

Abstract
Understanding how molecules in self-assembled soft-matter nanostructures are organized is essential for improving the design of next-generation nanomaterials. Imaging these assemblies can be challenging and usually requires processing, e.g., staining or embedding, which can damage or obscure features. An alternative is to use bioinspired mineralization, mimicking how certain organisms use biomolecules to template mineral formation. Previously, we have reported the design and characterization of Self-Assembled peptide caGEs (SAGEs) formed from de novo peptide building blocks. In SAGEs, two complementary, 3-fold symmetric, peptide hubs combine to form a hexagonal lattice, which curves and closes to form SAGE nanoparticles. As hexagons alone cannot tile onto spheres, the network must also incorporate nonhexagonal shapes. While the hexagonal ultrastructure of the SAGEs has been imaged, these defects have not been observed. Here, we show that positively charged SAGEs biotemplate a thin, protective silica coating. Electron microscopy shows that these SiO2-SAGEs do not collapse, but maintain their 3D shape when dried. Atomic force microscopy reveals a network of hexagonal and irregular features on the SiO2-SAGE surface. The dimensions of these (7.2 nm ± 1.4 nm across, internal angles 119.8° ± 26.1°) are in accord with the designed SAGE network and with coarse-grained modeling of the SAGE assembly. The SiO2-SAGEs are permeable to small molecules (<2 nm), but not to larger biomolecules (>6 nm). Thus, bioinspired silicification offers a mild technique that preserves soft-matter nanoparticles for imaging, revealing structural details <10 nm in size, while also maintaining desirable properties, such as permeability to small molecules.
Keywords: atomic force microscopy, bioinspired nanoparticles, biomineralization, coiled-coil peptides, electron microscopy, peptide design
In nature, organisms construct soft-matter nanostructures to define and stabilize the form and function of the cell. Natural self-assembled functional soft-matter architectures include coat proteins that protect viral genetic cargo and facilitate cell penetration for replication; protein and lipid structures that are used to compartmentalize, manufacture, and transport materials in bacterial and mammalian cells; and combinations of carbohydrate-based materials and lipid–protein membranes that define bacterial and plant cell walls. Biomolecular engineers take inspiration from these natural systems to design nanostructured materials that assemble from the bottom up. The self-assembling building blocks used can be based on natural, engineered, or completely de novo biomolecules, including peptides,1,2 proteins,3,4 lipids,5 polymers,6 and DNA.7 In turn, these materials have been adapted for use in a wide range of medical and biotechnological applications.3,5,7
Imaging the organization of bioinspired self-assembled materials at the molecular or nanoscale is essential for both confirming the targeted designs and advancing the understanding of how to assemble and apply these materials in the future. However, imaging nanoscale biomolecular structures is challenging, as they are small, flexible, typically not very electron dense, and assembled via weak noncovalent interactions. Staining, embedding, coating, or cross-linking can all help to tackle these challenges and aid the visualization of soft structures.8 Nonetheless, such processing can also introduce unwelcome artifacts or disrupt these delicate architectures.8 Thus, simple, nondestructive processing methods that provide contrast are still required. Here we take inspiration from nature and demonstrate the use of bioinspired mineralization to preserve the 3D structure of soft, peptide-based, self-assembled nanoparticles.
In biomineralization biomolecules direct the precipitation of minerals to produce hierarchical composite tissues.9,10 Over 60 biominerals occur in natural biological systems,11 including calcium carbonate,12 calcium phosphate,13,14 magnetite,15 and silica.16−19 For example, diatoms are unicellular eukaryotic algae that construct intricate silicified cell walls, called frustules, using biomolecules such as long-chain polyamines20 and proteins such as arginine- and lysine-rich silaffins,21,22 cingulins,23 and phosphorylated silicidins.24 These biomolecules are thought to direct silica to precipitate from soluble silicic acid in vivo.21,22 Silicification has been studied in vitro using positively charged bioinspired molecules, such as lysine-rich polypeptides22,25 and other polyamines.26 The size of the biotemplated silica structures usually correlates with their presilicified size in solution.27 As such, silica coating has been used to preserve soft-matter nanostructures faithfully.27−31 In pioneering work from the Brinker lab, this has been extended to coat cells32,33 and even whole organisms.34 This has allowed features to be clearly imaged in 3D down to ∼10 nm.31 Permeable biomineral coatings have also been used to protect soft-matter structures for use in medicine35,36 and catalysis.37,38 In these ways, bioinspired silicification is able to provide contrast for nanoscale imaging and stabilize the function of soft-matter structures.
Protein cages are specifically addressable soft-matter nanostructures with potential biomedical applications in drug targeting and delivery,39−42 vaccination,4,43 and biotechnology as nanoreactors.44−46 An example is the Self-Assembled peptide caGEs (SAGEs, Figure 1).1 SAGEs are rationally designed1 from fully characterized de novo peptide components.47 Specifically, a parallel α-helical coiled-coil peptide homotrimer, CC-Tri3, is linked via a disulfide bond and back-to-back to one of two obligate heterodimers, CC-Di-A or CC-Di-B,1 making two complementary hubs (hub A and hub B) each with 3-fold symmetry. When these hubs are mixed, the heterodimers are formed, and these drive the self-assembly of what is intended to be a hexagonal network. Surprisingly, when imaged by electron microscopy (EM) and using in-liquid atomic force microscopy (AFM), closed, spherical particles are observed. This is rationalized by in silico molecular-dynamics simulations of small patches of the SAGE network, which consistently indicate that these multipeptide structures curve in one direction.1 However, hexagons alone cannot tile on a sphere and are unable to form closed, convex, regular Platonic solids.48 For example, C60 (buckminsterfullerene)49 combines 20 hexagons with 12 pentagons to form a closed Archimedean solid structure.48 Thus, the SAGE network must incorporate irregularities to form spherical nanoparticles. It is not known if these consist of holes in the lattice or if nonhexagonal or irregular polygons close the structures. To date, such features on the SAGE surface have not been observed. Therefore, we sought a method that could reveal how the peptides are organized in SAGE particles.
Figure 1.

Schematic for Self-Assembled peptide caGE (SAGE)1 design and silicification. A homotrimerizing coiled-coil peptide (CC-Tri3, green) is joined back-to-back via a disulfide bond to one of two heterodimeric sequences, namely, a negatively charged CC-Di-A peptide (red) or a positively charged CC-Di-B peptide (blue). These give hub A (CC-Tri3-CC-Di-A) and hub B (CC-Tri3-CC-Di-B), respectively. When mixed, the hubs are posited to form a regular hexagonal array that closes to form SAGEs. To ensure that silica precipitation is localized onto the SAGEs when immersed in silicic acid, the N termini or C termini of the homotrimers are decorated with positively charged peptides.
Here we describe the application of bioinspired silicification to form a stabilizing permeable coating on the SAGEs. We show that this allows detailed imaging of their surfaces. AFM of the silica-coated SAGEs (SiO2-SAGEs) reveals a hexagonal network as designed. Moreover, with ∼7 nm vertex-to-vertex lengths, the size of these polygons is fully consistent with the designed underlying peptide network. The network predominantly comprises regular hexagons, but other shapes, which are required to form a closed object from a hexagonal lattice, are also apparent. This concurs with coarse-grained computational modeling that we have recently performed.50 As the silicified SAGEs can be imaged clearly using standard EM and AFM techniques, bioinspired silicification offers a facile technique for preserving soft-matter nanostructures in 3D so that their self-organization can be studied. In addition, silicification offers a route to stabilizing soft, self-assembled materials for use in bionanotechnology and synthetic biology.35,36,51
Results and Discussion
The N and C Termini of the SAGE Homotrimer Can Be Decorated with Positive Charge
Molecular dynamics (MD) simulations of a hexagonal peptide network designed for the “parent” SAGE indicate that the arrays curve.1 In the MD, the long axes of the trimeric coiled-coil units remain perpendicular to the plane of the array, whereas the dimeric components rotate freely about the two disulfide bonds. Moreover, the direction of curvature is consistent across the simulations, with the N termini of the homotrimers present on the convex (i.e., external) face of the SAGEs, while the C termini are on the concave interior. This is our working model for SAGE structure, and it provides a basis for further design.
For this study, tetra-arginine (R4) or tetra-lysine (K4) peptides were appended to either the N or C terminus of the homotrimer (CC-Tri3, Figure 1, Table 1, and Tables S1–S3). The aim being that, when assembled into SAGEs, these constructs would present positively charged residues on the external or internal surfaces of SAGEs for N-terminal or C-terminal appendages, respectively. In turn, these should act as templates to localize and precipitate silica from aqueous silicic acid onto the peptide cages. In this way, we aimed to create SiO2-SAGE particles. As controls, negatively charged tetra-glutamate (E4) variants were also generated, which should be poor at biotemplating silica.25
Table 1. Summary of the Main Peptide Sequences Used to Assemble Decorated SAGEs for Silicificationa.

