 0.7 | 0.9 |
| 10 | 22.8 ± 1.0 | 22.6 ± 1.6 | 0.8 |
| CA-4 | 4.7 ± 0.2 | 5.7 ± 0.4 | 1.2 |
| Paclitaxel | 1.6 ± 0.2 | 7.7 ± 0.2 | 4.7 |
Compound 3, the most potent microtubule depolymerizing agent in the current study, was selected by the National Cancer Institute for evaluation in the NCI 60 cell line panel. It showed potent GI50s against most of the NCI 60 cancer cell lines (Table 7). Compound 3 is equipotent with paclitaxel across the panel and thus has the potential for further preclinical development and would have advantages over paclitaxel in resistant tumors.
Table 7.
Cancer cell inhibitory activity GI50 of 3.
| Panel/Cell line | GI50 (nM) | Panel/Cell line | GI50 (nM) | Panel/Cell line | GI50 (nM) | Panel/Cell line | GI50 (nM) |
|---|---|---|---|---|---|---|---|
| NSCLC | Renal Cancer | Ovarian cancer | Prostate Cancer | ||||
| A549/ATC C | <10 | 786 – 0 | <10 | IGROV1 | <10 | PC-3 | <10 |
| EKVX | <10 | A498 | <10 | OVCAR-3 | <10 | DU-145 | <10 |
| HOP-62 | <10 | ACHN | <10 | OVCAR-4 | <10 | Breast Cancer | |
| HOP-92 | <10 | CAKI-1 | <10 | OVCAR-5 | >10000 | MCF7 | <10 |
| NCI-H226 | <10 | RXF 393 | <10 | OVCAR-8 | <10 | MDA-MB-231/ATCC | <10 |
| NCI-H23 | <10 | SN 12C | <10 | NCI/ADR-RES | <10 | HS 578T | <10 |
| NCI-H322M | >10000 | TK-10 | SK-OV-3 | <10 | BT-549 | <10 | |
| NCI-H460 | <10 | UO-31 | Melanoma | MDA-MB-468 | <10 | ||
| NCI-H522 | <10 | Colon Cancer | LOX IMVI | <10 | Leukemia | ||
| CNS Cancer | COLO 205 | <10 | MALME-3M | CCRF-CEM | <10 | ||
| SF-268 | <10 | HCC-2998 | <10 | M14 | <10 | HL-60 | <10 |
| SF-295 | <10 | HCT-116 | <10 | MDA-MB-435 | <10 | K-562 | <10 |
| SF-539 | <10 | HCT-15 | <10 | SK-MEL-2 | <10 | MOLT-4 | <10 |
| SNB-19 | <10 | HT29 | <10 | SK-MEL-28 | >10000 | RPMI-8226 | <10 |
| SNB-75 | <10 | KM12 | <10 | SK-MEL-5 | <10 | SR | <10 |
| U251 | <10 | SW-620 | <10 | UACC-62 | <10 |
5. Metabolite Identification Study of 3
In order to study its metabolic stability, 3 (Table 1) was incubated with human liver microsomes (from both genders) for 60 min. Aliquots were examined by LC/MS/MS and the results show that 49.5% of 3 was oxidized to 5 (M2) (Figure 4 and Table 8). The structure was validated by MS/MS (Supporting information Figure S1). The sulfone (M1) and presumptive hydroxyl (M4 and M5) and dihydroxyl (M3) metabolites are minor, each of which consist of less than 2.2% of the overall metabolite pool. The MS 302.1306 ([M+H]+ of 5) (Supporting information Figure S1) and MS/MS 239.1340 (mass of the demethylsulfinyl fragmentation product of 5) clearly illustrated that M2 was 5. Similarly, MS 318.1272 ([M+H]+ of 6) and MS/MS 239.1340 (mass of the demethylsulfonyl fragmentation product of 6) clearly proved that M1 was 6. Hence, 5 was the primary metabolite, and 6 was a minor metabolite of 3.
Figure 4.

Identification the metabolites of 3 by LC/MS/MS. HPLC spectra of 3 incubated with human liver microsomes at 0 min and 60 min. Metabolites M1–M5 and parent 3 were illustrated on the spectra. Some peaks (retention times larger than compound 3) in 60 min sample have different absorbance compared to that in 0 min sample, which may due to metabolic changes of the microsomes initiated by compound 3.
Table 8.
Summary of the metabolites identified by LC/MS/MS.
| Metabolite | RT (min) | Metabolite [M+H]+ (m/z) | Met ID | MS Peak Area in 60 min sample | |
|---|---|---|---|---|---|
| HPLC area | Percentage (%) | ||||
| 3 | 17.4 | 286 | Parent | 2,061,000 | 46.4 |
| M1 (6) | 7.9 | 318 | sulfone | 98,900 | 2.2 |
| M2 (5) | 9.6 | 302 | sulfoxide | 2,199,500 | 49.5 |
| M3 | 9.9 | 318 | Di-Hydroxy | 57,156 | 1.3 |
| M4 | 15.5 | 302 | Hydroxy | 81,795 | 1.8 |
| M5 | 16.0 | 302 | Hydroxy | 92,546 | 2.1 |
6. Conformational study of 3
We36 reported that potent furo[2,3-d]pyrimidine based microtubule targeting agents have (in DMSO-d6 solution) a stable conformation, in which the aniline ring resides above the furo ring. It was of interest to determine the stable conformation of compound 3. We used N4-desmethyl compound 3a as a comparison. The chemical shifts of the 5-position hydrogens are 2.76 ppm for 3a, and 1.84 ppm for 3 (Figure 5). The shielding effect in 1H NMR of the 5-position hydrogens in 3 demonstrated that in DMSO solution the aniline ring does reside above the cyclopenta ring. This stable conformation of compound 3 was further validated by NOE signal between phenyl protons and 5-CH2 in NOESY experiment (Supporting information Figure S4).
Figure 5.

1H NMR of N4-desmethyl compound 3a and compound 3. In 3a, the 5-position hydrogens have a chemical shift 2.76 ppm. In compound 3, the 5-position hydrogens have a chemical shift 1.84 ppm. This is the result of the shielding effect of the aniline moiety. (400 MHz, solvent: DMSO-d6).
