

Abstract
N-methyl-d-aspartate receptors (NMDARs) are an important receptor in the brain and have been implicated in multiple neurological disorders. Many non-selective NMDAR-targeting drugs are poorly tolerated, leading to efforts to target NMDAR subtypes to improve the therapeutic index. We describe here a series of negative allosteric NMDAR modulators with submaximal inhibition at saturating concentrations. Modest changes to the chemical structure interconvert negative and positive modulation. All modulators share the ability to enhance agonist potency and are use-dependent, requiring the binding of both agonists before modulators act with high potency. Data suggest that these modulators, including both enantiomers, bind to the same site on the receptor and share structural determinants of action. Due to the modulator properties, submaximal negative modulators in this series may spare NMDAR at the synapse, while augmenting the response of NMDAR in extrasynaptic spaces. These modulators could serve as useful tools to probe the role of extrasynaptic NMDARs.
Research organism: None
eLife digest
The neurons in the brain form networks that can change in response to experience, causing new connections to form between certain neurons and breaking the connections between others. This remodeling process underlies learning and memory. However, in certain neurological disorders, such as schizophrenia and epilepsy, these networks are disrupted and no longer work correctly.
A receptor protein called the NMDA receptor plays an important role in reshaping the networks of neurons. Chemicals called neurotransmitters that are released by one neuron bind to and activate NMDA receptors on a neighbouring neuron to communicate with it. This activation encourages new connections to form between neurons.
Drugs that alter the activity of the NMDA receptor could potentially act as treatments for neurological conditions that disrupt how the networks of neurons work. However, few have been approved for use in patients because most of the potential drug compounds investigated so far produce severe side effects.
Perszyk et al. have now identified a new group of compounds that can potentially alter the activity of NMDA receptors without fully blocking the response, potentially eliminating the unwanted side effects. The compounds were tested on frog egg cells and human embryonic kidney cells that had been engineered to produce NMDA receptors. Small changes to the chemical structure of these compounds could switch their effect from increasing to decreasing the activity of the receptor. The compounds only interact with active receptors that have a neurotransmitter bound to them, and compete with each other to bind to the receptors.
Perszyk et al. also found that some compounds behaved differently depending on how active the NMDA receptor was. These compounds could potentially be used to sense the activity in a network of neurons, which opens up new options for treating neurological conditions that affect the networks.
Further experiments are now required to see how these compounds affect the activity of NMDA receptors in neurons and living animals. The range of effects produced by the compounds studied by Perszyk et al. suggests that other related compounds may have different effects on receptor activity. Future work could investigate the properties of these compounds to see if they could treat a different set of neurological disorders.
Introduction
N-methyl-d-aspartate receptors (NMDARs) are a subtype of ionotropic glutamate receptors that are broadly expressed in the brain and are important for normal development, neuronal plasticity and memory formation (Traynelis et al., 2010; Paoletti et al., 2013). NMDARs contribute a slow, Ca2+ permeable component to fast excitatory neurotransmission that is voltage dependent by virtue of its sensitivity to pore block by extracellular Mg2+ (Traynelis et al., 2010). The typical NMDAR is comprised of 2 GluN1 and 2 GluN2 subunits, creating the potential for diversity given that there are eight splice variants of GluN1 and 4 independent genes encoding GluN2 subunits (A-D) (Traynelis et al., 2010). The regulation and functional properties of the NMDAR are controlled by the subunits incorporated into the receptor (Monyer et al., 1994; Vicini et al., 1998; Vance et al., 2012). NMDARs are expressed at most excitatory synapses, and are also found in peri- and extrasynaptic locations (Sans et al., 2000; Steigerwald et al., 2000; Groc et al., 2006; Traynelis et al., 2010). Although NMDARs have been studied extensively, there are still important questions about the different roles that NMDARs may play given differences in subunit composition and synaptic localization (Hardingham and Bading, 2010; Gladding and Raymond, 2011; Paoletti et al., 2013). Pharmacological approaches can be useful for probing these questions, but tool compounds to differentiate these NMDAR subtypes have been limited (Ogden and Traynelis, 2011; Monaghan et al., 2012; Santangelo et al., 2012; Strong et al., 2014; Zhu and Paoletti, 2015). Whereas NMDAR receptors have been implicated in many neurological diseases, there remains a dearth of clinically-approved drugs that target NMDARs (Kalia et al., 2008; Traynelis et al., 2010; Paoletti et al., 2013; Strong et al., 2014).
Multiple endogenous and exogenous modulatory sites in the NMDAR recently have been described. In addition, nearly complete NMDAR structures obtained using crystallographic approaches are now available (Karakas and Furukawa, 2014; Lee et al., 2014), and these data, together with recent cryo-EM structures in the receptor super family (Twomey and Sobolevsky, 2018; Twomey et al., 2017a; Twomey et al., 2017b), illustrate the overall topography of the NMDAR and suggest a mechanism for some allosteric modulators, such as GluN2B-selective ifenprodil (Tajima et al., 2016; Zhu et al., 2016). However, a complete understanding of how other modulatory sites operate and can be exploited has been elusive. Advances in the understanding of specific roles of particular NMDAR subunits have led to renewed interest in targeted therapeutic intervention, and recent work has yielded a growing tool box of novel ligands that act on NMDARs with diverse sites and mechanisms (Ogden and Traynelis, 2011; Monaghan et al., 2012; Hackos and Hanson, 2017). To date, there are positive and negative modulators that bind to the amino-terminal domain (ATD), agonist binding domain (ABD), or transmembrane domain (TMD). Each new class of compounds discovered enriches our understanding of the function of NMDAR and how these receptors can be modulated. The information gained from these modulators has the potential to provide insight into both NMDAR function and therapeutic strategies to treat complex neurological diseases.
This study describes a series of compounds that includes both positive allosteric modulators (PAMs) as well as negative allosteric modulators (NAMs) of NMDAR function (Katzman et al., 2015). A set of aliphatic substitutions off an ester linkage to the scaffold interconverts these compounds between positive and negative allosteric modulators. Remarkably, the difference between the positive and negative modulation to the same chemical scaffold was influenced by the addition or removal of individual methyl groups. These modulators appear to bind to a shared site to bring about opposing actions, and share mechanistic features, such as agonist dependence and enhancement of agonist potency. The spectrum of properties in this series of modulators could serve as useful tool compounds for probing the role of NMDARs in circuits in both healthy brain and in neuropathological situations.
Results
Identification of a new class of positive allosteric modulators of NMDAR function
We have previously described the structure-activity relationship (SAR) of a series of negative allosteric pan-NMDAR modulators that contained an amidothiophene core, the most potent possessing a tetrahydrobenzothiophene, with different alkyl and aryl substitutions connected via an ester linkage at the 3-position (Katzman et al., 2015). These compounds inhibited the response to saturating concentration of co-agonists at NMDARs expressed in Xenopus laevis oocyte experiments, typified by EU1794-2 (compound 4 in Katzman et al., 2015). We describe here a new set of closely related analogues that can either potentiate or inhibit responses depending on subtle changes in structure and agonist concentration. As shown in Figure 1 and Table 1, EU1794-4 contained an ethyl ester similar to EU1794-2 and lacked a methyl substituent on the tetrahydrobenzothiophene core. EU1794-4 potently inhibited GluN1/GluN2D with a substantial residual current remaining at saturating concentrations (Figure 1B,C, Table 1). Interestingly, ester substitutions that had a larger calculated functional group volume than the ethyl ester in EU1794-4 potentiated NMDAR responses to saturating concentrations of glutamate and glycine. For example, the isopropyl ester (EU1794-5) potentiated GluN1/GluN2D responses to nearly 200% of control (Figure 1A–C). Restoration of the methyl to the tetrahydrobenzothiophene core restored negative allosteric modulation (EU1794-19, Figure 1B,C). Thus, the direction of modulation (positive or negative) could be determined by the size of the alkyl ester and substitution to the tetrahydrobenzothiophene core.
Figure 1. The EU1794 series of NMDAR NAMs can be converted to PAMs with subtle structural modifications.

