

Abstract
Conformational changes of NADPH-cytochrome P450 oxidoreductase (CYPOR) associated with electron transfer from NADPH to electron acceptors via FAD and FMN have been investigated through structural studies of the 4-electron-reduced NADP+-bound enzyme and kinetic and structural studies of mutants affecting the conformation of the mobile Gly631-Asn635 loop (Asp632 loop). The structure of 4-electron-reduced, NADP+-bound wild type CYPOR shows the plane of the nicotinamide ring positioned perpendicular to the FAD isoalloxazine with its carboxamide group forming H-bonds with N1 of the flavin ring and the Thr535 hydroxyl group. In the reduced enzyme, the C8-C8 atoms of the two flavin rings are ~1 Å closer compared to the fully oxidized and 1-electron-reduced structures, which suggests that flavin reduction facilitates interflavin electron transfer. Structural and kinetic studies of mutants, Asp632Ala, Asp632Phe, Asp632Asn, and Asp632Glu demonstrate that the carboxyl group of Asp632 is important for stabilizing the Asp632 loop in a retracted position that is required for the binding of the NADPH ribityl-nicotinamide in a hydride-transfer-competent conformation. Structures of the mutants and reduced wild type CYPOR permit us to identify a possible pathway for NADP(H) binding/release to/from CYPOR. Asp632 mutants unable to form stable H-bonds with the backbone amides of Arg634, Asn635, and Met636, exhibit decreased catalytic activity and severely impaired hydride transfer from NADPH to FAD, but leave interflavin electron transfer intact. Intriguingly, the Arg634Ala mutation slightly increases the cytochrome P450 2B4 activity. We propose that Asp632 loop movement, in addition to facilitating NADP(H) binding and release, participates in domain movements modulating interflavin electron transfer.
Keywords: diflavin oxidoreductase, NADPH-cytochrome P450 oxidoreductase, microsomal electron transport system, hydride transfer, electron transfer
Graphical Abstract
For Table of Contents use only - The structures of reduced NADPH cytochrome P450 oxidoreductase and mutants of conserved acidic residue Asp632, which regulates NADPH binding and hydride transfer
Introduction
The microsomal NADPH-cytochrome P450 oxidoreductase (CYPOR, also referred to as CPR or POR) is a ~78kDa membrane-bound protein and the first and prototypic member of the diflavin oxidoreductase family of enzymes that contain one molecule each of FAD and FMN in a single polypeptide chain. (Reviewed in Iyanagi, T., et al (2012) Arch Biochem Biophys, 72–89 and Kim and Waskell (2015))1,2 Its FAD receives two electrons in the form of a hydride from the obligate 2-electron donor, NADPH, and transfers an electron one at a time to the FMN. The FMN hydroquinone, but not the semiquinone, then transfers the electrons to physiologic acceptor proteins such as the cytochromes P450 (cyt P450 or P450), cytochrome b5, squalene monooxygenase, 7-dehydrocholesterol reductase, and heme oxygenase, as well as nonphysiologic acceptors such as cytochrome c (cyt c) and ferricyanide [Fe(CN)6]3−. In humans, the ubiquitous microsomal cytochromes P450 are responsible for the metabolism of at least one step in the biodegradation of the vast majority of drugs and are also involved in biosynthesis and biodegradation of the myriad of essential endogenous compounds, including steroid hormones, vitamins, and eicosanoids (3–7). Because CYPOR is the exclusive electron donor to microsomal P450s, it is critical to the function of the large number of physiologic processes regulated by P450s.
The P450s require the delivery of two electrons (delivered one at a time) provided by CYPOR for catalytic function. The first electron is required to reduce ferric substrate-bound P450 to ferrous form, while the second electron reduces the oxyferrous P450, which rapidly and irreversibly proceeds through the catalytic cycle to yield an oxidized substrate and a water molecule.8,9 This electron transfer from CYPOR to the cytochromes P450 must proceed in a controlled fashion resulting in substrate turnover without unwanted side products. During P450 catalysis, CYPOR cycles between the three, two, and one electron-reduced states. The one electron-reduced state is the stable blue semiquinone.10 Crystallographic studies have revealed the structure of the so-called “closed conformation” where the two flavin rings are juxtaposed, whereby FMN receives electrons from FAD, as well as “open” conformation that allows the FMN hydroquinone to transfer an electron to P450.11 How the chemistry and protein dynamics of reduction of CYPOR by NADPH and oxidation by P450 are coordinated, regulated, and controlled has been extensively investigated.12–17 Nevertheless many of the molecular details are lacking at this time. A greater understanding of the molecular mechanism of NADPH binding to and reduction of FAD as well as coordination of the dynamics of electron transfer from CYPOR to P450 is essential. In particular, more insight into whether the separation of the FAD and FMN domains, necessary for reduction of cyt P450 by the FMN hydroquinone, is coordinated by the oxidation state of the cofactors or is stochastic would be highly valuable. Results described in this manuscript suggest that movement of residues Gly631 to Asn635, termed the Asp632 loop, plays a role in this process.
Comparison of structures of wild type (WT) and mutant CYPORs, crystallized in the presence or absence of NADP+, have demonstrated the requirement for domain movements during catalysis and identified structural rearrangements relevant to cofactor binding,7,11,18–20 most notably movements of Trp677 and the Asp632 loop. In the closed conformation seen in the wild type oxidized NADP+-bound reductase structure, the two flavins are in van der Waals contact, and the nicotinamide moiety of NADP+ is blocked from interaction with FAD by Trp677, whose indole ring stacks against the re-face of the isoalloxazine ring of FAD.18 In the absence the indole side chain, (pdb codes IJA0 and IJ9Z), the nicotinamide moiety of NADP+ is observed almost parallel to and in van der Waals contact to the isoalloxazine ring,19 indicating that the Trp677 ring must move away from the isoalloxazine ring of the FAD to allow the nicotinamide ring to interact with the FAD and donate a hydride ion. In the “147CC514” (3OJW) mutant structure which lacks a bound NADP+ the Asp632 loop adopts a conformation that would sterically clash with the pyrophosphate-ribityl moiety of NADPH and thereby hinder its binding [pdb code 3OJX].7 In contrast, most structures of CYPOR that have been reported so far have a bound NADP+, and the Asp632 loop is folded away from the site that the pyrophosphate-ribityl moiety of NADP(H) would occupy. In its folded retracted position, the carboxyl of the Asp632 side chain forms three hydrogen bonds with the backbone amides of Arg634, Asn635, and Met636, suggesting a pivotal role in stabilization of this loop. It has also been demonstrated that the residue in human CYPOR corresponding to Asp632 is important for catalytic activity.21 To investigate how the aspartic acid at position 632 influences the association and dissociation of the nicotinamide cofactor and the CYPOR structure, we constructed four mutants: Asp632Ala, Asp632Glu, Asp632Phe, and Asp632Asn. Sequence alignment of the Asp632 loop of the diflavin reductase family (Table S1) reveals that Asp632 is conserved in all members of the diflavin reductase family, but not in NR1. The glutamic acid mutant will demonstrate the importance of the size of the acidic residue side chain whereas the asparagine mutant will provide insight into the function of the acidic group in forming three hydrogen bonds to the main chain amides of Arg634, Asn635, and Met636. Further structural and functional information about the ideal size of a side chain at position 632 will be gleaned from substituting the aspartic acid side chain with a smaller (alanine) and larger hydrophobic residue (phenylalanine).
With the exception of eNOS and nNOS isozymes, and cyt P450 BM3, the diflavin reductase family members have a basic residue two amino acids downstream of the conserved aspartic acid. Lysine is the basic residue in all except CYPOR, which has an Arg at this position and cyt P450 BM3 where the residue is a serine. Previous studies demonstrated that the mutation of Arg636 in human CYPOR corresponding to Arg634 in rat CYPOR to either alanine or serine slightly increased activity with both cyt c and ferricyanide compared to the wild type protein.21 We constructed the Arg634Ala mutant to determine whether the Arg634 was also important for activity with its physiologic redox partner cyt P450. Overall, our results demonstrate that the carboxyl group of Asp632 is important for stabilizing a retracted Asp632 loop which allows for the binding of the nicotinamide ring of NADPH in an electron transfer competent conformation and suggest that Arg634 exerts a slight inhibitory effect on the activity of the wild type protein.
EXPERIMENTAL PROCEDURES
Materials
NADPH, sodium dithionite, FMN, dithiothreitol, Triton-X 100, benzphetamine, horse heart cytochrome c, DEAE-Sepharose Fast Flow resin, and Octyl-Sepharose CL-4B were purchased from Sigma-Aldrich (St. Louis, MO). Potassium phosphate, glycerol, tryptone, sodium chloride, and yeast extract were purchased from Fisher Scientific. Hydroxyapatite and Bio-Beads were purchased from Bio-Rad. The Complete Mini Protease Inhibitor cocktail tablets and redistilled glycerol were purchased from Roche Diagnostics Corp (Indianapolis). Lucigen OverExpress Chemically Competent Cells and C41 (DE3) E. coli cells were purchased from Avidis (France). Isopropyl-β-D-thiogalactopyranoside (IPTG) and carbenicillin (disodium salt) were purchased from Research Products International Corp. Dilauroylphosphatidylcholine (DLPC) was purchased from Doosan Serdary Research Laboratory (Toronto, Canada). YM30 membrane was from Millipore Corporation (Billerica, MA). The Pierce BCA Assay Kit was purchased from Thermo Scientific (Rockford, IL).
Site-directed Mutagenesis of full length CYPOR
The site-directed mutants of the full-length Asp632Ala, Asp632Glu, Asp632Phe, Asp632Asn, and Arg634Ala rat CYPOR mutants were prepared by the PCR method using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). The oligonucleotides used to generate the reductase mutants were designed according to the guidelines from QuikChange II XL site-directed mutagenesis kit and were synthesized by Integrated DNA Technologies (Coralville, IA). Table S2 shows the oligonucleotides used to mutate full length CYPOR. The mutated plasmids from site-directed mutagenesis were transformed into competent C41 cells, and the transformants were grown in LB media overnight. The plasmids were extracted and purified using the QIAprep Mini-Kit (Qiagen, Chatsworth, CA). Following mutagenesis, the nucleotide sequences of the entire mutated reductase was determined at the University of Michigan DNA Sequencing Core Facility to confirm the presence of the desired mutation and the absence of any unwanted base changes.
Expression and Purification
Expression and purification of the full-length (no histidine tag) Asp632Ala, Asp632Glu, Asp632Phe, Asp632Asn, and Arg634Ala mutants and WT CYPOR were performed in freshly transformed competent C41 (DE3) Escherichia coli22 and grown in 1L of LB media containing 0.24 M carbenicillin, 0.2% (w/v) glucose, and 5.3 nM riboflavin as described previously for wild type. The purification procedure is described in detail in the supplement of the reference.11 Briefly, following induction with 0.4 mM IPTG for 24 h, the cells were harvested and purified according to the protocols reported in detail for the full-length WT CYPOR. The mutant proteins were purified in the same manner as the wild type protein. The average yield of the purified WT and mutant reductases was 18–25 mg/L of pure protein from 1 L cell culture. The amount of the diflavin protein was determined using ε454nm= 21.4 mM−1cm−1 and the Pierce BCA protein assay (Thermo Scientific, Rockford, IL). All the purified CYPOR mutants and wild type CYPOR ran as a single band on SDS-PAGE.
Determination of CYPOR Activity with Cytochrome c and Ferricyanide
The potential of the mutants to reduce cytochrome c or ferricyanide was measured in 270 mM potassium phosphate buffer, pH 7.7 at 30°C, as according to the published protocols.11 Cytochrome c (65 μM final concentration) in 270 mM potassium phosphate buffer pH 7.7 was incubated in a cuvette for 5 min at 30°C. After 5 min, CYPOR was added to cyt c, and the reaction was immediately initiated with NADPH (50 μM final concentration). The initial rate of cyt c reduction was followed at 550 nm. Reduction of ferricyanide was measured by preincubating a solution containing 10 nM CYPOR with 500 μM oxidized ferricyanide for 5 min at 30°C. The reaction was initiated by addition of NADPH to a final concentration of 100 μM. The initial rate of ferricyanide reduction was followed at 420 nm. Rates of cyt c and ferricyanide reduction were determined using Δε = 21.1 mM−1cm−1 at 550 nm and 1.02 mM−1cm−1 at 420 nm, respectively. The KmNADPH was determined according the published procedures23 by varying the concentration of NADPH (0.5 μM – 100 μM) and maintaining the concentration of cyt c constant (65 μM). Triplicate measurements were obtained on two separate days, and the values reported are an average of six measurements. The kinetic parameters were determined by fitting the data to the Michaelis-Menten equation using OriginLab (version 9.0, Northampton, MA).
Studies of the Effect of pH on CYPOR Cytochrome c Reductase Activity
To ensure the accuracy of the pH at which the cytochrome c reductase activity was measured, four different buffers were employed between pH 5.5–9.5: 1) MES [2-(N-morpholino) ethanesulfonic acid], pKa 6.15; 2) HEPES N-2-hydroxyethylpiperazine-N′-2 ethanesulfonic acid, pKa 7.55 at 20°C; 3) TES 2-{[tris-(hydroxymethyl)methyl]amino} ethanesulfonic acid, pKa 7.5 at 20°C; 4) Tricine N-[tris-(hydroxymethyl)methyl] glycine, pKa 8.15 at 20°C. The specified pH ranges at 20°C for the buffers were: MES (5.5–6.5), HEPES (6.7–7.5), TES (7.5–8.5), Tricine (8.5–9.56). The pH values of the buffers were corrected for the experimental temperature of 30°C. The reactions were performed at a constant ionic strength of 525 mM adjusted with KCl. The final concentrations in the reaction mixture were 100 mM of one of the four buffers: cyt c (65 μM), NADPH (50 μM), and either WT or Asp632Ala CYPOR (0.9 nM). Reactions were started by addition of enzyme. The initial rate of reduction of cyt c was monitored at 550 nm using an extinction coefficient of 21 mM−1 cm−1 for cyt c reduction after zeroing the absorbance at time zero.24
Measurement of Benzphetamine Metabolism by Cyt P450 2B4
Wild type cyt P450 2B4 was expressed and purified as previously described.25,26 The metabolism of benzphetamine at 30°C under steady-state conditions was determined by measuring formaldehyde formation using Nash’s reagent according to the published procedures. The reaction mixture contained 0.2 μM cyt P450 2B4, CYPOR, and cytochrome b5, and 45 uM dilauroylphosphatidyl choline in 50 mM potassium phosphate buffer pH7.4.25,27
Kinetics of the Reduction of WT CYPOR and Asp632 mutants by NADPH
The kinetics of reduction of WT CYPOR and the Asp632 mutants by NADPH were carried out at 25°C under anaerobic conditions using a Hi-Tech SF61DX2 stopped-flow spectrophotometer equipped with a temperature-controlled circulating water bath, housed in an anaerobic Belle Technology glove box (Hi-Tech, Salisbury, England) as previously described.11 The buffer contained 100 mM potassium phosphate pH 7.4 and 10% (v/v) glycerol. Briefly, the wild type or mutant reductase (10 μM in 100 mM potassium phosphate buffer) was loaded into one syringe, and another syringe was filled with either a 1- or 10-fold molar excess NADPH in 100 mM potassium phosphate buffer. The contents of the two syringes were rapidly mixed together in the stopped-flow spectrophotometer at 25°C, and the kinetics of flavin reduction were monitored at 450 nm (measures overall reduction) and 585 nm (measures neutral blue semiquinone formation). The kinetic traces were fitted to a biphasic equation using KinetAsyst2 software (Hi-Tech).
