

Abstract
Obesity is associated with poor prognosis in triple-negative breast cancer (TNBC). Preclinical models of TNBC were used to test the hypothesis that increased leptin signaling drives obesity-associated TNBC development by promoting cancer stem cell (CSC) enrichment and/or epithelial-to-mesenchymal transition (EMT). MMTV-Wnt-1 transgenic mice, which develop spontaneous basal-like, triple-negative mammary tumors, received either a control diet (10% kcal from fat) or a diet-induced obesity regimen (DIO, 60% kcal from fat) for up to 42 weeks (n=15/group). Mice were monitored for tumor development and euthanized when tumor diameter reached 1.5 cm. Tumoral gene expression was assessed via RNA sequencing (RNA-seq). DIO mice had greater body weight and percent body fat at termination than controls. DIO mice, versus controls, demonstrated reduced survival, increased systemic metabolic and inflammatory perturbations, upregulated tumoral CSC/EMT gene signature, elevated tumoral aldehyde dehydrogenase (ALDH) activity (a CSC marker), and greater leptin signaling. In cell culture experiments using TNBC cells (murine: E-Wnt and M-Wnt; human: MDA-MB-231), leptin enhanced mammosphere formation, and media supplemented with serum from DIO versus control mice increased cell viability, migration, invasion, and CSC- and EMT-related gene expression, including Foxc2, Twist2, Vim, Akt3, and Sox2. In E-Wnt cells, knockdown of leptin receptor ablated these pro-cancer effects induced by DIO mouse serum. These findings indicate that increased leptin signaling is causally linked to obesity-associated TNBC development by promoting CSC enrichment and EMT.
Keywords: Breast cancer, obesity, leptin, cancer stem cells, epithelial-to-mesenchymal transition
INTRODUCTION
The prevalence of obesity, an established risk factor for several breast cancer subtypes, including triple-negative breast cancer (TNBC), is over 40% in US women (1). Obesity is also associated with higher breast cancer-specific and overall mortality in patients with breast cancer, regardless of subtype (2-4). Moreover, obese patients do not respond as well as normoweight patients to several classes of chemotherapy (5-9). Unfortunately, the mechanisms underlying the obesity-TNBC link are incompletely understood, hampering efforts to decrease the burden of breast cancer in obese women.
One possible mediator of obesity-associated TNBC development, progression and chemotherapy resistance is cancer stem cell (CSC) enrichment. CSCs are an intratumoral subpopulation of cancer cells with stem cell-like properties, including self-renewal and multipotent differentiation, and are posited as primary drivers of tumor initiation and progression (10, 11). CSC enrichment is strongly associated with poor prognosis in patients with breast cancer (10). Obesity in preclinical transplant models of TNBC increases tumor growth, CSC-related gene expression, and markers of epithelial-to-mesenchymal transition (EMT), a key developmental program linked with CSC enrichment and metastasis (11). The procancer effects of obesity in these models are associated with increased circulating leptin levels (12). Leptin, an adipokine produced primarily by white adipose tissue, is positively correlated with body fat levels and regulates food intake, inflammation, cell differentiation and proliferation. Elevated levels of systemic leptin, intratumoral leptin, and leptin receptor expression confer a worse breast cancer prognosis (13-16).
The MMTV-Wnt-1 transgenic mouse is an attractive, relevant model of basal-like breast cancer, the most common form of TNBC. Although Wnt-1 is not implicated in human breast tumorigenesis, activation of the Wnt/βcatenin pathway is required for the tumorigenic behavior of basal-like breast cancer cells (17), and spontaneous tumors in MMTV-Wnt-1 mice display pathological and transcriptional characteristics of human basal-like TNBC (11). Using MMTV-Wnt-1 mice and in vitro models of basal-like breast cancer, we tested the hypothesis that increased leptin signaling drives obesity-associated TNBC development by promoting CSC enrichment and EMT.
MATERIALS AND METHODS
In vivo MMTV-Wnt-1 transgenic mouse study
Animal studies and procedures were approved and monitored by the University of Texas Institutional Animal Care and Use Committee. Female MMTV-Wnt-1 mice on a C57BL/6 background (n=30) were obtained from the Hursting laboratory’s breeding colony and ovariectomized at 12 weeks of age. Following a 1-week recovery period, the mice were randomized to receive one of two diets, both fed ad libitum: a control regimen or a diet-induced obesity (DIO) regimen (n=15/diet group) as previously described (18). Mice were followed for up to 42 weeks and euthanized when tumor diameter was >1.5 cm in any direction or at study termination, whichever occurred first. Mice that died or were euthanized for non-tumor related cause prior to study termination were censored from the survival analysis. Mice were palpated weekly for mammary tumors, and tumors were measured (2 dimensions) with calipers twice weekly after detection. Mice were fasted for 4-6 hours before blood collection. Tumor (when present), distal mammary fat pad, and blood were collected, processed, and stored as previously described (19). Body fat and lean mass were assessed after euthanization using a Lunar PIXImus Dual Emission X-Ray Absorptiometer (GE Medical Systems, Ontario, CA). One DIO mouse and 4 control mice died from nontumor-related causes, were censored at the time of death in body weight and survival analyses, and excluded from serum and tissue analyses.
Serum hormone, adipokine, and cytokine analyses
Analyses of serum levels of several obesity-associated hormones, adipokines, and cytokines were performed on 10 randomly selected mice per group. Serum IGF-1 was measured via a Mouse Magnetic Luminex® Screening Assay (R&D Systems, Minneapolis, MN, USA). Other serum proteins were measured via Bio-Plex Pro™ Multiplex Immunoassays (Bio-Rad, Inc., Hercules, CA, USA). All assays were analyzed on a Bio-Plex® MAGPIX™ Multiplex Reader (Bio-Rad).
Immunohistochemical analyses
Paraffin-embedded tumor tissue from 6 randomly selected mice per group was cut into 4 μm thick sections for immunohistochemical analysis. Tumor tissue was stained, processed and analyzed as previously described (11) with the following primary antibodies: Ki67 (Bethyl Laboratories #IHC-00375), CD31 (BD Pharmingen #550274), E-cadherin (Santa Cruz #sc-7870), and vimentin (Abcam #ab92547).
RNA sequencing and Ingenuity Pathway Analysis
Total RNA was isolated from flash-frozen tumor samples using TRIzol® Reagent (Sigma-Aldrich, St. Louis, MO, USA) according to manufacturer’s instructions. RNA quality was assessed by Agilent 2100 Bioanalyzer (Santa Clara, CA, USA), and randomly selected samples from control (n=5) and DIO (n=6) mice were prepared for sequencing using the Illumina TruSeq RNA Library Preparation Kit. RNA libraries were then sequenced on the Illumina HiSeq 2000 instrument (Illumina Inc., San Diego, CA, USA). Sequencing reads were quality assessed and trimmed for any remaining sequencing adaptor, and reads smaller than 50 bp were removed. RNA sequencing reads were aligned to Mouse Genome Ensembl GRCm38 using TopHat (version 2.1.0), and the differential levels of transcripts were quantified by Cuffdiff/Cufflink 2.2.1. Normalized fragments per kilobase of exon per million fragments mapped (FPKM) were generated by Cufflinks as representative of gene expression level. With principle component analysis, 3 outlier samples (1 control and 2 DIO) were removed, and 4 samples per group were subjected to further differential expression and pathway enrichment analysis. Genes with an adjusted two-tailed P-value of <0.05 and greater than 20.75-fold change (~1.7 fold) in expression were considered differentially expressed. These genes were then entered into Ingenuity Pathway Analysis (Qiagen, Germantown, MD, USA) to identify modulated signaling pathways and potential upstream regulators. The RNA sequencing data has been deposited in Sequence Read Archive (accession numbers SRX3558172-9).
Quantitative RT-PCR analyses
Total RNA was isolated from tumor and distal mammary pad tissues, reverse transcribed, and assayed in triplicate as previously described (19).
Aldehyde dehydrogenase (ALDH) activity assay
A colorimetric ALDH Activity Assay Kit (Abcam, Cambridge, UK) was used to assess tumor ALDH enzymatic activity in 6 randomly selected mice per group. Levels of NADH (produced by ALDH oxidation of acetaldehyde) were normalized to protein concentration of the tumor lysate, measured via Bradford protein assay, to determine ALDH activity.
Cell lines
Two mouse mammary tumor cell lines isolated from MMTV-Wnt-1 mice in 2010 by the Hursting laboratory and found to cluster with the basal-like (E-Wnt) and claudin-low (M-Wnt) breast cancer subtypes (11), and human MDA-MB-231 breast cancer cells (ATCC #HTB-26, obtained in 2011), which cluster with the claudin-low subtype, were used in in vitro studies. The cell lines were maintained in RPMI 1640 media (GIBCO Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, 10 mM HEPES buffer, and 2 mM L-glutamine (complete media). All experiments were performed on cells maintained at <30 passages and within 10 passages of cell line removal from liquid nitrogen storage. The cell lines were found to be negative for mycoplasma by the University of North Carolina’s Tissue Culture Facility and were authenticated via species identification and karyotyping analysis at the T.C. Shu Molecular Cytogenetics Core at the University of Texas MD Anderson Cancer Center in 2016.