(A) Representative concentration-response experiments for a negative (EU1794-2, above) and positive (EU1794-5, below) allosteric modulator using two-electrode voltage-clamp recordings of GluN1/GluN2D NMDARs activated by saturating concentrations of agonist. 100/30 µM glutamate/glycine application is represented by the grey bar and increasing concentrations of modulator are shown by grey scale boxes (0.3, 1, 3, 10, 30 µM for EU1794-2 and 1, 3, 10, 30, 100 µM for EU1794-5). (B) Chemical structures of the NAMs EU1794-2, EU1794-4, EU1794-19 and PAM EU1794-5 are given, which show all permutations of two methyl substitutions. This series illustrates how subtle changes in chemical structure interconvert modulator activity. (C) The concentration-response curves for the compounds shown in (B) acting on GluN1/GluN2D NMDARs activated by maximally effective concentrations of co-agonists and fitted by the Hill equation. (D) Chemical structures of EU1794 PAMs. (E) Concentration-response curves for EU1794 PAMs on the responses of GluN1/GluN2D NMDARs to maximally effective concentrations of co-agonists. See Table 1 for fitted EC50 values at all diheteromeric NMDARs. Data represent 4–18 oocytes recorded in at least two independent experiments.
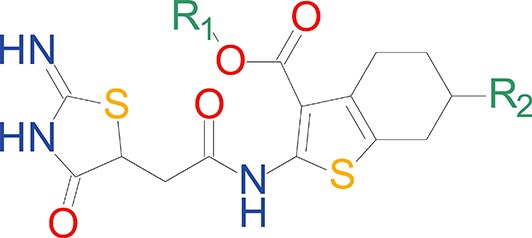
Table 1. The effect of alkyl ester and tetrahydrobenzothiophene ring substitutions on the potency and efficacy of EU1794 analogues.
 |
EC50 (µM) [conf. int.]* Maximal Degree of Modulation (% of control)† |
||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R1 volume (Å3) | R2 | GluN2A | GluN2B | GluN2C | GluN2D |
| EU1794-4 | Et | 45.15 | H | 2.2 [1.8, 2.8] 32 ± 3% |
2.6 [1.4, 4.8] 67 ± 4% |
0.42 [0.28, 0.61] 52 ± 2% |
0.36 [0.29, 0.45] 51 ± 3% |
| EU1794-2 | Et | 45.15 | Me | 0.60 [0.44, 0.82] 6 ± 2% |
1.2 [0.8, 1.9] 10 ± 3% |
0.26 [0.21, 0.31] 14 ± 1% |
0.20 [0.17, 0.25] 14 ± 1% |
| EU1794-5 | iPr | 62.16 | H | 18 [9.0, 36] 36 ± 5% |
2.5 [1.4, 4.4] 71 ± 4% |
4.0 [3.5, 4.6] 140 ± 3% |
9.3 [8.1, 11] 169 ± 7% |
| EU1794-19 | iPr | 62.16 | Me | 1.1 [0.5, 2.3] 19 ± 6% |
1.1 [0.9, 1.3] 43 ± 2% |
0.74 [0.57, 0.96] 32 ± 3% |
0.43 [0.32, 0.59] 46 ± 2% |
| EU1794-27 | tBu | 79.23 | H | 7.4 [5.3, 10] 52 ± 7% |
1.4 [1.2, 1.7] 130 ± 3% |
2.8 [2.5, 3.2] 230 ± 10% |
2.4 [1.8, 3.3] 250 ± 8% |
| EU1794-25 | Bn | 98.87 | H | - | 1.0 [0.9, 1.2] 220 ± 13% |
0.77 [0.56, 1.0] 180 ± 12% |
1.0 [0.95, 1.1] 250 ± 19% |
| EU1794-29 | n-Bu | 79.12 | H | 3.7 [1.7, 8.0] 59 ± 5% |
1.4 [1.1, 1.8] 140 ± 3% |
3.8 [2.6, 5.7] 130 ± 7% |
5.7 [5.1, 6.4] 166 ± 8% |
*EC50 values for potentiation of responses to saturating glutamate and glycine (100 μM, 30 μM) were obtained by least-squares fitting of data from individual experiments by the Hill equation. EC50 values are given as the mean with the 95% confidence interval determined from log(EC50).
†The maximal degree of modulation is given as mean ±SEM. For all compounds, data are from 4 to 18 oocytes recorded in at least two independent experiments. Data were not fit (shown as -) if the response recorded at 30 µM of test compound did not differ by more than 15% from control.
The size of the ester-linked substituent controlled the extent and potency of positive modulation. The t-butyl ester EU1794-27 potentiated GluN1/GluN2D NMDARs with greater efficacy and higher potency, increasing responses to maximally effective glutamate and glycine to 250% of control with an EC50 value of 2.4 µM (Figure 1D,E). The benzyl ester (EU1794-25) also strongly potentiated responses with a potency similar to EU1794-27 (Figure 1D,E). However, EU1794-29, which had a longer substitution, n-butyl, reduced both the maximal potentiation and potency, suggesting an optimal substituent size and shape (Figure 1D,E).
Allosteric modulation of agonist potency by NAMs and PAMs
The tetrahydrobenzothiophene-containing NAMs reported here inhibit all diheteromeric NMDARs without substantial subunit selectivity, similar to those that we previously described (Katzman et al., 2015). By contrast, the novel PAMs described here show distinct GluN2 dependence (Table 1). All PAMs are active at GluN1/GluN2C and GluN1/GluN2D, but do not enhance the response of GluN1/GluN2A to maximally effective concentrations of agonist, in some cases resulted in inhibition of these responses. Most PAMs were also capable of potentiating GluN1/GluN2B (Table 1, Figure 2A). As seen with other series of NMDAR PAMs (Malayev et al., 2002; Horak et al., 2004; Horak et al., 2006; Hackos et al., 2016; Wang et al., 2017), allosteric modulation can be dependent on agonist concentrations. Thus, we assessed the ability of EU1794-27 to modulate NMDAR responses activated by sub-saturating co-agonist concentrations, clearly exemplified by modulation of GluN1/GluN2C (Figure 2B, Table 2). In these conditions, EU1794-27 positively modulated all NMDAR subtypes and enhanced previously potentiated subtypes to a greater extent than in saturating agonist (compare Figure 2A and C). We hypothesized that this agonist-dependence was due to EU1794-27 altering the agonist potency. Therefore, we determined the glutamate and glycine EC50 values in the absence and presence of EU1794-27 (Figure 3A,B, Table 3, Figure 3—figure supplement 1). EU1794-27 produced modest but significant decreases (higher potency) in the EC50 values for both glutamate and glycine at all NMDARs (Table 3, Figure 3—figure supplement 1). Given this effect by the PAMs in this series, we subsequently considered whether the NAMs in the series shared this mechanism. We selected EU1794-4 for use in evaluating actions on agonist potency since it retains a large steady-state current even for receptors that have bound EU1794-4 (saturated inhibition is between 40–70% of control, Figure 2D). We co-applied EU1794-4 during responses stimulated by sub-saturating concentrations of agonist (glutamate and glycine concentrations that resulted approximately in a EC20 response), which resulted in positive modulation (Figure 2E,F, Table 2). Furthermore, the negative modulator EU1794-4 enhanced the glutamate and glycine potencies (Figure 3C,D, Table 4, Figure 3—figure supplement 1). Interestingly, positive modulation elicited by 10 µM EU1794-4 is dependent on the sub-saturating agonist concentrations used (Table 5). EU1794-4 inhibited GluN1/GluN2C responses to saturating concentrations of agonist to 54% of control, and positively modulated equivalent EC30 responses (equal effective concentrations of glutamate and glycine concentrations that when applied resulted in a 30% of a maximal response) to 140% of control, and positively modulated average equivalent EC3 responses by 660% (Table 5). We did not observe augmentation of sub-saturating agonist responses by EU1794-2, most likely due to its greater extent of inhibition (Figure 2—figure supplement 1). However, the degree of inhibition produced by EU1794-2 was reduced on sub-saturating responses at GluN1/GluN2C from 16% to 45% of control (Table 5).
Figure 2. Modulation of both EU1794 PAMs and NAMs is dependent on agonist concentration.
(A) The concentration-response relationship for EU1794-27 at diheteromeric NMDARs activated by 100/30 µM glutamate/glycine (data for GluN1/GluN2D shown in Figure 1E is included here for clarity). GluN1/GluN2C and GluN1/GluN2D (which are superimposed) are potentiated to a greater extent than GluN1/GluN2B; there appears to be slight inhibition of GluN1/GluN2A. (B) A representative concentration-response recording of EU1794-27 at GluN1/GluN2B activated by sub-saturating concentrations of agonist. The initial response was activated using saturating 100/30 µM glutamate/glycine (black box) followed by 1/0.3 µM glutamate/glycine (grey box, roughly resulting in an activity state that was 20% of the maximal response) administered in the absence and presence of increasing concentrations of EU1794-27 (0.3, 1, 3, 10, 30 µM). (C) Concentration-response relationship for EU1794-27 at diheteromeric NMDARs activated by sub-saturating concentration of agonist (glutamate/glycine concentrations used for GluN1/GluN2A were 2/0.6 µM, GluN1/GluN2B and GluN1/GluN2C were 1/0.3 µM, and GluN1/GluN2D were 0.6/0.2 µM). Note potentiation of all subunits. (D–F) Similar experiments as in A-C, but using the NAM EU1794-4 at saturating (D) or sub-saturating (E,F) concentrations of agonist (glutamate/glycine concentrations used for GluN1/GluN2A, GluN1/GluN2B and GluN1/GluN2C were 1/0.3 µM, and GluN1/GluN2D were 0.3/0.09 µM); data for GluN1/GluN2D presented in Figure 1C are included here for comparison. Note the inhibition by the NAM at saturating agonist concentration (D) compared to potentiation in submaximal concentrations of agonist in (E,F). Data represent 4–13 oocytes recorded in two independent experiments.

Figure 2—figure supplement 1. EU1794-2 actions possess modest agonist concentration-dependence.