Crystallization and Data Collection
Prior to crystallization setup, wild type or CYPOR mutant proteins were dialyzed and concentrated in 50 mM HEPES (pH 7.5) buffer to ~15 mg/mL. All samples also contained 110% molar ratio of NADP+ to protein. In the case of the 2′-AMP-bound Asp632Ala mutant, instead of adding extra NADP+, 0.3 mM (final) 2′-AMP was added into 0.03 mM dialyzed Asp632Ala mutant (~1 mg of protein in 0.5 mL), then concentrated to about 50 μL. The concentrated sample was diluted back in 0.5 mL HEPES buffer and another 0.3 mM 2′-AMP was added, and then concentrated again to 15 mg/ml for crystallization set up. Crystals were grown using the hanging drop method28 by mixing 5 μL of the 15 mg/mL protein solution and 2 μL of reservoir solution containing 150 mM HEPES, pH 7.0, 150 mM NaAc2 and 20% polyethyleneglycol (PEG) 4000. For wild type CYPOR, large crystals grew in 7 to 10 days. The crystals were first transferred to a well solution containing 25% PEG 4000 (cryo solution 1), and then transferred to another well solution containing 30% PEG 4000 (cryo solution 2), before they were flash frozen in liquid nitrogen. For the NADP+-bound Asp632Ala mutant case, only very small crystals appeared, and most of them had high mosaicity and low resolutions. When left to grow further, the crystals became round-shaped (like lemon drops) and lost diffraction within 1 to 2 days after they first appeared. Therefore, these NADP+-bound mutant crystals were flash frozen for x-ray screening soon after they appeared. In order to obtain 4e-reduced, wild type NADP+-bound crystals, the preformed, oxidized crystals were soaked for one minute in cryo solution 1, and then transferred to a freshly made solution containing 150 mM HEPES, pH 7.0, 150 mM Na2S2O4, 150 mM NaAc2 and 30% PEG 4000 and kept for about 30 minutes. During this reduction process, crystals cracked to small pieces and most of them lost their diffraction quality. These colorless pieces of broken crystals were flash frozen in liquid nitrogen and screened for diffraction quality using our in-house R-AXIS system. Data for wild type CYPOR crystals were collected at Brookhaven National Laboratory National Synchrotron Light Source. Beamline X26C Data for the Asp632Ala mutant crystals were collected at 21ID-D beamline at the Advanced Photon Source, Argonne National Laboratory. Data for NADP+-bound Asp632Phe crystals were collected using our in-house R-AXIS IV++ detector system with a Micromax 007 generator. All data processing was done using the program HKL2000.29
Structure Determination
Initial phases were calculated using the published high resolution rat CYPOR mutant structure (Protein Data Bank accession code, 1JA1). Multiple rounds of reciprocal space refinement were carried out using CNS 1.3,30 and subsequent manual fitting with program COOT31 was performed using 2Fo-Fc and Fo-Fc maps. To refine the local structure near the Asp632 loop, omit maps were used that were calculated from the CYPOR model omitting residues of Ala625-Gly652, Tyr672-Ser678, FAD, and NADP+. Water molecules were added according to Fo-Fc map in the program COOT, followed by manual addition or deletion of water molecules until Rfree convergence was achieved. Data collection and the refinement statistics are given in Table 1.
Table 1.
Data Collection and Refinement Statistics
| Proteins | WTox/NADP+ | WTred/NADP+ | D632A/NADP+ | D632F/NADP+ | D632A/2′-AMP |
|---|---|---|---|---|---|
| PDB Code | 5URD | 5URE | 5URH | 5URG | 5URI |
| Data Collection | NSLS(X26C)a | NSLS(X26C) | APS(LS-21)b | R-AXIS IV++(in house) | APS(LS-21) |
| Resolution (Å) | 50–1.9 (1.93–1.90) | 50–2.3(2.34–2.30) | 50–2.5(2.54–2.50) | 50–2.3(2.38–2.30) | 50–2.7(2.75–2.70) |
| No. of Measured Reflections | 1096331 | 253880 | 428795 | 458490 | 263957 |
| No. of Unique Refs. | 107662 | 61912 | 50250 | 61571 | 39088 |
| Completeness (%) | 100(99.9)c | 97.2(71.2) | 100(99.6) | 96.7(99.8) | 99.9(100) |
| Redundancy | 10.2(9.8) | 4.2(2.3) | 8.5(5.4) | 7.5(6.6) | 6.8(6.8) |
| I/σI | 47(3.8) | 21(2.0) | 25(2.5) | 34(3.9) | 19(3.5) |
| Space Group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Unit Cell Dimensions (Å) | |||||
| a | 101.18 | 102.10 | 101.68 | 101.35 | 101.28 |
| b | 114.93 | 114.98 | 116.21 | 117.40 | 115.81 |
| c | 117.61 | 118.13 | 118.90 | 118.99 | 117.99 |
| R_symm | 0.083 | 0.096 | 0.086(0.583) | 0.049(0.484) | 0.099(0.554) |
| Refinementd | |||||
| Rcrystal | 0.205 | 0.199 | 0.204 | 0.212 | 0.203 |
| Rfree | 0.239 | 0.249 | 0.251 | 0.264 | 0.264 |
| No. of protein atoms | 9674 | 9725 | 9682 | 9736 | 9649 |
| No. of ligand atoms | 243 | 274 | 258 | 264 | |
| No. of water atoms | 723 | 483 | 336 | 426 | 229 |
| B-factor Analysis (Å2) | |||||
| MolA(FMN/FAD/all)e | 35.1/36.2/35.9 | 36.6/38.1/37.7 | 42.6/40.1/40.8 | 49.5/43.7/45.3 | 36.0/35.5/35.7 |
| MolB(FMN/FAD/all)e | 77.6/36.9/48.2 | 75.6/41.3/51.0 | 81.6/43.4/54.2 | 66.8/44.7/50.6 | 87.3/38.1/50.7 |
| Protein/Ligand/Water | 42.0/37.9/43.7 | 44.3/43.7/40.9 | 47.5/46.1/38.2 | 48.0/50.4/44.1 | 43.5/42.5/30.0 |
| Ramachandran Analysis | |||||
| Most Favored | 97.0% | 96.3% | 95.3% | 95.8% | 93.2% |
| Generously Allowed | 2.7% | 3.4% | 4.4% | 3.9% | 6.4% |
| Outliers | 0.3% | 0.3% | 0.3% | 0.3% | 0.4% |
NLSL: National Synchrotron Light Source, Brookhaven National Laboratory
Advanced Photon Source, Argonne National Laboratory
Values in parentheses are those for the highest resolution shell.
Refinement with bond length RMSD≤;0.007 Å and bond angle RMSD<1.31
MolA(FMN/FAD/all): Average B-factors for Molecule A in the asymmetric unit, values for the FMN domain/FAD domain/All atoms./MolB(FMN/FAD/all): Same as above, but for Molecule B in the asymmetric unit.
RESULTS
The Structure of Oxidized Wild Type CYPOR with Bound NADP+ at 1.9 Å Resolution
Although many higher resolution structures of CYPOR molecules have been determined, only one structure of NADP+-bound, wild type rat CYPOR has been reported, which is of a relatively low resolution (1AMO, 2.6 Å resolution). Thus, we set out to determine the structure of the wild type at a higher resolution (1.9 Å) that can be directly compared with the reduced structure. As expected, the overall fold of the resulting structure is the same as other previously determined oxidized structures, including the NADP+-bound (1AMO) and the 2′-AMP–bound (4YAF) structures. As with previously determined structures, the new higher resolution structure did not reveal the exact position and conformation of the ribityl-nicotinamide moiety of the bound NADP+ cofactor (Figures S1 and 1), confirming the bipartite-binding nature of NADPH to the enzyme, with the nicotinamide searching for a suitable binding site (see below for details).
The 4-Electron Reduced Structure of Wild Type with Bound NADP+ at 2.3 Å Resolution
Although structures of the oxidized form of wild type and mutant CYPOR with bound NADP+ have been determined, no structure of the NADP+-bound reduced state has been obtained. Here, we report the novel structure of the four-electron-reduced wild type CYPOR with bound NADP+. Unlike the 2′-AMP-bound CYPOR crystals (pdb code 4YAL),32 NADP+-bound CYPOR crystals were much more difficult to reduce with sodium dithionite. When the wild type NADP+-bound crystals were soaked in sodium dithionite-containing cryo-solution (see Methods section), the crystals bleached within a few minutes, but cracked into small pieces, and lost their diffraction quality, suggesting that there were some significant structural re-arrangements upon reduction.
However, after numerous trials, a data set from a dithionite-reduced crystal with bound NADP+ was obtained that diffracted to 2.3 Å resolution. As expected, the overall structure, including the Asp632 loop conformation forming a single turn of a π-helix (Asp632-Asn635) (hereafter referred to as “retracted” conformation), is the same as in the oxidized NADP+-bound structure18 and the current higher resolution structure (Table 1) (Figure 1). However, upon careful inspection, there are many significant differences from the oxidized structure. In the FMN domain they include: 1) the Gly141-Glu142 peptide bond in the FMN domain adopts an O-up conformation forming an H-bond with the protonated flavin N5, and 2) the phenolic side chain of Tyr140 rotates, breaking an H-bond between the phosphate of FMN and its hydroxyl group, adopting a conformation approximately parallel to the FMN ring. These features in the FMN domain are exactly the same as those observed in the reduced 2′-AMP-bound structure32 (pdb code: 4YAL). The movement of Tyr140 is of interest, as this residue has been suggested to influence the rate of FMN reduction.33 In addition, the movement of the Gly141 loop together with that of Tyr140 contributes to the tighter interaction between the two flavin domains in the reduced enzyme (see Discussion). As for the FAD domain: 3) the indole ring of Trp677 stacks onto the FAD isoalloxazine ring, but with the long axes of both rings in a parallel conformation instead of the perpendicular orientation observed in the oxidized structure (Figures 1, S1, S2, and S3). 4) The side chain of Asp675, previously shown to be essential for hydride transfer, forms a hydrogen bond with the Nε1 of Trp677 (Figures 1, S1, and S2), and 5) the C-terminal carboxylate of Ser678 is shifted toward the FMN domain, making H-bonds with the side chain of Tyr178, which stacks against the si face of the FMN ring (Figure S3A).
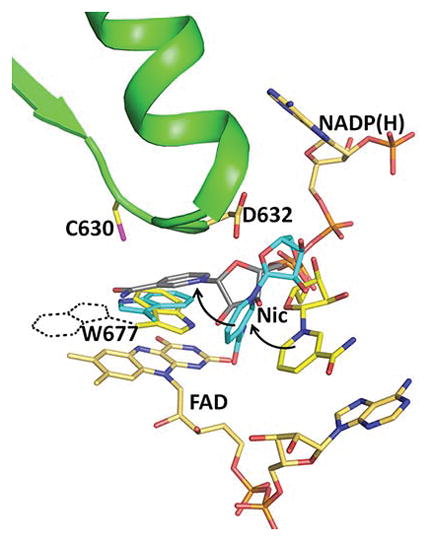
Figure 1.

Structures of oxidized and reduced wild type CYPOR in complex with NADP+ in the vicinity of the Asp632 loop and FAD. Oxidized (A, gold), reduced (B, grey), and overlay (C). The notable differences in the FAD domain are the conformations of Trp677 and the nicotinamide moiety of NADP+. In the oxidized structure, the planes of the Trp677 indole ring and the FAD ring are stacking onto each other, but not completely overlapping, and the long axes of their planes are perpendicular. In the reduced structure, the two planes also stack and overlap, and their long axes are parallel. In the oxidized structure, the ribose-nicotinamide moiety is disordered (possible location shown with thin sticks, taken from the structure of the oxidized triple mutant structure, Ser457Ala/Cys630Ala/Asp675Asn (1JA1)19. In the reduced structure, the nicotinamide is well defined and the carboxamide group of NADP+ forms two H-bonds (dotted lines), the amide group with the negatively charged N1 of the FAD isoalloxazine ring and the carbonyl group with the hydroxyl of the Thr535 side chain, which in turn makes an H-bond with a pyrophosphate oxygen. In the reduced structure, the plane of the nicotinamide ring is perpendicular to both the Trp677 indole ring and the FAD ring, both of which are stacked on each other.