An Ob-R shRNA Plasmid (sc-36116-SH, Santa Cruz Biotechnology, Dallas, TX) was transfected into E-Wnt cells using FuGENE 6 to generate Lepr knockdown E-Wnt cell lines. Stably transfected cells were selected based on growth in media with 5 μg/ml puromycin dihydrochloride (Sigma-Aldrich). Two clones with the lowest Lepr expression (EWnt-L1 and EWnt-L2), as determined by quantitative RT-PCR, were maintained in complete media plus 2.5 μg/ml puromycin dihydrochloride. An E-Wnt cell line stably transfected with the scrambled Control shRNA Plasmid-A (sc-108060, Santa Cruz Biotechnology) was also generated (EWnt-S) and maintained via the same methodology. The parental E-Wnt cell line (EWnt-P) used to generate these 3 cell lines was also utilized for comparison in the in vitro experiments.
Mammosphere assays
E-Wnt, M-Wnt, and MDA-MB-231 cells were plated for the mammosphere assays as described previously (12). Serial passage of the mammospheres, a common approach in the CSC field (20), was performed to assess whether leptin treatment further promotes mammary CSC self-renewal in secondary spheres. For propagation 1 (P1), cells were treated with vehicle (sterile H2O) or leptin (100, 200, and 400 ng/ml, final concentrations) for 7 days, then the number of spheres/well quantified. Previous reports examining the impact of leptin on CSC markers in multiple types of cancer cells have used leptin concentrations in this range (12, 21-23). Propagation 2 (P2) was then initiated by dissociating the mammospheres grown under each treatment condition and replating the cells in new media with the same treatments for an additional 7 days. The number of spheres was quantified at the end of P2, and representative images (x10 magnification) were captured on an EVOS FL Auto 2 Cell Imaging System (Invitrogen). Mammosphere assays of EWnt-P, EWnt-S, EWnt-L1, and EWnt-L2 cells used the same plating methodology and media, but only one propagation period.
Generation and characterization of mouse serum for in vitro studies
Female 8-week old wild-type C57BL/6 mice (n=202) were acclimated on the control diet regimen for 1 week, then randomized to remain on control diet (n=110) or change to the DIO regimen (n=92). The mice were maintained on these diets for 14 weeks, then euthanized. Blood was collected by cardiac puncture and serum isolated. The serum samples were pooled by diet group, and levels of serum hormones, adipokines, and cytokines measured in each pool as described above. The remaining serum was aliquoted and stored at −80°C for use in the in vitro experiments.
In vitro gene expression analyses
To assess basal gene expression in vitro, EWnt-P, EWnt-S, EWnt-L1, and EWnt-L2 cells were maintained in complete media before harvesting. To evaluate DIO-induced gene expression in vitro, E-Wnt, M-Wnt, MDA-MB-231, EWnt-S, and EWnt-L2 cells were serum-starved for 18 hours, then exposed to media containing 2% control or 2% DIO mouse serum in serum-free media (SFM) for 24 hours. RNA was isolated and reverse transcribed, and quantitative RT-PCR completed as described above.
In vitro metastatic phenotype assays
For the cell viability assay, all cell lines were seeded at a density of 5×103 in 96-well plates. After 24 hours, cells were serum-starved for 18 hours then continuously exposed to 2% control or 2% DIO mouse serum in SFM for 48 hours. MTT reagent was used to assess cell viability levels as previously described (24), with the absorbance read at 570 nm on a Cytation 3 Cell Imaging Multi-Mode Reader (BioTek Instruments Inc., Winooski, VT, USA).
To assess cell migration, all cell lines were seeded in 2-Well Culture-Inserts (ibidi, Munich, GER) in 24-well plates with 7.5×104 cells per insert well. After 24 hours, cells were serum-starved for 18 hours, then inserts removed and cells continuously exposed to 2% control or 2% DIO mouse serum in SFM for 6 hours. The EVOS FL Auto 2 Cell Imaging System (Invitrogen) was used to capture images (x10 magnification) of the wound diameter at baseline and at 6 hours.
To measure invasive capacity, 1.0×104 cells were seeded in SFM in each 24-well Corning Matrigel® Invasion Chamber (8.0 micron; Corning, NY, USA), and 2% control or 2% DIO mouse serum in SFM was placed beneath the chambers. After 24 hours, the invading cells were fixed in 100% methanol for 1 minute, then stained for 30 minutes with 0.5% crystal violet in 50% methanol. The EVOS FL Auto 2 Cell Imaging System (Invitrogen) was used to capture images (x10 magnification) of the stained cells.
Statistical analyses
Animal study data is presented as mean ± SD and in vitro data as mean ± SEM. All in vitro data shown represents the average of at least 3 independent experiments. For all statistical tests, GraphPad Prism software was used (GraphPad Software Inc., La Jolla, CA, USA), and P<0.05 was considered significantly different. Differences between animals or cells exposed to 2 different experimental conditions were analyzed using Student’s t test. Kaplan-Meier survival curves were plotted, and between-group difference in survival analyzed using the log-rank (Mantel-Cox) test. Differences between cells exposed to more than 2 experimental conditions were analyzed using one-way ANOVA, followed by Tukey’s post hoc test. Data from experiments with more than one independent variable was analyzed using two-way ANOVA, followed by Tukey’s post hoc test.
RESULTS
DIO regimen promotes excess body weight, excess body fat, and systemic metabolic and inflammatory perturbations
Female ovariectomized MMTV-Wnt-1 transgenic mice fed a DIO versus control diet regimen had significantly greater body weight from week 2 through week 35 on diet (P<0.05; Supplementary Figure 1a) and significantly greater body weight and percent body fat at final assessment (week 42 or when tumor diameter was >1.5 cm, whichever occurred first; P<0.01 for each comparison; Supplementary Figure 1b–c). At final assessment, DIO mice, versus controls, also had significantly elevated serum insulin, leptin, resistin (each P<0.001), gastric inhibitory peptide (P<0.01), glucagon-like peptide 1, tumor necrosis factor alpha (TNFα), and interleukin (IL)-17A (each P<0.05; Table 1). Serum IGF-1 (P=0.06), plasminogen activator inhibitor-1 (P=0.09), and IL-6 (P=0.09) were elevated in DIO mice relative to controls, but these differences did not reach statistical significance.
Table 1.
Serum hormones, adipokines, and cytokines in MMTV-Wnt-1 mice on control or DIO diet regimens.
| Analyte | Control | DIO | p-value |
|---|---|---|---|
| Insulin (ng/mL) | 2.90 (0.53) | 6.30 (2.28) | 0.0004 |
| IGF-1 (ng/mL) | 59.9 (13.0) | 85.0 (35.2) | 0.06 |
| Leptin (ng/mL) | 5.29 (3.15) | 31.0 (20.3) | 0.0009 |
| Adiponectin (μg/mL) | 5.27 (0.75) | 4.84 (1.17) | 0.37 |
| Resistin (ng/mL) | 80.7 (41.2) | 216 (96.1) | 0.0007 |
| PAI-1 (ng/mL) | 1.78 (0.31) | 4.55 (4.58) | 0.09 |
| GIP (pg/mL) | 131 (49.1) | 279 (139) | 0.008 |
| GLP-1 (pg/mL) | 35.7 (13.4) | 50.7 (14.8) | 0.04 |
| Ghrelin (ng/mL) | 39.4 (2.3) | 39.9 (3.6) | 0.91 |
| Glucagon (pg/mL) | 154 (20.4) | 186 (25.2) | 0.33 |
| IL-1β (ng/mL) | 0.97 (0.05) | 1.05 (0.07) | 0.36 |
| IL-6 (pg/mL) | 157 (36.1) | 192 (49.6) | 0.09 |
| IL-10 (pg/mL) | 501 (33.2) | 578 (70.4) | 0.34 |
| IL-17A (ng/mL) | 1.21 (0.43) | 1.64 (0.46) | 0.05 |
| IFNγ (pg/mL) | 195 (25.0) | 241 (32.2) | 0.28 |
| MCP-1 (ng/mL) | 1.37 (0.08) | 1.38 (0.10) | 0.92 |
| TNFα (ng/mL) | 4.31 (1.25) | 5.81 (1.68) | 0.04 |
Multiplex immunoassay analyses performed on serum collected at euthanization (n=10/group). Standard deviations shown in parentheses. Abbreviations: IGF-1, insulin-like growth factor-1; PAI-1, plasminogen activator inhibitor-1; GIP, gastric inhibitory polypeptide; GLP-1, glucagon-like peptide-1; IL-1β, interleukin 1 beta; IL-6, interleukin 6; IL-10, interleukin 10; IL-17A, interleukin 17A; IFNγ, interferon gamma; MCP-1, macrophage chemoattractant protein 1; TNFα, tumor necrosis factor alpha.