7. Summary
We bioisoterically replaced the oxygen in 4′-methoxy group in lead compound 1 (O-methyl), which could be used to evaluate its hydrogen bonding effect. We designed and synthesized the S-methyl analog 3 and the N-dimethyl analog 4, both of which demonstrated high antiproliferative potency (MDA-MB-435 cells: IC50 = 4.6 nM for 3; IC50 = 8.2 nM for 4), strong antitubulin activities and ability to overcome drug resistance mechanisms. Interestingly, in the cellular microtubule depolymerization assays 3 was more potent than 4 and it was a more effective inhibitor of colchicine binding at both 1 and 5 μM. A possible metabolite of lead compound 1, the 4′-hydroxyl analog 2 had low potency (MDA-MB-435 cells: IC50 = 405 nM). The liver microsomal metabolite of 3, the methyl sulfinyl analog 5 however, also exhibited high antiproliferative potency (MDA-MB-435 cells: IC50 = 7.9 nM), strong activities against tubulin, and ability to overcome drug resistance mechanisms. Compound 3 emerged as the lead compound for further preclinical study due to its high potency and its active metabolite. Probable metabolites of compound 4, the 4′-mono methylamino analog 7 and the 4′-amino analog 8, were less active (MDA-MB-435 cells: IC50 = 20 nM for 7; IC50 = 1223 nM for 8). Conformational restriction of 4′-methoxy with a dihydrofuran in 9 or an ortho-methyl in 10 decreased their potency (MDA-MB-435 cells: IC50 = 12.1 nM for 9; IC50 = 18.4 nM for 10). Homologation of the 4-anilino moiety, as in 12–14, resulted in decrease or loss of activities. In the absence of an x-ray crystal structure, and assuming that the binding of these compounds did not induce any major changes to the tubulin protein, the activities can be explained in part by molecular docking studies, comparing the complementarity of electrostatic surface area of compounds to the colchicine site on tubulin as well as conformational analysis.
8. Experimental Section
8.1 Synthesis
Analytical samples were dried in vacuum (0.2 mm Hg) in a CHEM-DRY drying apparatus over P2O5 at 50 °C. Melting points were determined on a digital MEL-TEMP II melting point apparatus with a FLUKE 51K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for protons (1H NMR) were recorded on a Bruker Avance II 400 (400 MHz) or on a 500 (500 MHz) NMR system. The chemical shift values are expressed in ppm (parts per million) relative to tetramethylsilane as an internal standard: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; br, broad singlet. The coupling constants are measured by the software MestreC. Thin-layer chromatography (TLC) was performed on Whatman Sil G/UV254 silica gel plates with a fluorescent indicator, and the spots were visualized under 254 and 365 nm illumination. Proportions of solvents used for TLC are by volume. Column chromatography was performed on a 230–400 mesh silica gel (Fisher Scientific) column. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Elemental compositions are within ± 0.4% of the calculated values and indicate > 95% purity of the compounds. Fractional moles of water or organic solvents found in some analytical samples could not be prevented despite 24 – 48 h of drying in vacuo and were confirmed where possible by their presence in the 1H NMR spectra. HPLC-MS was analyzed with an Acquity system. A linear gradient of 90% of 0.1% formic acid in water, 10% of 0.1% formic acid in ACN over 10 min, and then 100% ACN for 5 min. All final compounds were > 95% pure as established by elemental analysis, HPLC, or both. Optical rotation was measured on a Rudolph polarimeter. All solvents and chemicals were purchased from Sigma-Aldrich or Fisher Scientific and were used as received.
8.1.1 4-Methylthio-N-methylphenylamine (19)
Compound 18 (0.695 g, 5 mmol) and NaH (60% dispersed in mineral oil, 0.12 g, 7.5 mmol) were dissolved in THF at 0–5 °C. To this solution CH3I (1.06 g, 7.5 mmol) was added, and the mixture was stirred at 0–5 °C for an additional 3 h. The reaction mixture was carefully quenched with water and was diluted with EtOAc and water. The organic layer was separated and washed with water (2 × 10 mL). After drying with anhydrous Na2SO4, the organic solvent was concentrated in a rotary evaporator to less than 1 mL. This residue was placed on top of a silica gel column and eluted with hexane and EtOAc. Fractions containing the product (TLC) were pooled and evaporated to afford a yellow oil, compound 19, in 75% yield. TLC Rf 0.52 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ2.43 (s, 3H, NCH3), 2.85 (s, 3H, SCH3), 3.75 (br, 1H, exch, NH), 6.58 (d, J = 8.51 Hz, 2H, ArH), 7.28 (d, J = 8.55 Hz, 2H, ArH); HPLC > 95%; ESIMS m/z [M+H]+, calcd 154.06, found 154.06.
8.1.2 4-Methylaminophenyl methyl sulfoxide (20)
Compound 19 (0.507 g, 3 mmol) and mCPBA (0.564 g, 3.3 mmol) were dissolved in ACN at 0–5 °C and stirred for 1 h. The reaction mixture was diluted with EtOAc and water. The organic layer was separated and washed with water (2 × 10 mL). After drying with anhydrous Na2SO4, the organic solvent was concentrated by a rotary evaporator to less than 1 mL. This residue was placed on top of a silica gel column and eluted with CHCl3 and MeOH. Fractions containing the product (TLC) were pooled and evaporated to afford compound 20 as a yellow oil in 67% yield. TLC Rf 0.35 (CHCl3: MeOH, 20:1); 1H NMR (CDCl3) δ2.71 (s, 3H, NCH3), 2.90 (d, J = 4.79 Hz, 3H, SCH3), 4.15 (m, 1H, exch, NH), 6.69 (d, J = 8.66 Hz, 2H, ArH), 7.50 (d, J = 8.64 Hz, 2H, ArH); ESIMS m/z [M+H]+, calcd 170.06, found 170.00.
8.1.3 4-Methylaminophenyl methyl sulfone (21)
Compound 20 (0.169 g, 1 mmol) and mCPBA (0.376 g, 2.2 mmol) were dissolved in ACN at 0–5 °C and stirred for 1 h. The reaction mixture was diluted with EtOAc and water. The organic layer was separated and washed with water (2 × 10 mL). After drying with anhydrous Na2SO4, the organic solvent was concentrated by a rotary evaporator to less than 1 mL. This residue was placed on top of a silica gel column and eluted with hexane and EtOAc. Fractions containing the product (TLC) were pooled and evaporated to afford compound 21 as an off-white solid in 59% yield. mp 127.7–129.0 °C; TLC Rf 0.37 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ2.93 (d, J = 4.79 Hz, 3H, SCH3), 3.03 (s, 3H, NCH3), 4.34 (m, 1H, exch, NH), 6.64 (d, J = 8.78 Hz, 2H, ArH), 7.74 (d, J = 8.77 Hz, 2H, ArH); HPLC > 95%; ESIMS m/z [M+H]+, calcd 186.05, found 186.13.
8.1.4 N-Methyl-2,3-dihydrobenzofuro-5-amine (23)
Compound 23 was synthesized from 22, following the procedure of 19, and it was obtained as an off-white solid in 68% yield. The color of this compound slowly turned dark when exposed to air, and it was directly used for the next step. mp 118.3–121.3 °C; TLC Rf 0.47 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ2.82 (s, 3H, NCH3), 3.18 (t, J = 8.56 Hz, 2H, CH2), 3.43 (br, 1H, exch, NH), 4.50 (t, J = 8.58 Hz, 2H, CH2), 6.42 (dd, J = 2.47 Hz, 8.43 Hz, 1H, ArH), 6.58 (d, J = 2.25 Hz, 1H, ArH), 6.67 (d, J = 8.43 Hz, 1H, ArH); ESIMS m/z [M+H]+, calcd 150.08, found 150.09.