Systematic names of the trimer components used to make the hubs, their amino-acid sequences, and their overall charge as calculated using pepcalc.52 See Supporting Information for further details of the naming system (Table S1), mixing peptide hubs to form SAGEs (Table S2), and the full amino-acid sequences for all peptides used in this study (Table S3).
Peptides were synthesized, purified, and assembled into SAGE peptide hubs (Figure S1, Table 1, and Tables S1–S4 for sequences, naming, and characterization). For the C-terminally decorated trimers, CC-Tri3-K4 and CC-Tri3-R4, circular dichroism (CD) spectra gave α helicities similar to the undecorated parent homotrimer (CC-Tri3) and as predicted based on the peptide sequences (Figures S2 and S3 and Table S5). Moreover, these three constructs had very similar thermal unfolding transitions, with midpoints (TM’s) of 54, 57, and 56 °C, respectively (Figure S2 and Table S5). By contrast, appending positive charge to the N termini reduced the α helicities and also the TM values to 33 and 34 °C for the K4 and R4 variants, respectively (Figures S2 and S3 and Table S5). CC-Tri3 variants with negatively charged E4 blocks at the N and C termini showed the opposite behavior: the former was as folded and stable as the parent (TM = 65.5 °C), whereas the latter was as destabilized (TM = 31 °C) as the N-terminal K4 and R4 variants, Figures S2 and S3 and Table S5. These changes in stability of CC-Tri3 when charge is appended mirror a recent study of free-standing α-helical peptides.53 They can be explained in terms of repulsive interactions between the blocks and the partial charges associated with the helical backbone, which are positive and negative for the N and C termini, respectively.53 Analytical ultracentrifugation (AUC) sedimentation velocity (SV) and sedimentation equilibrium (SE) experiments conducted at room temperature with the parent and decorated homotrimers returned molecular weights consistent with trimeric coiled-coil assemblies for each construct in solution, Figure S4 and Table S6.
Together, these data for the modified CC-Tri3 peptides show that they are stably folded and trimeric at room temperature, albeit with some of the thermal stabilities compromised. Therefore, we continued with these variants, using them to construct decorated hubs for the assembly of modified SAGE particles.
Positive Charge Localizes Silica Biomineralization onto SAGEs
Herein, we refer to the unmodified parent assemblies as SAGE, Figure 2a, and the SAGE variants as follows: E4-SAGE, K4-SAGE, and R4-SAGE for the N-terminally decorated particles, Figure 2e, and SAGE-E4, SAGE-K4, and SAGE-R4 for the C-terminally decorated structures, Figure 2i. Acidification of sodium silicate forms a mixture of metastable ortho- and oligosilicic acids.54 For simplicity, we refer to this as “silicic acid”. Polymerization of these mixtures was monitored by fluorescence of 2-(4-pyridyl)-5-((4-(2-dimethylaminoethylaminocarbamoyl)methoxy)phenyl)oxazole (PDMPO) without and with the various SAGE particles (Figure S5). The PDMPO indicated that the different SAGEs did not appear to alter the amount or rate of bulk silica precipitation. The silicification of SAGEs to form discrete biomineralized particles was optimized to form electron dense particles (Figures 2 and S6).
Figure 2.

Representative transmission electron microscopy (TEM) images of silicic-acid-treated SAGE particles and controls. (a, e, i) Cartoons showing peptide modules (left) and assembled SAGEs (right). (b–d) Controls showing (b) silica precipitated in the absence of peptide, (c) with parent SAGE, and (d) parent SAGE without silicic acid added. (f–h) Silicic-acid-treated SAGE with N-terminally appended tetrapeptides (f) E4-SAGE, (g) K4-SAGE, and (h) R4-SAGE. (j–l) Same as in panels f–h, but for C-terminally decorated SAGEs. Samples were prepared at a concentration of 2 μM peptide, 24 mM silicic acid, phosphate (PI) buffer (10 mM potassium phosphate (8.02 mM dibasic K2HPO4, 1.98 mM monobasic KH2PO4), pH 7.4), 24 h, 20 °C. Scale bars are 200 nm.
As controls, silicification of the peptide hubs alone, i.e., negatively charged hub A or positively charged hub B, formed mesh networks with poor contrast, Figure S7, and similar precipitates were observed with short positively charged peptides, GYGKKKK and GYGRRRR, Figure S8. By comparison, with both hub A and hub B present, i.e., with SAGEs formed, silica particles were observed, and these were particularly clear with excellent contrast for the K4- and R4-decorated SAGEs, Figures 2, S6, and S7.
In more detail, the C-terminal tetra-arginine decoration, SAGE-R4, gave large, slightly interconnected, and polydispersed spheres (305 ± 120 nm). The tetra-lysine variants, K4-SAGE and SAGE-K4, assembled into an interconnected mesh and smaller, slightly interconnected spheres (Figures 2g and k and S6e and h). The most monodisperse and unconnected nanospheres were formed by N-terminal tetra-arginine-decorated R4-SAGEs, Figures 2h and S6f. These were 213 ± 84 nm in diameter, Table S7, Figure 2h, and Figures S6h and S9. This is a significant increase in size and decrease of the distribution compared with unmineralized R4-SAGEs (109 ± 84 nm), which were difficult to measure, as they gave poor contrast (Figure S10). Similarly, the silicified R4-SAGEs gave better quality scanning electron microscope (SEM) images, revealing clearer particles when compared to unmineralized R4-SAGEs (Figure S11). Energy dispersive X-ray (EDX) spectra confirmed the presence of silicon (Si Kα = 1.74 keV) and oxygen (O Kα = 0.53 keV) in the SiO2-R4-SAGEs (Figure S12). The absence of any coherent electron diffraction patterns, however, indicated that the silica coatings are likely amorphous rather than a crystalline polymorph of silica.
In summary to this section, a combination of TEM, SEM, and EDX demonstrates that SAGEs can be modified with cationic tetra-peptides to generate discrete, spherical, silicified nanoparticles, which we refer to as SiO2-SAGEs. The best of these preparations were made from R4-SAGEs, which we used next to optimize conditions for preparing SiO2-SAGEs.
Formation of Discrete SiO2-SAGEs Can Be Optimized
R4-SAGEs were mineralized with a range of silicic acid concentrations (0–36 mM), imaged (Figure S13), and grain-sized (Table S7). Without silicic acid or at low concentrations (6 mM), it was difficult to discern structures by TEM, and the grains were polydisperse. At 36 mM silicic acid, large interconnected SiO2-R4-SAGE particles were clear (217 ± 58 nm). We propose that high silicic acid concentrations deposit a thicker layer of SiO2, leading to particle aggregation. Optimal mineralization occurred at 12–24 mM silicic acid, leading to discrete SiO2-R4-SAGE spheres with sizes distributed around ∼200 nm in diameter (Figure 2h).
Next, we varied the peptide concentration between 2 and 25 μM peptide for K4 and R4 SAGE variants and mineralized these with 24 mM silicic acid at 20 °C (Figure S14). At low peptide concentrations (2–5 μM), the K4- and R4-SAGE constructs formed discrete mineralized spheres similar to those noted above. At the higher peptide concentrations (10–25 μM), much larger micrometer-sized mesh and network structures were observed, which is consistent with our observations that unmineralized SAGEs aggregate at high μM concentrations.
The SAGE system is modular, which means that the hub modules can be mixed and matched to tune the SAGE properties. Thus far, both hub A and hub B carried the E4, K4, or R4 decorations; that is, they were 100% decorated. To vary this, first we prepared SAGEs with undecorated parent hub A plus hub B fully decorated with K4 or R4 (Figure S15). These were silicified using standard conditions of 2 μM peptide, 24 mM silicic acid, and 10 mM phosphate (PI) buffer pH 7.4, 20 °C, 24 h. Compared with 100% decoration, these 50% decorated SAGEs form smaller particles, particularly for the R4 variants. R4-SAGEs displaying between 50% and 100% tetra-arginine were prepared by combining R4-hub B and mixtures of hub A with and without the R4 decoration (Figure S16). The less highly charged SAGEs, i.e., 50–80% R4-SAGE, formed smaller interconnected mineralized silica particles, with average diameters of 60–90 nm, Table S8, whereas the 90% and 100% R4-SAGEs formed the larger but nonetheless discrete structures noted above.