DIO promotes MMTV-Wnt-1 mammary tumor development
DIO mice had significantly reduced survival compared with controls (P<0.05; Figure 1a). One DIO mouse and 4 control mice remained alive and tumor-free at 42 weeks. Mammary tumors from DIO mice, relative to controls, expressed significantly higher levels of the proliferation marker Ki67 (P<0.01) and endothelial cell marker CD31 (P<0.05; Figure 1b). Gross necropsy detected no liver or lung metastases in either diet group.
Figure 1.
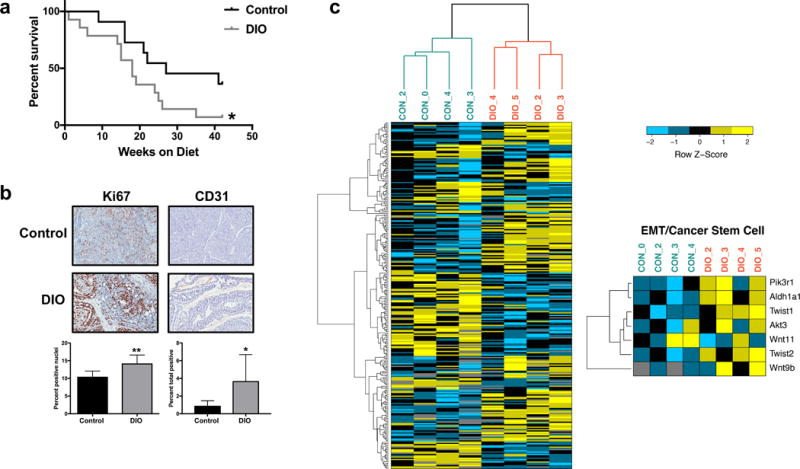
DIO decreases survival in MMTV-Wnt-1 mice. (a) Kaplan-Meier survival curves for DIO and control mice. (b) Immunohistochemical staining for tumor Ki67 and CD31 expression in DIO and control mice. Representative images shown at x20 magnification. (c) Tumor expression of CSC/EMT-related genes is modulated by DIO in MMTV-Wnt-1 mice. Heatmap illustration of differentially expressed genes in the tumors of DIO and control mice. Genes with an adjusted two-tailed P-value of <0.05 and greater than 20.75-fold change (~1.7 fold) in expression were considered differentially expressed. Differential expression of CSC/EMT-related genes is shown in the smaller heatmap on the right. *P<0.05, **P<0.01
DIO modulates CSC- and EMT-related tumoral gene and protein expression
Using RNA sequencing and Qiagen’s Ingenuity Pathway Analysis (IPA), we characterized differential mammary tumor gene expression in randomly selected tumors from DIO versus control mice. Genes differentially expressed (P<0.05; Figure 1c) were evaluated using IPA. This analysis indicated significant upregulation of protumorigenic pathways (associated genes listed parenthetically) characterized as ‘regulation of EMT’ (AKT3, PIK3R1, TWIST1, TWIST2, WNT11, WNT9B; P=0.003), ‘human embryonic stem cell pluripotency,’ (AKT3, PIK3R1, WNT11, WNT9B; P=0.026), and ‘Wnt/β-catenin signaling’ (AKT3, WNT11, FRAT1, WNT9B; P=0.038), and identified Lep as a highly significant (P<0.001) upstream master regulator with the highest Z-score (Z=2.06). Using quantitative RT-PCR we verified diet group-dependent differences in tumoral expression of CSC- and EMT-related genes identified by our RNA sequencing analysis (Figure 2a) or previous studies (11) in mammary tumor transplant models (Figure 2b). Tumors from DIO mice versus controls displayed significantly upregulated Aldh1a1 (P<0.01), Akt3, Pik3r1, Twist1, and Twist2 (each P<0.05; Figure 2a) and Pou5f1 (P<0.01), Foxc2, and Vim (each P<0.05; Figure 2b) as well as downregulated Cdh1 (P<0.05; Figure 2b). No between-group differences were detected in tumoral expression of Nanog, Notch1, Snai1, Snai2, Tgfb, and Zeb1 (data not shown). Tumor ALDH activity, a measure of CSC enrichment, was greater in DIO mice than controls (P<0.01; Figure 2c). In addition, tumors in DIO mice versus controls had significantly elevated protein expression of the mesenchymal marker vimentin (both P<0.05), but not lower protein expression of the epithelial marker E-cadherin (Figure 2d), as assessed by immunohistochemistry.
Figure 2.
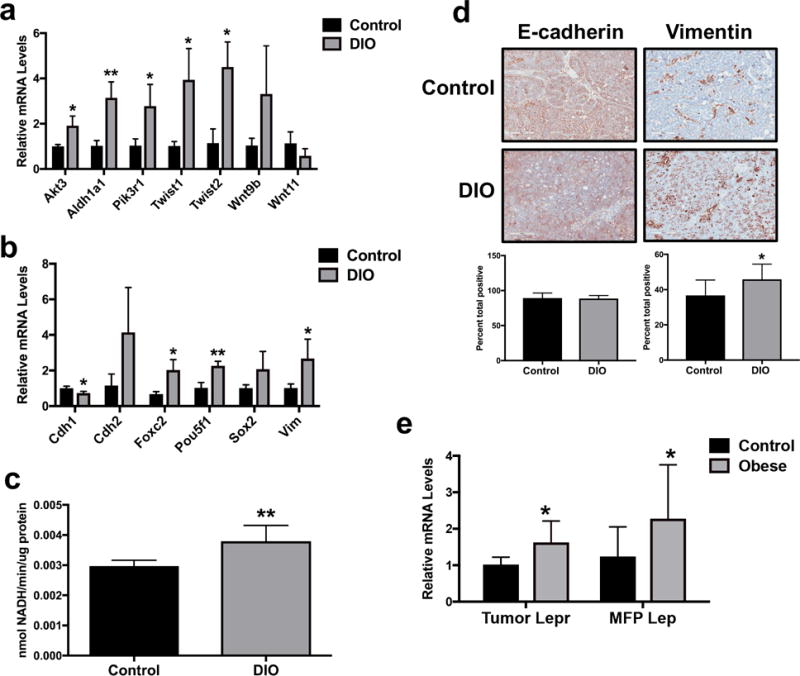
CSC/EMT markers are elevated in tumors from DIO mice in association with increased local leptin signaling. (a) Tumor expression of several CSC/EMT-related genes in DIO and control mice identified by RNA sequencing analysis was verified via quantitative RT-PCR. (b) Tumor expression in DIO and control mice of previously established CSC/EMT-related genes (11) not identified by RNA sequencing was measured by quantitative RT-PCR. (c) ALDH activity, another marker of CSC enrichment, was quantified in DIO and control mouse tumors. (d) Immunohistochemical staining for tumor E-cadherin and vimentin expression in DIO and control mice. Representative images shown at x20 magnification. (e) Tumor expression of Lepr as well as mammary fat pad (MFP) expression of Lep in DIO and control mice were measured via quantitative RT-PCR. *P<0.05, **P<0.01
We examined whether local signaling of leptin (an IPA-identified master regulator) is increased in the tumor microenvironment of DIO mice versus controls. Tumor Lepr and mammary fat pad Lep expression were significantly higher in DIO mice compared with controls (P<0.05 for both; Figure 2e). No between-group differences in tumor expression of Lep were found (data not shown).
Leptin signaling promotes CSC phenotype in triple-negative mammary tumor cells in vitro
The in vitro effect of leptin (100, 200, and 400 ng/ml, final concentrations) on mammary CSC enrichment was examined in two murine (E-Wnt and M-Wnt) and one human (MDA-MB-231) triple-negative mammary tumor cell lines by quantifying mammospheres formed during two sequential propagations (P1 and P2). Leptin, compared with vehicle, significantly increased mammosphere formation in E-Wnt cells at 400 ng/ml leptin in P2 (P<0.05; Figure 3a), in M-Wnt cells at each leptin concentration in P2 (100 ng/ml, P<0.05; 200 and 400 ng/ml, P<0.001; Figure 3b), and in MDA-MB-231 cells at each leptin concentration in P1 (100 ng/ml, P<0.01; 200 and 400 ng/ml, P<0.001) and P2 (100 ng/ml, P<0.001; 200 and 400 ng/ml, P<0.0001; Figure 3c). In each cell line, there were more mammospheres in P2 than P1 for each leptin concentration (E-Wnt: 100 and 200 ng/ml, P<0.05; 400 ng/ml, P<0.001; M-Wnt: 100 ng/ml, P<0.01; 200 and 400 ng/ml, P<0.05; MDA-MB-231: 400 ng/ml, P<0.001; 100 and 200 ng/ml, P<0.0001; Figure 3a–c).