8.1.5 N,3-Dimethyl-4-methoxyaniline (25)
Compound 25 was synthesized from 24, following the procedure of 19, and it was obtained as an off-white solid in 65% yield. The color of this compound slowly turned dark when exposed to air, and it was directly used for the next step. mp 109.6–111.3 °C; TLC Rf 0.44 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ2.22 (s, 3H, CH3), 2.83 (s, 3H, NCH3), 3.39 (br, 1H, exch, NH), 3.79 (s, 3H, OCH3), 6.47 (dd, J = 8.50 Hz, 2.43 Hz, 1H, ArH), 6.52 (d, J = 2.13 Hz, 1H, ArH), 6.74 (d, J = 8.51 Hz, 1H, ArH); ESIMS m/z [M+H]+, calcd 152.11, found 152.17.
8.1.6 N,N′-(1,4-Diphenylene)diformamide (27a)
Formic acid (0.46 g, 10 mmol) and acetic anhydride (0.54 g, 9 mmol) were stirred in CH2Cl2 (5 mL) at 0 °C for 0.5 h. 1,4-Phenyldiamine (26a, 0.54 g, 5 mmol) was added to the solution, and the mixture was stirred at rt overnight. The reaction mixture was diluted with EtOAc (50 mL), and washed with aqueous NaHCO3 (2 × 10 mL) and water (10 mL). The organic layer was dried with 4 Å MS (molecular sieves) and concentrated with a rotary evaporator to give a yellow solid. The crude product was purified by a silica gel column and eluted with CHCl3 and MeOH. Fractions containing the product (TLC) were pooled and evaporated to afford an off-white solid 27a in 79% yield. mp 210.3–213.5 °C (lit37 210–212 °C); TLC Rf 0.30 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ7.52 (s, 4H, ArH), 8.23 (d, J = 1.91 Hz, 2H, exch, CHO), 10.15 (s, 2H, exch, NH).
8.1.7 N-[4-(Formylamino)phenyl]-N-methyl-formamide (27b)
Compound 27b was synthesized from 26b following the procedure of 27a, and it was obtained as an off-white solid in 78% yield. mp 136.7–138.2 °C (lit37 mp 138–141 °C); TLC Rf 0.44 (CHCl3: MeOH, 20:1); 1H NMR (CDCl3) δ3.33 (s, 3H, NCH3), 7.17–7.28 (m, 4H, ArH), 7.46 (br, 1H, exch, NH), 8.47 (s, 2H, exch, CHO).
8.1.8 N,N′-Dimethyl-phenyl-1,4-diamine (28a)
Compound 27a was dissolved in anhydrous THF (15 mL) under N2. To this solution was slowly added LiAlH4 (5 mL, 2 N in hexane and THF). The resulting mixture was stirred at 60 °C for 4 h. The reaction mixture was placed in an ice bath and quenched with NaOH (6 N) dropwise. The resulting suspension was filtered and washed with EtOH (5 mL). The filtrate was concentrated and diluted with EtOAc (15 mL) and H2O (5 mL). The organic layer was separated and washed with H2O (2 X 5 mL), dried with Na2SO4 and concentrated with 1 g silca gel to afford a dry plug. The plug was placed on top of a silica gel column and eluted with hexane and EtOAc. Fractions containing the product (TLC) were pooled and evaporated to afford a yellow solid 28a in 72% yield. mp 111.3–113.6 °C (lit38 mp 111–114 °C); TLC Rf 0.28 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ2.44 (s, 3H, NCH3), 2.85 (s, 3H, NCH3), 3.75 (br, 2H, exch, NH), 6.58 (d, J = 8.51 Hz, 2H, ArH), 7.28 (d, J = 8.55 Hz, 2H, ArH).
8.1.9 N,N,N′-Trimethylphenyl-1,4-diamine (28b)
Compound 28b was synthesized from 27b following the procedure of 28a, and it was obtained as a brown oil in 63% yield. The color of this compound slowly turned into dark when exposed to air, and it was directly used for the next step. TLC Rf 0.45 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ1.60 (br, 1H, exch, NH), 2.85 (s, 9H, NCH3), 6.63 (d, J = 7.94 Hz, 2H, ArH), 6.78 (d, J = 6.63 Hz, 2H, ArH).
8.1.10 Benzyl (trans-4-hydroxycyclohexyl)carbamate (30)
Compound 29 (1.15 g, 10 mmol), CBzCl (2.14 g, 12 mmol) and TEA (1.23 g, 12 mmol) were dissolved in ACN at 0–5 °C and stirred for 1 h. The reaction mixture was diluted with EtOAc and water. The organic layer was separated and washed with water (2 × 10 mL). After drying with anhydrous Na2SO4, the organic layer was treated with 1 g of silica gel and evaporated under reduced pressure to give a dry plug. This plug was placed on top of a silica gel column and eluted with CHCl3 and MeOH. Fractions containing the product (TLC) were pooled and evaporated to afford a white solid 30 in 90% yield. mp 161.6–163.8 °C (lit39 163–165 °C); TLC Rf 0.49 (CHCl3: MeOH, 20:1); 1H NMR (CDCl3) δ1.25 (m, 2H, CH2), 1.39 (m, 2H, CH2), 1.50 (d, J = 4.00 Hz, 1H, exch, OH), 1.97 (m, 4H, CH2), 3.51 (m, 1H, NCH), 3.60 (m, 1H, OCH), 4.60 (br, 1H, exch, NH), 5.10 (s, 2H, CH2), 7.38 (m, 5H, ArH); HPLC > 95%; ESIMS m/z [M+H]+, calcd 250.14, found 250.22.
8.1.11 Benzyl N-methyl-(trans-4-methoxycyclohexyl)carbamate (31)
Compound 30 (0.75 g, 3 mmol) was dissolved in 12 mL THF at −5 °C. To the above solution, NaH (60% dispersed in mineral oil, 0.36 g, 9 mmol) and MeI (1.31 g, 9 mmol) were added and stirred at −5 °C for 3 h. The reaction was carefully quenched with water (30 mL), and EtOAc (60 mL) was added. The organic layer was separated and washed with water (3 × 20 mL), dried with Na2SO4 and concentrated with 1 g silca gel to afford a dry plug. The plug was placed on top of a silica gel column and eluted with CHCl3 and MeOH. Fractions containing the product (TLC) were pooled and evaporated to afford a white solid 31 in 68%. mp 53.6–58.4 °C; TLC Rf 0.52 (CHCl3: MeOH, 20:1); 1H NMR (CDCl3) δ1.35 (m, 2H, CH2), 1.53 (m, 2H, CH2), 1.78 (m, 2H, CH2), 2.16 (m, 2H, CH2), 2.81 (s, 3H, NCH3), 3.09 (m, 1H, NCH), 3.36 (s, 3H, OCH3), 4.15 (m, 1H, OCH), 5.16 (s, 2H, OCH2), 7.38 (m, 5H, ArH).