The 100% R4-SAGEs were also assembled and silicified at a range of temperatures (4–37 °C). This range should not significantly perturb the silicification process,55 but, as the midpoint unfolding temperature (TM) of CC-R4-Tri3 is 34 °C, this range could affect homotrimer folding and thus SAGE assembly (Figure S17). At 4 °C, the mineralized particles were small (77 ± 26 nm) interconnected spheres. At 20 °C, the particles were larger (213 ± 84 nm) and discrete and were larger still (330 ± 168 nm) and clumped together at 25 °C. At 37 °C, smaller (105 ± 42 nm) interconnected spheres were formed. Thus, the optimal temperature for assembling discrete mineralized R4-SAGEs is at ∼20 °C, where the CC-R4-Tri3 should be well folded, but there is enough thermal energy to avoid forming the small, interconnected structures assembled seen at lower temperatures.
Finally, the components for 100% R4-SAGEs were mixed at between pH 5.5 and 8.0, and the products silicified at the assembly pH. At low pH, silica formation is known to be slowed, but the 24 h mineralization used should be ample for silica formation.55 TEM images (Figure S18) reveal that silicification at neutral pH (7.0 and 7.4) led to discrete spherical particles, whereas at higher pH values (7.5–8.0), interconnected SAGEs resembling those formed at higher (36 mM) silicic acid concentrations were formed, which we attribute to rapid and more complete precipitation of silica. At slightly lower pH values (6.5 and 6.0) spheres were discernible, but these were embedded in meshes. Finally, at pH 5.5 only thin sheets were visible, which we ascribed to the precipitation of silica by largely unfolded aggregated peptide.
Thus, we find that for R4-SAGE particles, which provide the best substrates for silicification, there are optimal conditions for mineralization to produce SiO2-R4-SAGEs. These are summarized in Figure 3 and comprise 2–5 μM peptide, 90–100% R4-decorated hubs, 20 °C, and pH 7.0–7.4 PI buffer, mineralized with 12–24 mM silicic acid for 24 h.
Figure 3.
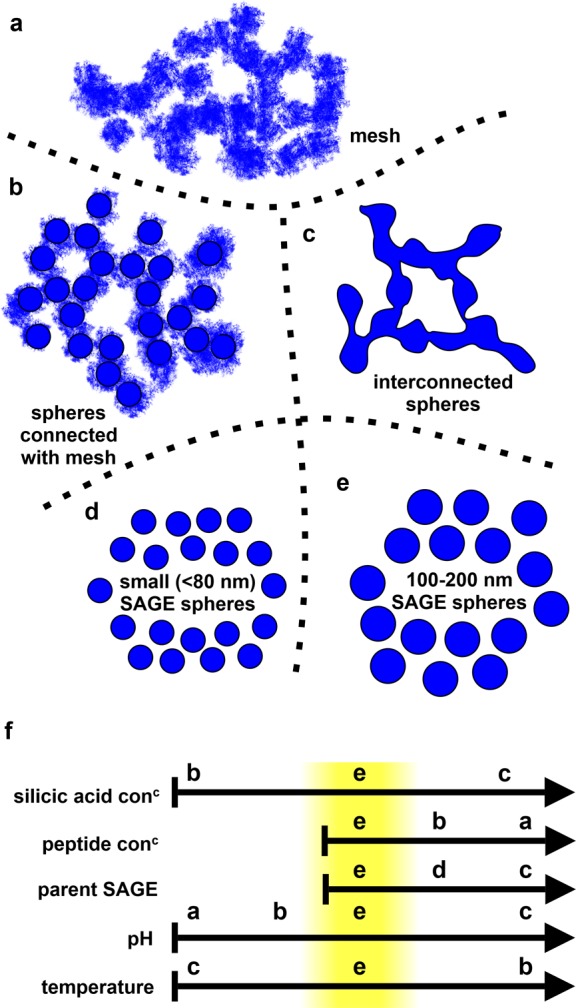
Summary of structures assembled during optimization of SiO2-R4-SAGE formation. (a) Fine network or mesh of disordered mineralized peptide, (b) spheres interconnected with fine mesh, and (c) spheres interconnected with assembled mineralized peptide. Individual SAGEs that are small (d) and regularly sized (e) (highlighted in yellow in panel f) can also be produced. (f) Summary indicating how changing silicic acid and peptide concentrations, proportion of parent SAGE, pH, and temperature alter the types of assemblies observed for the SiO2-R4-SAGEs. The “Goldilocks zone” for making individual, regularly sized, spherical SAGEs is highlighted in yellow. Arrows represent increasing values of the condition illustrated.
3D Structure of SiO2-SAGEs Can Be Visualized by EM Tomography and AFM
We recorded TEM tilt series for SiO2-R4-SAGEs and used these to construct tomograms and 3D models of the mineralized particles (Movies S1–S4, Figure S19). The tomograms showed that the particles did not collapse significantly when dried onto TEM grids; that is, dried SiO2-R4-SAGEs retained their 3D structure. SiO2-R4-SAGEs were also imaged by tapping-mode atomic force microscopy (TM-AFM) and PeakForce AFM (PF-AFM), Figure S20. AFM measurements of unmineralized SAGEs revealed significant collapse of the peptide particles upon drying to give an aspect ratio (AR) between the height z and the diameter in the x–y plane of ∼0.1.1 By contrast, the dried SiO2-R4-SAGEs had an aspect ratio of ∼0.7 in AFM. Thus, the silica coating reinforces SiO2-R4-SAGEs against collapse.
We were interested to see if silicification also preserved the structure of the peptide building blocks and pores between the hub subunits anticipated from the original design, Figure S21. PF-AFM was used to map variations in the height, adhesion, and dissipation properties of the surface of a cluster of SiO2-R4-SAGEs (Figure 4). Despite variations in the topography due to the near-spherical shape of the SiO2-R4-SAGEs, the surfaces of the nanoparticles could be imaged. This revealed a polygonal lattice texture on the surface of all the SiO2-R4-SAGEs imaged (Figure S22).
Figure 4.

PeakForce atomic force microscopy (PF-AFM) of SiO2-R4-SAGE particles on mica. 2D plots show variations recorded in (a) the height, (b) adhesion, and (c) dissipation properties of the SiO2-R4-SAGEs (upper parts of images) and mica (lower parts of images). (d) 3D plot of the topography. The mica surface is smooth, whereas the SiO2-R4-SAGE surface reveals texture, which is shown in greater detail in Figure 5. Silicification was performed with 2 μM peptide, 12 mM silicic acid, and PI buffer, 24 h, 20 °C.
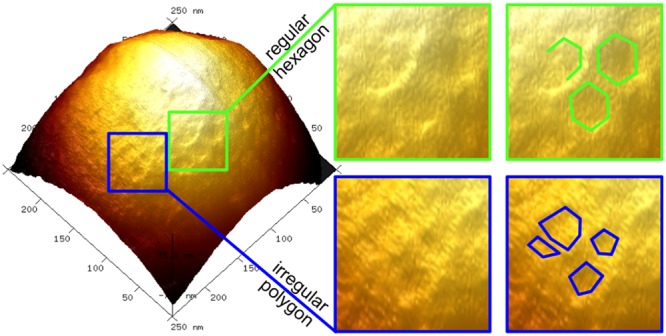
Interrogation of these patterns on individual SiO2-R4-SAGEs with PF-AFM identified polygons with vertex-to-vertex spacings of 7.2 ± 1.4 nm (Figure 5). This is entirely consistent with the SAGE design.1 Moreover, these features were apparent in the height, adhesion, and dissipation plots, with regions of increased height and adhesion colocalized, Figure 5e, h, and i. The network was also seen in phase contrast in TM-AFM (Figure S23f and i). Ridges, created by the peptide framework within the biotemplated silica, should present as raised lattice structures on the curved SAGE surface with increased height, nanoscale roughness, and flexibility when compared to a silica-filled pore (Figure S21). This was indeed observed in PF-AFM, with raised ridges corresponding to higher adhesion (roughness) and dissipation (flexibility) when compared to the pore centers, and confirmed by positive phase shifts on ridges in TM-AFM.
Figure 5.

PeakForce atomic force microscopy (PF-AFM) of an individual SiO2-R4-SAGE particle. 2D plots zoomed in on area shown in Figure 4, which map variation in (a) height, (b) adhesion, and (c) dissipation, and a plot showing (d) height contrast. (e) The same surface as shown in (d) with the corresponding adhesion map overlaid as a blue-scale color-map, which shows that variations in topography and adhesion colocalize. (f – i) 3× magnifications for (f and g) 3D height plots and (h and i) height-adhesion overlays, revealing (f and h) irregular polygons and (g and i) regular hexagons. (j–m) Annotated version of panels f–i with shapes drawn to guide the eye. Silicification was performed with 2 μM peptide, 12 mM silicic acid, and PI buffer, 24 h, 20 °C.