Figure 3.
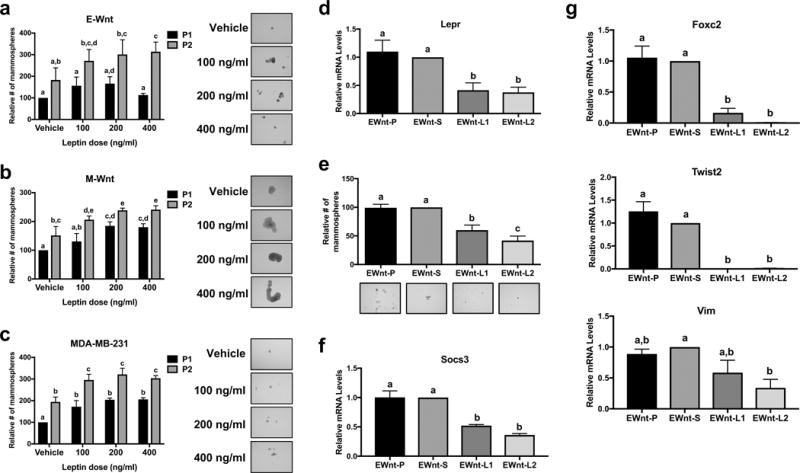
Leptin stimulates mammosphere formation in triple-negative mammary tumor cells. Mammosphere formation in (a) E-Wnt, (b) M-Wnt, and (c) MDA-MB-231 cells was assessed at the end of propagation 1 (P1), during which the cells were treated for 7 days with leptin, and following propagation 2 (P2), in which the spheres from P1 were dissociated and then replated with the same treatments for another 7 days. Representative images of mammospheres at the end of P2 are shown at x10 magnification. (d) Lepr expression in parental E-Wnt cells (EWnt-P) as well as E-Wnt cells stably transfected with a scrambled shRNA plasmid (EWnt-S) or shRNA to Lepr (EWnt-L1 and EWnt-L2) was measured by quantitative RT-PCR. (e) Mammosphere formation was assessed in EWnt-P, EWnt-S, EWnt-L1, and EWnt-L2 cells after a 7-day incubation in mammosphere media. Socs3 (f) and Foxc2, Twist2, and Vim (g) gene expression was measured by quantitative RT-PCR. Different letters indicate significant differences, P<0.05.
To establish the causal relationship between obesity-induced leptin signaling and CSC enrichment, we used shRNA to silence Lepr expression in E-Wnt cells. Specifically, we characterized leptin- and CSC/EMT-related gene expression and mammosphere formation capability of 2 stably transfected Lepr knockdown E-Wnt lines (EWnt-L1 and EWnt-L2) relative to E-Wnt cells stably transfected with a scrambled plasmid (EWnt-S) and parental E-Wnt cells (EWnt-P). Lepr expression in EWnt-L1 and EWnt-L2 was significantly reduced by 58.7±13.1% and 62.3±9.1%, respectively, compared with EWnt-S cells (P<0.001 for both), while there was no significant difference in Lepr expression between EWnt-S and EWnt-P cells (Figure 3d). In both EWnt-L1 and EWnt-L2 cells compared with EWnt-P and EWnt-S cells, mammosphere formation (each P<0.001) and expression of the leptin target gene Socs3 (each P<0.001) were significantly reduced (Figure 3e–f). In addition, EWnt-L1 and EWnt-L2 cells, relative to EWnt-P and EWnt-S cells, had significantly reduced expression of the CSC/EMT-related genes Foxc2 (P<0.001 for all) and Twist2 (EWnt-P and EWnt-S vs. EWnt-L1, P<0.01 and EWnt-L2, P<0.001), and Vim expression was significantly lower in EWnt-L2 than EWnt-S cells (P<0.05; Figure 3g). No significant differences between cell lines were noted in expression of Akt3, Aldh1a1, Cdh1, Cdh2, Pik3r1, Pou5f1, Sox2, Twist1, Wnt9b and Wnt11 (data not shown).
Obesity-associated circulating factors stimulate metastatic phenotype and CSC/EMT gene signature in triple-negative mammary tumor cells
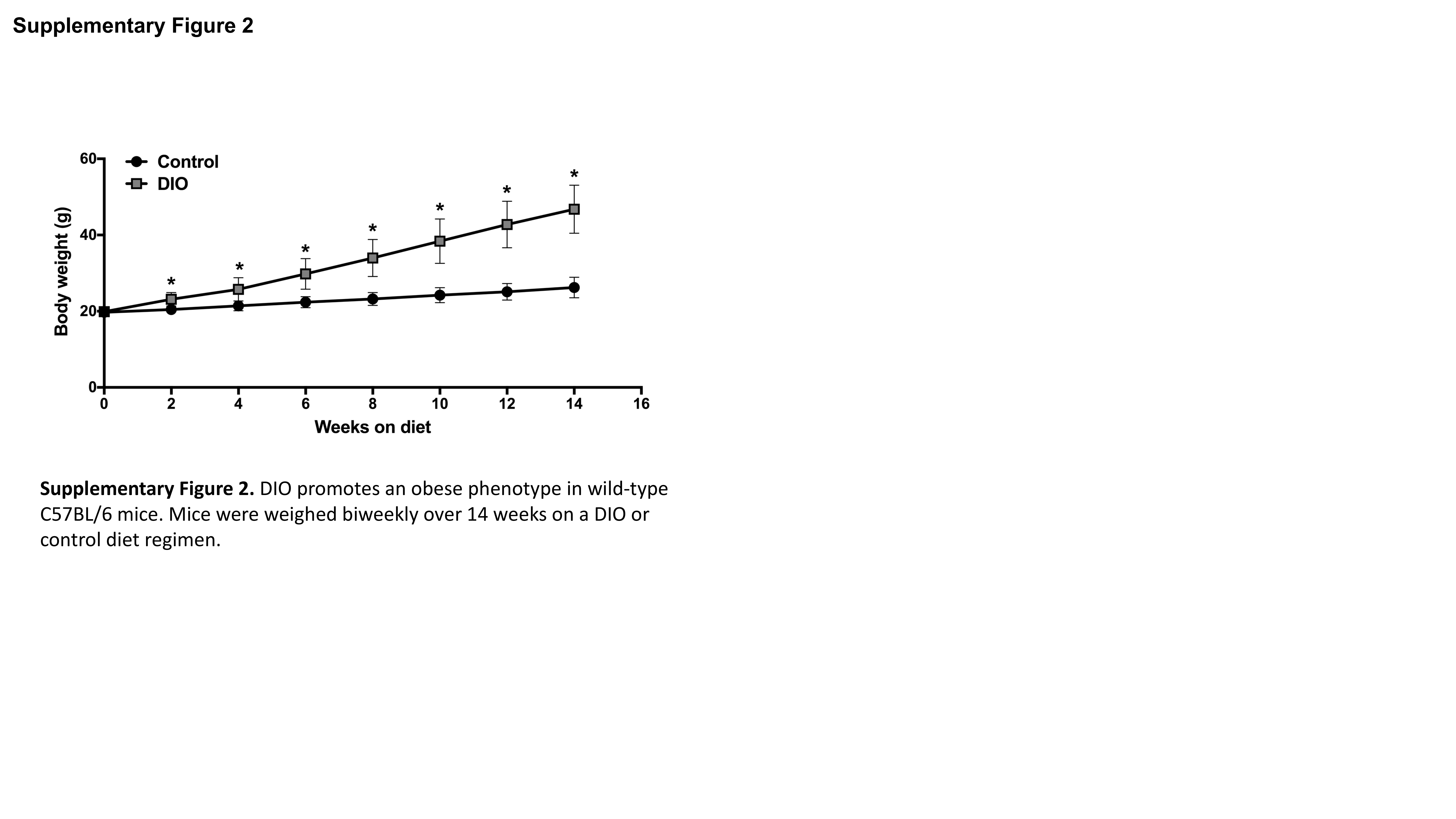
Because metastasis is closely linked with EMT and CSC enrichment, we examined the impact of obesity-associated circulating factors on in vitro measures of metastatic potential. Pooled serum isolated from female wild-type C57BL/6 mice maintained on the DIO (n=92) or control (n=110) diet regimens for 14 weeks was used to model an obese versus non-obese control state in vitro. Body weight in C57BL/6 mice on the DIO diet was significantly greater than controls during weeks 2-14 on diet (P<0.0001 for all weeks; Supplementary Figure 2). Serum collected from the DIO mice, compared with controls, had significantly elevated levels of several hormones, adipokines, and cytokines, including leptin (P<0.0001; Supplementary Table 1).