8.1.12 N-Methyl-trans-4-methoxycyclohexylamine (32)
Compound 31 (0.56 g, 2 mmol) was dissolved in 20 mL MeOH. The solution was added to Pd/C (10%, 0.05 g) in a hydrogenation flask under an Ar atmosphere. The flask was placed on a Parr shaker hydrogenation apparatus with 55 psi H2 for 2 h. The resulting mixture was filtered and concentrated to give compound 32 as a colorless oil in 92% yield. TLC Rf 0.38 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ1.21 (m, 2H, CH2), 1.42 (br, 1H, exch, NH), 1.66 (m, 2H, CH2), 2.12 (m, 2H, CH2), 2.28 (m, 2H, CH2), 2.69 (s, 3H, NCH3), 2.95 (m, 1H, NCH), 3.52 (s, 3H, OCH3), 4.15 (m, 1H, OCH); ESIMS m/z [M+H]+, calcd 144.13, found 144.13.
8.1.13 4-Methoxy-N-methyl-benzenemethylamine (35a)
Compound 35a was synthesized from 33a following the procedure of 27a and 28a and obtained as a light yellow oil in 67% yield. TLC Rf 0.40 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ 2.41 (s, 3H, NCH3), 2.59 (br, 1H, exch, NH), 3.12 (s, 2H, NCH2), 3.81 (s, 3H, OCH3), 6.76 (d, J = 8.66 Hz, 2H, ArH), 7.20 (d, J = 8.66 Hz, 2H, ArH).
8.1.14 4-Methoxy-N, α(αR)-dimethyl-benzenemethylamine (35b)
Compound 35b was synthesized from 33b following the procedure of 27a and 28a and obtained as a light yellow oil in 71% yield. TLC Rf 0.44 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ1.47 (d, J = 6.61 Hz, 3H, CH3), 2.33 (s, 3H, NCH3), 2.72 (br, 1H, exch, NH), 3.68 (m, 1H, CH), 3.82 (s, 3H, OCH3), 6.89 (d, J = 8.51 Hz, 2H, ArH), 7.28 (d, J = 8.19 Hz, 2H, ArH).
8.1.15 4-Methoxy-N, α (αS)-dimethyl-benzenemethylamine (35c)
Compound 35c was synthesized from 33c following the procedure of 27a and 28a and obtained as a light yellow oil in 68% yield. TLC Rf 0.44 (hexanes: EtOAc, 3:1); 1H NMR (CDCl3) δ1.47 (d, J = 6.61 Hz, 3H, CH3), 2.33 (s, 3H, NCH3), 2.72 (br, 1H, exch, NH), 3.68 (m, 1H, CH), 3.82 (s, 3H, OCH3), 6.89 (d, J = 8.51 Hz, 2H, ArH), 7.28 (d, J = 8.19 Hz, 2H, ArH).
8.1.16 General Procedure for compounds 3–7, 9–14
A solution of 2-methyl-4-chlorocyclopenta[d]pyrimidine (17, 1 eq.) and appropriate anilines or benzylamines (1.05 eq.) were placed in dioxane in a microwave vial. To the solution, HCl (2 N in dioxane, 1–2 drops) was added. The reaction mixture was set in a Biotage microwave initiator at 120 °C for 3–6 h with pressure less than 4 bar. The reaction was cooled and transferred to a flask, and the solvent was evaporated at reduced pressure. The residue was diluted with EtOAc, neutralized with NH4OH, and then washed with water (2 × 10 mL). After drying with anhydrous Na2SO4, the organic solvent was concentrated in a rotary evaporator to less than 1 mL. This residue was placed on top of a silica gel column and eluted with hexane and EtOAc or with CHCl3 and MeOH. Fractions containing the product (TLC) were pooled and evaporated to afford pure compounds 3–7, 9–14.
8.1.17 General Method to Prepare the HCl Salt from Base
HCl in ether solution (2 N) was added dropwise to the solution of the free base in methyl tert-butyl ether (MtBE) until no further solid precipitated. The solid was collected by filtration and dried to give the HCl salt.
8.1.18 N-(4-Methiophenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (3)
Compound 3 was synthesized from 17 and 19 following the general procedure above to afford an off-white solid in 68% yield. mp 84.2–85.1 °C; TLC Rf 0.44 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.65 (quint, J = 7.23 Hz, 2H, CH2), 1.86 (t, J = 7.15 Hz, 2H, CH2), 2.39 (s, 3H, CH3), 2.49 (s, 3H, CH3), 2.61 (t, J = 7.71 Hz, 2H, CH2), 3.40 (s, 3H, NCH3), 7.16 (dd, J = 6.59 Hz, 1.95Hz, 2H, ArH), 7.26 (dd, J = 6.66 Hz, 2.05 Hz, 2H, ArH); Anal. calcd. for (C16H20ClN3S· 0.55H2O) C, H, N, S, Cl; HPLC > 95%; ESIMS m/z [M+H]+, calcd 286.13, found 286.19.
8.1.19 N-(4-Dimethylaminophenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (4)
Compound 4 was synthesized from 17 and 31b following the general procedure above to provide an off-white solid in 75% yield. mp 126.0–126.9 °C; TLC Rf 0.42 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.78 (quint, J = 7.39 Hz, 2H, CH2), 1.95 (t, J = 7.22 Hz, 2H, CH2), 2.49 (s, 3H, CH3), 2.70 (t, J = 7.68 Hz, 2H, CH2), 2.99 (s, 6H, NCH3), 3.47 (s, 3H, NCH3), 6.77 (dd, J = 6.80 Hz, 2.15 Hz, 2H, ArH), 7.07 (dd, J = 6.78 Hz, 2.21 Hz, 2H, ArH); Anal. calcd. for (C17H22N4· 0.25EtOAc) C, H, N; HPLC > 95%; ESIMS m/z [M+H]+, calcd 283.18, found 283.24.
8.1.20 N-(4-Methylsulfinylphenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (5)
Compound 5 was synthesized from 17 and 20 following the general procedure above to provide an off-white solid in 72% yield. mp 64.7–64.9 °C; TLC Rf 0.32 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.69 (quint, J = 7.21 Hz, 2H, CH2), 1.83 (t, J = 7.21 Hz, 2H, CH2), 2.43 (s, 3H, CH3), 2.50 (s, 3H, NCH3), 2.63 (t, J = 7.71 Hz, 2H, CH2), 3.38 (s, 3H, SCH3), 7.18 (d, J = 8.49 Hz, 2H, ArH), 7.28 (d, J = 8.48 Hz, 2H, ArH); HPLC > 95%, ESIMS m/z [M+H]+, calcd 302.12, found 302.26.