Table 2. Effect of EU1794-4 and EU1794-27 at sub-saturated NMDAR responses.
| EC50 (µM) [conf. int.]* Maximal Degree of Modulation (% of control)† |
||||
|---|---|---|---|---|
| GluN2A | GluN2B | GluN2C | GluN2D | |
| EU1794-4 | 3.9 [0.78, 20] 500 ± 32% |
4.3 [1.3, 14] 210 ± 6.8% |
15 [6.5, 35] 220 ± 20% |
14 [6.5, 29] 540 ± 190% |
| EU1794-27 | 8.1 [5.7, 12] 340 ± 32% |
6.3 [5.0, 8.0] 400 ± 50% |
5.0 [3.7, 6.6] 680 ± 50% |
8.3 [5.3, 13] 580 ± 86% |
*EC50 values of modulator action on responses to sub-saturating glutamate/glycine concentrations were obtained by least-squares fitting of data from individual experiments by the Hill equation. For EU1794-4 modulation of GluN1/GluN2A, GluN1/GluN2B and GluN1/GluN2C, glutamate/glycine concentrations were 1/0.3 µM; for GluN1/GluN2D glutamate/glycine were 0.3/0.09 µM. For EU1794-27 potentiation, glutamate/glycine concentrations were 2/0.6 µM (GluN1/GluN2A), 1/0.3 µM (GluN1/GluN2B, GluN1/GluN2C), and 0.6/0.2 µM (GluN1/GluN2D). EC50 for potentiation values are given as the mean with the 95% confidence interval determined from log(EC50).
†The extent of modulation is given as a percent of the control response in the absence of test compound. The maximal degree of modulation is given as mean ±SEM. For all compounds, data are from 4 to 13 oocytes recorded in two independent experiments.
Figure 3. EU1794 PAMs and NAMs enhance glutamate and glycine potency.
(A) Representative glutamate (left) and glycine (right) concentration-response curves of oocytes expressing GluN1/GluN2C with and without EU1794-27 (10 µM, grey box). The glutamate concentration-response curve (0.1, 0.3, 1, 3, 10, 30 µM) was generated with 30 µM glycine and the glycine concentration-response curve (0.03, 0.1, 0.3, 1, 3, 10 µM) was recorded in 100 µM glutamate. (B) Mean concentration-response data for glutamate (left) and glycine (right) at GluN1/GluN2C (see Figure 2—figure supplement 1 for other subunits). Each curve was normalized to the maximal response in the absence of EU1794-27 to illustrate the actions of EU1794-27 on agonist EC50. (C,D) A similar set of experiments as in (A,B), but with EU1794-4 are shown (10 µM, grey box). Note the variation of the order of EU1794-4 application in experimental traces, which was selected randomly. The glutamate concentration-response curve (glutamate concentration are 0.03, 0.1, 0.3, 1, 3, and 100 µM and glycine concentrations are 10 and 30 µM) and the glycine concentration-response curve (glycine concentrations are 0.01, 0.03, 0.1, 0.3, 1, 30 µM, and glutamate concentration are 30 and 100 µM). Each curve was normalized to the maximal response from each oocyte in the absence of EU1794-4. Data are from 4 to 12 paired oocytes recordings from at least two independent experiments.
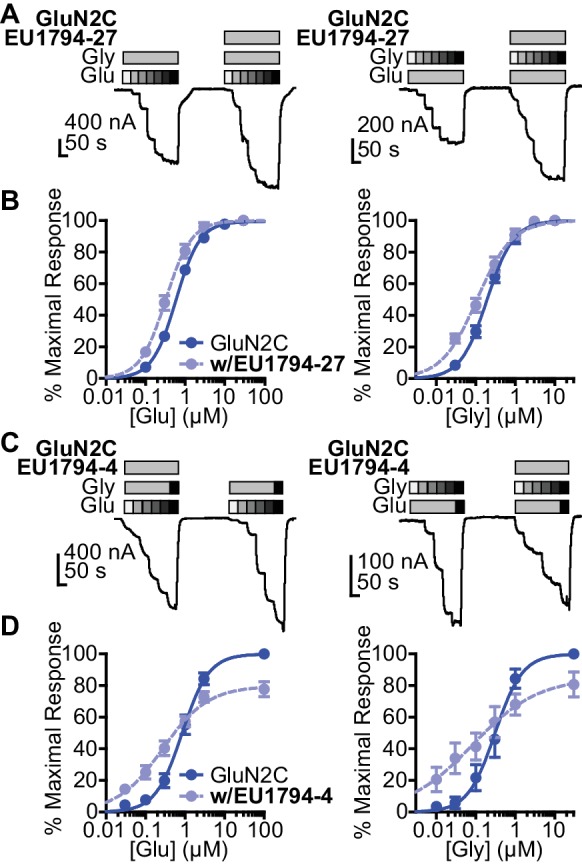
Figure 3—figure supplement 1. The effects of EU1794-27 and EU1794-4 on agonist potency for all diheteromeric NMDARs.

Table 3. EU1794-27 effects on glutamate and glycine EC50 values.
| EC50 (µM) [conf. int.]* | ||||||
|---|---|---|---|---|---|---|
| Control Glutamate | EU1794-27 Glutamate |
Fold Difference |
Control Glycine |
EU1794-27 Glycine |
Fold Difference |
|
| GluN2A | 2.9 [2.2, 4.0] |
1.1 [0.88, 2.1] |
2.3† | 0.57 [0.34, 0.96] |
0.38 [0.23, 0.64] |
1.6 |
| GluN2B | 1.2 [1.1, 1.3] |
0.65 [0.49, 0.85] |
2.0† | 0.28 [0.22, 0.36] |
0.14 [0.08, 0.24] |
2.9† |
| GluN2C | 0.67 [0.61, 0.73] |
0.22 [0.16, 0.31] |
3.3† | 0.23 [0.13, 0.40] |
0.10 [0.07, 0.15] |
2.4 |
| GluN2D | 0.21 [0.17,0.27] |
0.054 [0.028, 0.11] |
5.3† | 0.086 [0.077, 0.095] |
0.020 [0.013, 0.029] |
4.9† |
*Glutamate EC50 values (in the presence of 30 µM glycine) and glycine EC50 values (in the presence of 100 µM glutamate) were obtained by least-squares fitting of data from independent oocyte recordings by the Hill equation. EC50 values are given as the mean with the 95% confidence interval determined from log(EC50). Data in the absence and presence of EU1794-27 were obtained from the same oocyte. Data are from 5 to 12 paired oocytes recordings from at least two independent experiments.
†indicates paired measurements with non-overlapping confidence intervals.
Table 4. EU1794-4 effects on glutamate and glycine EC50 values.
| EC50 (µM) [conf. int.]* | ||||||
|---|---|---|---|---|---|---|
| Control Glutamate |
EU1794-4 Glutamate |
Fold Difference |
Control Glycine |
EU1794-4 Glycine |
Fold Difference |
|
| GluN2A | 3.6 [3.3, 4.0] |
1.5 [1.2, 2.1] |
2.4† | 0.89 [0.72, 1.1] |
0.35 [0.17, 0.70] |
2.8† |
| GluN2B | 1.5 [1.2, 2.0] |
0.8 [0.48, 1.3] |
2.0† | 0.46 [0.37, 0.56] |
0.14 [0.12, 0.17] |
3.3† |
| GluN2C | 0.84 [0.57, 1.2] |
0.29 [0.15, 0.55] |
3.2† | 0.31 [0.15, 0.62] |
0.08 [0.024, 0.25] |
4.6† |
| GluN2D | 0.63 [0.41, 0.95] |
0.17 [0.065, 0.43] |
3.9† | 0.15 [0.089, 0.24] |
0.016 [0.007, 0.038] |
8.9† |
*Glutamate EC50 values (in the presence of 10 μM glycine) and glycine EC50 values (in the presence of 30 μM glutamate) were obtained by least-squares fitting of data by the Hill equation. EC50 values are given as the mean with the 95% confidence interval determined from log(EC50). Data in the absence and presence of EU1794-4 were obtained from the same oocyte. Data are from 4 to 8 paired oocytes recordings from two independent experiments.
†indicates paired measurements with non-overlapping confidence intervals.
Table 5. Comparison of EU1794-27, EU1794-4, and EU1794-2 effects at GluN1/GluN2C NMDAR responses to sub-saturating agonist.
| GluN2C | Glu/Gly (µM) |
Relative agonist Response* (% of maximal response) |
Modulation by 10 µM†
(% of control) |
|---|---|---|---|
| EU1794-27 | 100/30 1/0.3 |
- 20 ± 1.6 |
190 ± 10% 480 ± 31%‡ |
| EU1794-4 | 100/30 1/0.3 0.3/0.09 |
- 27.7 ± 3.3 2.8 ± 0.7 |
54 ± 1.6% 140 ± 12%‡ 660 ± 110%‡ |
| EU1794-2 | 100/30 0.6/0.2 |
- 21.5 ± 6.8 |
16 ± 1.0% 43 ± 2.1%‡ |
The data presented are mean ±SEM.
*Reported average responses to sub-saturating agonist were normalized to a 100/30 µM glutamate/glycine response.
†The degree modulation is reported as a percent of the control response to sub-saturating agonist. Some of these data points correspond to those used to calculated EC50 values for each modulator in Table 1, Table 2 or Supplementary file 1. Data are mean ±SEM.
‡p<0.05 as compared to 100/30 µM glutamate/glycine response, determined by an unpaired t-test, n = 4–10 cells.
PAMs and NAMs display both glutamate and glycine dependence
We studied NMDARs expressed in HEK293 cells to investigate the time course of modulator action. Concentration-dependent association of EU1794-2, EU1794-4 and EU1794-27 and concentration-independent disassociation were evaluated by co-applying modulator with glutamate and glycine (Figure 4A,B). These modulators were tested on GluN1/GluN2A and GluN1/GluN2D to see whether there were differences in modulation given the distinct properties of these NMDAR subtypes. We analyzed the concentration-dependence of the exponential time course describing the onset of action during co-application with saturating concentrations of glutamate plus glycine to determine modulator association and dissociation rates. From these we calculated the kinetically-determined affinity constant (Kd), which we found to be similar to the EC50 values determined from concentration-response experiments. EU1794-2 Kd was 1.1 µM at GluN1/GluN2D (Figure 4A,B). Complex actions of EU1794-4 and EU1794-27 were observed, with two temporally-distinct phases of modulation evident for the association of these modulators (Figure 4A). Both compounds produced a rapid inhibition followed by a slowly developing potentiating phase for EU1794-27. The rapid association rate determined during the rapid inhibition produced by EU1794-4 was approximately three times faster than EU1794-2; similarly, the dissociation rate was also faster for EU1794-4 than EU1794-2. Quantitative analysis of the slower phases was challenging due to its lower signal-to-noise ratio. Likewise, the rapid inhibitory phase was also difficult to measure for EU1794-27 because its rapid time course was convolved with the potentiation time course. Additionally, the depotentiation time course is preceded by a transient enhancement of the current response, followed by a relaxation to the pre-modulation level. The time course for potentiation and depotentiation were best fit with an exponential function summed with an additional linear component after the rapid phase subsided. Kd determined from the association and dissociation rates of EU1794-4 was 4.2 µM and EU1794-27 was 4.3 µM (Figure 4B). All modulatory effects were independent of voltage (Figure 4—figure supplement 1A). The steady state modulator responses from HEK293 cells approximately match the concentration-response relationship determined from TEVC recordings from X. laevis oocytes (Figure 4A, inset graphs). The time-course of modulator binding of EU1794-2, EU1794-4 and EU1794-27 was also determined for GluN1/GluN2A (Figure 4C,D). Higher Kd values (lower potency) were determined for each molecule at GluN1/GluN2A as compared to GluN1/GluN2D that parallel EC50 values determined using X. laevis oocytes. Similar to oocyte data, robust inhibition was produced by EU1794-2, modest inhibition was produced by EU1794-4, and transient inhibition and recovery was observed by the application of EU1794-27. Interestingly, the extent of steady-state modulation for this series appeared to be dependent on the level of desensitization of the receptors at the time of modulator application. When classifying the cells as either having high or low levels of desensitization (using 35% steady-state/peak response as a cut-off), the extent of modulation by 10 µM was significantly different for EU1794-27 (p=0.02, unpaired t-test, N = 4,2, respectively). The low desensitized group (40% average desensitization) was modulated by 107%, whereas the high desensitized group (10% average desensitization) was modulated to 247% of control by 10 µM EU1794-27 (Figure 4C and Figure 4—figure supplement 1F).
Figure 4. The association and dissociation time course of EU1794-2, EU1794-4 and EU1794-27 differ at GluN1/GluN2A and GluN1/GluN2D.
(A) Representative whole-cell current recordings of GluN1/GluN2D NMDARs exemplifying the modulation time course of EU1794-2, EU1794-4 and EU1794-27 (1, 3, 10 µM). Responses were evoked by 100/30 µM glutamate/glycine and were normalized for display. Insets show the steady state modulation extent from the HEK cell experiments fit by the Hill equation (Hill slope fixed to 1). (B) Analysis of the time course for the onset of PAM and NAM action as a function of concentrations for EU1794-2 (left), EU1794-4 (left) and EU1794-27 (right) used to determine the association rate kON (black circles) and dissociation rate kOFF (grey squares), from which we can calculate Kd for each modulator. Data are from at least 3 cells for each concentration of modulator. For EU1794-27 the association and dissociation rate linear component was also found to be concentration dependent (mon = −0.98 pA/(ms*µM), moff = 0.62 pA/(ms*µM)). (C,D) Similar set of panels as A,B but examining EU1794-2, EU1794-4 and EU1794-27 modulation of GluN1/GluN2A. In example recordings, all modulator concentrations are 10 µM. Data are from at least 3 cells for each concentration of modulator. For EU1794-27 time-course was fitted with the sum of an exponential and linear function, and both the time constant and slope were concentration dependent (mon = −0.33 pA/(s*µM), moff = 0.16 pA/(s*µM)).