Cell viability was significantly higher in E-Wnt (P<0.01), M-Wnt, and MDA-MB-231 (both P<0.05) cells following a 48-hour incubation in media containing serum from DIO versus control mice (Figure 4a). DIO mouse serum also promoted significantly greater cell migration, relative to control, in E-Wnt, M-Wnt, and MDA-MB-231 cells during a 6-hour incubation (all P<0.001; Figure 4b). Furthermore, DIO mouse serum stimulated significantly greater invasive activity in all 3 cell lines, as a higher number of E-Wnt, M-Wnt, and MDA-MB-231 cells invaded over 24 hours through Matrigel membrane towards media containing serum from DIO versus control mice (all P<0.001, Figure 4c).
Figure 4.
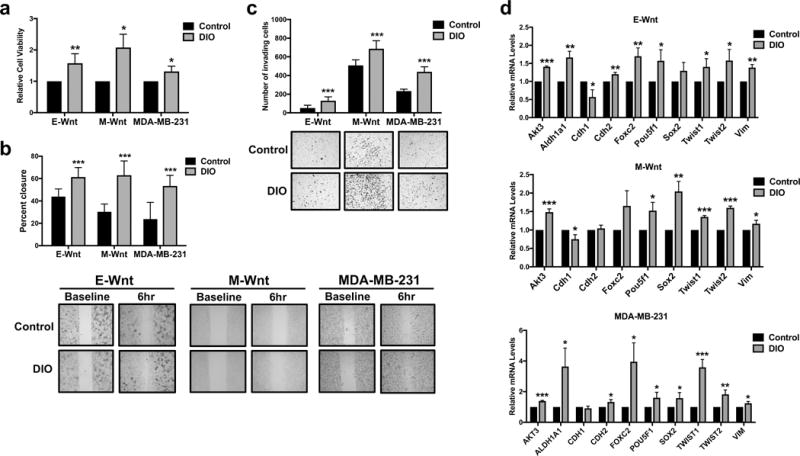
Obesity-associated circulating factors promote triple-negative mammary tumor cell viability, migration, invasion, and a CSC/EMT genotype. (a) E-Wnt, M-Wnt, and MDA-MB-231 cell viability following a 48-hour exposure to media containing 2% DIO or control mouse serum was assessed by MTT assay. (b) Migration of E-Wnt, M-Wnt, and MDA-MB-231 cells during a 6-hour exposure to media containing 2% DIO or 2% control mouse serum was measured by wound healing assay. Representative images of cells at baseline and 6 hours are shown at x10 magnification. (c) The invasive capacity of E-Wnt, M-Wnt, and MDA-MB-231 cells in response to chemoattraction with media containing 2% DIO or 2% control mouse serum over 24 hours was measured using Matrigel invasion chambers. Representative images of invading cells are shown at x10 magnification. (d) Expression of CSC/EMT-related genes in E-Wnt, M-Wnt, and MDA-MB-231 cells following a 24-hour exposure to media containing 2% DIO or 2% control mouse serum was measured by quantitative RT-PCR. *P<0.05, **P<0.01, ***P<0.001
A 24-hour incubation of triple-negative mammary tumor cells in media containing DIO mouse serum, relative to control serum, resulted in significantly increased (or decreased where noted) expression in E-Wnt cells of the CSC- and EMT-related genes Akt3 (P<0.001), Aldh1a1, Cdh2, Foxc2, Vim (each P<0.01), Cdh1 (decreased), Pou5f1, Twist1, and Twist2 (each P<0.05); in M-Wnt cells of Akt3, Twist1, Twist2 (each P<0.001), Sox2 (P<0.01), Cdh1 (decreased), Pou5f1, and Vim (each P<0.05); and in MDA-MB-231 cells of AKT3, TWIST1 (both P<0.001), TWIST2 (P<0.01), ALDH1A1, CDH2, FOXC2, POU5F1, SOX2, and VIM (each P<0.05; Figure 4d). Aldh1a1 was not detectable in M-Wnt cells. No differences were observed in Wnt9b, Wnt11, or Pik3r1 expression in the 3 cell lines (data not shown).
Leptin signaling contributes to metastatic phenotype and CSC/EMT gene signature in an in vitro model of obesity and triple-negative breast cancer
To examine the causal role of leptin signaling in the metastatic phenotype observed following mammary tumor cell exposure to DIO mouse serum, we assessed cell viability, migration, and invasion capability in Lepr knockdown E-Wnt cell lines following exposure to DIO mouse and control serum. Relative to control, DIO mouse serum promoted significantly greater cell viability in EWnt-S cells (P<0.05) but not EWnt-L1 or EWnt-L2 cells. Cell viability was significantly lower in EWnt-L2 versus EWnt-S cells exposed to DIO serum (P<0.05) and did not significantly differ between cell lines exposed to control mouse serum (Figure 5a). DIO mouse serum, relative to control serum, significantly increased migration of EWnt-P (P<0.05) and EWnt-S (P<0.01) cells but not EWnt-L1 or EWnt-L2 cells. Migration was significantly reduced in EWnt-L2 cells, relative to EWnt-P, EWnt-S, and EWnt-L1 cells, when cultured in DIO mouse serum (all P<0.05), and did not differ between cell lines when cultured in control mouse serum (Figure 5b). DIO mouse serum, relative to control, significantly increased invasion in Lepr-proficient cells (EWnt-P and EWnt-S, both P<0.0001) and to a lesser extent Lepr-knockdown cells (EWnt-L1 and EWnt-L2, both P<0.01). When stimulated with DIO mouse serum, the number of invading cells was significantly lower for EWnt-L2 versus EWnt-P (P<0.0001) and EWnt-S (P<0.01) cells, while EWnt-L1 cells demonstrated intermediate invasion that was significantly lower than the EWnt-P cells (P<0.05). Invasion did not significantly differ between cell lines when exposed to control mouse serum (Figure 5c).
Figure 5.
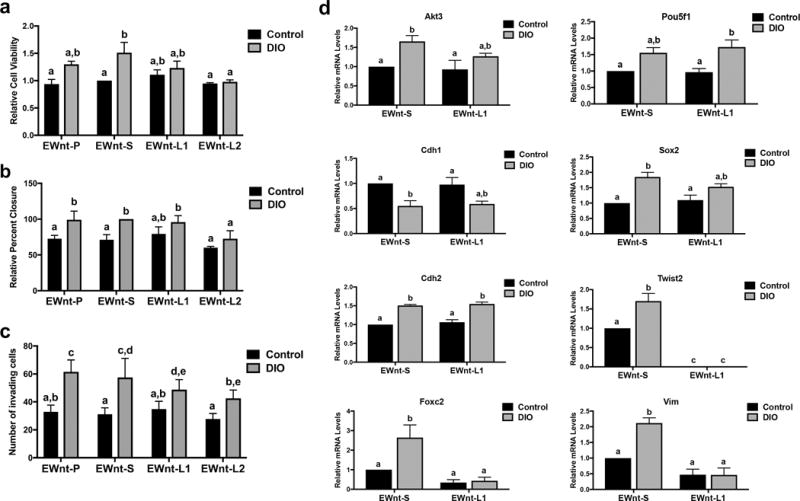
Leptin contributes to an obesity-associated metastatic phenotype and CSC/EMT genotype. (a) Cell viability in parental E-Wnt cells (EWnt-P) as well as E-Wnt cells stably transfected with a scrambled shRNA plasmid (EWnt-S) or shRNA to Lepr (EWnt-L1 and EWnt-L2) was measured by MTT assay following a 48-hour exposure to media containing 2% DIO or control mouse serum. (b) Migration of EWnt-P, EWnt-S, EWnt-L1, and EWnt-L2 cells during a 6-hour exposure to media containing 2% DIO or control mouse serum was measured by wound healing assay. (c) The invasive capacity of EWnt-P, EWnt-S, EWnt-L1, and EWnt-L2 cells in response to chemoattraction with media containing 2% DIO or control mouse serum over 24 hours was measured using Matrigel invasion chambers. (d) Expression of CSC/EMT-related genes in EWnt-S and EWnt-L2 cells following a 24-hour exposure to media containing 2% DIO or control mouse serum was measured by quantitative RT-PCR. Different letters indicate significant differences, P<0.05.