The most prominent and exciting feature of the NADP+-bound, reduced wild type structure is that the ribityl-nicotinamide moiety of NADP+ is clearly visible (Fig. 1 and S2), in contrast to the oxidized structure (Figure S1), in which the ribityl-nicotinamide ring is completely disordered. In the reduced structure, the plane of the nicotinamide ring is perpendicular to the FAD isoalloxazine ring as well as Trp677, which is stacked on the flavin ring. The carboxamide of the nicotinamide ring forms two H-bonds, one with the carboxamide nitrogen to the unprotonated negatively charged N1 of the isoalloxazine ring, and a second with the carbonyl oxygen of the carboxamide to the hydroxyl group of Thr535. The Asp632 loop is in the retracted, H-bonded conformation as in the oxidized 2′-AMP structure. Since NADP+ cannot be reduced by sodium dithionite at neutral pH, this reduced structure with the combination of the FAD hydroquinone and NADP+ may be mimicking either the structure of ribityl-nicotinamide moiety of NADPH approaching the re face of the FAD isoalloxazine ring after securely anchoring the 2 ′-AMP-5′PPi half of the NADPH molecule prior to hydride transfer, or the structure of the enzyme immediately after hydride transfer from NADPH to FAD, forming NADP+ and reduced FAD (FADH2). Since the catalytic cycle in microsomes starts with the 1e-reduced air-stable blue semiquinone form (FADox-FMNsq)1, the enzyme after hydride transfer would be the 3e-reduced form (FADhq-FMNsq). Although we do not have the structure of the 3e-reduced form, it will very likely be the same as our observed 4 e-reduced structure, because the structures of the FMN domain at 1e-reduced and 2e-reduced states would be the same, as it is in the case of flavodoxins.34,35
These observations are consistent with the notion that NADPH binding to the enzyme involves two steps: first the 2′-AMP-PPi moiety binds tightly by making salt bridges and hydrogen bonds with Arg597 and Lys602 (with 2′-AMP), and Arg297 and Arg567 (with PPi), followed by the ribityl-nicotinamide moiety searching for and finally landing onto the re-face of the FAD ring, replacing the indole ring of Trp677 (Figure 2). Figure 2 shows an overlay of three structures (the wild type oxidized, reduced, and the Trp677G mutant) in the vicinity of the Asp632 loop. The Trp677G structure shows the A side of the nicotinamide ring facing the re-face of the FAD isoalloxazine ring and the two rings stacked onto each other, poised for the hydride transfer reaction.19 Relatively minor changes of the phosphodiester torsional angles and of the glycosidic bond in the ribityl-nicotinamide moiety of NADPH can easily convert the NADP(H) conformation in the reduced structure to that of the hydride transfer-competent mode (Figure 2).
Figure 2.
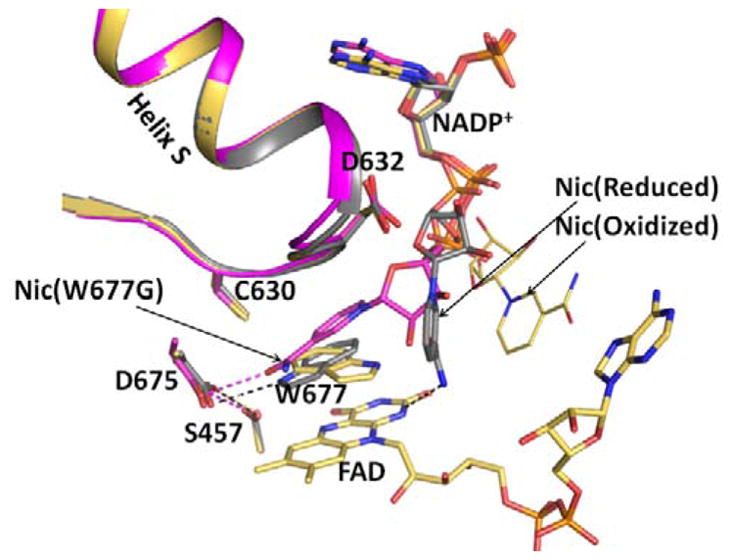
Structures of the oxidized triple mutant (Ser457Ala/Cys630Ala/Asp675Asn) CYPOR, (1JA1) (gold), dithionite-reduced CYPOR (grey), and oxidized Trp677Gly mutant (1J9Z, magenta) were superimposed. They reveal three conformations of the ribityl nicotinamide moiety of NADP+. In the oxidized wild type structure, the indole ring of Trp677 stacks onto the FAD ring and the ribityl nicotinamide (Nic-ox) is flexible (estimated occupancy, ~<50%) with no defined interactions with FAD and Trp677. In the reduced structure, the indole ring also stacks onto the flavin with the long axes of both rings being parallel. There is an H-bond between the Nε1 of Trp677 and Asp675 side chain. In addition, the carboxamide of the ribityl nicotinamide ring (Nic-reduced) forms hydrogen bonds with the negative N1 of the flavin and the Thr535 side chain hydroxyl. The face of the nicotinamide ring interacts with the edge of the Trp677 indole ring. In the Trp677Gly structure, the nicotinamide ring (Nic-W677G) stacks against the re-face of the isoalloxazine ring and is poised for hydride transfer. The three different ribityl-nicotinamide ring conformations may represent the nicotinamide binding pathway into the active site for hydride transfer, after securely anchoring the 2′, 5′-ADP half of NADP+. For clarity, only FAD of the oxidized structure is shown.
Another significant difference between the structures of the oxidized and reduced enzymes is the closer distance between the two flavins. The area of the interface between the two flavin domains in the reduced structure is about 10% greater than that of the oxidized structure for both molecules in the asymmetric unit (Mol A/Mol B = 1030/970 Å2 for the oxidized structure and 1120/1080 Å2 for the reduced structure), resulting in shorter distances between the two flavin rings (C8–C8 distance in Mol A/Mol B = 3.8/5.4 Å for the oxidized; 3.6/4.6 Å for the reduced structure). In Mol B, where crystal packing is not as tight, the two flavin domains are closer together, and their C8-C8 atom distance is smaller by ~1 Å. The closer interdomain distance in Mol B of the reduced protein is likely the result of the following changes that occur when CYPOR is reduced: 1) the FMN 140s loop converts to a “up” conformation, 2) the phenolic ring of Tyr140 rotates and breaks an H-bond with the phosphate of FMN, and 3) the Arg514 side chain rotates to form a salt bridge with Asp147 thereby stabilizing the more closed conformation (Figure S3B).
In the structure of dithionite-reduced, 2′-AMP bound wild type CYPOR, the two flavin domains are also more closed and the interflavin distance is about 1 Å shorter than the oxidized structure (pdb code, 4YAF and 4YAL32). These are not large differences, but indicate that the two flavin domains (and thus two flavins) are more closed when the enzyme is reduced, especially in solution where there is no influence of crystal packing. A shorter distance between the two flavins in the reduced structure is reasonable, since the interflavin electron transfer immediately (30–60 s−1) follows the hydride transfer.36,37
Crystal structure of the NADP+-bound Asp632Phe mutant protein
The crystal structure of the Asp632Phe mutant provides additional support for the positioning of the nicotinamide ring of NADPH observed in the reduced enzyme.
The mutant structure revealed that the Asp632 loop adopts the down and extended conformation, the same conformation as that seen in the NADP+-free CYPOR structures, but with a bound NADP+ (Figure 3). More surprisingly, unlike most of the NADP+-bound CYPOR structures, in which the ribityl-nicotinamide moiety of NADP+ is disordered with no or only very weak electron densities, the Asp632Phe structure shows the position of the ribityl-nicotinamide moiety of the bound NADP+, although with a lower occupancy compared to the 2′,5′-ADP moiety of NADP+ (Figure S4). The Phe632 phenyl ring is situated between the edge of Trp677 and the face of the nicotinamide ring, which lies on the surface of the protein.38 Interestingly, the orientation of the Trp677 indole ring is similar to that of the reduced wild type structure. The indole ring is stacked on top of the isoalloxazine ring with their long axes parallel, with a distance of ~3.4 Å between the flavin N5 and Cε3 atom of Trp677 and the Trp677 indole nitrogen atom is hydrogen bonded to the side chain of the conserved Asp675. The exact conformation of the nicotinamide ring cannot be determined due to the weak electron density for the nicotinamide ring. It is most likely that the nicotinamide ring is oscillating about 10° along its glycosidic bond. This bulky triple ring arrangement is possible because the current ribityl-nicotinamide binding site is completely exposed to the surface of the enzyme molecule and contributes to the stabilization of the phenyl ring of Asp632Phe despite the Asp632 loop adopting the extended conformation that has previously been observed only in the NADP+-free structures. The other important factor for the stabilization of the phenyl position is its interaction with the Trp677 indole, by making a T-shaped structure between the two rings.39
Figure 3.
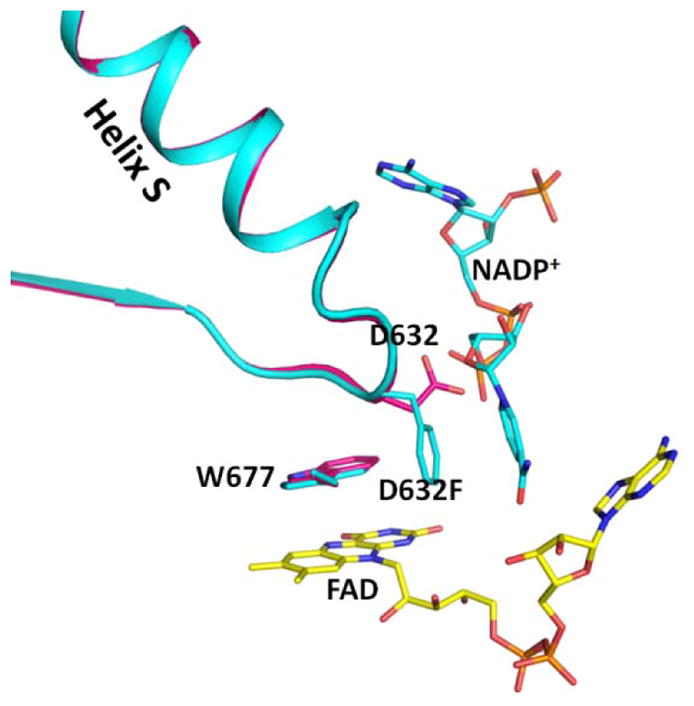
Comparison of the structures of the crosslinked NADP+-free structure (3OJW, magenta) and the Asp632Phe mutant structure (cyan) by superimposing the FAD-binding domains. The Trp indole ring conformations in the two structures are the same. The Asp632 loop conformations in the two structures are almost identical, except for the side chains of Asp632 and Phe632. In the Asp632Phe structure, the phenyl side chain is oriented parallel to the nicotinamide ring but both are perpendicular to the indole ring of Trp677 and the flavin ring.
Although the triple ring arrangement permits NADP+ binding to the Asp632Phe mutant, this is not a productive binding mode for the nicotinamide ring. The mutant is almost completely inactive (Table 2), since the phenyl side chain and Trp677 block the nicotinamide ring’s path toward the isoalloxazine ring for hydride transfer. However, the binding mode of the ribityl-nicotinamide of NADP+ observed in the Asp632Phe structure is similar to that observed in the 4-elecron-reduced wild type structure and may mimic the path of the ribityl-nicotinamide of NADPH approaching the re-face of the FAD ring, after anchoring the 2′-AMP moiety of NADPH, consistent with our interpretation of the structure of the 4-electron reduced wild type structure (see above). Figure 4 shows a comparison of the binding mode of NADP+ observed in the Asp632Phe structure with that in the 4-electron reduced structure of wild type CYPOR. The similarity between the two nicotinamide binding modes is intriguing. In addition, the orientations of the Trp677 indole ring in the two structures are also identical. The main differences are that the conformations of Asp632 loops are different and the nicotinamide ring is displaced away from the flavin toward the protein surface in the Asp632Phe structure, due to the presence of the Phe632 phenyl ring. Unlike the retracted conformation seen in the wild type reduced structure, the loop in the mutant structure is extended. As in the reduced structure, the conformation of the bound ribityl nicotinamide moiety in the Asp632Phe structure can be easily converted to the productive conformation by adjusting the dihedral angles of the bonds from the C4′ atom of the ribose to the phosphodiester bonds and the glycosidic bond (see Figure 1C).
Table 2A.
Kinetics of Reduction of cytochrome P450 reductase by 1 Molar Equivalent of NADPH at 25° C
| Enzyme | λ = 452 nm | λ= 585 nm | ||
|---|---|---|---|---|
|
|
|
|||
| k1obs/s−1 (amplitude) | k2obs/s−1 (amplitude) | k1obss−1 (amplitude) | k2obs/s−1 (amplitude) | |
| WT CYPOR | 42.1 ± 0.4 (84.5±0.6%) | 2.4 ± 0.1 (15.6±0.6%) | 38.3 ± 2.2 (79.9±0.4%) | 1.3 ± 0.5 (20.2±0.4%) |
| Asp632Ala | 2.54 ± 0.13 (69.8±1.0%) | 0.28 ± 0.02 (30.2±1.0%) | 3.68 ± 0.92 (53.8±1.3%) | 0.46 ± 0.05 (46.2±1.3%) |
| Asp632Glu | 32.60 ± 1.56 (74.8±0.3%) | 3.17 ± 0.12 (25.2±0.3%) | 32.8 ± 1.5 (66.2±2.0%) | 1.8 ± 0.6 (33.8±2.0%) |
| Asp632Phe | 0.135 ± 0.002 (100%) | nd | nd | nd |
| Asp632Asn | 15.7 ± 0.1 (78.6±0.2%) | 0.94 ± 0.02 (21.4±0.2%) | 16.1 ± 0.2 (69.6±0.5%) | 0.84 ± 0.06 (30.4±0.5%) |
nd = not detected
Figure 4.