8.1.21 N-(4-Methylsulfonylphenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (6)
Compound 6 was synthesized from 17 and 21 following the general procedure above to provide an off-white solid in 75% yield. mp 163.5–164.7 °C; TLC Rf 0.52 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.81 (m, 2H, CH2), 2.00 (m, 2H, CH2), 2.49 (s, 3H, CH3), 2.74 (m, 2H, CH2), 3.23 (s, 3H, NCH3), 3.52 (s, 3H, SCH3), 7.32 (s, 2H, ArH), 7.89 (d, J = 3.49 Hz, 2H, ArH); HPLC > 95%; ESIMS m/z [M+H]+, calcd 318.12, found 318.29.
8.1.22 N-(4-Methylaminophenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (7)
Compound 7 was synthesized from 17 and 31a following the general procedure above to provide an off-white solid in 70% yield. mp 153.6–154.6 °C; TLC Rf 0.40 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.74 (quint, J = 7.41 Hz, 2H, CH2), 1.99 (t, J = 7.23 Hz, 2H, CH2), 2.15 (s, 3H, CH3), 2.50 (s, 3H, CH3), 2.70 (t, J = 7.71 Hz, 2H, CH2), 2.91 (d, J = 4.84 Hz, 3H, CH3), 5.26 (br, 1H, exch, NH), 6.96 (d, J = 8.55 Hz, 2H, ArH), 7.08 (d, J = 8.57 Hz, 2H, ArH); Anal. calcd. for (C16H20N4· 0.32EtOAc) C, H, N.
8.1.23 N-(2,3-Dihydrobenzofuran-5-yl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (9)
Compound 9 was synthesized from 17 and 23 following the general procedure above to provide an off-white solid in 70% yield. mp 100.3–100.9 °C; TLC Rf 0.49 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.64 (quint, J = 7.16 Hz, 2H, CH2), 1.84 (t, J = 6.76 Hz, 2H, CH2), 2.41 (s, 3H, CH3), 2.61 (t, J = 7.50 Hz, 2H, CH2), 3.19 (t, J = 8.51 Hz, 2H, CH2), 3.34 (s, 3H, NCH3), 4.57 (t, J = 8.60 Hz, 2H, CH2), 6.75 (d, J = 8.27 Hz, 1H, ArH), 6.96 (d, J = 7.64 Hz, 1H, ArH), 7.14 (s, 1H, ArH); Anal. calcd. for (C17H19N3O· 0.15EtOAc) C, H, N; HPLC > 95%; ESIMS m/z [M+H]+, calcd 282.15, found 282.21.
8.1.24 N-(3-Methyl-4-methoxylphenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (10)
Compound 10 was synthesized from 17 and 25 following the general procedure above to provide an off-white solid in 79% yield. mp 116.6–117.2 °C; TLC Rf 0.52 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.64 (quint, J = 7.60 Hz, 2H, CH2), 1.78 (t, J = 7.19 Hz, 2H, CH2), 2.15 (s, 3H, ArCH3), 2.41 (s, 3H, CH3), 2.50 (s, 3H, NCH3), 2.59 (t, J = 7.79 Hz, 2H, CH2), 3.81 (s, 3H, OCH3), 6.92 (d, J = 9.27 Hz, 1H, ArH), 7.03 (t, J = 2.61 Hz, 1H, ArH), 7.05 (s, 1H, ArH); Anal. calcd. for (C17H21N3O) C, H, N; HPLC > 95%; ESIMS m/z [M+H]+, calcd 284.17, found 284.24.
8.1.25 N-(4-Methoxycyclohexyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (11)
Compound 11 was synthesized from 17 and 35 following the general procedure above to provide an off-white solid in 68% yield. mp 54.1–55.1 °C; TLC Rf 0.49 (CHCl3: MeOH, 20:1); 1H NMR (MeOD-d4) δ1.36 (m, 2H, CH2), 1.71 (m, 2H, CH2), 2.03 (m, 2H, CH2), 2.23 (m, 2H, CH2), 2.40 (s, 3H, ArCH3), 2.79 (m, 2H, CH2), 3.01 (s, 3H, NCH3), 3.12 (m, 3H, NCH, CH2), 3.22 (m, 2H, CH2), 3.38 (s, 3H, OCH3), 4.43 (m, 1H, OCH); Anal. calcd. for (C16H25N3O·0.35H2O) C, H, N; ESIMS m/z [M+H]+, calcd 276.20, found 276.26.
8.1.26 N-(4-Methoxylphenyl)methyl-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-ammonium chloride (12·HCl)
Compound 12·HCl was synthesized from 17 and 28a following the general procedure above and the salt formation procedure above to provide an off-white solid in 71% yield. mp 151.6–152.9 °C; TLC Rf 0.44 (CHCl3: MeOH: TEA, 20:1:0.5); 1H NMR (DMSO-d6) δ1.89(m, 2H, CH2), 2.34 (s, 3H, ArCH3), 2.51 (m, 2H, CH2), 3.00 (m, 2H, CH2), 3.06 (s, 3H, NCH3), 3.72 (s, 3H, OCH3), 4.73 (m,2H, NCH2), 6.88 (d, J = 8.64 Hz, 2H, ArH), 7.16 (d, J = 8.60 Hz, 2H, ArH); HPLC > 95%; ESIMS m/z [M+H]+, calcd 284.17, found 284.24.
8.1.27 N-[1(1S)-(4-Methoxylphenyl)ethyl]-N,2-dimethyl-6,7-dihydro-5H-yclopenta[d]pyrimidin-4-amine (13)
Compound 13 was synthesized from 17 and 28b following the general procedure above to provide an off-white solid in 75% yield. mp 53.2–53.5 °C; TLC Rf 0.46 (CHCl3: MeOH, 20:1); −204 (c 0.1, MeOH); 1H NMR (DMSO-d6) δ1.50 (d, J = 6.96 Hz, 3H, CH3), 2.04 (m, 2H, CH2), 2.43 (s, 3H, ArCH3), 2.87 (m, 2H, CH2), 2.85 (s, 3H, NCH3), 3.13 (m, 2H, CH2), 3.79 (s, 3H, OCH3), 6.13 (m, 1H, CH), 6.93 (d, J = 8.75 Hz, 2H, ArH), 7.24 (d, J = 8.49 Hz, 2H, ArH); Anal. calcd. for (C18H23N3O·0.17EtOAc) C, H, N; HPLC > 95%; ESIMS m/z [M+H]+, calcd 298.18, found 298.23.