Lastly, we assessed CSC/EMT gene expression in EWnt-S and EWnt-L2 cell lines when exposed for 24 hours to DIO mouse serum, compared with control serum (Figure 5d). In EWnt-S cells, exposure to DIO versus control serum significantly modulated Cdh2 (P<0.001), Cdh1, Sox2, Twist2, Vim (each P<0.01), Akt3, and Foxc2 (both P<0.05) expression (as seen previously in parental E-Wnt cells, Figure 4d). In EWnt-L2 cells, knockdown of Lepr eliminated the difference between the DIO and control mouse serum conditions for Foxc2, Twist2, and Vim expression. These genes also exhibited reduced expression in EWnt-L2 cells compared with EWnt-S cells, particularly in cells exposed to DIO mouse serum (DIO mouse serum-treated EWnt-S vs. EWnt-L2 cells: Foxc2, P<0.01; Twist2, P<0.0001; and Vim, P<0.001). Furthermore, differences between the serum conditions in Akt3 and Sox2 expression were attenuated with Lepr knockdown in the EWnt-L2 cells.
DISCUSSION
Although obesity is associated with a poor TNBC prognosis (3), mechanisms mediating this link are not fully understood and the current standard of TNBC care does not differ for obese patients. Our findings suggest a leptin-dependent mechanism involving CSC enrichment and EMT, which could lead to increased tumor development and metastasis. Specifically, we report that: a) obesity decreases tumor-free survival and promotes a CSC/EMT gene signature in the MMTV-Wnt-1 transgenic mouse model of TNBC; b) tumors from DIO mice have higher expression of genes that promote stem cell pluripotency and EMT; and c) an IPA-predicted upstream regulator of these genes is leptin. Given the significant elevation of systemic leptin in DIO mice, we focused on defining the role of leptin signaling in obesity-induced CSC enrichment and EMT. Utilizing in vitro models of obesity and TNBC, we found that leptin is a key contributor to obesity’s effects, specifically promoting a CSC/EMT phenotype and upregulating the expression of multiple CSC/EMT-related genes, including Foxc2, Twist2, Vim, and to a lesser extent Akt3 and Sox2 (Figure 6). To our knowledge, this is the first study to characterize the impact of DIO on global gene expression in MMTV-Wnt-1 tumors and the first to identify Foxc2, Twist2, and Akt3 as leptin-regulated targets in tumors.
Figure 6.
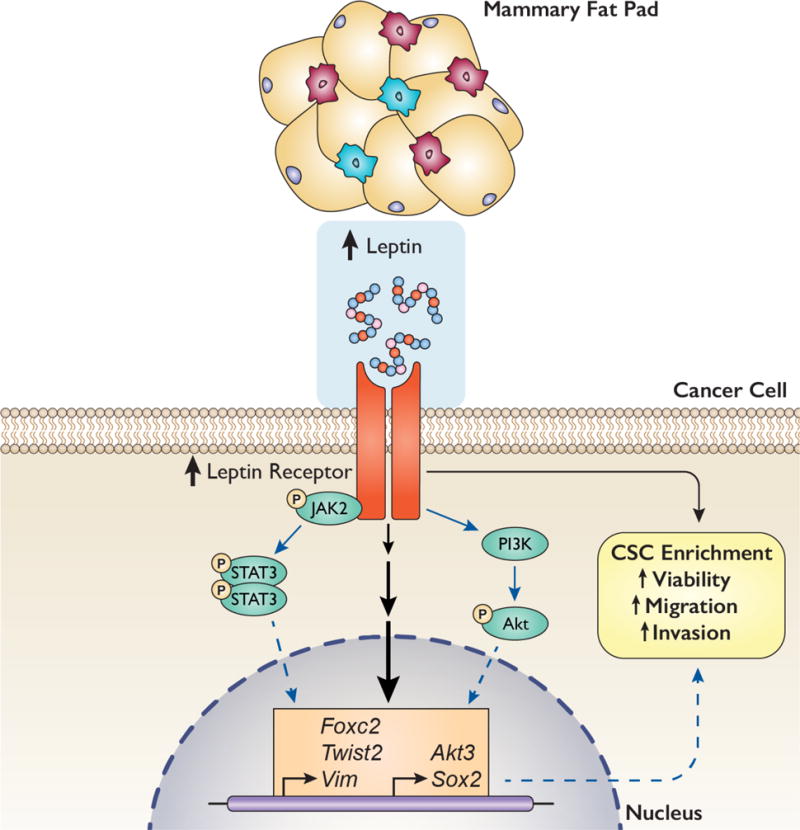
Proposed model illustrating leptin-mediated upregulation in CSC/EMT-related genes and phenotype. Our findings suggest that obesity in MMTV-Wnt-1 mice promotes both an excess of leptin production in the tumor microenvironment (normal mammary tissue) and an upregulation in tumor expression of the leptin receptor and CSC/EMT-related genes. They also indicate that leptin signaling promotes a CSC/EMT-related phenotype, including increased CSC enrichment and cell viability, migration, and invasion, and specifically regulates the expression of Foxc2, Twist2, Vim, Akt3, and Sox2 in triple-negative mammary tumor cells. We hypothesize that these genes may mediate the observed leptin-induced CSC/EMT-related phenotype and that leptin regulates these genes via stimulation of the JAK2/STAT3 and/or PI3K/Akt pathways. Black arrows indicate effects observed in this study, solid blue arrows indicate pathways known from the literature, and dotted blue arrows indicate hypothesized mechanisms.
We and others have previously explored leptin’s impact on CSC enrichment and/or EMT in breast and other cancers. We established that: a) the growth of orthotopically transplanted MMTV-Wnt-1 tumors is enhanced in genetically obese db/db mice, which have a compensatory elevation in leptin production due to a Lepr gene mutation; and b) MMTV-Wnt-1 tumor growth is reduced in the genetically obese ob/ob model, which lacks functional Lep expression (12). These findings suggested that leptin signaling may contribute to obesity’s pro-tumor effects on TNBC. In a follow-up study in non-obese C57BL/6 and nude mice, we also showed Lepr silencing in M-Wnt cells inhibits transplanted tumor growth and decreases Nanog expression, while Lepr silencing in human MDA-MB-231 cells decreases NANOG, POU5F1, SOX2, and VIM and increases CDH1 expression (25). Chang et al. (21) also found that leptin induces an EMT phenotype, mammosphere formation, and LEPR expression via STAT3 and miR-200c in pre-clinical models of normal breast epithelial cells (MCF12A) and luminal A breast cancer (MCF7). In addition, sorted LEPRHI primary human breast cancer cells have elevated sphere-forming capacity and CSC/EMT markers, including higher NANOG, SOX2, and POU5F1 (21). Others have found similar leptin-mediated increases in CSC/EMT markers in models of both luminal A and triple-negative breast cancer, suggesting that leptin’s effects are likely not specific to one breast cancer subtype (26-32). Feldman et al. (22) further demonstrated that leptin treatment stimulates liver CSC Sox2 and Pou5f1 (but not Nanog) expression via STAT3, and Lepr expression is induced by Sox2 and Oct4 (the protein encoded by Pou5f1). Leptin treatment also upregulates EMT and/or CSC markers in ovarian (23) and pancreatic (33) cancer cell lines and promotes the self-renewal of normal human breast epithelial stem cells (34). These data suggest that leptin not only promotes CSC enrichment and EMT in multiple types of cancer but also induces CSC Lepr expression to maintain the pluripotent state.
Lepr knockdown in E-Wnt cells indicated that only Foxc2, Twist2, Vim, Akt3, and Sox2 are regulated by leptin signaling in that cell line. The lack of a leptin-mediated effect on Nanog, Pou5f1 and Cdh1 expression contrasted with others’ findings (21, 22, 25) and may be related to differences between cell lines used. Zheng et al (25) utilized M-Wnt and MDA-MB-231 cell lines, both of which cluster with claudin-low breast cancer subtype and have a more mesenchymal phenotype, while we knocked down Lepr in the E-Wnt cell line, which clusters genetically with basal-like breast cancer (11). We chose to utilize E-Wnt cells for our mechanistic studies because they are more representative of the majority of the tumor cells that comprise spontaneous basal-like triple-negative MMTV-Wnt-1 tumors (11). In addition, given that they are more epithelial-like and less aggressive than the M-Wnt cell line, we were particularly interested in examining whether leptin and other obesity-associated factors could induce a more mesenchymal phenotype and genotype in these cells.