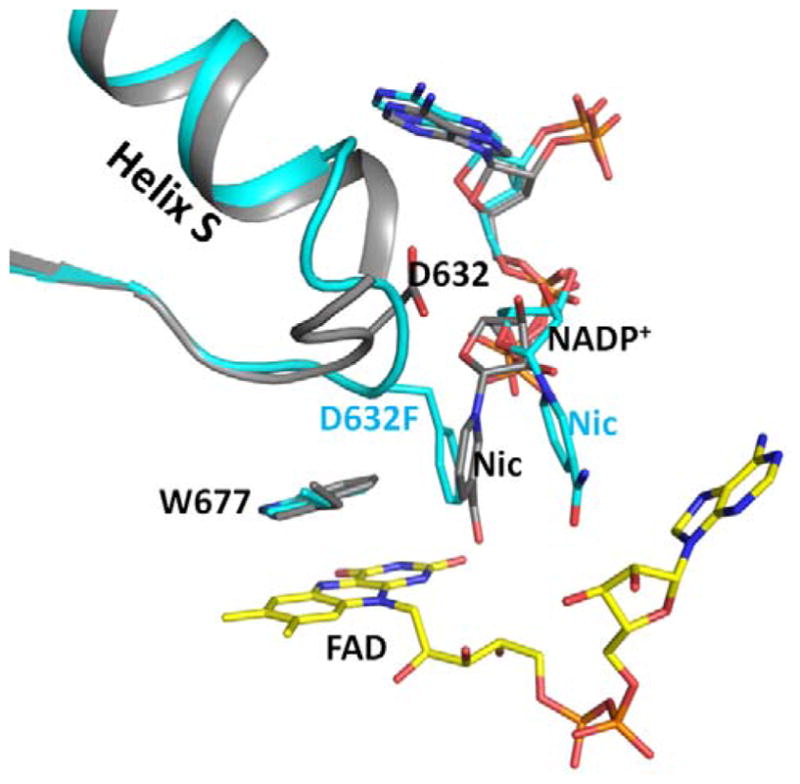
Superimposed structures of the reduced wild type (grey) and Asp632Phe mutant (cyan). The Asp632 loop conformations are different. The loop adopts the tight, retracted conformation in the reduced wild type structure, and a relaxed extended conformation in the Asp632Phe mutant structure. In the Asp632Phe structure, the side chain of Phe632 occupies the position of the nicotinamide ring of the reduced structure, and is almost perpendicular to the Trp677 indole ring in an edge to face conformation. The nicotinamide ring is essentially parallel to the phenyl ring in the Asp632Phe structure.
Another striking feature of the Asp632Phe structure is the relative positions of the FAD and FMN domains. In contrast to the reduced NADP+-bound wild type structure, the FMN domain of the oxidized NADP+-bound Asp632Phe has moved about 2 Å away from the FAD domain compared to that observed in the wild type oxidized NADP+-bound structure (Figure 5). When aligned by superimposing their FAD/NADPH domains (residues from Arg243 to Ser678), the rmsd of the alpha carbons between the FMN domains of the Asp632Phe mutant and oxidized wild type is 2.2 Å.
Figure 5.

Demonstration of the 2 Å-greater separation of the FAD and FMN domains of the Asp632Phe structure compared to wild type. Superposition of the structures of oxidized wild type (gold) and oxidized Asp632Phe mutant (cyan). The FAD/NADPH-binding domains (residues Arg243-Ser678) were superimposed. Black labels indicate the wild type and cyan labels indicate the Asp632Phe mutant. Arg634 of the wild type structure is shown as black thin sticks and the position of Arg634 in the Asp632Phe mutant structure is shown as thick cyan sticks for carbon atoms. Right panel: enlarged view of the rectangular dashed area of the left panel. In the Asp632Phe structure the FMN has moved about 2 Å from FAD compared to that observed in the oxidized wild type.
It appears that the long positive side chain of Arg634 might be partially responsible for this domain separation. Although the Arg634 side chain in the Asp632Phe mutant is relatively flexible (~50% occupancy), it is directed toward the FMN domain where it forms a salt bridge with the C-terminal carboxylate (Ser678), which is also located at the interface of the two flavin domains. Therefore, if the FMN domain remains at the “normal” closed position, the Arg634 side chain would cause steric hindrance with Thr177 and Thr178 (Figure S4). This is consistent with a proposed role of Arg634 at least partly involved in facilitating the domain opening and closing.
Crystal structures of oxidized Asp632Ala with NADP+, and with 2′-AMP
Replacement of Asp632 with alanine examines the relative contributions of hydrogen bonding and electrostatic interactions on loop movement and NADP+ binding. While the absence of the Asp632 carboxylate group would not interfere with the binding of the NADP+ pyrophosphate and 2′ AMP groups, lack of H-bonds between Ala632 and the main chain amide groups would mean that the Asp632 (i.e., Ala632) loop would only form an extended loop. The overall structure of the oxidized NADP+-bound Asp632Ala is the same as those of the previously determined wild type and mutant rat CYPOR with either NADP+- or 2′-AMP-bound. Unlike the Asp632Phe structure, whose FMN and FAD domains are further separated by about 2 Å, the structure of NADP+- bound Asp632Ala is of the normal closed conformation, with the FMN-FAD (C8-C8) distance of ~4 Å.
As expected, the Ala632 loop in the NADP+-bound Asp632Ala mutant adopts an extended loop conformation (3OJW),7 except that the, Ala 633 and Arg634 residues, including their main chain atoms, are invisible in the electron density map (Figure S5), while the electron densities for Ala632 (Asp632 in the wild type) and Asn635 are clearly shown. Thus, the middle part of the loop (residues Ala633-Arg634) appears to be highly flexible, alternating between different conformations and resulting in an unstable extended Asp632 loop, partially blocking the passage to the hydride-transfer-competent binding of NADPH. The nicotinamide ring is also disordered. The positioning of Trp677 in this mutant, however, differs from that of other structures with the extended loop. Unlike the crosslinked 3OJW and Asp632Phe, where the Trp677 indole ring’s long axis is parallel to the long axis of the FAD ring and the nitrogen atom of the indole ring makes an H-bond with Asp675 carboxylate, Trp677 in the Asp632Ala structure is positioned in a perpendicular orientation; thus there is no H-bond between Asp675 and Trp677. This highly unstable loop structure is consistent with the peculiar behavior of the crystals of the Asp632Ala mutant with bound NADP+. The mutant crystals are generally much smaller and have very high mosaicity. In addition, they don’t “age” well. In fact, even a two-day-old crystal could not be indexed, even if it had a reasonable diffraction quality, suggesting that the crystal is no longer a single crystal. Thus, numerous attempts to reduce the crystalline protein with dithionite were not successful.
Interestingly, crystals of Asp632Ala complexed with 2′-AMP are stable and behave more like the wild type crystals. However, the 2′-AMP-bound Asp632Ala crystal structure (Figure S6) shows that the Asp632 loop appears as flexible as that in the NADP+-bound structure. In fact, both Ala633 and Arg634 residues are completely disordered, including their main chain atoms, even though the 2′-AMP-bound crystals are stable. This suggests that the instability of both the oxidized and especially the dithionite-reduced crystals with NADP+-bound, is due to the presence of ribityl nicotinamide conformations that disrupt crystal formation, suggesting that the ribose-nicotinamide moiety of NADP+ in the Asp632Ala mutant must adopt a conformation(s) that cannot be stably accommodated in the Asp632Ala mutant structure, and even more so in its reduced structure (see Figure 6). The inability to obtain reduced, NADP+-bound Asp632Ala crystals indicates that, in the reduced Asp632Ala structure, the compact structure of the “closed” conformation of the CYPOR molecule (Figure 6) cannot accommodate the conformations of the mutant Asp 632 loop, reduced FAD (FADH-), Trp677, and the nicotinamide ring of NADP+. More likely, the two flavin domains must move apart from each other to accommodate the conformational changes caused by the mutation in the Asp632 loop and reduction of the flavins, especially in the presence of NADP(H).
Figure 6.
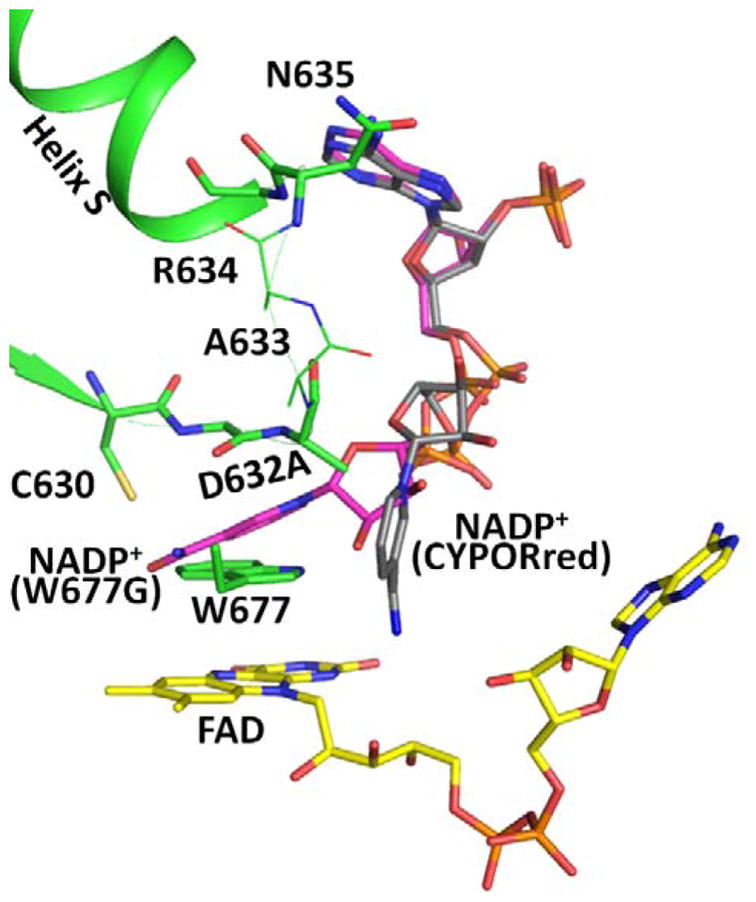
Comparison of the structures of the Asp632Ala mutant with bound NADP+ (green; for clarity, no NADP+ is shown), Trp677Gly (pdb code, 1J9Z, magenta), and reduced wild type CYPOR (grey). For clarity, only the Asp632 loop in the Asp632Ala structure (green) is shown. Nicotinamide rings in the Trp677Gly structure [magenta carbons, NADP+ (W677G)] and in the reduced wild type structure [(grey carbons, NADP+ (CYPORred)] are shown. Ala633 and Arg634 are disordered and their possible positions are shown as thin sticks.
UV-visible Spectra of Oxidized WT CYPOR and the Asp632 Mutants
The UV-Visible absorption spectra of the oxidized forms of Asp632Ala, Asp632Glu, and Asp632Asn reductase mutants are similar to that of wild type CYPOR (Figure 7). In contrast, the Asp632Phe mutant spectrum exhibits a significant increase in the intensity of the 380 nm band. However, the wavelength of the absorbance maximum at 380 nm or 454 nm is not altered in either the mutant or the wild-type reductase. The increase in intensity of the 380 nm peak in the Asp632Phe variant likely reflects the more hydrophobic environment of the FAD isoalloxazine ring as a result of its interactions with Phe632 (Figure 3, Figure S4).40,41
Figure 7.
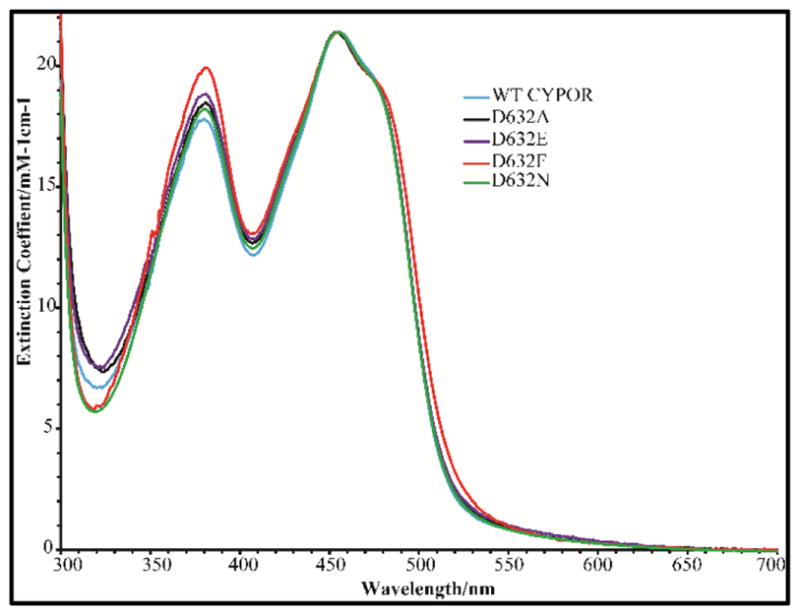
UV visible absorption spectra of oxidized wild type and Asp632 mutants of CYPOR.
Kinetics of the Pre-Steady- State Reduction of WT and Mutant CYPOR by 1-Molar Equivalent of NADPH
In an effort to determine whether FAD reduction and interflavin electron transfer were perturbed by the mutations in the Asp632 loop, the pre-steady-state kinetics of reduction of the wild type and Asp632 mutants by 1-molar equivalent of NADPH under anaerobic conditions was investigated. Figure 8 and Table 2A show the rate of reduction of the wild type and the Asp632 mutants with 1-molar equivalent of NADPH. The absorbance changes during anaerobic reduction of wild type CYPOR and the Asp632 mutants were monitored at 452 nm and 585 nm for wavelengths associated with overall flavin reduction and blue semiquinone formation, respectively.
Figure 8.
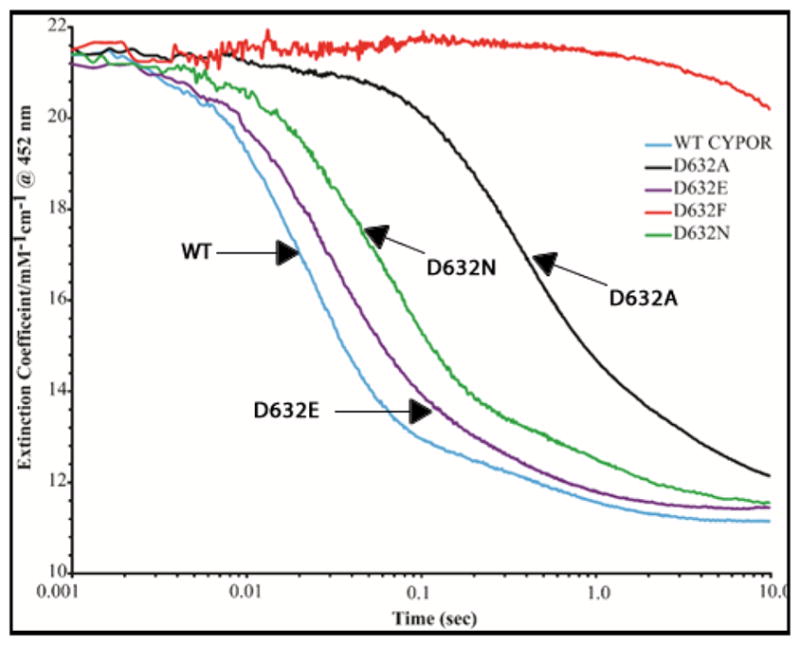
Kinetics traces at 452 nm demonstrating the reduction of the FAD of wild type and Asp632 mutants of CYPOR by 1 molar equivalent of NADPH under anaerobic conditions at 25°C.