Interestingly, the AFM data also revealed variations in the originally intended hexagonal network (Figure 5j and l). To help quantify the extent of these variations, a model for a smooth nonsymmetric convex surface was fitted to the height data to flatten the dome shape of the particle (Figure S24b). This generated a topology map of the particle, highlighting variance due to surface features (i.e., pores and ridges). The internal angles of the polygonal structures from unflattened images were distributed around 120° (119.4° ± 26.9°, Figure S24d). Nonetheless, other angles, e.g., for pentagons (108.0°), heptagons (128.6°), and irregular polygons, also fell comfortably within the distribution.
The internal angles measured from coarse-grained modeling of SAGE assembly (see Mosayebi etal.50 and Figure S24c and d) showed a major peak centered on hexagonal polygons (122.0° ± 25.0°), which agrees well with the experimental data. However, there was also a small peak for squares at 89.6° ± 8.6°, which is not observed experimentally. The squares are likely to be due to the simulated network, which comprises obligate heteromeric pairings of pure hubs (i.e., hub A and hub B only), not being able to access odd-sided polygons, e.g., pentagons, to close the SAGEs. In the experimental system, however, nonhexagonal polygons could form by noncognate peptide interactions (i.e., CC-Di-B to CC-Di-B or CC-Di-A to CC-Di-A), especially as hubs of the same type are brought into close proximity during assembly, significantly increasing the local concentration of heterodimer. Alternatively, as the peptide hubs are inherently dynamic, the homotrimers may exchange to form mixed hubs, which would also increase the chances of incorporating nonhexagonal structures. The current simulations do not allow for subunit exchange in hubs and penalize homomerization of the heterodimer components, both of which would facilitate the formation of the odd-sided polygons by SAGE hubs during assembly. This will be explored in future coarse-grained models.
These observations indicate how the peptide lattice adapts to close and leads us to refine our initial hypothesis for SAGE assembly and structure, which centered on the formation and propagation of hexagonal peptide arrays, Figure 1.1 The inclusion of nonhexagonal and irregular shapes, as revealed herein using the SiO2-SAGE system and supported by coarse-grained computational modeling of SAGE assembly (vide infra),50 presents a clear mechanism for closure. The variations that we see are not evenly spaced on the surface (Figures 5f–m, S24) as required in Archimedean solids,48 so the SAGE peptides self-assemble to form more complex, irregular polyhedra.
SiO2-SAGEs Are Permeable to Small Molecules and Peptides, but Not to Proteins
Permeable biomineral coatings have been used to protect enzymes,37,38 drug cargos,56 and vaccines.35,36,51 With a view to using SiO2-SAGEs in these contexts, we tested their permeability to small molecules and proteins. To do this, we set up a reporter system, which uses the modularity of the SAGE system to introduce a fluorophore into the peptide fabric of the SAGEs, Figure 6a. An R4-SAGE preparation was doped with 2.5% of hub B harboring carboxyfluorescein (hub B-fl). When silicified, these R4-SAGE-fls formed particles indistinguishable from R4-SAGEs by TEM, Figure 6b. Moreover, the particles were fluorescent, and correlative light and electron microscopy (CLEM)57,58 showed that this fluorescence colocalized with the particles observed by TEM, Figures 6c and d and S25. Consistent with this, prior to silicification R4-SAGE-fls were difficult to visualize by TEM, but fluorescent particles were clear by light microscopy (LM), Figure S26. We found that higher concentrations of silicic acid (36 mM) reduced detection of the fluorescence signal, and, therefore, we used intermediate silicic acid concentrations (12–24 mM) to form thinner silica coatings.
Figure 6.

Correlative light and electron microscopy (CLEM) and fluorescence microscope images of R4-SAGE-fl fluorescent SAGEs. (a) Schematic showing the peptide modules used to assemble R4-SAGE-fls (2 μM peptide, PI buffer, 1 h, 20 °C). (b) High-magnification TEM image of a cluster of SiO2-R4-SAGE-fls (mineralization conditions: 24 mM silicic acid, 24 h, PI buffer, 20 °C). (c and d) TEM images superimposed with fluorescence microscope images of the same area to produce CLEM images. In (c) and (d), red boxes highlight the zoomed area for the previous panel. (e–p) Fluorescence microscope images of R4-SAGE-fls (scale bars 10 μm): (e–h) unmineralized; mineralized with (i–l) 12 mM silicic acid and (m–p) 24 mM silicic acid. Separate bright-field and overlaid images are given in Figure S26. R4-SAGE-fls without additives (e, i, m) and after treatment with (f, j, n) Trypan blue; (g, k, n) tris(2-carboxyethyl)phosphine hydrochloride (TCEP); and (h, l, p) trypsin.
To monitor small-molecule ingress into SiO2-R4-SAGE-fls, we used the fluorescence quencher Trypan blue, which must contact the fluorophore to quench it.59,60 Trypan blue is small (∼1.5 × 0.5 × 0.5 nm) and was able to quench the fluorescence of both R4-SAGE-fls and SiO2-R4-SAGE-fls, Figure 6f, j, and n. The SAGE structures were still visible in bright-field LM images (Figure S26), indicating that Trypan blue can traverse the silica coating to effect quenching.
For delivery applications, small-molecule cargos must be able to exit the host vehicle. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) reduces the disulfide bonds used to construct the SAGE hubs and so disassembles SAGE particles.1 TCEP treatment reduced the number and intensity of fluorescent SiO2-R4-SAGE-fls particles, Figure 6g, k, and o, consistent with disruption of SAGEs and the release of peptides from the biomineralized structures (CC-Tri3 is ∼3.0 × 1.5 × 1.5 nm when folded). Nonetheless, particles were still visible by bright-field LM and TEM after this treatment, Figures S26 and S27, demonstrating that the biomineral replicas remain intact after TCEP treatment to remove the peptides.
Finally, to test if larger proteins penetrated the silica barrier, R4-SAGE-fls and SiO2-R4-SAGE-fls were treated with the protease trypsin. For the former, nonmineralized particles’ fluorescent puncta were lost, Figure 6h, indicating proteolysis of the SAGE peptides and release of the fluorophores. However, with the SiO2-R4-SAGE-fls these objects remained visible, indicating that the silica coat protected the peptides from proteolysis by trypsin, Figure 6l and p. The trypsin may be too large (∼7.0 × 6.0 × 5.5 nm) to penetrate the silicified SAGEs or may adhere to the silica coat becasue it has a pI of ∼10. However, as the pI of CC-Tri3 (∼9) and trypsin are similar and the smaller CC-Tri3-fl does appear to traverse the silica coating, it is unlikely that charge alone is responsible for preventing proteolysis of the SiO2-SAGEs by trypsin. Together, these data indicate that SiO2-SAGEs are permeable to small molecules (<2 nm across), including fluorescence quenchers, reducing agents, and the peptide building blocks of the SAGEs themselves, but not to larger (∼6 nm across) protein molecules such as trypsin.
Conclusions and Future Directions
Herein we demonstrate that SAGE particles can be decorated with positive charge, assembled, and controllably silicified under mild conditions in aqueous buffer. We find that an N-terminal tetra-arginine tag, to give R4-SAGEs, forms discrete silica-coated particles. We have tuned the particle size between ∼50 and 200 nm by controlling the proportion of tetra-arginine in the SAGE particles, by altering silicic acid concentration, and also by optimizing the conditions for SAGE assembly (peptide concentration, temperature, and pH). We envisage this ability to control particle size and functionalization could allow silicified SAGE particles to be tailored for use in a range of biotechnological applications. We find that SiO2-SAGEs particles do not form below pH 6.0 and that they aggregate above pH 7.5. This suggests that SAGE particles assembled at or near neutral pH could then be disassembled under acidic conditions. Such pH-triggered disassembly could be used to release encapsulated cargos controllably within target cells, as acidification during endosomal trafficking should lead to disassembly and thus delivery.61
SiO2-SAGEs maintain their 3D structure when dried, which facilitates imaging of the underlying polygonal peptide network. The network shows predominantly hexagonal shapes ∼7 nm across, consistent with our original design and in silico models.1,50 However, and interestingly, it also reveals the presence of nonhexagonal and irregular polygons with internal angles distributed evenly about the hexagonal 120°. These are necessary to close the network, as hexagons alone cannot tile on a sphere. These experimental measurements largely concur with a coarse-grained computational model for SAGE assembly that we are developing,50 which shows the dominance of a hexagonal arrangement of hubs and highlights the importance of nonhexagonal polygons to achieve closure. However, the simulations suggest that square arrangements of hubs should be more abundant than we observe experimentally. This current model disfavors mechanisms that can access odd-sided polygons in the lattice, e.g., homotypic association of the two heterodimer components or exchange within the homotrimer core to form mixed hubs. The AFM data show that the peptide network within SiO2-R4-SAGEs is able to form internal angles consistent with odd-sided polygons rather than closing using squares. In turn, this indicates that defect formation via one of the above two or other mechanisms must be in operation during SAGE assembly. These concepts and the experimental data from the SiO2-SAGEs are being incorporated into a revised coarse-grained model to explore the formation of odd- and even-sided hub assemblies.