Our finding that leptin signaling regulates mammary cancer cell Foxc2 and Akt3 expression is novel. In addition, while previous studies have demonstrated that Twist expression is increased with leptin treatment in several breast cancer cell lines (27, 28), we found that leptin regulates the expression of Twist2, but not Twist1. The function and regulation of the third Akt isoform remains poorly characterized in comparison to Akt1/2, but Akt3 signaling is known to stimulate CSC enrichment in breast tumor models (35, 36). Others have demonstrated that Akt3 amplification is more frequent in TNBC versus estrogen receptor-positive tumors and is negatively correlated with recurrence-free survival in TNBC (37, 38). The transcription factors Foxc2 and Twist2 are both closely linked to stem cell-like properties as well as EMT and metastatic capability in breast and other cancers (39-44). In addition, Foxc2 mediates resistance to multiple forms of chemotherapy, including paclitaxel, epirubicin, 5-fluorouracil, and cisplatin in breast cancer cells, in correlation with an EMT phenotype (41, 45). Furthermore, Foxc2 is enriched in claudin-low breast tumors in comparison to other subtypes, which links this gene with the most aggressive form of breast cancer (32). We previously demonstrated that obesity promotes greater Foxc2 expression in orthotopically transplanted E-Wnt and M-Wnt cells (11), but we are unaware of any studies linking obesity with greater tumoral Twist2 or Akt3 expression. Here we report that obesity upregulates Foxc2, Twist2, and Akt3 expression in spontaneous MMTV-Wnt-1 tumors in association with greater local and systemic leptin signaling (Figure 2a–b). Moreover, Lepr knockdown ablates E-Wnt expression of Foxc2, Twist2, and Vim, while partially attenuating Akt3 and Sox2 expression, in response to DIO mouse serum (Figure 5d). Consequently, Foxc2, Twist2, Vim, Akt3, and Sox2 may be mediators of obesity’s pro-CSC/EMT effects that are all induced at least in part by leptin.
This study used global gene expression data from a spontaneous model of TNBC to drive the generation of hypotheses regarding potential mechanisms mediating the reduced survival in obese MMTV-Wnt-1 mice. In addition, the use of mouse sera to model the obese versus non-obese tumor microenvironment in vitro allowed us to examine the combined impact of multiple obesity-associated systemic factors as well as the relative contribution of leptin. However, this in vitro approach to assessing the effects of obesity in mammosphere assays was precluded because serum inhibits sphere formation, so we were limited to treating with leptin alone. Our in vitro model of obesity using DIO mouse serum was also limited by its inability to account for intercellular crosstalk in the tumor microenvironment. Given that TNBC is a highly heterogeneous disease that includes multiple distinct molecular subtypes (46, 47), our use of the basal-like E-Wnt cell line alone for the mechanistic experiments is an additional study limitation. We did not mechanistically link leptin signaling to an EMT phenotype and CSC/EMT-related gene expression in the two claudin-low cell lines used (M-Wnt and MDA-MB-231), hindering our ability to extend our conclusions beyond the basal-like subtype. In addition, we are not able to conclude that the observed effects are limited to TNBC, as we did not assess leptin’s effects on models representing other breast cancer subtypes. Finally, while this study provides a broad characterization of leptin’s impact on CSC/EMT-related phenotypes and genes in TNBC cells, we did not pursue definition of the exact molecular pathways mediating the observed downstream effects. We can therefore only hypothesize that these effects are likely mediated through one or both of the two major pathways implicated by others in leptin’s pro-CSC/EMT effects: JAK2/STAT3 and PI3K/Akt (21-23, 25-28, 30) (Figure 6).
Given the continuing increase in global obesity rates, it is critical that we improve our understanding of mechanisms mediating the obesity-breast cancer link. Our findings indicate that increased leptin signaling is causally linked to obesity-associated TNBC development by promoting CSC enrichment and EMT. Future studies will further analyze the leptin targets we identified (Foxc2, Twist2, and Akt3) to define their role in obesity-induced CSC enrichment, EMT, and chemotherapy resistance in TNBC. In addition, the contribution of other adipokines and cytokines to this obesity-associated CSC/EMT genotype and phenotype will be examined, as our in vitro analyses indicate that leptin signaling does not fully account for all of the observed effects of DIO on TNBC. Greater understanding of the signals impacting CSC and EMT may provide new targets and intervention strategies for decreasing TNBC burden in obese women.
Supplementary Material
{kind=link}
{kind=link}
Implications.
Leptin-associated signals impacting CSC and EMT may provide new targets and intervention strategies for decreasing TNBC burden in obese women.
Acknowledgments
This study was supported by grants from the National Institutes of Health (R35 CA197627 to S.D. Hursting) and Breast Cancer Research Foundation (S.D. Hursting). L.W. Bowers and E.L. Rossi were supported by fellowships from the National Cancer Institute (R25 CA057726), and S.B. McDonell was supported by a fellowship from the National Institutes of Health (T32 LM12420-2).
Financial Support: This study was supported by grants from the Breast Cancer Research Foundation (S.D. Hursting) and the National Institutes of Health (R35 CA197627, S.D. Hursting). L.W. Bowers and E.L. Rossi were supported by fellowships from the National Cancer Institute (R25 CA057726), and S.B. McDonell was supported by a fellowship from the National Institutes of Health (T32 LM12420-2).
Footnotes
Conflicts of interest: The authors disclose no potential conflicts of interest.
References
- 1.Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307(5):491–7. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- 2.Protani M, Coory M, Martin JH. Effect of obesity on survival of women with breast cancer: systematic review and meta-analysis. Breast Cancer Res Treat. 2010;123(3):627–35. doi: 10.1007/s10549-010-0990-0. [DOI] [PubMed] [Google Scholar]
- 3.Bao PP, Cai H, Peng P, Gu K, Su Y, Shu XO, Zheng Y. Body mass index and weight change in relation to triple-negative breast cancer survival. Cancer Causes Control. 2016;27(2):229–36. doi: 10.1007/s10552-015-0700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Widschwendter P, Friedl TW, Schwentner L, DeGregorio N, Jaeger B, Schramm A, Bekes I, Deniz M, Lato K, Weissenbacher T, Kost B, Andergassen U, Jueckstock J, Neugebauer J, Trapp E, Fasching PA, Beckmann MW, Schneeweiss A, Schrader I, Rack B, Janni W, Scholz C. The influence of obesity on survival in early, high-risk breast cancer: results from the randomized SUCCESS A trial. Breast Cancer Res. 2015;17:129. doi: 10.1186/s13058-015-0639-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Litton JK, Gonzalez-Angulo AM, Warneke CL, Buzdar AU, Kau SW, Bondy M, Mahabir S, Hortobagyi GN, Brewster AM. Relationship between obesity and pathologic response to neoadjuvant chemotherapy among women with operable breast cancer. J Clin Oncol. 2008;26(25):4072–7. doi: 10.1200/JCO.2007.14.4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen S, Chen CM, Zhou Y, Zhou RJ, Yu KD, Shao ZM. Obesity or overweight is associated with worse pathological response to neoadjuvant chemotherapy among Chinese women with breast cancer. PLoS One. 2012;7(7):e41380. doi: 10.1371/journal.pone.0041380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwase T, Sangai T, Nagashima T, Sakakibara M, Sakakibara J, Hayama S, Ishigami E, Masuda T, Miyazaki M. Impact of body fat distribution on neoadjuvant chemotherapy outcomes in advanced breast cancer patients. Cancer Med. 2016;5(1):41–8. doi: 10.1002/cam4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pajares B, Pollan M, Martin M, Mackey JR, Lluch A, Gavila J, Vogel C, Ruiz-Borrego M, Calvo L, Pienkowski T, Rodriguez-Lescure A, Segui MA, Tredan O, Anton A, Ramos M, Camara Mdel C, Rodriguez-Martin C, Carrasco E, Alba E. Obesity and survival in operable breast cancer patients treated with adjuvant anthracyclines and taxanes according to pathological subtypes: a pooled analysis. Breast Cancer Res. 2013;15(6):R105. doi: 10.1186/bcr3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Del Fabbro E, Parsons H, Warneke CL, Pulivarthi K, Litton JK, Dev R, Palla SL, Brewster A, Bruera E. The relationship between body composition and response to neoadjuvant chemotherapy in women with operable breast cancer. Oncologist. 2012;17(10):1240–5. doi: 10.1634/theoncologist.2012-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei W, Lewis MT. Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr Relat Cancer. 2015;22(3):R135–55. doi: 10.1530/ERC-14-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunlap SM, Chiao LJ, Nogueira L, Usary J, Perou CM, Varticovski L, Hursting SD. Dietary energy balance modulates epithelial-to-mesenchymal transition and tumor progression in murine claudin-low and basal-like mammary tumor models. Cancer Prev Res (Phila) 2012;5(7):930–42. doi: 10.1158/1940-6207.CAPR-12-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Q, Dunlap SM, Zhu J, Downs-Kelly E, Rich J, Hursting SD, Berger NA, Reizes O. Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocr Relat Cancer. 2011;18(4):491–503. doi: 10.1530/ERC-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Taylor SK, Hood N. Insulin- and obesity-related variables in early-stage breast cancer: correlations and time course of prognostic associations. J Clin Oncol. 2012;30(2):164–71. doi: 10.1200/JCO.2011.36.2723. [DOI] [PubMed] [Google Scholar]