As shown in Figure 8, the kinetics of reduction of WT CYPOR are biphasic at 452 nm with an initial fast phase (k1 ~42.1 s−1) and a slower second phase (k2 ~2.4 s−1) with amplitudes of 84% and 15%, respectively (Table 3). The first phase in wild type CYPOR represents the reduction of FAD by NADPH and rapid intramolecular electron transfer from FADH2 to FMN which, at equilibrium, results in a mixture of about 66% of the FMN and FAD disemiquinone (FADsq-FMNsq) and 33% of the FMN hydroquinone form (FADox-FMNhq) of the CYPOR. The rate of reduction of Asp632Glu is slightly less than that of WT CYPOR (k1=32.6 s−1 vs k1=42.1 s−1 for WT CYPOR), indicating that the Asp632Glu mutation did not significantly alter the rate of reduction of FAD (Table 2). Reduction of the FAD of the Asp632Ala, Asp632Phe, and Asp632Asn mutants by a 1-molar equivalent of NADPH, when monitored at 452 nm and 585 nm, is significantly slower than with the WT (Table 2A and Figures 8 and 9). The rate of reduction of the Asp632Ala mutant is decreased by ~95% compared to that of wild type CYPOR (k1=2.54 s−1 versus k1=42.1 s−1), whereas the Asp632Phe mutant has only been minimally reduced by NADPH (k1- undetermined) after 10 sec (Table 2, Figures 8 and 9). The rate of reduction of Asp632Asn, when monitored at 452 nm, is ~62% slower than that of WT CYPOR (k1=15.7 s−1 versus k1=42.1 s−1).
Table 3.
The activity of wild type and mutant reductases with their redox partners, cytochrome c, cyt P450 2B4 and ferricyanide was determined at 30°C as described in the experimental section.
| CYPOR reductase mutant/WT | Fe3+ cytochrome c | cyt P450 2B4 | Fe(CN)63− | |
|---|---|---|---|---|
| nmol cyt c reduced/s/nmol CYPOR | KmNADPH μM |
nmol CH2O/s/nmol CYPOR | nmol reduced/s/nmol CYPOR | |
| WT CYPOR | 87.1±1.7 (100%) | 6.95±0.62 | 0.68±0.002 (100%) | 131.9±5.9 (100%) |
| Asp632Ala | 2.5±0.1 (2.9%) | 0.67±0.07 | 0.13±0.003 (19%) | 5.3±2.8 (4%) |
| Asp632Glu | 76.3±2.4 (88%) | 16.3±0.60 | 0.56±0.003 (82%) | 120.6±0.6 (92%) |
| Asp632Phe | < 0.1 | nda | < 0.01 | < 0.1 |
| Asp632Asn | 10.7±0.2 (12.3%) | 1.40±0.14 | 0.33±0.003 (48.5%) | 17.5±0.7 (13%) |
| Arg634Ala | 91±9 (100%) | nd | 1.02±0.02 (150%) | nd |
nd means not determined.
Figure 9.
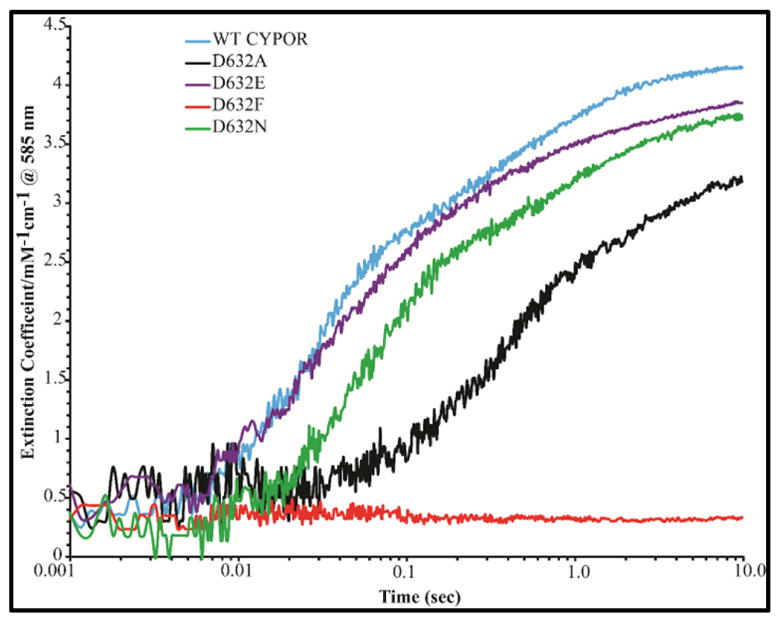
Kinetic traces at 585 nm demonstrating the rate of formation of the blue semiquinones of wild type and Asp632 mutants of CYPOR by 1 molar equivalent of NADPH under anaerobic conditions at 25°C.
Electron transfer from FAD to FMN was monitored by following the formation of the blue semiquinone (FMNsq/FADsq), as indicated by the absorbance increase at 585 nm. Semiquinone formation occurs rapidly in the presence of a 1-molar equivalent of NADPH for the Asp632Glu mutant and WT CYPOR (k1=32.8 s−1versus k1=38.3 s−1), moderately slower for Asp632Asn (k1=16.1 s−1), and much slower (10-fold decrease) for Asp632Ala mutant (k=3.7 s−1) (Figure 9, Table 2A). In all cases, the rates and amplitudes of FMN semiquinone formation are similar to that of FAD reduction, indicating that electron transfer to FMN occurs almost simultaneously with FAD reduction. This demonstrates intramolecular interflavin electron transfer from FAD to FMN is intact and was not affected by the mutations. Semiquinone formation was not detected in the Asp632Phe mutant since only a trace amount of FAD was reduced during the experiment (Table 2A, Figures 8 and 9)
Kinetics of the Pre-Steady- State Reduction of WT and Mutant CYPOR by 10-Molar Equivalents of NADPH
Further characterization of the electron transfer properties of the WT and mutants was undertaken by investigating the kinetics of reduction with a 10-fold molar excess of NADPH (Table 2B and Figures 10 and 11). The kinetics of reduction by a 10-fold molar excess of NADPH are more complicated than reduction by 1-molar equivalent but provide additional information.7 The rate of reduction of FAD is slightly faster in the presence of a 10-fold excess of NADPH for the WT and the Asp632Glu mutant. Interestingly, at the higher NADPH concentration, Asp632Glu is reduced at a rate virtually the same as the wild type (Table 2B and Figure 10). This contrasts with the somewhat slower reduction of Asp632Glu compared to WT by the 1-molar equivalent of NADPH. The rates of reduction of the Asp632 Ala, Asn, and Phe mutants are retarded (30-fold, 3-fold, and ~400-fold, respectively) regardless of the NADPH concentration.
Table 2B.
Kinetics of reduction of cytochrome P450 reductase by 10 Molar Equivalents of NADPH at 25° C
| Enzyme | λ = 452 nm | λ= 585 nm | ||
|---|---|---|---|---|
|
|
|
|||
| k1obs/s−1 (amplitude) | k2obs/s−1 (amplitude) | k1obss−1 (amplitude) | k2obs/s−1 (amplitude) | |
| WT CYPOR | 60.7±1.7 (67.3±1.0%) | 6.4±0.4 (32.7±1.0%) | 54.9±3.3 | 7.2±0.8 (Decay) |
| Asp632Ala | 1.95±0.01 (71.7±0.4%) | 0.40±0.01 (28.3±0.4%) | 3.48±0.37 | 0.44±0.02 (Decay) |
| Asp632Glu | 54.17±1.62 (59.8±1.2%) | 16.0±0.30 (40.2±1.2%) | 51.3±4.46 | 17.0±0.38 (Decay) |
| Asp632Phe | 0.146±0.003 (100%) | nd | nd | nd |
| Asp632Asn | 17.8±0.8 (61.4±2.5%) | 2.5±0.4 (38.6±2.5%) | 26.1±0.3 | 2.01±0.01 |
nd = not detected
Figure 10.
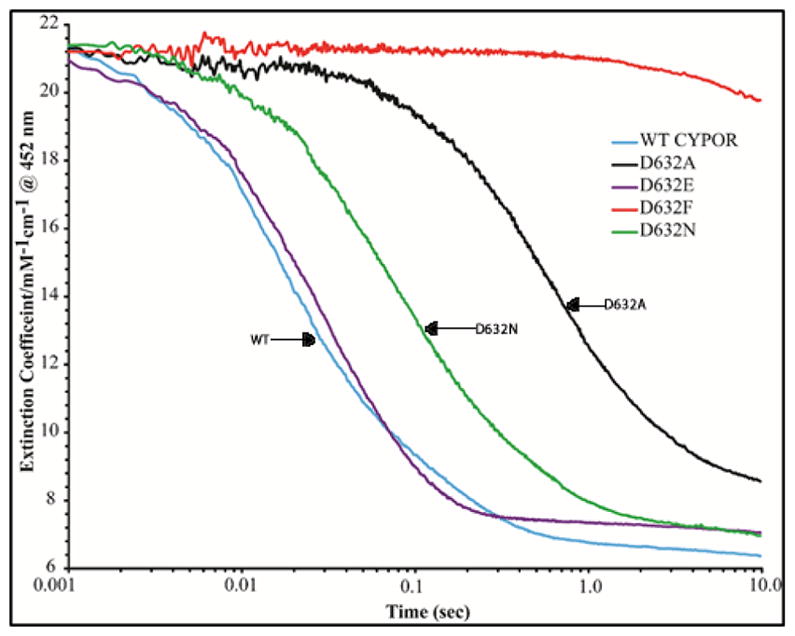
Kinetic traces at 452 nm demonstrating the reduction of the FAD of wild type and the Asp632 mutants of CYPOR by 10 molar equivalents of NADPH under anaerobic conditions at 25°C.
Figure 11.
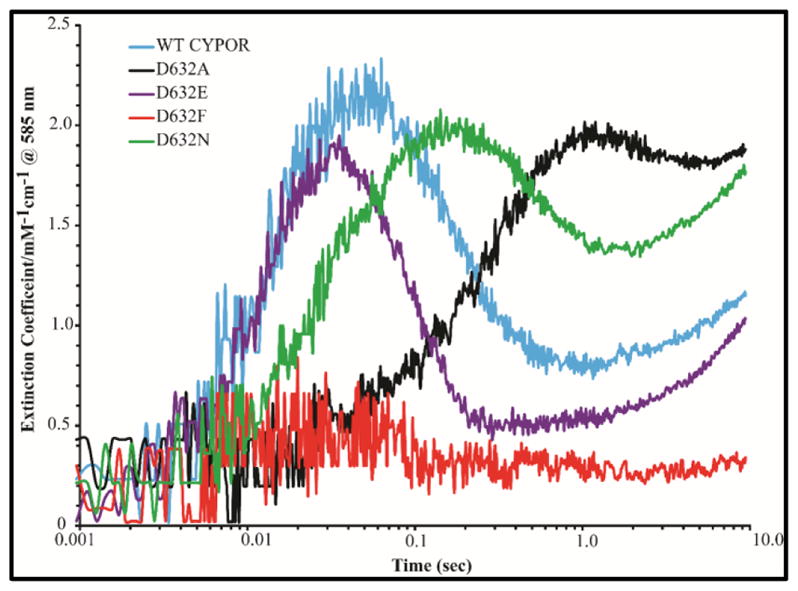
Kinetic traces at 585 nm demonstrating the rate of formation and decay of the blue semiquinones of wild type and the Asp632 mutants of CYPOR by 10 molar equivalents of NADPH under anaerobic conditions at 25°C.
As with 1-molar equivalent of NADPH, appearance of the neutral blue flavin semiquinone at 585 nm (Table 2B, Figure 11) occurred at the same rate as the reduction of FAD. Semiquinone formation was not detected in the Asp632Phe mutant. This is consistent with the conclusion that the Asp632Ala and Asp632Asn mutations impair hydride transfer but do not perturb interflavin electron transfer.
With a 10-fold molar excess of NADPH, the concentration of the blue semiquinone peaks at ~35 ms for the WT and Asp632Glu proteins, while the maximum of the blue semiquinone occurs later for Asp632Ala and Asp632Asn (Figure 11) as a result of the slower rate of reduction of FAD by the first and second molecules of NADPH. The absorbance at 585 nm decays when a second molecule of NADPH transiently generates the four-electron reduced reductase (FADhq-FMNhq) by reducing the 2-electron reduced form of the enzyme (FADox-FMNhq). The slower rate of reduction of CYPOR by a second molecule of NADPH is partially accounted for by a decreased thermodynamic driving force for electron transfer from FAD hydroquinone to the FMN semiquinone. A second consideration is the lower concentration of oxidized FAD (~30%) in the two-electron reduced enzyme compared to 100% in the completely oxidized reductase.11 Although the WT and Asp632Glu proteins are reduced by the first NADPH molecule at the same rate, reduction by the second NADPH molecule occurs ~2-fold faster in the Asp632Glu mutant (k=17±0.38, vs WT 7.2±0.8 s−1) perhaps because NADP+ is released faster when a bulkier glutamic acid is at position 632 rather than the smaller aspartic residue. As expected, the bleaching of absorbance at 585 nm of Asp632Ala and Asp632Asn by the second molecule of NADPH is also slow (Table 2B, Figure 11). It is noteworthy that after the maximum decrease in the 585 nm absorbance, it again slowly increases, reflecting more blue semiquinone formation. Since thermodynamic considerations predict that a 10-fold molar excess of NADPH (Em=-320 mV) will not reduce 100% of the reductase molecules in the reaction mixture, we hypothesize that the FAD hydroquinone disproportionates and reduces the FAD in a second reductase molecule forming two FADH• semiquinones (Em FADH• = -290 mV).10 During the first 10 sec of the reaction between Asp632Phe and NADPH, semiquinone formation at 585 nm could not be detected. Our results with a 10-fold excess of NADPH and reduction of FAD (452 nm) and the Asp632Asn and Asp632Ala mutants are in agreement with previous results in the human reductase.21 The slow semiquinone formation is a consequence of slow FAD reduction, not impaired interflavin electron transfer.