Figure 4—figure supplement 1. The actions of EU1794 modulators are voltage-independent and are influenced by the desensitization level.

Given that other NMDAR modulators have displayed agonist-dependence (Petrovic et al., 2005; Acker et al., 2011; Hansen and Traynelis, 2011; Borovska et al., 2012; Vyklicky et al., 2015; Wang et al., 2017), we performed rapid solution exchange experiments to examine if these modulators had different affinities at agonist-bound and apo receptors (Figure 5A,B). The instantaneous current response to a rapid step into glutamate plus glycine (from glycine alone) gives an estimate of whether a modulator was pre-bound to receptors with only glycine (but not glutamate) bound. The immediate NMDAR activation after a rapid switch to a solution containing glutamate and glycine should occur faster than modulator binding. We chose to test for agonist-dependence using GluN1/GluN2D since they lack desensitization, which would complicate interpretation of this modulator property. We found that the peak current was similar when cells were preincubated in glycine with or without 3 µM EU1794-2, suggesting that EU1794-2 does not bind appreciably to the receptor in the absence of glutamate (Figure 5A,C). After the rapid step into glutamate where glycine and modulator were pre-exposed, we observed a relaxation to a new response level that was similar in amplitude to that observed with steady state co-application of glutamate plus glycine and modulator (Figure 5A,C). Interestingly, we also observed a similar effect for the converse experiment, in which we pre-applied glutamate with or without EU1794-2, followed by a rapid step into glutamate plus glycine and EU1794-2 (Figure 5B,C). We interpret these data to suggest that EU1794-2 associates with the receptor with higher affinity after glutamate and glycine binding.
Figure 5. The EU1794 series has agonist-dependence that alters the response time-course.
(A) Representative whole-cell recordings of GluN1/GluN2D NMDARs exemplifying the EU1794-2 dependence on glutamate binding. The cell was exposed continuously to 30 µM glycine with/without 3 µM EU1794-2, and then was rapidly stepped into a solution additionally containing 100 µM glutamate. The right panel shows an expanded view of the rise of the receptor response and the rapid time course of EU1794-2 action. (B) Representative whole-cell current recordings of GluN1/GluN2D NMDARs exemplifying the EU1794-2 dependence on glycine binding. The cell was exposed continuously to 100 µM glutamate with/without 3 µM EU1794-2, and then was rapidly stepped into a solution additionally containing 30 µM glycine. The right panel shows an expanded view of the rise of the receptor response and the rapid time course of EU1794-2 action. (C) Quantification of the modulation of the immediate peak response and steady state data (shown as a percentage EU1794-2/control for each phase) from agonist-dependence experiments for EU1794-2 (3 µM, Figure 5A,B and S4B). * signifies p<0.05 as determined by a Bonferroni's multiple comparison test where only EU1794-2 vs control was compared for both peak and steady state conditions after a significant repeat measures ANOVA (F(3,3) = 7.66 for pre-applied glycine and F(3,7) = 12.4 for pre-applied glutamate). (D) Representative whole-cell current recordings of GluN1/GluN2D NMDARs exemplifying the EU1794-27 dependence on glutamate binding. The cell was exposed continuously to 30 µM glycine with/without 3 µM EU1794-27, and the solution was rapidly changed to one additionally containing 100 µM glutamate. The right panel is an expanded view of the activation kinetics of the responses and the onset of modulation. (E) Quantification of the modulation of the immediate peak and steady state response (shown as a percentage EU1794-27/control for each phase) from agonist-dependence experiments for EU1794-27 (3 µM, Figure 5D and S4A). * signifies p<0.05 from the post hoc Bonferroni's multiple comparison test (F(3,3) = 17.8 for pre-applied glycine and F(3,2) = 13.5 for pre-applied glutamate).

Figure 5—figure supplement 1. EU1794 modulators are dependent on co-agonist binding.