- 14.Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10(13):4325–31. doi: 10.1158/1078-0432.CCR-03-0749. [DOI] [PubMed] [Google Scholar]
- 15.Garofalo C, Koda M, Cascio S, Sulkowska M, Kanczuga-Koda L, Golaszewska J, Russo A, Sulkowski S, Surmacz E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clin Cancer Res. 2006;12(5):1447–53. doi: 10.1158/1078-0432.CCR-05-1913. [DOI] [PubMed] [Google Scholar]
- 16.Miyoshi Y, Funahashi T, Tanaka S, Taguchi T, Tamaki Y, Shimomura I, Noguchi S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int J Cancer. 2006;118(6):1414–9. doi: 10.1002/ijc.21543. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Prosperi JR, Choudhury N, Olopade OI, Goss KH. beta-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One. 2015;10(2):e0117097. doi: 10.1371/journal.pone.0117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Flanagan CH, Rossi EL, McDonell SB, Chen X, Tsai YH, Parker JS, Usary J, Perou CM, Hursting SD. Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer. 2017;3:26. doi: 10.1038/s41523-017-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi EL, Dunlap SM, Bowers LW, Khatib SA, Doerstling SS, Smith LA, Ford NA, Holley D, Brown PH, Estecio MR, Kusewitt DF, deGraffenried LA, Bultman SJ, Hursting SD. Energy Balance Modulation Impacts Epigenetic Reprogramming, ERalpha and ERbeta Expression, and Mammary Tumor Development in MMTV-neu Transgenic Mice. Cancer Res. 2017;77(9):2500–11. doi: 10.1158/0008-5472.CAN-16-2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–30. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang CC, Wu MJ, Yang JY, Camarillo IG, Chang CJ. Leptin-STAT3-G9a Signaling Promotes Obesity-Mediated Breast Cancer Progression. Cancer Res. 2015;75(11):2375–86. doi: 10.1158/0008-5472.CAN-14-3076. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 22.Feldman DE, Chen C, Punj V, Tsukamoto H, Machida K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc Natl Acad Sci U S A. 2012;109(3):829–34. doi: 10.1073/pnas.1114438109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato S, Abarzua-Catalan L, Trigo C, Delpiano A, Sanhueza C, Garcia K, Ibanez C, Hormazabal K, Diaz D, Branes J, Castellon E, Bravo E, Owen G, Cuello MA. Leptin stimulates migration and invasion and maintains cancer stem-like properties in ovarian cancer cells: an explanation for poor outcomes in obese women. Oncotarget. 2015;6(25):21100–19. doi: 10.18632/oncotarget.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowers LW, Cavazos DA, Maximo IX, Brenner AJ, Hursting SD, deGraffenried LA. Obesity enhances nongenomic estrogen receptor crosstalk with the PI3K/Akt and MAPK pathways to promote in vitro measures of breast cancer progression. Breast Cancer Res. 2013;15(4):R59. doi: 10.1186/bcr3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Q, Banaszak L, Fracci S, Basali D, Dunlap SM, Hursting SD, Rich JN, Hjlemeland AB, Vasanji A, Berger NA, Lathia JD, Reizes O. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr Relat Cancer. 2013;20(6):797–808. doi: 10.1530/ERC-13-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Tang C, Cao H, Li K, Pang X, Zhong L, Dang W, Tang H, Huang Y, Wei L, Su M, Chen T. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol Ther. 2015;16(8):1220–30. doi: 10.1080/15384047.2015.1056409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan D, Avtanski D, Saxena NK, Sharma D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires beta-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J Biol Chem. 2012;287(11):8598–612. doi: 10.1074/jbc.M111.322800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei L, Li K, Pang X, Guo B, Su M, Huang Y, Wang N, Ji F, Zhong C, Yang J, Zhang Z, Jiang Y, Liu Y, Chen T. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J Exp Clin Cancer Res. 2016;35(1):166. doi: 10.1186/s13046-016-0446-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiagarajan PS, Zheng Q, Bhagrath M, Mulkearns-Hubert EE, Myers MG, Lathia JD, Reizes O. STAT3 activation by leptin receptor is essential for TNBC stem cell maintenance. Endocr Relat Cancer. 2017;24(8):415–26. doi: 10.1530/ERC-16-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mishra AK, Parish CR, Wong ML, Licinio J, Blackburn AC. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS One. 2017;12(5):e0178454. doi: 10.1371/journal.pone.0178454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giordano C, Chemi F, Panza S, Barone I, Bonofiglio D, Lanzino M, Cordella A, Campana A, Hashim A, Rizza P, Leggio A, Gyorffy B, Simoes BM, Clarke RB, Weisz A, Catalano S, Ando S. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget. 2016;7(2):1262–75. doi: 10.18632/oncotarget.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strong AL, Ohlstein JF, Biagas BA, Rhodes LV, Pei DT, Tucker HA, Llamas C, Bowles AC, Dutreil MF, Zhang S, Gimble JM, Burow ME, Bunnell BA. Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res. 2015;17:112. doi: 10.1186/s13058-015-0622-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harbuzariu A, Rampoldi A, Daley-Brown DS, Candelaria P, Harmon TL, Lipsey CC, Beech DJ, Quarshie A, Ilies GO, Gonzalez-Perez RR. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget. 2017;8(5):7740–52. doi: 10.18632/oncotarget.13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esper RM, Dame M, McClintock S, Holt PR, Dannenberg AJ, Wicha MS, Brenner DE. Leptin and Adiponectin Modulate the Self-renewal of Normal Human Breast Epithelial Stem Cells. Cancer Prev Res (Phila) 2015;8(12):1174–83. doi: 10.1158/1940-6207.CAPR-14-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S, Patel SH, Ginestier C, Ibarra I, Martin-Trevino R, Bai S, McDermott SP, Shang L, Ke J, Ou SJ, Heath A, Zhang KJ, Korkaya H, Clouthier SG, Charafe-Jauffret E, Birnbaum D, Hannon GJ, Wicha MS. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet. 2012;8(6):e1002751. doi: 10.1371/journal.pgen.1002751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014;74(3):964–73. doi: 10.1158/0008-5472.CAN-13-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Hurley G, Daly E, O’Grady A, Cummins R, Quinn C, Flanagan L, Pierce A, Fan Y, Lynn MA, Rafferty M, Fitzgerald D, Ponten F, Duffy MJ, Jirstrom K, Kay EW, Gallagher WM. Investigation of molecular alterations of AKT-3 in triple-negative breast cancer. Histopathology. 2014;64(5):660–70. doi: 10.1111/his.12313. [DOI] [PubMed] [Google Scholar]
- 38.Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, Weigel RJ, Roth RA. Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J Biol Chem. 1999;274(31):21528–32. doi: 10.1074/jbc.274.31.21528. [DOI] [PubMed] [Google Scholar]
- 39.Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104(24):10069–74. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paranjape AN, Soundararajan R, Werden SJ, Joseph R, Taube JH, Liu H, Rodriguez-Canales J, Sphyris N, Wistuba I, Miura N, Dhillon J, Mahajan N, Mahajan K, Chang JT, Ittmann M, Maity SN, Logothetis C, Tang DG, Mani SA. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene. 2016;35(46):5963–76. doi: 10.1038/onc.2015.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, Herschkowitz JI, Guerra R, Chang JT, Miura N, Rosen JM, Mani SA. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013;73(6):1981–92. doi: 10.1158/0008-5472.CAN-12-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 2011;30(47):4707–20. doi: 10.1038/onc.2011.181. [DOI] [PubMed] [Google Scholar]
- 43.Liu AY, Cai Y, Mao Y, Lin Y, Zheng H, Wu T, Huang Y, Fang X, Lin S, Feng Q, Huang Z, Yang T, Luo Q, Ouyang G. Twist2 promotes self-renewal of liver cancer stem-like cells by regulating CD24. Carcinogenesis. 2014;35(3):537–45. doi: 10.1093/carcin/bgt364. [DOI] [PubMed] [Google Scholar]
- 44.Wang T, Li Y, Wang W, Tuerhanjiang A, Wu Z, Yang R, Yuan M, Ma D, Wang W, Wang S. Twist2, the key Twist isoform related to prognosis, promotes invasion of cervical cancer by inducing epithelial-mesenchymal transition and blocking senescence. Hum Pathol. 2014;45(9):1839–46. doi: 10.1016/j.humpath.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Cai J, Tian AX, Wang QS, Kong PZ, Du X, Li XQ, Feng YM. FOXF2 suppresses the FOXC2-mediated epithelial-mesenchymal transition and multidrug resistance of basal-like breast cancer. Cancer Lett. 2015;367(2):129–37. doi: 10.1016/j.canlet.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–90. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abramson VG, Mayer IA. Molecular Heterogeneity of Triple Negative Breast Cancer. Curr Breast Cancer Rep. 2014;6(3):154–8. doi: 10.1007/s12609-014-0152-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.