8.1.28 N-[1(1R)-(4-Methoxylphenyl)ethyl]-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (14)
Compound 14 was synthesized from 17 and 28c following the general procedure above to provide an off-white solid in 68% yield. mp 54.1–54.9 °C; TLC Rf 0.47 (CHCl3: MeOH, 20:1); 196 (c 0.1, MeOH); 1H NMR (DMSO-d6) δ1.48 (d, J = 6.95 Hz, 3H, CH3), 1.89 (t, J = 7.60 Hz, 2H, CH2), 2.39 (s, 3H, ArCH3), 2.68 (t, J = 7.91 Hz, 2H, CH2), 2.70 (s, 3H, NCH3), 3.07 (t, J = 8.06 Hz, 2H, CH2), 3.74 (s, 3H, OCH3), 6.02 (m, 1H, CH), 6.91 (d, J = 8.69 Hz, 2H, CH2), 7.22 (d, J = 8.61 Hz, 2H, CH2); Anal. calcd. for (C18H23N3O·0.18EtOAc) C, H, N; HPLC > 95%; ESIMS m/z [M+H]+, calcd 298.18, found 298.24.
8.1.29 N-(4-Hydroxylphenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (2)
Compound 1 (0.27 g, 1 mmol) was dissolved in 5 mL HBr (48% aq.) and stirred at 80 °C for 3 h. The reaction mixture was cooled to rt and placed in an ice bath, and the pH was adjusted to 11 with concentrated NH4OH (20%). EtOAc (20 mL) was added, and the organic layer was separated and washed with brine (3 X 5 mL), dried and concentrated to 1 mL. The concentrated solution was directly added to a silica gel column and eluted with CHCl3 and MeOH. The portion with the desired compound (TLC) was pooled and concentrated to give 0.18 g of a white solid 2, 72% yield. mp 209.3–210.4 °C; TLC Rf 0.49 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.61 (quint, J = 7.66 Hz, 2H, CH2), 1.79 (t, J = 7.49 Hz, 2H, CH2), 2.42 (s, 3H, ArCH3), 2.58 (t, J = 7.58 Hz, 2H, CH2), 3.31 (s, 3H, NCH3), 6.75 (d, J = 7.53 Hz, 2H, ArH), 7.03 (d, J = 7.72 Hz, 2H, ArH), 9.62 (s, 1H, exch, OH); HPLC > 95%; ESIMS m/z [M+H]+, calcd 256.14, found 256.26.
8.1.30 N-(4-Nitrophenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (36)
Compound 36 was synthesized from 17 and N-methyl-4-nitroaniline following the general procedure above to provide an off-white solid in 79% yield. mp 151.9–152.4 °C; TLC Rf 0.52 (CHCl3: MeOH, 20:1); 1H NMR (DMSO-d6) δ1.85 (quint, J = 8.00 Hz, 2H, CH2), 2.14 (t, J = 7.69 Hz, 2H, CH2), 2.49 (s, 3H, ArCH3), 2.77 (t, J = 7.60 Hz, 2H, CH2), 3.55 (s, 3H, NCH3), 7.20 (d, J = 8.80 Hz, 2H, ArH), 8.18 (t, J = 8.60 Hz, 2H, ArH); Anal. calcd. for (C15H16N4O2·0.25H2O) C, H, N.
8.1.31 N-(4-Aminophenyl)-N,2-dimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amine (8)
Compound 36 (0.56 g, 2 mmol) was dissolved in 20 mL MeOH. The solution was added to Pd/C (10%, 0.05 g) in a hydrogenation flask under an Ar atmosphere. The reaction was placed on a Parr shaker hydrogenation apparatus with 55 psi H2 for 2 h. The mixture was filtered, concentrated and followed by two repeated columns to remove most of the residual Pd to give compound 8 as a white solid in 69% yield. mp 170.1–170.9 °C; TLC Rf 0.21 (CHCl3: MeOH, 20:1); 1H NMR (CDCl3) δ1.60 (quint, J = 8.00 Hz, 2H, CH2), 1.84 (t, J = 7.70 Hz, 2H, CH2), 2.38 (s, 3H, ArCH3), 2.58 (t, J = 8.00 Hz, 2H, CH2), 3.29 (s, 3H, NCH3), 5.22 (br, 2H, exch, NH2), 6.54 (dd, J = 2.00Hz, 4.40 Hz, 2H, ArH), 6.88 (dd, J = 2.00 Hz, 4.42Hz, 2H, ArH); Anal. calcd. for (C15H18N4·0.45H2O) C, H, N; HPLC > 95%; ESIMS m/z [M+H]+, calcd 255.15, found 255.26.
8.2 Molecular Modeling
Docking of compounds 1–14 was carried out in the published x-ray crystal structure of colchicine bound to tubulin (PDB: 4O2B, 2.3 Å using Molecular Operating Environment (MOE 2016.08). The crystal structure was obtained from the protein database, imported into MOE 2016.08 and was then prepared using the QuickPrep function and the Amber10:EHT forcefield for energy minimization under default settings. Structure preparation using QuickPrep function utilizes interactive ligand modification and energy minimization in the active site of the selected flexible receptor. It deletes distant solvents, adds hydrogens, installs tethers, calculates charges and performs refinement of the system. Ligands were sketched using the builder function in MOE and minimized using the Amber10:EHT forcefield. Placement was carried using Triangle Matcher and scored using London dG. Refinement was carried out using Induced Fit, the side chain was set free, cutoff was increased to 8 Å and radius offset was set to 0.6. For preparation of tubulin, prior to setting up the protein for QuickPrep, chains C, D, E and F were deleted to reduce the time for protein preparation. After the preparation of the protein, Ca2+, Mg2+, GDP, GTP and all other bound ligands except for colchicine were deleted. To validate our docking studies, colchicine was re-docked into the binding site using the same set of parameters as described above. The rmsd of the best docked pose of colchicine in tubulin was 0.358 Å, thus validating docking using MOE.
The conformational search in MOE was carried out using default settings.
8.3 Cellular Studies
8.3.1 Effects on Cellular Microtubules
A-10 cells were used to evaluate the phenotypic effects of the compounds on microtubules using indirect immunofluorescence techniques. These cells arrest in interphase in response to tubulin targeting agents and thus are useful to compare the effects compounds on interphase microtubules. EC50 values were calculated as previously described40 and represent a minimum of three independent experiments.
8.3.2 SRB Assay
The antiproliferative and cytotoxic activities of the compounds were evaluated using the SRB assay as previously described.40 The IC50 values’ represent an average of at least three independent experiments, each conducted in triplicate and are expressed plus or minus the standard deviation.