We repeated these use-dependent experiments with EU1794-27, which yielded a similar result, although a slightly different experimental design was required for consistent responses. The pre-application of glycine with EU1794-27 produced a similar level of immediate activation following glutamate application (Figure 5D,E). This was then followed by the complex actions observed when EU1794-27 was applied to steady-state responses of NMDARs (Figure 5D, right panel). Using the same protocol to pre-apply glutamate became problematic for some cells due the degree to which EU1794-27 enhances agonist potency, a feature exemplified by the prolongation of deactivation. This action of EU1794-27 was able to render nanomolar contaminate levels of glycine (when present) more active, which necessitated a different experimental design (Figure 5—figure supplement 1A). When EU1794-2 was used in this experimental paradigm, inhibition of the contaminate level of activity was observed upon modulator application (Figure 5—figure supplement 1B). Nevertheless, we still observed a similar result with pre-application of glycine and EU1794-27, with the instantaneous response reaching the control level, followed by a slow relaxation to a new potentiated level that reflected the time course for association of EU1794-27 after binding of both glutamate and glycine (Figure 5E). This result illustrates a requirement for glycine to be bound to the receptor for high-affinity binding of EU1794-27. To circumvent any ambiguity associated with glycine contamination, we utilized another experimental design described by Vyklicky et al. (2015). Glutamate was pre-applied with 7-CKA (100 µM) to antagonize the glycine site, blocking any occupancy by contaminant glycine. The receptor was activated by switching to a solution that lacked 7-CKA and contained glycine plus glutamate (with or without modulator, Figure 5—figure supplement 1C). To test the ability of EU1794-27 to bind to the receptor with the GluN1 ABD bound to antagonist, EU1794-27 (3 µM) was added to the solution containing 7-CKA. Upon the switch from the 7-CKA/EU1794-27 solution containing glutamate to a solution just containing glutamate and glycine, no detectable change in the response rise time or peak amplitude was observed. If EU1794-27 bound during the 7-CKA phase prior to agonist binding, we would have expected an increased instantaneous peak current upon switching to glutamate plus glycine given that the dissociation of EU1794-27 is slower than 7-CKA (compare the steady state response and the 7-CKA response in Figure 5—figure supplement 1C). Additionally, comparable experiments were performed with 100 µM APV with similar results, as APV stabilizes the GluN2 agonist binding domain (ABD) open-cleft conformation, and thereby prevents the pre-binding of EU1794-27 (Figure 5—figure supplement 1D). When EU1794-27 (3 µM) was included in all solutions, potentiation was observed but APV unbinding was a required prior to modulation (Figure 5—figure supplement 1D). Together these data suggest that the EU1794 series is capable of high affinity binding only when glutamate and glycine are bound to the receptor.
Evaluation of interactions with known modulatory sites
To identify the molecular determinants of action for the EU1794 series, we evaluated the ability of GluN2 ATD deletion, GluN1 ATD splice variants, and co-application of known modulators to alter the effects of the EU1794 modulators. The ATD harbors the binding site for the GluN2B-selective negative allosteric modulator ifenprodil. Deletion of the ATD from GluN2A, GluN2B, or GluN2C had no effect on the actions of the PAM EU1794-27; deletion of the ATD from GluN2D reduced but did not eliminate potentiation (Figure 6—figure supplement 1A). Similarly, inclusion of 21 residues in the ATD encoded by alternatively spliced GluN1 exon5 only slightly altered the extent of potentiation of EU1794-27, but was without effect on EC50 for potentiation of GluN2B- and GluN2D-containing NMDARs activated by saturating agonist (Figure 6—figure supplement 1B). We previously described a similar result for the negative allosteric modulator EU1794-2 (Katzman et al., 2015). These data are consistent with minimal involvement of the GluN1 or GluN2 ATD in the actions of EU1794 modulators.
We next screened for interaction with known modulators to focus our search for the molecular determinants of EU1794 series modulation (Mullasseril et al., 2010; Acker et al., 2011; Hansen and Traynelis, 2011; Hansen et al., 2012; Khatri et al., 2014; Ogden et al., 2014; Hackos et al., 2016; Tajima et al., 2016; Yi et al., 2016) (Figure 6—figure supplement 2A). In these experiments, a known positive or negative modulator was co-applied with either EU1794-2 or EU1794-27, with each pair always containing one PAM and one NAM. If there is no interaction between paired modulators, their combined activity should be predicted by multiplying the extent of their independent actions. Co-application of the modulator pairs ifenprodil/EU1794-27, EU1794-2/CIQ and EU1794-2/PYD-106 produced levels of modulation that largely could be predicted from their independent actions (Figure 6—figure supplement 2B, top row). Modest differences from predictions were observed with modulators that bind to the ABD interface, TCN-201/EU1794-27, EU1794-2/GNE-6901 and EU1794-2/GNE-0723 (Figure 6—figure supplement 2B, bottom left). Co-application of the GluN2C/GluN2D-selective negative allosteric modulators QNZ46 and DQP-1105 paired with EU1794-27 resulted in the greatest divergences from predictions, raising the possibility that these modulators have partially overlapping binding sites (Figure 6—figure supplement 2B, bottom right) or similar downstream mechanisms.
We also examined the ability of EU1794-2 and EU1794-27 to modulate NMDARs harboring mutations within the structural determinants for other known allosteric modulators. Mutations in GluN1 (I519A, R755A) and GluN2A (L780A, G786A) that block TCN-201 inhibition were evaluated for effects on EU1794 series of modulators (Hansen et al., 2012). NMDARs that contained GluN1/GluN2A(L780A), GluN1/GluN2A(G786A), GluN1(R755A)/GluN2D, GluN1(I519A)/GluN2D were equally sensitive as wild type receptors to inhibition by EU1794-2 or potentiation by EU1794-27 (Figure 6—figure supplement 3A). GluN1/GluN2A(E530A), GluN1/GluN2A(V783W), GluN1(Y535W)/GluN2A, GluN1(Y535V)/GluN2D and GluN1(Y535W)/GluN2D, which reduce the actions of GNE-6901 and GNE-0723 (Hackos et al., 2016), produced no significant effects on inhibition by EU1794-2 or potentiation by EU1794-27 (Figure 6—figure supplement 3B). Inhibition by EU1794-2 of GluN1/GluN2C(K470G) and GluN1/GluN2C(S472T), which block PYD-106 potentiation of GluN1/GluN2C (Khatri et al., 2014), was similar to wild type NMDARs (Figure 6—figure supplement 3C). Interestingly, inhibition by EU1794-2 and potentiation by EU1794-27 was not significantly changed by GluN1/GluN2D(Q701Y) and GluN1/GluN2D(L705F), mutations that appear to confer subunit selectivity for GluN2C/D over GluN2A/B for QNZ-46 and DQP-1105 (Figure 6—figure supplement 3D) (Acker et al., 2011; Hansen and Traynelis, 2011).
Mutagenesis suggests shared structural determinants of action for PAMs and NAMs
The result obtained with the QNZ-46/DQP-1105 interaction test suggested that the two residues we evaluated for QNZ-46 and DQP-1105 insufficiently probed the structural determinants of action for these compounds. We therefore examined a GluA2 AMPA receptor structure bound to CP-465,022, which shares a core scaffold with QNZ modulators (Yelshanskaya et al., 2016). Residues identified as being important for CP-465,022 binding in Yelshanskaya et al., 2016 were aligned to the GluN1 and GluN2D subunits to map this modulatory site onto the NMDAR subunits, in addition to critical residues for GNE-9278 in this same region (Figure 6A, Wang et al., 2017). This broader range of residues were located on the pre-M1, M3, and pre-M4 regions of both GluN1 and GluN2D, which have previously been suggested to cooperate to control gating (Ogden and Traynelis, 2013; Alsaloum et al., 2016; Chen et al., 2017; Ogden et al., 2017; Yelshanskaya et al., 2017). These residues were suggested by Yelshanskaya et al., 2016 to constitute a binding site in homomeric GluA2 AMPARs. However, mapping the homologous residues onto NMDAR structures yields two pockets given the multimeric subunit architecture, one of which consists of the residues of GluN1 and the other of residues of GluN2. Certain residues of M3, depending on their position on the helix, could point towards either pocket, rendering the two pockets to be lined by a mixture of GluN1 and GluN2 residues. 15 GluN1 and 15 GluN2D residues in these two regions that probed these pockets were identified and mutated to allow a test of the contribution of each residue to EU1794-2 inhibition and EU1794-27 potentiation. Inhibition by EU1794-2 was significantly altered by substitutions at six residues in GluN1 and 2 GluN2D residues (Figure 6B), which were found on pre-M1 and M3 regions of both GluN1 and GluN2D. Potentiation by EU1794-27 was more labile, being altered in mutations at 9 GluN1 residues and 6 GluN2D residues (Figure 6B). Residues that perturbed the actions of EU1794-27 were spread across all regions tested except for the GluN2D pre-M1. Modulation was observed to be altered in three different ways by the mutations studied here: activity could be reduced, increased, or inverted.
Figure 6. Distinct and overlapping pattern of residues contribute to the actions of EU1794 modulators with opposing effects.
(A) Ribbon representation (left) of a GluN1/GluN2D model based on published crystal structures of GluN1/GluN2B and GluA2 (Karakas and Furukawa, 2014; Lee et al., 2014; Yelshanskaya et al., 2016). The GluN1 subunits are grey and the GluN2D are purple; surface shell indicate residues of interest in GluN1 and GluN2D, including residues that are homologous to those identified in recent studies investigating transmembrane domain interacting modulators of NMDAR and AMPAR (Yelshanskaya et al., 2016; Wang et al., 2017). These GluN1 and GluN2D residues were changed to alanine (or tyrosine in two cases, as previously reported) and tested for effects on the NMDAR sensitivity to EU1794-2 and EU1794-27. Top down (right top) and side (right bottom) view of the model TMD and linker segments highlight two potential pockets with GluN1 and GluN2 M3 helices in the background. One pocket is primarily associated with GluN1 and the other GluN2. (B) The responses (mean ±99% CI) in EU1794-2 (5 µM, left) and EU1794-27 (10 µM, right) are shown as a % of the mutant receptor response to 100 µM glutamate and 30 µM glycine in the absence of the test compound. The wild type mean response is shown by the dashed line, surrounding dotted lines, which indicate the 99% confidence interval. Residues with non-overlapping confidence intervals with the wild type are colored with magenta indicating apparent loss of activity, yellow indicating augmented activity, and green indicating robust inverted modulation. Data for the mutations shown represent 4–18 oocytes from at least two independent experiments and the wild type response shown represent 83 oocytes for EU1794-2 and 76 oocytes for EU1794-27, recorded each experimental day to ensure consistency. We were not able to test all mutant receptors due to low expression for some (NT, not tested). (C) The residues (shown by the space-filling shell) identified in (B) that altered the effects of EU1794-2 (left) and/or EU1794-27 (right) were mapped onto the model with colors as described in (B). Views are the same as (A), with the all residues probed shown as grey transparent shell representation.

Figure 6—figure supplement 1. EU1794 series effects are not altered by ATD perturbations.

Figure 6—figure supplement 2. Compound competition screen of the EU1794 series with NMDAR modulators highlights potential interactions with the ABD and the TMD.

Figure 6—figure supplement 3. Residues in known modulator binding sites do not perturb actions of the EU1794 series.