As the SAGEs are closed by small shape defects rather than by large holes in the lattice, any larger cargos contained within SAGEs should not leak out. This bodes well for using them to support active proteins and enzymes;45,46 for drug encapsulation, targeting, and delivery;42,62 and as antigen presentation vaccine platforms.3,4 Biomineral coatings are currently used to protect drug cargos,56,63 vaccines,36,51 and enzymes37,64 by stabilizing biomolecules against thermal and other degradation. Thus, bioinspired silicification of SAGEs functionalized for use in medical or biotechnological applications could afford similar protection to these particles. SAGEs could also be designed to template other materials.65,66 For example, SAGEs displaying antigens could be coated with adjuvantive51 and/or protective36 calcium phosphate to create stabilized vaccine formulations with improved efficacies and shelf-lives. SAGEs displaying ligands for cell-surface receptors and disease treatment could also be decorated with gold nanoparticles to combine targeting, imaging, and treatment of diseased cells,67 or SAGEs coated with platinum nanoparticles could evolve hydrogen more effectively than disordered Pt nanoparticles.68 The SAGE system has clear attributes to facilitate such applications, including its modularity, our ability to redesign it, and the ease with which the SAGE components can be functionalized. Therefore, by mixing and matching appropriately modified hub components, there is considerable potential to develop a range of tailored multifunctional organic, inorganic, and/or biologically decorated SAGEs for future applications.
Materials and Methods
For extended materials and methods, see the Supporting Information.
Peptides were synthesized using Fmoc solid-phase synthesis69 on Rink amide resin in a Liberty microwave peptide synthesizer on 0.1 mM scales (see Tables S1–S3). Each amino acid was coupled (5 equiv of Fmoc amino acid, 4.5 equiv of hydroxybenzotriazole (HOBt), and 10 equiv of N,N′-diisopropylcarbodiimide (DIC) in 7 mL of dimethylforamide (DMF) with 25 W microwave irradiation at 50 °C for 5 min), washed (5 × 7 mL of DMF), deprotected (20% (v/v) morpholine in DMF, with 20 W microwave irradiation at 75 °C for 5 min), and then washed before the next amino acid was added. Assembled peptides were acetylated on resin (3 equiv of acetic anhydride and 4.5 equiv of N,N-diisopropylethlamine (DIPEA) in 7 mL of DMF for 30 min). Peptides were cleaved from the resin with 94% (v/v) trifluoroacetic acid (TFA), 2% (v/v) 1,2-ethanedithiol (EDT), 2% (v/v) water, and 2% (v/v) triisopropylsilane (TIPS) for 3 h. Peptides were filtered, precipitated using diethyl ether (Et2O), pelleted (4000g, 10 min, 4 °C), and freeze-dried.
Peptides were purified using reverse-phase high-pressure liquid chromatography (RP-HPLC) on a Kromatek (semimicro, 5 μm, 100 Å, 10 mm i.d. × 150 mm L) C18 column, using a linear gradient of buffer A (0.1% (v/v) TFA in water) and 20–80% buffer B (0.1% (v/v) TFA in MeCN). Purified peptide peaks were analyzed by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF) and analytical RP-HPLC. For 2,2′-dipyridyldisulfide (DPDS) thiol activation70 of heterodimer A or B, ∼5 mg of heterodimer was dissolved in 5 mL of water or phosphate-buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer, pH 7.4). Ten equivalents of DPDS in 1 mL of methanol (MeOH) was added. After 1 h, excess DPDS was removed using 3 × 30 mL of Et2O, and the activated peptide was freeze-dried, purified using RP-HPLC, characterized (MALDI-TOF and analytical RP-HPLC), and freeze-dried. To form hubs, each homotrimer was mixed with an activated heterodimer in equimolar amounts in PBS (∼1 mg mL–1 peptide) and mixed for 3 h at 20 °C. Hubs were purified by RP-HPLC and characterized (MALDI-TOF and analytical RP-HPLC, Figure S1 and Table S4).
Circular dichroism (CD) spectra were recorded at between 5 and 90 °C using a JASCO J-810 spectropolarimeter fitted with a Peltier temperature controller. A 1 mm path length quartz cuvette containing 50 μM peptide in phosphate buffer (PI, 10 mM potassium phosphate (8.02 mM dibasic K2HPO4, 1.98 mM monobasic KH2PO4) pH 7.4), 25 mM NaCl and 250 μM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was heated at 40 °C h–1. The mean residue ellipticity (MRE) at 222 nm was recorded every 1 °C, and full CD spectra (190–260 nm) were recorded every 5 °C. Analytical ultracentrifugation was performed on each homotrimer (325 μM peptide in PI, 1.625 mM TCEP, and 25 mM NaCl). Sedimentation velocity and sedimentation equilibrium experiments were conducted using Beckman Optima XL-A and XL-I analytical ultracentrifuges with An-60 and An-50 rotors, respectively, and at 20 °C. For SV, samples were spun at 60 krpm with 120 scans collected between 5.8 and 7.3 cm, and data fitted using SEDFIT.71 For SE, data were collected at six rotor speeds between 30 and 48 krpm and fitted with a single ideal species model using Ultrascan II.72 The partial specific volumes (v̅) for each peptide, buffer densities, and viscosities were calculated using SEDNTERP73 (Table S6).
Typically for silicification, SAGEs were assembled (2 μM hub A + 2 μM hub B) in PI buffer for 1 h at 20 °C; then 500 μL of these samples were mixed with 500 μL of PI buffer containing 0–36 mM freshly prepared silicic acid74 and washed 3× with water after 24 h. A 0.5 μL volume of 2-(4-pyridyl)-5-((4-(2-dimethylaminoethylaminocarbamoyl)methoxy)phenyl)oxazole (PDMPO, LysoTracker)75 was added to 100 μL of mineralization solution to monitor the polymerization of silica in solution by fluorescence emission.
For SEM, samples were dried on silicon wafers fixed to an aluminum stub. Unmineralized samples were coated with ∼5 nm Au–Pd using an Emtech 575X sputter coater. Stubs were imaged in a FEI Quanta FEG-SEM. Samples were dried on carbon-coated copper grids and imaged on a Tecnai 12–FEI 120 kV BioTwin Spirit transmission electron microscope. Tilt series between angles of ∼70° and ∼−70° for tomographic reconstructions were collected using a Tecnai 20–FEI 200 kV Twin Lens scanning transmission electron microscope (STEM). Images were collected using a FEI Eagle 4k × 4k CCD camera. Grain sizes of SiO2-SAGEs were measured from representative TEM images using ImageJ76,77 and distributions fitted in Origin 2015 64 bit (eq S3). Tilt series were aligned and reconstructed using eTomo in the IMOD78,79 software package (University of Colorado, Boulder). 3D reconstructions of tomograms were rendered in Amira 3D (FEI software). Movie files of the tomographic reconstruction software outputs were compiled in ImageJ76 and compressed using HandBrake. EDX spectra were recorded using a JEOL 2100F STEM and a Gatan Orius 11 megapixel 832 camera. X-rays were detected using an 80 mm2 AZtec detector and processed using AZtecTEM software. For CLEM,57,58 fluorescent R4-SAGE-fls were imaged first using a Leica DMI4000 B inverted epifluorescence microscope, then using a TEM (see above). Fluorescence was overlain with TEM images using the TurboReg plug-in in Fiji.80
TM-AFM was performed on a Multimode microscope with a Quadrexed Nanoscope III controller using Scout-Beta cantilevers (NuNano, Bristol, UK). PF-AFM was conducted on a Multimode VIII microscope with Nanoscope V controller and a fast scan head unit in combination with SCANASYST-AIR-HR cantilevers (Bruker, CA, USA). Data were analyzed using Nanoscope Analysis 1.5 software (Bruker, CA, USA), and scripts written in Matlab (Mathworks). Design and parametrization of the coarse-grained computational modeling are described in Mosayabi etal.50
The permeability of particles to small molecules was tested using silica-coated and unsilicified fluorescent R4-SAGE-fls. A 1 mL mineralization of silicified R4-SAGE-fls was washed as above, pelleted (17000g, 5 min, 20 °C) and resuspended in 100 μL of PI buffer, and 10 μL was added to 30 μL of PI buffer. As a control, 40 μL of unmineralized fluorescent SAGE was used. Samples were made up to a total volume of 50 μL by PI buffer (control, no additive) or PI buffer plus a fluorescence quenching molecule (10 μL 0.4% (w/v) Trypan blue, 2 h at 20 °C), a reducing agent (10 μL of TCEP, 2 h at 20 °C), or a protease (trypsin, 5 μL 1 mg mL–1, 2 h at 37 °C). Samples were transferred to slides and imaged on a fluorescence microscope (detailed above).