8.3.3 Quantitative Tubulin Studies
Purified bovine brain tubulin was used in these studies to determine IC50 values for tubulin polymerization and compound inhibition of colchicine binding. The techniques used in both assays have previously been described in detail41,42 are summarized below. In the polymerization assay, 10 μM (1.0 mg/mL) tubulin was preincubated for 15 min at 30 °C with varying compound concentrations in 0.8 M monosodium glutamate, taken from a 2.0 M stock solution adjusted to pH 6.6 with HCl. Reaction mixtures also contained 4% (v/v) dimethyl sulfoxide (compound solvent). Following the preincubation, reaction mixtures were placed on ice, and 10 μL of 10 mM GTP (0.4 mM final concentration) was added to each mixture. Reaction volume was 0.24 mL prior to GTP addition, and all concentrations are in terms of the final reaction volume of 0.25 mL. Reaction mixtures were transferred to 0 °C cuvettes in Beckman DU7400/7500 recording spectrophotometers equipped with electronic temperature controllers. Polymer formation was measured turbidimetrically at 350 nm. After baselines were established at 0 °C, temperature was rapidly jumped to 30 °C (less than 1 min), and the IC50 was defined as the compound concentration inhibiting the extent of assembly by 50% after 20 min at 30 °C. In the colchicine binding assay,42 0.1 mL reaction volumes contained 1.0 μM tubulin, 5.0 μM [3H]colchicine (from Perkin-Elmer), and potential inhibitors at 1.0 or 5.0 μM, together with reaction components that strongly stabilize the colchicine binding activity of tubulin. Reaction mixtures also contained 5% dimethyl sulfoxide (compound solvent). Tubulin was the last component added to the reaction mixtures, which were prepared on ice. Binding of colchicine was initiated by transferring the reaction mixtures to a 37 °C water bath, and the mixtures were incubated for 10 min (a time when the reaction without inhibitor is 40–60% complete). Reactions were stopped with ice water, and the diluted reaction mixtures were filtered through a stack of two Whatman DEAE-cellulose filters obtained from GE Healthcare Life Sciences. Radiolabel bound to the filters was quantitated by scintillation counting.
8.4 Metabolite Identification
Briefly, 1.0 mg/mL of microsomes were added to potassium phosphate buffer containing MgCl2 and NADPH. Sample (propranolol as control) sets were pre-incubated at 37 °C for 5 min, and the metabolism reaction was initiated by the addition of 3 at 20 μM or propranolol 5 μM. Aliquots for LC/MS/MS analysis were withdrawn at 0 and 1 h after initiation of the metabolism reaction.
8.5 Incubation Procedure
Stock solutions of compound 3 (20 mM) and propranolol (1 mM) were prepared in DMSO. Incubation plate wells or tubes contained 100 mM pH 7.4 PBS, 3 mM MgCl2, 10 mM NADPH, and microsomes (20 mg/mL). The incubation vessels were preincubated for 3 min in a 37 °C water bath. For metabolite identification reactions, 1 μL of 20 mM 3 was added to the incubation vessels to yield a final concentration of 20 μM. For control reactions, 1 μL of 1 mM propranolol was added to the incubation vessels to yield a final concentration of 5 μM. Each metabolite identification reaction was initiated by adding 999 μL of the microsome mix to the incubation vessel and mixing with a pipette. A 200 μL aliquot was removed from each metabolism reaction at 0 and 60 min and the reactions were terminated by adding the aliquot to a vial containing 400 μL of ice-cold ACN. Terminated reactions were centrifuged at 14,000 g for 5 min. The supernatant (600 μL) of each sample was transferred to a siliconized microfuge tube and evaporated to approximately 75 μL in a Speed-Vac.
8.6 Analysis Method
Shimadzu VP System HPLC with mobile phase of A-0.2% formic acid in water and B-0.18% formic acid in methanol. The samples were run on the following column: 2 × 30 mm Higgins Targa-C18 5 μm. Injection volume was 50 μL, with gradient (5–75% B in 30 min) and a flow rate of 200 μL/min. Mass of each component was analyzed using an Applied Biosystems/MDS SCIEX QSTAR mass spectrometer, with IonSpray (ESI) split at ~1:10 interface. The parent ion scan was TOF positive from 300–1200 amu, and the product Ion Scan was the TOF product ion from 200–1200 amu.
Supplementary Material
Acknowledgments
The work was supported, in part, by a grant from the National Institutes of Health, National Cancer Institute, R01 CA142868 (AG and SLM) and by the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (AG). We thank the National Cancer Institute for performing the in vitro antitumor evaluation in their 60 tumor preclinical screening program.
Abbreviations
- TBAs
tubulin binding agents
- HB
hydrogen bond
- Pgp
P-glycoprotein
- ACN
acetonitrile
- Rr
resistance ratio
- TLC
thin layer chromatography
Footnotes
Disclaimer
The content of this paper is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Notes
The authors declare no competing financial interest.
Supplementary data associated with this article can be in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borisy G, Heald R, Howard J, Janke C, Musacchio A, Nogales E. Microtubules: 50 years on from the discovery of tubulin. Nat Rev Mol Cell Biol. 2016;17(5):322–328. doi: 10.1038/nrm.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9(10):790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saez-Calvo G, Sharma A, Balaguer FA, et al. Triazolopyrimidines are microtubule-stabilizing agents that bind the vinca inhibitor site of tubulin. Cell chemical biology. 2017;24(6):737–750. e736. doi: 10.1016/j.chembiol.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 4.Madiraju C, Edler MC, Hamel E, et al. Tubulin assembly, taxoid site binding, and cellular effects of the microtubule-stabilizing agent dictyostatin. Biochemistry. 2005;44(45):15053–15063. doi: 10.1021/bi050685l. [DOI] [PubMed] [Google Scholar]
- 5.Jang SH, Wientjes MG, Au JLS. Determinants of paclitaxel uptake, accumulation and retention in solid tumors. Invest New Drugs. 2001;19(2):113–123. doi: 10.1023/a:1010662413174. [DOI] [PubMed] [Google Scholar]
- 6.Prota AE, Bargsten K, Northcote PT, et al. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew Chem Int Ed Engl. 2014;53(6):1621–1625. doi: 10.1002/anie.201307749. [DOI] [PubMed] [Google Scholar]
- 7.Prota AE, Bargsten K, Diaz JF, et al. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc Natl Acad Sci U S A. 2014;111(38):13817–13821. doi: 10.1073/pnas.1408124111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Y, Chen J, Xiao M, Li W, Miller DD. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012;29(11):2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10(3):194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Wang Q, Li W. Recent advances in heterocyclic tubulin Inhibitors targeting the colchicine binding site. Anticancer Agents Med Chem. 2016;16(10):1325–1338. doi: 10.2174/1871520616666160219161921. [DOI] [PubMed] [Google Scholar]