Figure 6—figure supplement 4. Mutated residues that impact EU1794 actions have lower open probability.
The distributions of residues that altered modulation by EU1794-2 and EU1794-27 shows clear overlap (Figure 6C). Inhibition by EU1794-2 was altered primarily by mutations in GluN1, whereas potentiation by EU1794-27 was perturbed by residues both in GluN1 and in the M3 helix of GluN2D. Interestingly, the mutations that invert activity of EU1794-2 and EU1794-27 were distinct, but in some cases were in close proximity. For example, EU1794-2 was inverted by GluN1-F654A, whereas mutation of the adjacent residue GluN1-L655A/Y inverted the modulatory action of EU1794-27 (Figure 6D). EU1794-27 potentiation was also converted to inhibition by four mutations at residues residing on the 3 areas of GluN1 that were investigated (pre-M1, M1, M3) and also on the M3 GluN2D helix, which were in close proximity to each other (Figure 6D). EU1794-27 actions on GluN1-L655A/Y and GluN2D-M678Y, which are homologous residues immediately downstream of the SYTANLAAF motif, resulted in opposite effects (GluN1-L655A/Y converts EU1794-27 to an inhibitor and GluN2D-M678Y increases the potentiation of EU1794-27). We interpret these results to suggest that the activity of both PAMs and NAMs of the EU1794 series is dependent on multiple residues in the GluN1 subunit, some of which are overlapping. Furthermore, potentiation by EU1794-27 is dependent on a wider range of GluN1 and GluN2 residues. One possible way to account for this would be if both modulators bound near the GluN1 pre-M1 helix, with potentiator actions dependent on the nearby GluN2 M3 residues associated with the GluN1 pocket (Figure 6A,C).
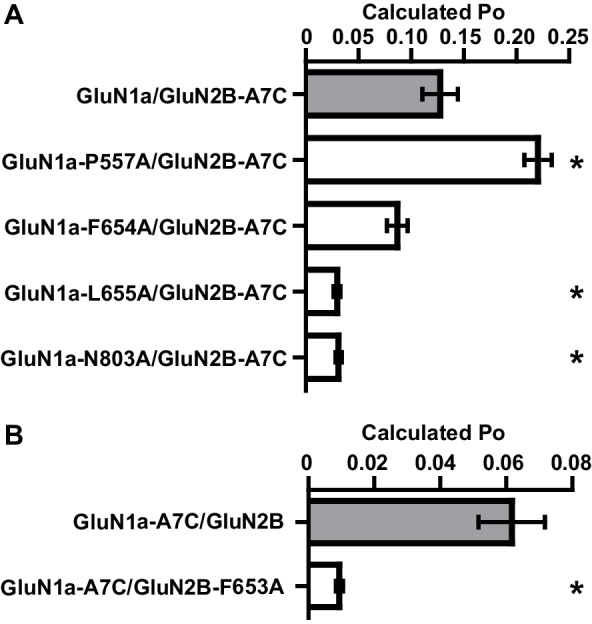
To assess whether mutated residues impacted EU1794 series modulation through altering receptor open probability, we estimated mutant open probability using MTSEA modification of an introduced cysteine residue (Jones et al., 2002; Yuan et al., 2005). The open probability of GluN2D-containing receptors is very low and may not allow for precise determination of the effects of mutations, thus we used GluN2B-containing receptors for comparison. GluN1 mutations were expressed with a GluN2B A7C construct (See Figure 6—figure supplement 4 legend) and GluN2 mutations were expressed with a GluN1 A7C construct. Several residues were assayed that resulted in reversed, enhanced, or blocked modulator activity (Figure 6—figure supplement 4A). GluN1-P557A, which blocked activity of both EU1794-2 and EU1794-27, had an increased open probability as compared to wild type. GluN1-F654A, which converted EU1794-2 into a potentiator and enhanced EU1794-27 potentiation, had a similar open probability as compared to wild type (Figure 6—figure supplement 4A). Interestingly, mutation of the homologous GluN2 residue (GluN2-F653A), which reduced EU1794-27 potentiation, also lowered open probability as compared to wild type (Figure 6—figure supplement 4B). Figure 6—figure supplement 4 shows that GluN1-L665A and GluN1-N803A, which both converted EU1794-27 into an inhibitor, had significantly lower open probabilities as compared to wild type NMDARs (Figure 6—figure supplement 4A). These data suggest that altered EU1794 modulator action by mutated residues is not directly dependent on alterations in open probability.
PAMs and NAMs exert their opposing effects via a shared binding site on NMDARs
Given the similarity in chemical structure between positive and negative modulators in this series, we hypothesized that they might have overlapping binding sites. In order to conduct a detailed functional analysis of competition between allosteric modulators, we first used modeling to examine a receptor’s functional response to co-application of positive and negative modulators acting at the same or different sites. We evaluated two models of modulator binding built into a scheme first proposed by (Lester and Jahr, 1992). Model 1 describes receptors with a single binding site that can accommodate either PAM or NAM, but not both. Model 2 describes a receptor to which both modulators can bind at the same time in different sites (Figure 7A, Figure 7—figure supplement 1). We evaluated hypothetical responses from these models (see Materials and methods, Figure 7—figure supplement 1A–C). Responses to maximally effective concentrations of agonist were simulated at various concentrations of the positive and negative modulators. When the modulators compete for the same binding site, they act similarly to stated theory about competitive antagonists (Arunlakshana and Schild, 1959; Christopoulos and Kenakin, 2002), with the PAM causing a rightward shift of the NAM IC50 and generating a linear relationship in dose ratio analysis (Figure 7B, Figure 7—figure supplement 1D,E, Model 1). By contrast, there is no apparent EC50 shift when the two modulators are capable of binding simultaneously (Figure 7C, Figure 7—figure supplement 1D,E, Model 2). Additional simulations showed the reciprocal effect of NAM on PAM EC50 (Figure 7—figure supplement 1F,G). We subsequently performed competition experiments to test the hypothesis that this series of PAMs and NAMs compete for a mutually exclusive modulatory pocket (Figure 7D). We observed that fixed concentrations of the PAM EU1794-27 caused parallel shifts in the concentration-response curve of the NAM EU1794-2 (Figure 7E, Table 6). A similar phenomenon was shown for the reverse, as fixed concentrations of EU1794-2 caused parallel shifts in the EU1794-27 concentration-response curves (Table 6). These results closely match Model 1, where PAMs and NAMs bind in a mutually exclusive fashion. This suggests that the opposing modulators in the EU1794 series share overlapping binding sites instead of the coincidence that the subtle chemical differences confer unique binding sites.
Figure 7. EU1794 PAMs and NAMs act in a competitive manner that matches receptor models with mutually exclusive modulator binding.
(A) Cartoon models show two potential modulator binding schemes. Model 1 possesses mutually exclusive modulator binding whereas Model 2 can bind either or both modulators. Ligands are denoted by [N] (NAM) and [P] (PAM). (B,C) Dose-ratio plots are shown for simulated concentration-response curves of the NAM in various concentration of the PAM for Model 1 (B) and Model 2 (C). IC50’ is the observed NAM IC50 in the presence of a fixed concentration PAM. (D) A representative TEVC recording (fixed 3 µM EU1794-27 and 0.3, 1, 3, 10, 30 µM EU1794-2) is shown for GluN1/GluN2D NMDARs (top). (E) The mean steady state responses in (D) were fitted with the Hill equation. Concentration-response data in the absence of EU1794-27 are included for comparison from Figure 1C. (F) Dose-ratio analysis of experimental data illustrates the shift in the IC50 of the NAM as a function of PAM concentration. Data are from at least six oocytes evaluated from two independent experiments.