Acknowledgments
J.M.G., L.R.H., J.M.M., W.-F.X., P.V., and D.N.W. are supported by the BBSRC (BB/M002969/1). P.C. and L.S. are supported by the European Research Council (282101). D.N.W. holds a Royal Society Wolfson Research Merit Award (WM140008). P.C., P.V., T.B.L., M.M., and D.N.W. are members of BrisSynBio, a BBSRC/EPSRC funded Synthetic Biology Research Centre (BB/L01386X/1). PF-AFM was carried out in the Chemical Imaging Facility, University of Bristol, with equipment funded by the EPSRC (EP/K035746/1). Mass spectra were recorded using the EPSRC-funded equipment (EP/K03927X/1), School of Chemistry Mass Spectrometry Facility, University of Bristol. The authors would like to thank, at the University of Bristol, Jon A. Jones for recording EDX spectra and the associated TEM images, Sarah Ratcliffe for determining the concentration of the sodium silicate stock using ICP-AES, and James Ross for help with PyMOL images.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b07785.
Extended methods, supplementary notes, figures and tables that include analytical RP-HPLC, MALDI-TOF, AUC data, CD data, PDMPO fluorescence spectra, TEM images, grain sizing, SEM images, AFM plots, CLEM images, bright-field and fluorescence LM images, and diagrams (PDF)
Tomograms and reconstruction movies:
Tilt series 675 29,000× magnification, S1 29kx tomo.avi (AVI)
50 000× magnification, S2 50kx tomo.avi (AVI)
3D reconstructions of the 50 000× magnification series rotating equatorially, S3 50kx 3D1.avi (AVI)
Polar, S4 50kx 3D2.avi (AVI)
Author Contributions
J.M.G., J.M.F., P.C., and D.N.W. conceived the project, designed the experiments, and drafted the manuscript. J.M.G. and L.S. performed the mineralization experiments, and J.M.G. recorded TEM and SEM images and reconstructed tomograms with J.M.M., J.C., and L.R.H. J.M.G. performed grain sizing. J.M.G. and J.L.B. synthesized the peptides and conducted the solution-phase biophysics and fluorescence microscopy. J.M.G., J.L.B., and L.R.H. recorded fluorescence and TEM images for CLEM with P.V. and J.C. J.M.G. and R.L.H. recorded the AFM data and analyzed them with W.-F.X. J.M.G. and G.G.R. recorded and analyzed the AUC data. M.M., N.L., and T.B.L. performed the coarse-grained computational modeling. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Fletcher J. M.; Harniman R. L.; Barnes F. R. H.; Boyle A. L.; Collins A.; Mantell J.; Sharp T. H.; Antognozzi M.; Booth P. J.; Linden N.; Miles M. J.; Sessions R. B.; Verkade P.; Woolfson D. N. Self-Assembling Cages from Coiled-Coil Peptide Modules. Science 2013, 340, 595–599. 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya M. J.; Spooner G. M.; Sunde M.; Thorpe J. R.; Rodger A.; Woolfson D. N. Sticky-End Assembly of a Designed Peptide Fiber Provides Insight into Protein Fibrillogenesis. Biochemistry 2000, 39, 8728–8734. 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]
- Wen A. M.; Steinmetz N. F. Design of Virus-Based Nanomaterials for Medicine, Biotechnology, and Energy. Chem. Soc. Rev. 2016, 45, 4074–4126. 10.1039/C5CS00287G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz B.; Douglas T. Development of Virus-like Particles for Diagnostic and Prophylactic Biomedical Applications. Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2015, 7, 722–735. 10.1002/wnan.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh D.; Ross A. H.; Hale A. H.; Baltimore D.; Eisen H. N. Synthetic Phospholipid Vesicles Containing a Purified Viral Antigen and Cell Membrane Proteins Stimulate the Development of Cytotoxic T Lymphocytes. J. Exp. Med. 1979, 150, 1067–1074. 10.1084/jem.150.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad Z.; Shah A.; Siddiq M.; Kraatz H.-B. Polymeric Micelles as Drug Delivery Vehicles. RSC Adv. 2014, 4, 17028–17038. 10.1039/C3RA47370H. [DOI] [Google Scholar]
- Veneziano R.; Ratanalert S.; Zhang K.; Zhang F.; Yan H.; Chiu W.; Bathe M. Designer Nanoscale DNA Assemblies Programmed from the Top down. Science 2016, 352, 1534. 10.1126/science.aaf4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methods in Cell Biology. Vol. 88: Introduction to Electron Microscopy for Biologists, 1st ed.; Allen T. D., Ed.; Elsevier: Burlington, MA, USA, 2008. [Google Scholar]
- Mann S.Biomineralization: Principals and Concepts in Bioinorganic Materials Chemistry; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Rao A.; Cölfen H. On the Biophysical Regulation of Mineral Growth: Standing out from the Crowd. J. Struct. Biol. 2016, 196, 232–243. 10.1016/j.jsb.2016.03.021. [DOI] [PubMed] [Google Scholar]
- Metzler R. A.; Kim I. W.; Delak K.; Evans J. S.; Zhou D.; Beniash E.; Wilt F.; Abrecht M.; Chiou J.-W.; Guo J.; Coppersmith S. N.; Gilbert P. U. P. A. Probing the Organic-Mineral Interface at the Molecular Level in Model Biominerals. Langmuir 2008, 24, 2680–2687. 10.1021/la7031237. [DOI] [PubMed] [Google Scholar]
- Meldrum F. C.; Cölfen H. Controlling Mineral Morphologies and Structures in Biological and Synthetic Systems. Chem. Rev. 2008, 108, 4332–4432. 10.1021/cr8002856. [DOI] [PubMed] [Google Scholar]
- He G.; Dahl T.; Veis A.; George A. Nucleation of Apatite Crystals in Vitro by Self-Assembled Dentin Matrix Protein 1. Nat. Mater. 2003, 2, 552–558. 10.1038/nmat945. [DOI] [PubMed] [Google Scholar]
- Reid D. G.; Duer M. J.; Murray R. C.; Wise E. R. The Organic-Mineral Interface in Teeth Is like That in Bone and Dominated by Polysaccharides: Universal Mediators of Normal Calcium Phosphate Biomineralization in Vertebrates?. Chem. Mater. 2008, 20, 3549–3550. 10.1021/cm800514u. [DOI] [Google Scholar]
- Raschdorf O.; Forstner Y.; Kolinko I.; Uebe R.; Plitzko J. M.; Schüler D. Genetic and Ultrastructural Analysis Reveals the Key Players and Initial Steps of Bacterial Magnetosome Membrane Biogenesis. PLoS Genet. 2016, 12, e1006101. 10.1371/journal.pgen.1006101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand M. Diatoms, Biomineralization Processes, and Genomics. Chem. Rev. 2008, 108, 4855–4874. 10.1021/cr078253z. [DOI] [PubMed] [Google Scholar]
- Jeffryes C.; Agathos S. N.; Rorrer G. Biogenic Nanomaterials from Photosynthetic Microorganisms. Curr. Opin. Biotechnol. 2015, 33, 23–31. 10.1016/j.copbio.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Patwardhan S. V. Biomimetic and Bioinspired Silica: Recent Developments and Applications. Chem. Commun. 2011, 47, 7567–7582. 10.1039/c0cc05648k. [DOI] [PubMed] [Google Scholar]
- Kharlampieva E.; Tsukruk T.; Slocik J. M.; Ko H.; Poulsen N.; Naik R. R.; Kröger N.; Tsukruk V. V. Bioenabled Surface-Mediated Growth of Titania Nanoparticles. Adv. Mater. 2008, 20, 3274–3279. 10.1002/adma.200800054. [DOI] [Google Scholar]
- Kröger N.; Deutzmann R.; Bergsdorf C.; Sumper M. Species-Specific Polyamines from Diatoms Control Silica Morphology. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 14133–14138. 10.1073/pnas.260496497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröger N.; Lorenz S.; Brunner E.; Sumper M. Self-Assembly of Highly Phosphorylated Silaffins and Their Function in Biosilica Morphogenesis. Science 2002, 298, 584–586. 10.1126/science.1076221. [DOI] [PubMed] [Google Scholar]