Activity of the Mutant Reductases with Redox Partners, Cyt P450 2B4, Cytochrome c and Ferricyanide
Under steady state conditions the effect of the mutations of Asp632 on the reduction of cytochrome c and ferricyanide and the cyt P450 2B4-mediated metabolism of benzphetamine is shown in Table 3. The cytochrome c reductase activity of the Asp632Phe mutant is almost completely lost (≤;0.1%) compared to that of wild type reductase (Table 3), whereas the Asp632Ala mutants retains ≤; 3% of the WT activity. These rates are consistent with the observed crystal structures showing that hydrogen bonding interactions stabilize the Asp632 loop in a retracted position necessary for binding of NADPH in a physiologically relevant conformation (Figures 1–6). The Asp632Glu protein exhibited essentially wild type cyt c reductase activity (88% active), indicating the mutation did not significantly perturb the activity of the reductase. The Asp632Asn mutant exhibited only ~12% of WT cyt c reductase activity, indicating that replacing the negatively charged carboxylate at position 632 with a carboxamide group significantly decreases the activity of the reductase; however, the four-fold increased activity of Asp632Asn compared to Asp632Ala implies that the carboxamide of the asparagine side chain can participate in weak hydrogen bond formation with the backbone amides.
In view of the apparent nonphysiologic binding of NADPH to the protein, it was rather puzzling when the mutants exhibited a decreased Km for NADPH for cytochrome c reduction. Generally a decreased Km indicates tighter binding of a substrate to an enzyme. However, an anomalous decrease in Km for NADPH is consistent with a two-site, ping-pong kinetic mechanism “When a mutation in the binding site of one substrate causes a decrease in the catalytic velocity of one of the two half-reactions, there is a consequent reduction in the value of Km for the substrate of the half-reaction that becomes rate limiting.”7,42–45 The KmNADPH value for the Asp632Phe mutant could not be measured because the protein is inactive with cytochrome c.
The effects of mutation on enzyme activity were also measured by using potassium ferricyanide as electron acceptor, which is reduced directly by the FAD domain.46 As shown in Table 3, the Asp632Glu mutant exhibited WT ferricyanide activity. The activities of the Asp632Asn, Asp632Ala, and Asp632Phe mutants with ferricyanide were ~13%, ~ 3%, and < 0.1%, respectively, of that of WT CYPOR, indicating that hydride transfer from NADPH to FAD is impaired when Asp632 is replaced by a non- acidic residue. The activities of Asp632Ala and Asp632Asn with cytochrome c and ferricyanide agree with previously determined values for the corresponding human CYPOR mutants.21
The mutant enzymes were further characterized with respect to their ability to support catalysis by a physiologic redox partner, cyt P450 2B4 (Table 3). The Asp632Glu mutant was able to support the metabolism of benzphetamine by cyt P450 2B4 (82% active) essentially to the same extent as the WT. The Asp632Asn mutant had moderate activity (~49% active) whereas the Asp632Ala mutant retained only ~19% of WT activity. The Asp632Phe mutant is essentially inactive with cyt P450 2B4. The Arg634Ala mutant is about 50% more active in supporting P450-dependent benzphetamine demethylase activity than the wild type and has the same activity as wild type protein with cytochrome c (Table 3).
The Effect of pH on WT and Asp632Ala CYPOR is Similar
The pH dependence of CYPOR cytochrome c reductase activity has demonstrated the presence of two ionizable groups, one acidic and one basic, which are essential for catalysis.47,48 Maximum activity requires that the acid group be unprotonated, and the basic group protonated. To determine if Asp632 was the acidic residue, the pH dependence of Asp632Asn was determined at 30°C using four different buffers: HEPES, Tricine, MES, and TES in the pH range within ~0.5 pH units of their pKa’s at an ionic strength of 525 mM. Figure 12 shows that the pH dependence of cytochrome c reductase activity is nearly identical to that of the wild type protein. This indicates that Asp632 does not account for the acidic pKa of the wild type protein.
Figure 12.
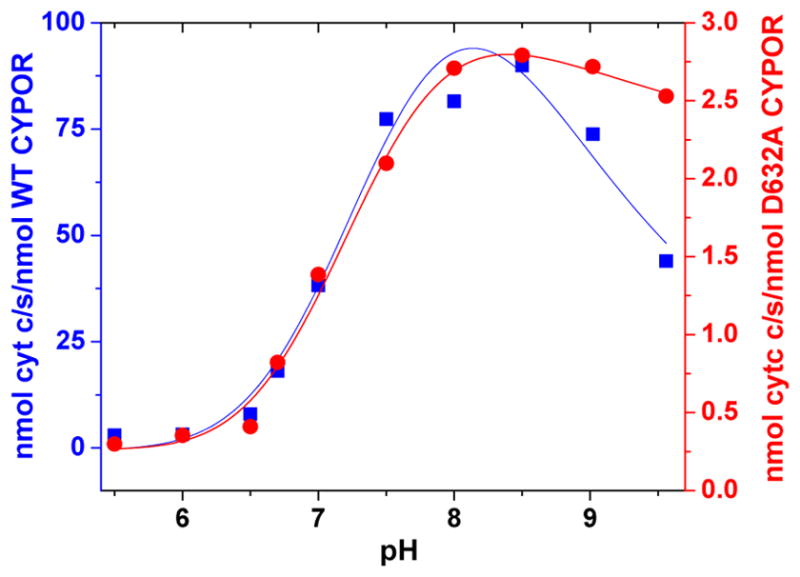
Comparison of the effect of pH on the cytochrome c activity of wild type (blue) and Asp632Ala (red) CYPOR at 30°C.
DISCUSSION
Importance of Asp632 Loop from Previous Studies
For many decades, several mutational studies have been conducted in order to obtain insights into the exact nature of binding NADPH to cyt P450 reductase. These studies have highlighted the importance of the 2′phosphate and the bipartite nature of cofactor binding.19,47,49,54 In an attempt to obtain further information on the mechanism of NADPH binding, we have compared and correlated structures and biochemical properties of mutants of Asp632 and Arg634, in the so-called “Asp632 loop,”7 as well as determining the structure of the NADP+-bound dithionite-reduced wild type CYPOR.
Evidence that the Asp632 loop is important in controlling the binding of NADPH and release of NADP+ first came from comparison of an NADP+-free reductase structure, 147CC514, (pdb code 3OJW) in which a disulfide bond was engineered between residues Asp147 (in the FMN domain) and Arg514 (FAD domain) with NADP+-bound structures, which showed that movement of the Asp632 loop correlated with NADP+ binding. In the NADP+-free structures reported to date [e.g., the 147C-C514 crosslinked (3OJW), yeast/human hybrid CYPOR (3FJO), and the FAD/NADP(H)-binding domain of human methionine synthase reductase (MSR, 2QTL) structures], the Asp632 loop forms an extended loop. The location of the Asp632 side chain near the would-be binding site of the NADP+ pyrophosphate group provides a feasible mechanism for assisting in NADP+ release by using the negative charge as well as the steric repulsion of the Asp632 carboxylate side chain. This is consistent with the fact that the 5′-PO4 group of NADP(H) provides a considerable binding affinity for NADP(H) to the reductase (Kd, ~20 uM for 2′-AMP vs. 0.5 uM for both 2′,5′-ADP and NADP+).16 When NADP(H) binds, this Asp632 side chain is forced away due to the charge and steric repulsion and forms three hydrogen bonds with the main chain amide groups of Arg634, Asn635, and Met636, resulting in a tight, retracted loop conformation (Figures S1 and 1). Thus, it was suggested that the Asp632 loop modulates NADPH binding and NADP+ release.7 Asp632 is conserved in the diflavin superfamily and effects of mutation of the corresponding residue in human CYPOR (Asp634, human CYPOR numbering),21 have confirmed the importance of Asp634 in the binding of NADPH.)
Mutants Support Hypothesis That Control of the Asp632 Loop Conformation is Essential for Catalysis
Substitution of Asp632 with alanine, phenylalanine, and asparagine resulted in significantly decreased catalytic activity of these reductase mutants compared to wild type reductase, whereas mutating Asp632 to glutamic acid minimally perturbed its catalytic properties. The decrease in catalytic activity for these mutants and the retention of the catalytic properties for Asp632Glu, and correlation with the structures in the vicinity of the Asp632 loop, show that the acidic residue (Asp632) is essential for control of the conformation of the Asp632 loop and for providing electrostatic and steric repulsion with the pyrophosphate of the NADP+, thereby contributing to the control of the hydride transfer, productive binding of NADPH, and possibly the release of NADP+.
Our results of steady state kinetic studies with the Asp632Asn and Asp632Ala mutants are largely in agreement with previous results in the human reductase. However, we do not observe the increase in cyt c reduction activity with our Arg634Ala mutant, reported by Mothersole and coworkers.21 A difference is that their cyt c rates were determined at low ionic strength (50 mM phosphate) while ours were conducted at a higher ionic strength (270 mM potassium phosphate). Studies with the pig liver CYPOR50 and with recombinant rat CYPOR24 have demonstrated the ionic strength dependence of cyt c reductase activity and rate-limiting NADP+ release at low ionic strength. This is not surprising considering the number of salt bridge and hydrogen bonding interactions identified in our structures. However, we do observe a 50% increase in cyt 2B4 activity with the Arg634Ala mutant, carried out in 50 mM potassium phosphate, compared to the wild type. The P450 activity of mutations of the Asp634 loop residues in human CYPOR were not tested in the studies by Wolthers and coworkers.21
Proposed Path of Entry and Exit of NADP(H) to CYPOR Active Site for Hydride Transfer
Elucidation of the structure of the 4-electron-reduced NADP+-bound wild type enzyme suggests for the first time a route of entry and exit of the nicotinamide and the function of the Asp632 loop in this process. The novel structures of the Asp632Phe and Asp632Ala mutants revealed that NADP(H) adopts various conformations on binding to CYPOR and, as a result, induces a variety of positions of the residues which it contacts.
The structures of the reduced NADP+-bound wild type and oxidized NADP+-bound Asp632Phe structures illustrate similar nicotinamide binding modes and Trp677 orientations in the presence of alternate Asp632 loop conformations. The loop adopts the retracted conformation in the reduced structure and the domains are in the closed conformation, while Phe632 loop is extended in the Asp632Phe structure and the domains are in an open conformation. However, the phenyl group in the Asp632Phe structure occupies the same position as that the nicotinamide ring occupies in the reduced structure (Figure 4). Therefore, we propose that, as in the case of the reduced wild type structure, the binding mode of the ribityl-nicotinamide of NADP+ observed in the Asp632Phe structure mimics the passage of the ribityl-nicotinamide portion of NADPH approaching the re-face of the FAD ring, after anchoring the 2′-AMP moiety of NADPH, which is consistent with our interpretation of the structure of the 4-electron reduced wild type structure (see above).
NADPH Binding Induces the Closed Conformation
In all previously reported three NADP+-free structures (3OJW, 3FJO, and 2QTL), the Asp632 loop adopts an extended loop, and the long Arg634 side chain extends out toward the FMN domain. However, none of these three NADP+-free structures has the FMN domain adjacent to the FAD domain. They are either the FAD domain only (MSR), the FMN/FAD domains are wide open (yeast-human chimera), or the two flavin domains are twisted (crosslinked structure), suggesting that NADP+ binding is associated with the closed conformation of the reductase. Interestingly, the recent structure of the Arabidopsis thaliana CYPOR2 (ATR2) has a very twisted arrangement of the two flavin domains (two C8 methyl groups of FMN and FAD distance, ~19 Å) and without NADP(H) bound.51 Surprisingly, the A. thaliana Asp636 loop (corresponding to Asp632 of rat CYPOR) in the ATR2 structure is in a tight retracted position. Nevertheless, evidence from these four structures is consistent with the notion that NADP(H) binding promotes the “normal” closed conformation for the two flavin domains.
Association of NADP(H) binding with a closed conformation is observed irrespective of flavin redox state. The newly determined structure of the NADP+-bound 4- electron reduced CYPOR reveals that the two flavin domains are even more closed and the distance between the C8-C8 atoms of the two flavin rings are closer by ~1 Å, compared to the fully oxidized enzyme, further facilitating interflavin electron transfer. This is more prominent in Mol B of the two molecules in the asymmetric unit, since Mol B is less constrained by the crystallographic packing. Binding of NADP(H) to the closed conformation is also consistent with observations of SAXS13 and SANS52 studies indicating formation of a more compact conformation in the presence of cofactor and with observations of increased rates of interflavin electron transfer in the presence of NADP+.53
Coordination of Trp677, Asp632 loop, and domain movements
The mechanisms by which NADP(H) binding and release and domain movement and electron transfer, including the role of the conserved aromatic residue near FAD in enabling NADPH binding, has been extensively studied in CYPOR, NOS, and cyt P450 BM3. Results from this and previous studies suggests that movements of Trp677, the Asp632 loop and the flavin domains are coordinated and we propose a mechanism for this process.
In CYPOR, a conserved Trp677, which is positioned in the would-be nicotinamide-binding site and covers the re-face of the FAD isoalloxazine ring, moves away to enable NADPH binding and hydride transfer.19 It was noted from our various structural analyses that the conformation of the Trp677 indole ring in oxidized NADP+-bound structures is different from that of oxidized NADP+-free structures. In both cases, the indole ring stacks onto the re-face of the FAD isoalloxazine ring. In most NADP+-bound structures, where the Asp632 loops are in the retracted conformation, the long axes of both rings are perpendicular to each other. In contrast, in NADP+-free structures, the Asp632 loop is extended, and the two rings’ long axes are parallel to each other, forming a parallel displaced π stacking, which is a highly stable ring arrangement.39 However, the wild type 4-electron reduced, NADP+-bound structure adopts a parallel arrangement of the long axes of the flavin and indole rings, even though the structure has a bound NADP+ and the Asp632 loop is retracted. In this structure and the Asp632Phe structure, the parallel stacking of the long axes of the tryptophan and flavin rings is further stabilized by an interaction between the edge of the tryptophan ring and the face of phenyl and nicotinamide ring, respectively. This parallel stacking arrangement of the indole and isoalloxazine rings results in a hydrogen bond between the Nε-atom of the Trp677 indole ring and the Asp675 carboxylate, a catalytic residue.