Figure 7—figure supplement 1. EU1794 PAMs and NAMs act in a competitive manner that suggests mutually exclusive modulator binding.

Table 6. The response to co-application of EU1794-2 and EU1794-27 suggest a common binding site.
| EU1794-2 IC50 (µM) [conf. int.]* | EU1794-27 EC50 (µM) [conf. int.]* | ||||||
|---|---|---|---|---|---|---|---|
| +EU1794-27 0 µM |
+EU1794-27 3 µM |
+EU1794-27 10 µM |
+EU1794-27 20 µM |
+EU1794-2 0 µM |
+EU1794-2 1 µM |
+EU1794-2 3 µM |
|
| GluN2A | 0.60†
[0.44, 0.82] |
2.5‡
[1.9, 3.1] |
3.6‡
[2.4, 4.8] |
8.0‡
[1.3, 15] |
-§ | -§ | -§ |
| GluN2B | 1.2†
[0.8, 1.9] |
NR | 3.5‡
[2.1, 4.9] |
12 [~, 26] |
1.4†
[1.2, 1.7] |
6.0‡
[4.6, 7.4] |
8.4 [~, 18] |
| GluN2C | 0.21†
[0.18, 0.25] |
1.2‡
[0.91, 1.4] |
2.4‡
[1.8, 3.0] |
6.0‡
[4.1, 7.8] |
2.8†
[2.5, 3.2] |
7.3‡
[5.9, 8.8] |
7.3 [2.1, 11.74] |
| GluN2D | 0.20†
[0.17, 0.25] |
1.4‡
[1.2, 1.6] |
2.7‡
[2.3, 3.2] |
7.3‡*
[4.6, 9.9] |
2.4†
[1.8, 3.3] |
7.5‡
[5.6, 9.3] |
7.8 [1.1, 14] |
A five point concentration response curve was obtained for modulator effects on responses to maximally effective concentrations of glutamate and glycine (100/30 µM) for EU1794-2 co-administered with increasing concentrations of EU1794-27, and for EU1794-27 in increasing concentrations of EU1794-2.
*IC50 and EC50 values were obtained by least-squares fitting of data by the Hill equation. IC50 and EC50 values are given as the mean with the 95% confidence interval determined from log(IC50) or log(EC50). Data are from at least six oocytes evaluated from two independent experiments for the EU1794-2 IC50 determinations and from at least three oocytes evaluated from one experiment for the EU1794-27 EC50 determinations.
NR, Given the low potency for modulators at GluN2B, the EU1794-2 concentration-response curve with 3 µM EU1794-27 determination was not recorded.
† Data from Table 1 is included here for clarity.
‡indicates non-overlapping 95% confidence interval with the EU1794-2 IC50 without EU1794-27 or EU1794-27 EC50 without EU1794-2.
§EU1794-27 does not potentiate GluN2A responses to maximally effective concentrations of glutamate and glycine.
~indicates that the confidence interval reached the theoretical limit (EC50 = 0).
The enantiomers of EU1794-27 were separated to determine whether the racemic activity reflected the actions of only one enantiomer (see Supplemental methods). (-)-EU1794-27 potentiated NMDAR responses similarly to the racemic mixture (Figure 8—figure supplement 1A, Table 7). By contrast, (+)-EU1794-27 exhibited only weak potentiation of GluN1/GluN2C and GluN1/GluN2D, which may be due to the purity achieved via chiral separation (Table 7). We previously reported (Katzman et al., 2015) that purified enantiomers were likely to racemize in aqueous solutions, which should proceed by a first-order reaction dependent on multiple factors such as ionic strength, pH, buffer, etc. (Smith et al., 1978). Thus, to quantitatively determine if racemization would impact our experiments using the enantiomers, we performed a functional assessment of this property in our standard experimental solution. We observed racemization of the enantiomers of EU1794-27 with a half-life of 197 min (Figure 8—figure supplement 1B, Table 7). Both enantiomers of EU1794-4 inhibited NMDAR responses, but with different potencies (Figure 8—figure supplement 1C, Supplementary File 2). A similar rate of racemization was observed for the enantiomers of EU1794-4 (half-life of 196 min, Figure 8—figure supplement 1D). All enantiomer studies, other than the racemization time-course, were performed rapidly to minimize any racemization, being completed in less than 80 min after making the aqueous solution of modulator. Similar to results in oocytes, when the enantiomers of EU1794-27 were applied to steady-state GluN1/GluN2D responses in HEK cells, (-)-EU1794-27 potentiated responses whereas (+)-EU1794-27 had a slight inhibitory effect (Figure 8A). In additional, 10 µM of both enantiomers of EU1794-4 had similar inhibitory actions on GluN1/GluN2D responses to maximally effective concentrations of glutamate and glycine (Figure 8B).
Table 7. Enantiomeric preference of EU1794-27.
| EC50 (µM) [conf. int.]* Maximal Modulation Extent (% of control) |
||||
|---|---|---|---|---|
| Compound | EU1794-27 | (-)-EU1794-27 | (+)-EU1794-27 | |
| GluN2A | 7.4 [5.3, 10]†
52 ± 7% |
5.6 [2.7, 12] 67 ± 5% |
- | ‡ |
| GluN2B | 1.4 [1.2, 1.7]†
130 ± 3% |
2.8 [2.0, 3.8] 170 ± 10% |
- | ‡ |
| GluN2C | 2.8 [2.5, 3.2]†
230 ± 10% |
5.1 [4.1, 6.4] 260 ± 24% |
7.1 [3.6, 14] 160 ± 6.2% |
‡ |
| GluN2D | 2.4 [1.8, 3.3]†
250 ± 8% |
8.2 [6.7, 9.9] 340 ± 21% |
7.4 [3.7, 15] 130 ± 4.7% |
‡ |
*EC50 values were obtained by least-squares fitting of data from individual experiments by the Hill equation. EC50 values are given as the mean with the 95% confidence interval determined from log(EC50); the maximal degree of modulation is given as mean ±SEM. Data are from 4 to 7 oocytes from at least two independent experiments. Data were not fitted (shown as -) if the response recorded at 30 µM of test compound did not differ by more than 15% from control.
†Data from Table 1 is included here for clarity.
‡indicates significant unpaired t-test between the percent modulation between 30 µM (-)-EU1794-27 and (+)-EU1794-27.
Figure 8. The enantiomers of EU1794-4 and EU1794-27 have different action.
(A) Representative modulation of GluN1/GluN2D responses by the enantiomers of EU1794-27 (both 10 µM, 100/30 µM glutamate/glycine). Similar results were observed in at least 3 cells to fresh modulator solution made less than one hour before experiment conclusion. (B) Representative modulation of GluN1/GluN2D responses by the enantiomers of EU1794-4 (both 10 µM, 100/30 µM glutamate/glycine). Similar results were observed in at least 3 cells to fresh modulator solution made less than one hour before experiment conclusion. (C) Single concentration competition experiments illustrating an interaction between both enantiomers of EU1794-4 (+and -) and the positive modulating enantiomer of EU1794-27 (-) at GluN1/GluN2D. The individual average effects of the pairs of modulators along with the co-applied effect are plotted, and the predicted mean net effect of the modulators is displayed as a dashed line (SEM, dotted lines, n = 10 from two independent experiments). * signifies p<0.05 as determined by a paired t-test comparing co-applied modulators and predicted net effect of modulator actions.
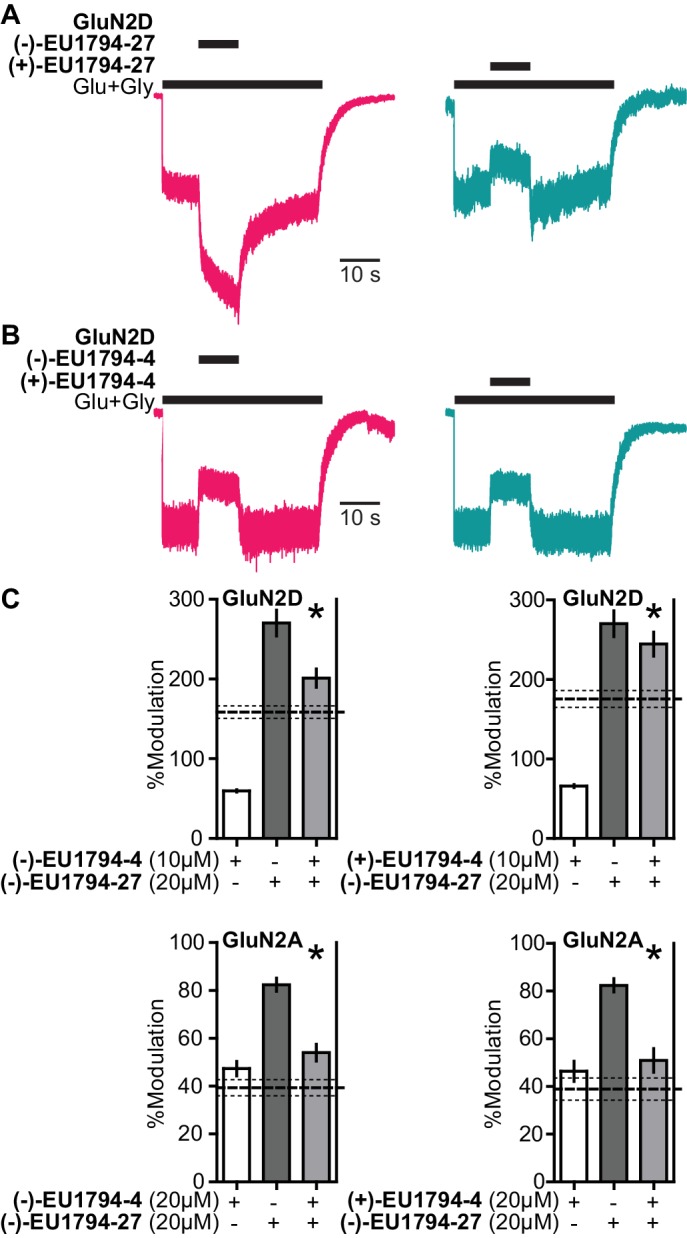
Figure 8—figure supplement 1. The concentration response and stability of the enantiomers of EU1794-27 and EU1794-4.