- 11.Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Molecular cancer therapeutics. 2014;13(2):275–284. doi: 10.1158/1535-7163.MCT-13-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gangjee A, Zhao Y, Raghavan S, Rohena CC, Mooberry SL, Hamel E. Structure–activity relationship and in vitro and in vivo evaluation of the potent cytotoxic anti-microtubule agent N-(4-methoxyphenyl)-N,2,6-trimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-aminium chloride and its analogues as antitumor agents. J Med Chem. 2013;56(17):6829–6844. doi: 10.1021/jm400639z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsourlakis MC, Weigand P, Grupp K, et al. βIII-Tubulin overexpression is an independent dredictor of prostate cancer progression tightly linked to ERG fusion status and PTEN deletion. Am J Pathol. 2014;184(3):609–617. doi: 10.1016/j.ajpath.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Xiao G. In adapalene. Vol. 3. John Wiley & Sons Ltd; 2014. [Google Scholar]
- 15.Palmer AM. New horizons in drug metabolism, pharmacokinetics and drug discovery. Drug News Perspect. 2003;16(1):57–62. [PubMed] [Google Scholar]
- 16.Hao M-H. Theoretical calculation of hydrogen-bonding strength for drug molecules. J Chem Theory Comput. 2006;2(3):863–872. doi: 10.1021/ct0600262. [DOI] [PubMed] [Google Scholar]
- 17.Zhou P, Tian F, Lv F, Shang Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins. 2009;76(1):151–163. doi: 10.1002/prot.22327. [DOI] [PubMed] [Google Scholar]
- 18.Böhm H-J, Brode S, Hesse U, Klebe G. Oxygen and nitrogen in competitive situations: which is the hydrogen-bond acceptor? Chem Eur J. 1996;2(12):1509–1513. [Google Scholar]
- 19.Wilkening S, Lin G, Fritsch ES, et al. An evaluation of high-throughput approaches to QTL mapping in Saccharomyces cerevisiae. Genetics. 2014;196(3):853–865. doi: 10.1534/genetics.113.160291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasahara K, Mashima A, Yoshida T, Chuman H. Molecular dynamics and density functional studies on the metabolic selectivity of antipsychotic thioridazine by cytochrome P450 2D6: Connection with crystallographic and metabolic results. Bioorg Med Chem. 2015;23(17):5459–5465. doi: 10.1016/j.bmc.2015.07.043. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Xiao Q, Humphreys WG, Dongre A, Shu Y-Z. Identification of human liver microsomal proteins adducted by a reactive metabolite using shotgun proteomics. Chem Res Toxicol. 2014;27(9):1537–1546. doi: 10.1021/tx500181p. [DOI] [PubMed] [Google Scholar]
- 22.Jang GR, Wrighton SA, Benet LZ. Identification of CYP3A4 as the principal enzyme catalyzing mifepristone (RU 486) oxidation in human liver microsomes. Biochem Pharmacol. 1996;52(5):753–761. doi: 10.1016/0006-2952(96)00357-7. [DOI] [PubMed] [Google Scholar]
- 23.Prota AE, Danel F, Bachmann F, et al. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J Mol Biol. 2014;426(8):1848–1860. doi: 10.1016/j.jmb.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 24.SYBYL-X 2.1.1. Tripos International; 1699 South Hanley Rd, St Louis, Missouri, 63144, USA: [Google Scholar]
- 25.Gangjee A, Zhao Y, Lin L, et al. Synthesis and discovery of water-soluble microtubule targeting agents that bind to the colchicine site on tubulin and circumvent Pgp mediated resistance. J Med Chem. 2010;53(22):8116–8128. doi: 10.1021/jm101010n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrov VA, Marshall W. Chemo- and stereo-selectivity in oxidation of fluorinated cyclic sulfides by m-chloroperoxybenzoic acid. J Fluorine Chem. 2015;169(0):6–11. [Google Scholar]
- 27.Zou CL, Ji H, Xie GB, Chen DL, Wang FP. An effective O-demethylation of some C19-diterpenoid alkaloids with HBr-glacial acetic acid. Journal of Asian natural products research. 2008;10(11–12):1063–1067. doi: 10.1080/10286020802280208. [DOI] [PubMed] [Google Scholar]
- 28.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. Journal of the National Cancer Institute. 1990;82(13):1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 29.Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Develop Res. 1995;34(2):91–109. [Google Scholar]
- 30.Molecular operating environment (MOE), 2016.08. Chemical Computing Group Inc; 1010 Sherbooke St West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2017. [Google Scholar]
- 31.Huggins DJ, Sherman W, Tidor B. Rational approaches to improving selectivity in drug design. J Med Chem. 2012;55(4):1424–1444. doi: 10.1021/jm2010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamel E, Lin CM, Flynn E, D’Amato RJ. Interactions of 2-methoxyestradiol, an endogenous mammalian metabolite, with unpolymerized tubulin and with tubulin polymers. Biochemistry. 1996;35(4):1304–1310. doi: 10.1021/bi951559s. [DOI] [PubMed] [Google Scholar]
- 33.Lin CM, Hamel E, Wolpert-DeFilippes MK. Binding of maytansine to tubulin: competition with other mitotic inhibitors. Research communications in chemical pathology and pharmacology. 1981;31(3):443–451. [PubMed] [Google Scholar]
- 34.Safa AR, Hamel E, Felsted RL. Photoaffinity labeling of tubulin subunits with a photoactive analogue of vinblastine. Biochemistry. 1987;26(1):97–102. doi: 10.1021/bi00375a014. [DOI] [PubMed] [Google Scholar]
- 35.Hamel E, Lin CM. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry. 1984;23(18):4173–4184. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Raghavan S, Ihnat M, et al. The design and discovery of water soluble 4-substituted-2,6-dimethylfuro[2,3-d]pyrimidines as multitargeted receptor tyrosine kinase inhibitors and microtubule targeting antitumor agents. Bioorg Med Chem. 2014;22(14):3753–3772. doi: 10.1016/j.bmc.2014.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirini F, Mazloumi M, Seddighi M. Acidic ionic liquid immobilized on nanoporous Na+-montmorillonite as an efficient and reusable catalyst for the formylation of amines and alcohols. Res Chem Intermediat. 2016;42(3):1759–1776. [Google Scholar]
- 38.Michlik S, Hille T, Kempe R. The iridium-catalyzed synthesis of symmetrically and unsymmetrically alkylated diamines under mild reaction conditions. Adv Synth Catal. 2012;354(5):847–862. [Google Scholar]
- 39.Büttner A, Cottin T, Xu J, Tzagkaroulaki L, Giannis A. Serotonin derivatives as a new class of non-ATP-competitive receptor tyrosine kinase inhibitors. Bioorgan Med Chem. 2010;18(10):3387–3402. doi: 10.1016/j.bmc.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Gangjee A, Zhao Y, Lin L, et al. Synthesis and discovery of water-soluble microtubule targeting agents that bind to the colchicine site on tubulin and circumvent Pgp mediated resistance. J Med Chem. 2010;53(22):8116–8128. doi: 10.1021/jm101010n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell biochemistry and biophysics. 2003;38(1):1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 42.Verdier-Pinard P, Lai JY, Yoo HD, et al. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Molecular pharmacology. 1998;53(1):62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.