Furthermore, the H-bond between the backbone amide of Trp677 and the carbonyl of Ala633 suggests a mechanism for coordination of retraction of the Asp632 loop and rotation of the Trp677 ring away from the flavin. A coordinated and concerted retraction of the Asp632 loop and displacement of the Trp677 from its stacked position on the flavin upon initial NADPH binding would ensure that NADPH binds to FAD in a hydride transfer-competent conformation. After hydride transfer, the concerted process would reverse and Trp677 would once again stack on the flavin, replacing the nicotinamide ring, and the Asp632 loop would extend facilitating expulsion of NADP+ from the active site.
Replacement of Trp677 with the aliphatic residue alanine, influenced the specificity for the reducing pyridine nucleotide in favor of NADH.49,54 The NADPH-dependent catalytic function of the Trp677Ala mutant enzyme is impaired because NADP+ is bound tightly, demonstrating that, both Trp677 and extension of the Asp632 loop are required for release of NADP+, consistent with our notion that the movements of Asp632 loop and Trp677 are coordinated.
A number of observations suggest that NADPH binding and FAD reduction trigger FMN and FAD domain separation and electron transfer to a redox partner.14–16, 53 Both the 4-electron reduced NADP+-bound and 4-electron-reduced 2′-AMP-bound wild type structures (above and Rwere et al32) demonstrate a more tightly closed conformation and shorter interflavin distance - consistent with SAXS/SANS solutions studies, which demonstrated a more compact conformation upon NADP+ binding.13,52 In the Asp632Ala mutant structures, Asp632 loop movement occurs even in the presence of bound NADP+ or 2′-AMP, and is associated with instability of the mutant crystals. Thus, increased mobility of the flavin domains as a result of increased Asp632 loop movement prevents formation of the closed NADP+-bound conformation required for stable crystal formation.
Domain movements of CYPOR in solution have been studied by various biophysical methods, including NMR.55 NMR coupled with SAXS,14 Forster Resonance Energy Transfer (FRET),12 small angle x-ray scattering (SAXS),13 and ELDOR/PELDOR methods.56 However, due to the complexity of the system (technical difficulties involving resolution of spectral overlaps, redox states, and presence or absence of cofactor binding) these studies have achieved only qualitative results. Recent studies by the Roberts group, using SANS52 methods, showed that only one or two major conformations exist at a given redox state in the presence or absence of NADP(H). Even though the structural resolution is very low, the SANS studies showed that major conformations existing in solution are the CLOSED (as observed in the crystal structure of oxidized wild type enzyme), OPEN (observed in the structure of ΔTGEE CYPOR), or a combination of both. In the reduced state, the enzyme is in a more open state. However, even in the same redox state, the nucleotide binding shifts the equilibrium toward the closed state. These results are in agreement with our studies using crystallographic methods.
Contacts between Arg634 and the FMN domain are observed in the triple mutant Ser457Ala/Cys630Ala/Asp675Asn (1JA1) and Trp677Gly mutant (1J9Z) structures, and the Asp632Phe structure. In the first two structures (1JA1 and 1J9Z)), Arg634 interacts with Asp209 and presumably stabilizes the closed positions; however, the Asp632Phe mutant structure shows that the long side chain of Arg634 is in a position to sterically push the domains apart (Figure 5). Taken together, it is reasonable to hypothesize the following sequence of events: Asp632 loop retraction →NADPH binding → hydride transfer →interflavin electron transfer →NADP+ release, would lead to the opening of the two flavin domains.
In Figure 13, we propose a model for the sequence of events governing nicotinamide binding, hydride transfer, and electron transfer. The elements of this model are as follows: The resting state is the physiologic NADPH-free, 1 electron-reduced air-stable FMN semiquinone of CYPOR, which exists in equilibrium between closed and open conformation, with the majority in an open conformation57 (state 1, 3ES9/3OJW). (Hereafter, only the major conformational state of CYPOR molecule is considered, unless otherwise stated.) Since the Gly141 loop conformation is the same in both FMNsq and FMNhq states,32 it remains in the “up” conformation throughout the entire catalytic cycle (i.e., states 1–9). Trp677 is coplanar with the FAD isoalloxazine, with the long axes of both rings in a parallel orientation.
Figure 13.

A) Conformational changes and the cofactor binding during the catalytic cycle of CYPOR - The FMN domain is in light blue and FAD domain in green. The symbols and redox states of the cofactors are shown in the figure, together with the conformations of the Asp632 loop. See text for detailed description of the catalytic cycle involving States 1–9. B) Redox cycling of CYPOR flavins (1:3:2:1 electron cycling model). The numbers in the parentheses indicate the related states shown in Figure 13A.
NADPH enters 1 electron-reduced (FAD/FMNH.) CYPOR when it is in the open conformation (state 1). The 2′AMP -PPi moiety binds first by interacting with Arg567 and Lys602, followed by pyrophosphate moiety binding to Arg567 and Arg290, thus securing the 2′-AMP-5′PPi half of the NADPH to the enzyme, and promotes retraction of the Asp632 loop and closure of the two flavin domains (state 2) by forming the Asp147-Arg514 salt bridge, while the ribityl-nicotinamide moiety searches for an appropriate binding site for hydride transfer. Retraction of the loop is also linked to rotation of the indole ring of Trp677 such that its long axis is perpendicular to that of the FAD ring (modeled after oxidized NADP+-bound structure, 1AMO and 3QE2 (human WT). Upon displacement of the indole ring of Trp677, the nicotinamide is now poised for hydride transfer (state 3, inferred from Trp677X, 1JA0 and Trp677Gly 1J9Z). Hydride transfer occurs; FAD is reduced, and NADPH becomes NADP+ (state 4, from pdb code 1JA0). States 1–4 complete the hydride transfer from NADPH to FADox at a rate of > 50s−1.36,37 Once the FAD is reduced, its isoalloxazine is negatively charged, while the nicotinamide ring is positively charged. The Trp677 indole ring displaces the nicotinamide ring from the re-face of the FAD ring and assumes a position with its long axis parallel to the long axis of the isoalloxazine ring (state 5, modeled after the reduced wild type structure). The outgoing path of NADP+ (presumably the same as its incoming path) shows the nicotinamide ring orienting itself perpendicular to the isoalloxazine ring by forming two hydrogen bonds from the carboxamide of NADP+ to N1 of FAD and the hydroxyl of Thr535. (modeled after the 4e-reduced, NADP+ bound structure). The Asp632 loop is retracted and the domains remain closed. Almost simultaneously, interflavin electron transfer occurs (~30–50/sec)36,37 resulting in the FADsq-FMNhq. The nicotinamide ring moves away from FAD and begins to probe for an exit pathway. The indole of Trp677 returns to the perpendicular orientation (state 6, oxidized wild type structure). The exact sequence of these events (i.e., interflavin electron transfer, indole movement, and nicotinamide movement) is not known, however most likely the interflavin electron transfer occurs first, as any protein movement would take a longer time. Note that, at this state, the 2′, 5′-ADP is still bound to the enzyme and the Asp632 loop is in the retracted position (modeled after 1AMO and 5URD).
In state7, the Asp632 loop has adopted the extended conformation, resulting in the expulsion of the adenosine PPi moiety, allowing the dissociation of the entire NADP+ molecule from the enzyme, which in turn opens the two flavin domains and permits binding of the acceptor protein P450 or cytochrome c. The role of Arg634 in facilitating the opening of the two domains is discussed below (modeled after 3ES9 for open domains and after 3OWG for the extended Asp632 loop). At this stage, the FMN hydroquinone of the 3e-reduced enzyme (FADsq-FMNhq) donates 1 electron to P450. The enzyme is now in the two-electron reduced state, FADsq – FMNsq. However, at this point, the enzyme closes again for a second interflavin electron transfer to yield FADox-FMNhq (state 8). Since domain movements in general are only slightly different in energy, the domains may reassociate by thermal fluctuations.58 Since no NADP+ is bound, the Asp632 loop is likely adopting the extended conformation, and the mechanism of the closing of the two domains at this stage must be different from that facilitated by the Asp632 loop movement as discussed in state 2 (modeled after Asp632Phe structure). The two flavin domains must now open in order to transfer a second electron from the reductase to a P450 (the same P450 or a different P450) (state 9). After electron transfer to the P450, the reductase returns to the 1-electron reduced (FADox-FMNsq), NADPH-free form and the cycle repeats (same model as state 7, 3ES9). Currently, the exact structural models for states 7–9 are not known.
A link between NADP+ release and domain movement is consistent with the observation that CYPOR displays very little NADPH oxidase activity in the absence of an electron acceptor.46 NADPH consumption is ~4,000 min−1 in the presence of cyt c, but in the absence of any acceptor, only 5 min−1. More recently it has been shown that the rate of formaldehyde formation with cyt P450 2B4 is 47 min−1,25 consistent with the hypothesis that the domain movement of the reductase is influenced by the presence of its electron acceptor. In addition, auto-oxidation rate of two-electron reduced CYPOR in the absence of acceptor is also very slow (turnover number, ~5 min−1). Furthermore, the rate of auto-oxidation of full length CYPOR is significantly slower than that with the isolated FMN domain (~20-fold), suggesting that the two flavin domains are predominantly in the closed conformation.32 In this scenario, absence of domain movement in the absence of an electron acceptor would be linked to positioning of the Asp632 loop in the retracted position, thus preventing NADP+ dissociation and freezing the reaction cycle at or prior to state 6. The hydrogen bonding interaction between Ala633 and Trp677 provides a mechanism linking rotation of Trp677 and Asp632 loop retraction. Evidence for a role for Arg634 in linkage of Asp632 loop movement to domain movement comes from inspection of the orientations of Arg634 in CYPOR structures in which the domains are open or closed and which suggest that Arg634 is also in a position to regulate interdomain distance. A role for Arg634 in linkage of Asp632 loop movement and domain movement is consistent with the kinetic properties of the Arg634 mutants, which exhibit decreased 2′5′-ADP binding affinity and increased rates of hydride transfer and cytochrome c reductase activity21 as well as increased cytochrome 2B4 activity (Table 3). Structural evidence includes observations of positioning of Arg634 in the domain interface of the Asp632Phe structure and observations of hydrogen bonding interactions with Asp209 (rat CYPOR, IJA1 and human CYPOR, 3QE2);19,20 however, the net effect of mutating Arg634 to alanine is that it increases the activity of the reductase toward cyt P450 2B4 and does not inhibit activity with cyt c, and is consistent with the observation that under conditions of low ionic strength, the Arg634Ala mutant is more active with cyt c than the wild type.21
In conclusion, we have characterized the kinetic properties and structures of a number of CYPOR mutants in the Asp632 loop that must undergo a large conformational change in order for NADPH to bind in a hydride-transfer competent conformation. In addition, the structure of the 4-electron reduced, NADP+-bound wild type structure together with various mutant structures enable us to propose a pathway for NADPH binding to and NADP+ release from CYPOR during the catalytic cycle. Finally, we propose a mechanistic scheme for coordination of NADPH binding and release and domain movements.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported in whole or in part by: National Institutes of Health Grants GM097031 (to JJK), CA22484 (to ALS) and GM035533 and GM094209 to LW, who also received funding from VA Merit review grant Project ID 1168720.
We thank the staff at the Advanced Photon Source beamlines SBC 19ID and at the National Synchrotron Light Source, Brookhaven National Laboratory X26C for their assistance in data collection. The atomic coordinates and structure factors have been deposited in the Protein Data Bank at http://www.pdb.org (PDB codes: 5URD, 5URE, 5URH, 5URG, and 5URI).
ABBREVIATIONS
- CYPOR
cytochrome P450 reductase
- cyt c
cytochrome c
- HEPES
N-2-hydroxyethylpiperazine-N′-2 ethanesulfonic acid
- MES
2-(N-morpholino) ethanesulfonic acid
- TES
2-{[tris-(hydroxymethyl)methyl]amino} ethanesulfonic acid
- Tricine
N-[tris-(hydroxymethyl)methyl] glycine
- WT
wild type
- P450 or cyt P450
cytochrome P450
Footnotes
Author Contributions
The manuscript was written through contributions of all authors./All authors have given approval to the final version of the manuscript./*C.X. and F.R. contributed equally.
Notes
The authors declare no competing financial interests.
Structural and kinetic studies of Asp632 mutants and fully-reduced NADPH-cytochrome P450 oxidoreductase define the role of Asp632 loop dynamics in control of NADPH binding and hydride transfer
Table S1: Structure-based amino acid sequence alignment for the Asp632 loop of the diflavin reductase family
Table S2: Oligonucleotide primer sequences used for generating mutant proteins
Figures S1 and S2: Structures of wild type oxidized (S1) and reduced (S2) CYPOR. Panel A: Ribbon and stick diagram and Panel B: Stereo diagram of an electron density map (2Fo-Fc) contoured at 1.5σ.
Figure S3: Structural differences observed in the oxidized structure (yellow carbon atoms and black dashed lines for H-bonds and salt bridges) and reduced wild type (grey carbons and red dashed lines for H-bonds and salt bridges) structures. Figures obtained by superimposing the FAD cofactor in Molecule B of the asymmetric unit. Panel A: in the vicinity of the FAD isoalloxazine ring and the C-terminus of the molecule, W677-S678. Panel B: at the FMN-FAD domain interface. In the oxidized structure, Y140 is parallel to the FMN ring and its hydroxyl group is H-bonded to an FMN phosphate oxygen and R514 makes a H-bond with H319. When reduced, Y140 rotates and makes a tilt with respect to the FMN ring, and R514 forms a salt bridge with D147, resulting in a slightly more closed FMN-FAD domain arrangement.