- Kröger N.; Deutzmann R.; Sumper M. Polycationic Peptides from Diatom Biosilica That Direct Silica Nanosphere Formation. Science 1999, 286, 1129–1132. 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]
- Scheffel A.; Poulsen N.; Shian S.; Kroger N. Nanopatterned Protein Microrings from a Diatom That Direct Silica Morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3175–3180. 10.1073/pnas.1012842108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richthammer P.; Börmel M.; Brunner E.; van Pée K.-H. Biomineralization in Diatoms: The Role of Silacidins. ChemBioChem 2011, 12, 1362–1366. 10.1002/cbic.201000775. [DOI] [PubMed] [Google Scholar]
- Cha J. N.; Stucky G. D.; Morse D. E.; Deming T. J. Biomimetic Synthesis of Ordered Silica Structures Mediated by Block Copolypeptides. Nature 2000, 403, 289–292. 10.1038/35002038. [DOI] [PubMed] [Google Scholar]
- Belton D. J.; Patwardhan S. V.; Annenkov V. V.; Danilovtseva E. N.; Perry C. C. From Biosilicification to Tailored Materials: Optimizing Hydrophobic Domains and Resistance to Protonation of Polyamines. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5963–5968. 10.1073/pnas.0710809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan S. V.; Clarson S. J. Silicification and Biosilicification. Part 4. Effect of Template Size on the Formation of Silica. J. Inorg. Organomet. Polym. 2002, 12, 109–116. 10.1023/A:1021257713504. [DOI] [Google Scholar]
- Niu L.; Jiao K.; Qi Y.; Yiu C. K. Y.; Ryou H.; Arola D. D.; Chen J.; Breschi L.; Pashley D. H.; Tay F. R. Infiltration of Silica inside Fibrillar Collagen. Angew. Chem., Int. Ed. 2011, 50, 11688–11691. 10.1002/anie.201105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmström S. C.; King P. J. S.; Ryadnov M. G.; Butler M. F.; Mann S.; Woolfson D. N. Templating Silica Nanostructures on Rationally Designed Self-Assembled Peptide Fibers. Langmuir 2008, 24, 11778–11783. 10.1021/la802009t. [DOI] [PubMed] [Google Scholar]
- Liu B.; Cao Y.; Huang Z.; Duan Y.; Che S. Silica Biomineralization via the Self-Assembly of Helical Biomolecules. Adv. Mater. 2015, 27, 479–497. 10.1002/adma.201401485. [DOI] [PubMed] [Google Scholar]
- Qiao Y.; Polzer F.; Kirmse H.; Kirstein S.; Rabe J. P. Nanohybrids from Nanotubular J-Aggregates and Transparent Silica Nanoshells. Chem. Commun. 2015, 51, 11980–11982. 10.1039/C5CC00901D. [DOI] [PubMed] [Google Scholar]
- Kaehr B.; Townson J. L.; Kalinich R. M.; Awad Y. H.; Swartzentruber B. S.; Dunphy D. R.; Brinker C. J. Cellular Complexity Captured in Durable Silica Biocomposites. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17336–17341. 10.1073/pnas.1205816109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Y.-R.; Kanninen L.; Kaehr B.; Townson J. L.; Niklander J.; Harjumäki R.; Brinker C. J.; Yliperttula M. Silica Bioreplication Preserves Three-Dimensional Spheroid Structures of Human Pluripotent Stem Cells and HepG2 Cells. Sci. Rep. 2015, 5, 13635 10.1038/srep13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townson J. L.; Lin Y.-S.; Chou S. S.; Awad Y. H.; Coker E. N.; Brinker C. J.; Kaehr B. Synthetic Fossilization of Soft Biological Tissues and Their Shape-Preserving Transformation into Silica or Electron-Conductive Replicas. Nat. Commun. 2014, 5, 5665. 10.1038/ncomms6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Wang H.-J.; Zhou H.; Nian Q.-G.; Song Z.; Deng Y.-Q.; Wang X.; Zhu S.-Y.; Li X.-F.; Qin C.-F.; Tang R. Hydrated Silica Exterior Produced by Biomimetic Silicification Confers Viral Vaccine Heat-Resistance. ACS Nano 2015, 9, 799–808. 10.1021/nn5063276. [DOI] [PubMed] [Google Scholar]
- Wang G.; Cao R.-Y.; Chen R.; Mo L.; Han J.-F.; Wang X.; Xu X.; Jiang T.; Deng Y.-Q.; Lyu K.; Zhu S.-Y.; Qin E.-D.; Tang R.; Qin C.-F. Rational Design of Thermostable Vaccines by Engineered Peptide-Induced Virus Self-Biomineralization under Physiological Conditions. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7619–7624. 10.1073/pnas.1300233110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase N. R.; Shian S.; Sandhage K. H.; Kröger N. Biocatalytic Nanoscale Coatings through Biomimetic Layer-by-Layer Mineralization. Adv. Funct. Mater. 2011, 21, 4243–4251. 10.1002/adfm.201101202. [DOI] [Google Scholar]
- Begum G.; Goodwin W. B.; DeGlee B. M.; Sandhage K.; Kroeger N. Compartmentalisation of Enzymes for Cascade Reactions through Biomimetic Layer-by-Layer Mineralization. J. Mater. Chem. B 2015, 3, 5232–5240. 10.1039/C5TB00333D. [DOI] [PubMed] [Google Scholar]
- Sanchez-Sanchez L.; Tapia-Moreno A.; Juarez-Moreno K.; Patterson D.; Cadena-Nava R.; Douglas T.; Vazquez-Duhalt R. Design of a VLP-Nanovehicle for CYP450 Enzymatic Activity Delivery. J. Nanobiotechnol. 2015, 13, 66. 10.1186/s12951-015-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qazi S.; Miettinen H. M.; Wilkinson R. A.; McCoy K.; Douglas T.; Wiedenheft B. Programmed Self-Assembly of an Active P22-Cas9 Nanocarrier System. Mol. Pharmaceutics 2016, 13, 1191–1196. 10.1021/acs.molpharmaceut.5b00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P.; O’Neil A.; Lin E.; Douglas T.; Holford M. Tailored Delivery of Analgesic Ziconotide across a Blood Brain Barrier Model Using Viral Nanocontainers. Sci. Rep. 2015, 5, 12497 10.1038/srep12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M.; Mirshekari H.; Moosavi Basri S. M.; Bahrami S.; Moghoofei M.; Hamblin M. R. Bacteriophages and Phage-Inspired Nanocarriers for Targeted Delivery of Therapeutic Cargos. Adv. Drug Delivery Rev. 2016, 106, 45–62. 10.1016/j.addr.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frietze K. M.; Peabody D. S.; Chackerian B. Engineering Virus-like Particles as Vaccine Platforms. Curr. Opin. Virol. 2016, 18, 44–49. 10.1016/j.coviro.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D. P.; Prevelige P. E.; Douglas T. Nanoreactors by Programmed Enzyme Encapsulation inside the Capsid of the Bacteriophage P22. ACS Nano 2012, 6, 5000–5009. 10.1021/nn300545z. [DOI] [PubMed] [Google Scholar]
- Patterson D. P.; Schwarz B.; Waters R. S.; Gedeon T.; Douglas T. Encapsulation of an Enzyme Cascade within the Bacteriophage P22 Virus-like Particle. ACS Chem. Biol. 2014, 9, 359–365. 10.1021/cb4006529. [DOI] [PubMed] [Google Scholar]
- Ross J. F.; Bridges A.; Fletcher J. M.; Shoemark D.; Alibhai D.; Bray H. E. V; Beesley J. L.; Dawson W. M.; Hodgson L. R.; Mantell J.; Verkade P.; Edge C. M.; Sessions R. B.; Tew D.; Woolfson D. N. Decorating Self-Assembled Peptide Cages with Proteins. ACS Nano 2017, 11, 7901–7914. 10.1021/acsnano.7b02368. [DOI] [PubMed] [Google Scholar]
- Fletcher J. M.; Boyle A. L.; Bruning M.; Bartlett G. J.; Vincent T. L.; Zaccai N. R.; Armstrong C. T.; Bromley E. H. C.; Booth P. J.; Brady R. L.; Thomson A. R.; Woolfson D. N. A Basis Set of de Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology. ACS Synth. Biol. 2012, 1, 240–250. 10.1021/sb300028q. [DOI] [PubMed] [Google Scholar]