Figure S4: Comparison of the structure of Asp632Phe and that of oxidized wild type in the vicinity of the Asp632 loop. Panel A: Ribbon and Stick diagram and Panel B: Stereo view of an electron density map contoured at 1.5 σ for the corresponding region shown in Panel A.
Figure S5: Structure of oxidized NADP+-bound Asp632A in the vicinity of the Asp632 loop. Panel A: Ribbon and stick diagram and Panel B: electron density map for the corresponding region.
Figure S6: Same as Figure 6, but for the structure of 2′-AMP bound oxidized Asp632A.
The Supporting Information is available free of charge on the ACS Publications website as a PDF file
References
- 1.Iyanagi T, Xia C, Kim JJ. NADPH-cytochrome P450 oxidoreductase: prototypic member of the diflavin reductase family. Arch Biochem Biophys. 2012;528(1):72–89. doi: 10.1016/j.abb.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim JJ, Waskell L. Electron Transfer Partners. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 4. Springer; Switzerland: 2015. pp. 33–68. [Google Scholar]
- 3.Paine MJ, Scrutton NS, Munro AW, Gutierrez A, Roberts GCK, Wolf CR. Electron Transfer Partners of Cytochrome P450. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 3. Kluwer Academic/Plenum Publishers; New York, Boston, Dordrecht, London, Moscow: 2005. pp. 115–148. [Google Scholar]
- 4.Ortiz de Montellano PR. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem Rev. 2010;110(2):932–48. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guengerich FP. Human Cytochrome P450 Enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 3. Vol. 1. Kluwer Academic/Plenum Publishers; New York, NY: 2005. pp. 377–463. [Google Scholar]
- 6.Guengerich FP. Human Cytochrome P450 Enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 4. Vol. 2. Springer International Publishers; Switzerland: 2015. pp. 523–785. [Google Scholar]
- 7.Xia C, Hamdane D, Shen AL, Choi V, Kasper CB, Pearl NM, Zhang H, Im SC, Waskell L, Kim JJ. Conformational changes of NADPH-cytochrome P450 oxido- reductase are essential for catalysis and cofactor binding. J Biol Chem. 2011;286(18):16246–60. doi: 10.1074/jbc.M111.230532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105(6):2253–77. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 9.Munro AW, Noble MA, Robledo L, Daff SN, Chapman SK. Determination of the redox properties of human NADPH-cytochrome P450 reductase. Biochemistry. 2001;40(7):1956–1963. doi: 10.1021/bi001718u. [DOI] [PubMed] [Google Scholar]
- 10.Oprian DD, Coon MJ. Oxidation-reduction states of FMN and FAD in NADPH-cytochrome P-450 reductase during reduction by NADPH. J Biol Chem. 1982;257(15):8935–44. [PubMed] [Google Scholar]
- 11.Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J Biol Chem. 2009;284(17):11374–84. doi: 10.1074/jbc.M807868200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedison TM, Hay S, Scrutton NS. Real-time analysis of conformational control in electron transfer reactions of human cytochrome P450 reductase with cytochrome c. FEBS J. 2015;282(22):4357–75. doi: 10.1111/febs.13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang WC, Ellis J, Moody Peter CE, Raven Emma L, Roberts Gordon CK. Redox-linked domain movements in the catalytic cycle of cytochrome P450 reductase. Structure. 2013;21(9):1581–1589. doi: 10.1016/j.str.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis J, Gutierrez A, Barsukov IL, Huang WC, Grossmann JG, Roberts GC. Domain motion in cytochrome P450 reductase: conformational equilibria revealed by NMR and small-angle x-ray scattering. J Biol Chem. 2009;284(52):36628–37. doi: 10.1074/jbc.M109.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pudney CR, Khara B, Johannissen LO, Scrutton NS. Coupled motions direct electrons along human microsomal P450 Chains. PLoS biology. 2011;9(12):e1001222. doi: 10.1371/journal.pbio.1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grunau A, Paine MJ, Ladbury JE, Gutierrez A. Global effects of the energetics of coenzyme binding: NADPH controls the protein interaction properties of human cytochrome P450 reductase. Biochemistry. 2006;45(5):1421–34. doi: 10.1021/bi052115r. [DOI] [PubMed] [Google Scholar]
- 17.Grunau A, Geraki K, Grossmann JG, Gutierrez A. Conformational dynamics and the energetics of protein--ligand interactions: role of interdomain loop in human cytochrome P450 reductase. Biochemistry. 2007;46(28):8244–55. doi: 10.1021/bi700596s. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci U S A. 1997;94(16):8411–6. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard PA, Shen AL, Paschke R, Kasper CB, Kim JJ. NADPH-cytochrome P450 oxidoreductase. Structural basis for hydride and electron transfer. J Biol Chem. 2001;276(31):29163–70. doi: 10.1074/jbc.M101731200. [DOI] [PubMed] [Google Scholar]
- 20.Xia C, Panda SP, Marohnic CC, Martasek P, Masters BS, Kim JJ. Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc Natl Acad Sci U S A. 2011;108(33):13486–91. doi: 10.1073/pnas.1106632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mothersole RG, Meints CE, Louder A, Wolthers KR. Role of active site loop in coenzyme binding and flavin reduction in cytochrome P450 reductase. Arch Biochem Biophys. 2016;606:111–9. doi: 10.1016/j.abb.2016.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260(3):289–98. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 23.Shen AL, Kasper CB. Differential contributions of NADPH-cytochrome P450 oxidoreductase FAD binding site residues to flavin binding and catalysis. J Biol Chem. 2000;275(52):41087–91. doi: 10.1074/jbc.M008380200. [DOI] [PubMed] [Google Scholar]
- 24.Sem DS, Kasper CB. Effect of ionic strength on the kinetic mechanism and relative rate limitation of steps in the model NADPH-cytochrome P450 oxidoreductase reaction with cytochrome c. Biochemistry. 1995;34(39):12768–74. doi: 10.1021/bi00039a037. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Im SC, Waskell L. Cytochrome b5 increases the rate of product formation by cytochrome P450 2B4 and competes with cytochrome P450 reductase for a binding site on cytochrome P450 2B4. J Biol Chem. 2007;282(41):29766–76. doi: 10.1074/jbc.M703845200. [DOI] [PubMed] [Google Scholar]
- 26.Saribas AS, Gruenke L, Waskell L. Overexpression and purification of the membrane-bound cytochrome P450 2B4. Protein Expr Purif. 2001;21(2):303–9. doi: 10.1006/prep.2000.1377. [DOI] [PubMed] [Google Scholar]
- 27.Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem. 1998;273(27):17036–49. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- 28.McPherson A. Crystallization of Biological Macromolecules. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1999. p. 586. [Google Scholar]
- 29.Otwinowski Z, Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2(11):2728–33. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Rwere F, Xia C, Im S, Haque MM, Stuehr DJ, Waskell L, Kim JJ. Mutants of cytochrome P450 reductase lacking either Gly-141 or Gly-143 destabilize its FMN semiquinone. J Biol Chem. 2016;291(28):14639–61. doi: 10.1074/jbc.M116.724625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem. 1989;264(13):7584–9. [PubMed] [Google Scholar]
- 34.Ludwig ML, Pattridge KA, Metzger AL, Dixon MM, Eren M, Feng Y, Swenson RP. Control of oxidation-reduction potentials in flavodoxin from Clostridium beijerinckii: the role of conformation changes. Biochemistry. 1997;36(6):1259–80. doi: 10.1021/bi962180o. [DOI] [PubMed] [Google Scholar]
- 35.Chang FC, Swenson RP. The midpoint potentials for the oxidized-semiquinone couple for Gly57 mutants of the Clostridium beijerinckii flavodoxin correlate with changes in the hydrogen-bonding interaction with the proton on N(5) of the reduced flavin mononucleotide cofactor as measured by NMR chemical shift temperature dependencies. Biochemistry. 1999;38(22):7168–76. doi: 10.1021/bi982203u. [DOI] [PubMed] [Google Scholar]
- 36.Bhattacharyya AK, Lipka JJ, Waskell L, Tollin G. Laser flash photolysis studies of the reduction kinetics of NADPH:cytochrome P-450 reductase. Biochemistry. 1991;30(3):759–765. doi: 10.1021/bi00217a027. [DOI] [PubMed] [Google Scholar]
- 37.Gutierrez A, Paine M, Wolf CR, Scrutton NS, Roberts GCK. Relaxation kinetics of cytochrome P450 reductase: internal electron transfer is limited by conformational change and regulated by coenzyme binding. Biochemistry. 2002;41(14):4626–4637. doi: 10.1021/bi0159433. [DOI] [PubMed] [Google Scholar]
- 38.Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271(5246):163–8. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 39.McGaughey GB, Gagne M, Rappe AK. pi-Stacking interactions. Alive and well in proteins. J Biol Chem. 1998;273(25):15458–63. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 40.Johansson LB, Davidsson A, Lindblom G, Naqvi KR. Electronic transitions in the isoalloxazine ring and orientation of flavins in model membranes studied by polarized light spectroscopy. Biochemistry. 1979;18(19):4249–53. doi: 10.1021/bi00586a033. [DOI] [PubMed] [Google Scholar]
- 41.Nishimoto K, Watanabe Y, Yagi K. Hydrogen bonding of flavoprotein. I. Effect of hydrogen bonding on electronic spectra of flavoprotein. Biochim Biophys Acta. 1978;526(1):34–41. doi: 10.1016/0005-2744(78)90287-5. [DOI] [PubMed] [Google Scholar]
- 42.Rescigno M, Perham RN. Structure of the NADPH-Binding Motif of Glutathione Reductase: Efficiency Determined by Evolution. Biochemistry. 1994;33(19):5721–5727. doi: 10.1021/bi00185a008. [DOI] [PubMed] [Google Scholar]
- 43.Matthews RG. Km Effects Associated with Site-directed Mutations that Reduce the Velocity of One Half Reaction of a Ping Pong Mechanism. In: Curti B, Ronchi S, Zanetti G, editors. Flavins and Flavoproteins. Walter de Gruyter & Co; Berlin, New York: 1991. pp. 593–597. [Google Scholar]
- 44.Sem DS, Kasper CB. Kinetic mechanism for the model reaction of NADPH- cytochrome P450 oxidoreductase with cytochrome c. Biochemistry. 1994;33(40):12012–21. doi: 10.1021/bi00206a002. [DOI] [PubMed] [Google Scholar]
- 45.Shen AL, Kasper CB. Role of Ser457 of NADPH-cytochrome P450 oxidoreductase in catalysis and control of FAD oxidation-reduction potential. Biochemistry. 1996;35(29):9451–9. doi: 10.1021/bi960587n. [DOI] [PubMed] [Google Scholar]
- 46.Vermilion JL, Coon MJ. Identification of the high and low potential flavins of liver microsomal NADPH-cytochrome P-450 reductase. J Biol Chem. 1978;253(24):8812–9. [PubMed] [Google Scholar]
- 47.Sem DS, Kasper CB. Enzyme-substrate binding interactions of nadph-cytochrome-P-450 oxidoreductase characterized with ph and alternate substrate inhibitor studies. Biochemistry. 1993;32(43):11539–11547. doi: 10.1021/bi00094a010. [DOI] [PubMed] [Google Scholar]
- 48.Shen AL, Sem DS, Kasper CB. Mechanistic studies on the reductive half-reaction of NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 1999;274(9):5391–8. doi: 10.1074/jbc.274.9.5391. [DOI] [PubMed] [Google Scholar]
- 49.Dohr O, Paine MJ, Friedberg T, Roberts GC, Wolf CR. Engineering of a functional human NADH-dependent cytochrome P450 system. Proc Natl Acad Sci U S A. 2001;98(1):81–6. doi: 10.1073/pnas.98.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yasukochi Y, Masters BS. Some properties of a detergent-solubilized NADPH-cytochrome c(cytochrome P-450) reductase purified by biospecific affinity chromatography. J of Biol Chem. 1976;251(17):5337–44. [PubMed] [Google Scholar]
- 51.Niu G, Zhao S, Wang L, Dong W, Liu L, He Y. Structure of the Arabidopsis thaliana NADPH-cytochrome P450 reductase 2 (ATR2) provides insight into its function. FEBS J. 2017;284(5):754–765. doi: 10.1111/febs.14017. [DOI] [PubMed] [Google Scholar]
- 52.Freeman SL, Martel A, Raven EL, Roberts GCK. Orchestrated Domain Movement in Catalysis by Cytochrome P450 Reductase. Scientific Reports. 2017;7(1):9741. doi: 10.1038/s41598-017-09840-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutierrez A, Munro AW, Grunau A, Wolf CR, Scrutton NS, Roberts GCK. Interflavin electron transfer in human cytochrome P450 reductase is enhanced by coenzyme binding. Relaxation kinetic studies with coenzyme analogues. European Journal of Biochemistry. 2003;270(12):2612–2621. doi: 10.1046/j.1432-1033.2003.03633.x. [DOI] [PubMed] [Google Scholar]
- 54.Elmore CL, Porter TD. Modification of the nucleotide cofactor-binding site of cytochrome P-450 reductase to enhance turnover with NADH in Vivo. J Biol Chem. 2002;277(50):48960–4. doi: 10.1074/jbc.M210173200. [DOI] [PubMed] [Google Scholar]
- 55.Vincent B, Morellet N, Fatemi F, Aigrain L, Truan G, Guittet E, Lescop E. The closed and compact domain organization of the 70-kda human cytochrome p450 reductase in its oxidized state as revealed by NMR. J Mol Biol. 2012;420:296–309. doi: 10.1016/j.jmb.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 56.Hay S, Brenner S, Khara B, Quinn AM, Rigby SE, Scrutton NS. Nature of the energy landscape for gated electron transfer in a dynamic redox protein. J Am Chem Soc. 2010;132:9738–9745. doi: 10.1021/ja1016206. [DOI] [PubMed] [Google Scholar]
- 57.Haque MM, Bayachou M, Tejero J, Kenney CT, Pearl NM, Im SC, Waskell L, Stuehr DJ. Distinct conformational behaviors of four mammalian dual-flavin reductases (cytochrome P450 reductase, methionine synthase reductase, neuronal nitric oxide synthase, endothelial nitric oxide synthase) determine their unique catalytic profiles. FEBS Journal. 2014;281(23):5325–5340. doi: 10.1111/febs.13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 2002;33(22):6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.