- Richardson D. S.Euler’s Gem: The Polyhedron Formula and the Birth of Topology; Princeton Univeristy Press: Princeton, NJ, USA, 2008. [Google Scholar]
- Kroto H. W.; Heath J. R.; O’Brien S. C.; Curl R. F.; Smalley R. E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. 10.1038/318162a0. [DOI] [Google Scholar]
- Mosayebi M.; Shoemark D. K.; Fletcher J. M.; Sessions R. B.; Linden N.; Woolfson D. N.; Liverpool T. B. Beyond Icosahedral Symmetry in Packings of Proteins in Spherical Shells. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9014–9019. 10.1073/pnas.1706825114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Sun C.; Li P.; Wu T.; Zhou H.; Yang D.; Liu Y.; Ma X.; Song Z.; Nian Q.; Feng L.; Qin C.; Chen L.; Tang R. Vaccine Engineering with Dual-Functional Mineral Shell: A Promising Strategy to Overcome Preexisting Immunity. Adv. Mater. 2016, 28, 694–700. 10.1002/adma.201503740. [DOI] [PubMed] [Google Scholar]
- Lear S.; Cobb S. L. Pep-Calc.com: A Set of Web Utilities for the Calculation of Peptide and Peptoid Properties and Automatic Mass Spectral Peak Assignment. J. Comput.-Aided Mol. Des. 2016, 30, 271–277. 10.1007/s10822-016-9902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker E. G.; Bartlett G. J.; Crump M. P.; Sessions R. B.; Linden N.; Faul C. F. J.; Woolfson D. N. Local and Macroscopic Electrostatic Interactions in Single α-Helices. Nat. Chem. Biol. 2015, 11, 221–228. 10.1038/nchembio.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton D. J.; Deschaume O.; Perry C. C. An Overview of the Fundamentals of the Chemistry of Silica with Relevance to Biosilicification and Technological Advances. FEBS J. 2012, 279, 1710–1720. 10.1111/j.1742-4658.2012.08531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaychuk P. A. The Revised Pourbaix Diagram for Silicon. Silicon 2014, 6, 109–116. 10.1007/s12633-013-9172-0. [DOI] [Google Scholar]
- Delalat B.; Sheppard V. C.; Rasi Ghaemi S.; Rao S.; Prestidge C. A.; McPhee G.; Rogers M.-L.; Donoghue J. F.; Pillay V.; Johns T. G.; Kroger N.; Voelcker N. H. Targeted Drug Delivery Using Genetically Engineered Diatom Biosilica. Nat. Commun. 2015, 6, 8791. 10.1038/ncomms9791. [DOI] [PubMed] [Google Scholar]
- de Boer P.; Hoogenboom J. P.; Giepmans B. N. G. Correlated Light and Electron Microscopy: Ultrastructure Lights Up!. Nat. Methods 2015, 12, 503–513. 10.1038/nmeth.3400. [DOI] [PubMed] [Google Scholar]
- Müller-Reichert T.; Verkade P.. Correlative Light and Electron Microscopy; Müller-Reichert T.; Verkade P., Eds.; Oxford Academic Press: Oxford, UK, 2012. [Google Scholar]
- Loike J. D.; Silverstein S. C. A Fluorescence Quenching Technique Using Trypan Blue to Differentiate between Attached and Ingested Glutaraldehyde-Fixed Red Blood Cells in Phagocytosing Murine Macrophages. J. Immunol. Methods 1983, 57, 373–379. 10.1016/0022-1759(83)90097-2. [DOI] [PubMed] [Google Scholar]
- Sahlin S.; Hed J.; Runfquist I. Differentiation between Attached and Ingested Immune Complexes by a Fluorescence Quenching Cytofluorometric Assay. J. Immunol. Methods 1983, 60, 115–124. 10.1016/0022-1759(83)90340-X. [DOI] [PubMed] [Google Scholar]
- Mura S.; Nicolas J.; Couvreur P. Stimuli-Responsive Nanocarriers for Drug Delivery. Nat. Mater. 2013, 12, 991–1003. 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- Rother M.; Nussbaumer M. G.; Renggli K.; Bruns N. Protein Cages and Synthetic Polymers: A Fruitful Symbiosis for Drug Delivery Applications, Bionanotechnology and Materials Science. Chem. Soc. Rev. 2016, 45, 6213–6249. 10.1039/C6CS00177G. [DOI] [PubMed] [Google Scholar]
- Zhang X. F.; Mansouri S.; Clime L.; Ly H. Q.; Yahia L. H; Veres T. Fe3O4-Silica Core-Shell Nanoporous Particles for High-Capacity pH-Triggered Drug Delivery. J. Mater. Chem. 2012, 22, 14450–14457. 10.1039/c2jm31749d. [DOI] [Google Scholar]
- Wang X.; Schröder H. C.; Müller W. E. G. Enzyme-Based Biosilica and Biocalcite: Biomaterials for the Future in Regenerative Medicine. Trends Biotechnol. 2014, 32, 441–447. 10.1016/j.tibtech.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Galloway J. M.; Staniland S. S. Protein and Peptide Biotemplated Metal and Metal Oxide Nanoparticles and Their Patterning onto Surfaces. J. Mater. Chem. 2012, 22, 12423–12434. 10.1039/c2jm31620j. [DOI] [Google Scholar]
- Galloway J. M.; Bramble J. P.; Staniland S. S. Biomimetic Synthesis of Materials for Technology. Chem. - Eur. J. 2013, 19, 8710–8725. 10.1002/chem.201300721. [DOI] [PubMed] [Google Scholar]
- Oh M. H.; Yu J. H.; Kim I.; Nam Y. S. Genetically Programmed Clusters of Gold Nanoparticles for Cancer Cell-Targeted Photothermal Therapy. ACS Appl. Mater. Interfaces 2015, 7, 22578–22586. 10.1021/acsami.5b07029. [DOI] [PubMed] [Google Scholar]
- Górzny M. Ł.; Walton A. S.; Evans S. D. Synthesis of High-Surface-Area Platinum Nanotubes Using a Viral Template. Adv. Funct. Mater. 2010, 20, 1295–1300. 10.1002/adfm.200902196. [DOI] [Google Scholar]
- Fields G. B.; Noble R. L. Solid Phase Peptide Synthesis Utilizing 9-Fluorenylmethoxycarbonyl Amino Acids. Int. J. Pept. Protein Res. 1990, 35, 161–214. 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- Ruiz-Gayo M.; Albericio F.; Pons M.; Royo M.; Pedroso E.; Giralt E. Uteroglobin-like Peptide Cavities I. Synthesis of Antiparallel and Parallel Dimers of Bis-Cysteine Peptides. Tetrahedron Lett. 1988, 29, 3845–3848. 10.1016/S0040-4039(00)82130-9. [DOI] [Google Scholar]
- Schuck P. Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys. J. 2000, 78, 1606–1619. 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UTHSCSA. Ultrascan http://ultrascan.uthscsa.edu/ (accessed Feb 22, 2017).
- Hurton T.; Wright A.; Deubler G.; Bashir B. SEDNTERP Daemon Version: 20120828 BETA http://sednterp.unh.edu/ (accessed Feb 22, 2017).
- Alexander G. B. The Preparation of Monosilicic Acid. J. Am. Chem. Soc. 1953, 75, 2887–2888. 10.1021/ja01108a029. [DOI] [Google Scholar]
- Shimizu K.; Del Amo Y.; Brzezinski M. A.; Stucky G. D.; Morse D. E. A Novel Fluorescent Silica Tracer for Biological Silicification Studies. Chem. Biol. 2001, 8, 1051–1060. 10.1016/S1074-5521(01)00072-2. [DOI] [PubMed] [Google Scholar]
- Abramoff M. D.; Magalhaes P. J.; Ram S. J. Image Processing with Image J. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Schindelin J.; Rueden C. T.; Hiner M. C.; Eliceiri K. W. The ImageJ Ecosystem: An Open Platform for Biomedical Image Analysis. Mol. Reprod. Dev. 2015, 82, 518–529. 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J. R.; Mastronarde D. N.; McIntosh J. R. Computer Visualization of Three-Dimensional Image Data Using IMOD. J. Struct. Biol. 1996, 116, 71–76. 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Mastronarde D. N. Dual-Axis Tomography: An Approach with Alignment Methods That Preserve Resolution. J. Struct. Biol. 1997, 120, 343–352. 10.1006/jsbi.1997.3919. [DOI] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J.-Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.