To assess whether both enantiomers bound to similar or distinct sites on the NMDAR, we performed single concentration competition experiments. Co-application of each enantiomer of EU1794-4 with the (-)-EU1794-27 resulted in a degree of modulation that was significantly different than that predicted for independent sites of action at both GluN1/GluN2A and GluN1/GluN2D (Figure 8C). Interestingly, the competition by EU1794-4 was not dependent on the direction of modulation by (-)-EU1794-27, which had a potentiating action at GluN1/GluN2D (Figure 8C, top panels) and an inhibitory effect at GluN1/GluN2A (Figure 8C, bottom panels). Although there remain potential caveats, the evidence suggests that the co-application of (-)-EU1794-27 with either enantiomer of EU1794-4 displays mutual exclusivity in modulator binding.
Discussion
This study highlights how subtle chemical variations in a series of NMDAR allosteric modulators can result in fundamentally different actions on NMDAR responses to maximally effective concentrations of agonists. This series of small molecule allosteric modulators show a spectrum of effects ranging from strong negative modulation to robust positive modulation. In addition, there are features that are shared across this series, including agonist-dependence and the ability to enhance agonist potency. Thorough analysis of the actions of this series illustrates a potential way forward in designing new analogues to achieve a wide range of activities. Additionally, the novel features found in this class of modulators suggest potential new strategies for targeting distinct populations of NMDARs, such as extrasynaptic receptors that typically are not activated by high concentrations of glutamate or distinct patterns of stimulation.
The site and mechanism of action for the EU1794 series
In the evaluation of the kinetic properties of modulator action, we observed complex actions of EU1794-4 and EU1794-27, which could arise from multiple binding sites, enantiomers of these compounds, or could reflect distinct modulator-dependent mechanistic actions from occupancy of a single binding site. The modulation by EU1794-4 and EU1794-27 is voltage independent, eliminating potential channel block within the ion channel pore by potential cationic species as a confounding site that contributes to the observed effects. Given that there is high homology between GluN1 and GluN2, especially in the ABD and the TMD, a reasonable hypothesis is that multiple binding sites exist for EU1794 series modulators in homologous regions on GluN1 and GluN2 subunits. However, the mutagenesis data argue against this idea, given that the residues at which mutations perturbed the actions of EU1794-2 and EU1794-27 overlapped and clustered around the pre-M1, M3, and M4 of the GluN1 subunit. Whereas there were a few residues of the GluN2 M3 helix that influenced EU1794-27 modulation, the structural NMDAR models suggest that these residues face the GluN1 M3 helix, and thus could interact with the pocket adjacent to the GluN1 pre-M1 helix. Additionally, it’s likely the M3 helixes of both GluN1 and GluN2, which are in close contact, act in concert with one another to control rapid pore opening or closing. If these residues identified by mutagenesis controlled conformational changes downstream of the EU1794 binding site(s), it would limit potential binding site candidates to the interface between the GluN1 and the GluN2 ABDs, which would then have only two identical binding sites in diheteromeric NMDARs. Therefore there would be less potential for non-identical binding sites that contribute to the mixed actions that are observed for EU1794-27 and EU1794-4. For these reasons, the idea that the positive and negative allosteric actions reflect modulator specific mechanisms from occupancy of a single site seems the most plausible interpretation.
Evaluation of the effects of enantiomers provides further insight into the binding site of the EU1794 series. EU1794-27 exhibits strong stereoselectivity, with the (-) enantiomer showing a typical potentiation time course and the (+) enantiomer producing inhibition. The enantiomers of EU1794-4 are both capable of producing inhibition but with the (-) enantiomer being more potent (3–10 fold) than the (+) enantiomer. Additionally, both enantiomers of EU1794-4 appear to compete with EU1794-27 for access to the binding site, suggesting that enantiomers may interact differently with the same binding site. An alternative explanation for lack of additivity of the effects of the two compounds could be that there are shared residues downstream of the PAM and NAM binding sites that mediate their effects; additional studies are required to evaluate this hypothesis. However, the idea that the enantiomers of the EU1794 series act at the same site suggests their complex actions on NMDARs expressed in HEK is a mixture of receptors bound to one or the other enantiomer of racemic EU1794-27 and EU1794-4. The available enantiomeric data further support the idea that positive and negative modulators within the EU1794 series share a single or overlapping binding site.
EU1794 series links positive and negative allosteric modulators that act at the TMD
An increasing number of NMDAR and AMPAR modulators have been identified with structural determinants of action that reside in transmembrane linker regions and extracellular portions of the transmembrane domain (Mullasseril et al., 2010; Acker et al., 2011; Hansen and Traynelis, 2011; Ogden and Traynelis, 2013; Yelshanskaya et al., 2016; Swanger et al., 2018; Wang et al., 2017). Other cell surface receptor families have bi-directional modulator pockets, including multiple GPCRs as well as the benzodiazepine binding site in GABA-A receptors (Barnard et al., 1998; Rudolph and Knoflach, 2011; Wootten et al., 2013). Among all ionotropic glutamate receptor modulators interacting with this region of the receptor, there are positive (CIQ, GNE-9278) and negative (DQP-1105, QNZ-46, CP-465,022, GYKI 52466) modulators with diverse scaffolds. However, the EU1794 compounds represent the first bidirectional NMDAR modulator series with structural determinants of activity within this region, low micromolar potency and with clear rules to control modulation. The similarity in structure of positive and negative modulators illustrates how subtle differences in the ligand can interconvert functional actions between inhibition and potentiation. It is unclear precisely how the different sized ester alkyl chain substitutions of these analogs bring about opposing actions, further investigation of the structure activity relationship may elucidate these details. The ability to potentiate NMDAR seems to be unique to (-)-EU1794-27, which may have a specific interaction with the receptor achieved only by its stereoselective active pose in the binding pocket. Work with this series may lead to an understanding of the mechanistic link between the PAMs and NAMs that interact in this portion of the receptor.
Differential actions on synaptic and extrasynaptic receptors by submaximal EU1794 analogues
The EU1794 series has a property of agonist-dependence, requiring both glutamate and glycine to be bound before the modulator binding site adopts a high potency orientation for members of the series. Relatedly, use-dependence is a property of open channel blockers (e.g. memantine, MK-801, ketamine, etc.) that inhibit the receptor through interactions within the ion permeation path, and thus rely on pore opening (Traynelis et al., 2010). Given that the EU1794 series are allosteric modulators are not voltage-dependent, the mechanism of their agonist-dependence is unclear. Previously, the NAMs QNZ-46, DQP-1105, and NAB-14 have been reported to show varying degrees of glutamate- but not glycine-dependence (Acker et al., 2011; Hansen and Traynelis, 2011; Swanger et al., 2018). Moreover, neurosteroid derivatives with NAM activity have also been shown to have glutamate- and glycine-dependence (Vyklicky et al., 2015), and the PAM GNE-9278 was reported to be glutamate-dependent (Wang et al., 2017). Thus, the EU1794 series is the first series of positive and negative modulators that has been shown to possess the property of being both glutamate- and glycine-dependent. Our working hypothesis is that the EU1794 series of modulators requires conformational changes in both GluN1 and GluN2 subunits that reflect pre-gating or gating transitions, after which its affinity for its binding site is increased. However, we cannot rule out at this time the possibility that the pore must open to increase modulator binding. Resolving the specific mechanism of the EU1794 may lead to a more complete understanding of the activation transitions NMDAR.
We believe that EU1794-4 highlights a novel sub-class of NMDAR modulators that have the capability to selectively act at extrasynaptic NMDARs based on the combinations of its properties. Extrasynaptic NMDAR are hypothesized to respond to glutamate spillover or glial release of glutamate (Rusakov and Kullmann, 1998; Haydon and Carmignoto, 2006; Sahlender et al., 2014). The potential ability to preferentially act at these non-synaptic sites arises from three mechanistic features of the allosteric mechanism: (1) the submaximal inhibitory effects at saturating concentrations of modulator and agonist, (2) the agonist-dependent and slow association rate, which might limit activity at synaptic receptors, and (3) the ability to enhance NMDAR responses to low agonist concentrations. A similar property has been described for the GluN2B-selective agent ifenprodil (Kew et al., 1996), although the extent to which EU1794-4 can enhance the response to low concentrations of agonist is amplified by the large degree of residual current at saturating levels of EU1794-4 (30–60%) compared to ifenprodil (~10%). There are numerous studies that suggest the importance of extrasynaptic NMDARs in normal biology but study of them requires complex experimental paradigms (Harris and Pettit, 2008; Paoletti et al., 2013; Papouin and Oliet, 2014). Alternatively, the agonist-dependence of EU1794-4 may alter this capability in instances of repeated stimulation. In either case, EU1794-4 is a unique compound that may act as a tool that could be used to probe the contribution of distinct types of NMDARs or their activity in circuit function. Further work is required to fully understand the utility of this modulator as a probe for extrasynaptic NMDARs.
Materials and methods
Key resources table.
| Reagent type (species) or resource |
Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| gene (Rattus norvegicus) | GluN1-1a | U11418 | ||
| gene (Rattus norvegicus) | GluN1-1b | U08263 | ||
| gene (Rattus norvegicus) | GluN2A | D13211 | ||
| gene (Rattus norvegicus) | GluN2B | U11419 | ||
| gene (Rattus norvegicus) | GluN2C | M91563 | ||
| gene (Rattus norvegicus) | GluN2D | L31611 | ||
| cell line (Xenopus laevis) | stage VI oocytes | Ecocyte Biosciences (Austin, TX); Xenopus 1 (Dexter, MI) |
NA; #10004 | |
| cell line (Homo sapiens) | HEK293 | ATCC (Manassas, VA) | CRL-1573 | |
| commercial assay or kit | QuikChange kit | Agilent Technologies (Santa Clara, CA) |
200524 | |
| commercial assay or kit | mMessage mMachine SP6 or t7 |
Ambion/ThermoFisher Scientific (Waltham, MA) |
AM1340 or AM1344 | |
| chemical compound, drug | EU1794-4 | PMID: 26525866 | SMILES: CCOC(=O)C1 = C(NC(=O) CC2SC(=N)NC2 = O)SC2 = C1CCCC2 |
|
| chemical compound, drug | EU1794-2 | PMID: 26525866 | SMILES: CCOC(=O)C1 = C(NC(=O) CC2SC(=N)NC2 = O)SC2 = C1CCC(C)C2 |
|
| chemical compound, drug | EU1794-5 | this paper | SMILES: CC(C)OC(=O)C1 = C(NC(=O) CC2SC(=N)NC2 = O)SC2 = C1CCCC2 |
See sumplemental information |
| chemical compound, drug | EU1794-19 | this paper | SMILES: CC(C)OC(=O)C1 = C (NC(=O)CC2SC(=N)NC2 = O) SC2 = C1CCC(C)C2 |
See sumplemental information |
| chemical compound, drug | EU1794-25 | this paper | SMILES: CC1CCC2 = C(C1)SC(NC(=O) CC1SC(=N)NC1 = O)=C2C(=O) OC1 = CC = CC=C1 |
See sumplemental information |
| chemical compound, drug | EU1794-27 | this paper | SMILES: CC(C)(C)OC(=O)C1 = C (NC(=O)CC2SC(=N)NC2 = O) SC2 = C1CCCC2 |
See sumplemental information |
| chemical compound, drug | EU1794-29 | this paper | SMILES: CCCCOC(=O)C1 = C(NC(=O) CC2SC(=N)NC2 = O)SC2 = C1CCC(C)C2 |
See sumplemental information |
| chemical compound, drug | βCD | Acros Organics or ChemCenter |
2-(hydroxypropyl)-β-cyclodextrin (CAS# 128446-35-5) |
|
| chemical compound, drug | APV | Tocris (Minneapolis, MN) | (2)-amino-5-phosphonovaleric acid (CAS# 76326-31-3) |
|
| chemical compound, drug | 7-CKA | Tocris (Minneapolis, MN) | 7-chlorokynurenic acid (CAS# 18000-24-3) | |
| chemical compound, drug | MTSEA | TRC Canada (Toronto, ON) | Methanethiosulfonate ethylammonium (CAS# 16599-33-0) |
|
| chemical compound, drug | CIQ | Tocris (Minneapolis, MN) | CAS# 486427-17-2 | |
| chemical compound, drug | Ifenprodil | Tocris (Minneapolis, MN) | CAS# 23210-56-2 | |
| chemical compound, drug | PYD-106 | PMID: 25205677 | ||
| chemical compound, drug | TCN-201 | Tocris (Minneapolis, MN) | CAS# 852918-02-6 | |
| chemical compound, drug | GNE-6901 | PMID: 26875626; Genentech (San Francisco, CA) |
generously provided by Jesse Hanson and Genentech |
|
| chemical compound, drug | GNE-0723 | PMID: 26919761; Genentech (San Francisco, CA) |
generously provided by Jesse Hanson and Genentech |
|
| chemical compound, drug | DQP-1105 | Tocris (Minneapolis, MN) | CAS# 380560-89-4 | |
| chemical compound, drug | QNZ-46 | Tocris (Minneapolis, MN) | CAS# 1237744-13-6 | |
| software, algorithm | Perszyk2018_ sim_Markov_ Modulator Competition |
this paper | Matlab code, see sumplemental information |
|
| software, algorithm | Axopatch and Clampfit |
Molecular Devices (San Jose, CA) |
||
| software, algorithm | Easy Oocyte | Custom written Software (Emory University, Atlanta, GA) |
||
| software, algorithm | Prism | GraphPad (La Jolla, CA) | ||
| software, algorithm | Matlab | Mathworks (Natick, MA) |
Molecular Biology
cDNAs for rat wild type NMDAR subunits GluN1-1a (GenBank U11418, U08261; hereafter GluN1 or GluN1a), GluN1-1b (U08263, hereafter GluN1b), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611, modified as described in Monyer et al., 1994) were provided by Drs. S. Heinemann (Salk Institute), S. Nakanishi (Kyoto University), and P. Seeburg (University of Heidelberg). Site-directed mutagenesis was conducted using the QuikChange kit (Agilent Technologies, Santa Clara, CA) following the recommended protocol. DNA sequencing was used to verify all mutations. The amino acid numbering system started with the initiating methionine as residue number one. For expression in Xenopus laevis oocytes, cDNA constructs were linearized by restriction enzymes, and subsequently used to synthesize in vitro cRNAs following the manufacturer’s protocol (mMessage mMachine, Ambion, ThermoFisher Scientific, Waltham, MA).
Two-electrode voltage-clamp recordings
Xenopus laevis stage VI oocytes (Ecocyte Biosciences, Austin, TX) were injected as previously described (Hansen et al., 2013) 2–7 days prior to recording with cRNA encoding the NMDAR subunits and stored at 15°C in media containing (in mM) 88 NaCl, 2.4 NaHCO3, 1 KCl, 0.33 Ca(NO3)2, 0.41 CaCl2, 0.82 MgSO4, 5 Tris-HCl; pH was adjusted to 7.4 with NaOH and the solution supplemented with 1 U/mL penicillin, 0.1 mg/mL gentamicin sulfate, and 1 μg/mL streptomycin. Two-electrode voltage-clamp recordings were performed at room temperature (21–23°C) using different internal solutions for voltage and current electrodes (0.3 M and 3.0 M KCl, respectively). All extracellular solutions were made from a solution containing (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, 0.01 EDTA (pH 7.4 with NaOH). Experiments were performed using an eight port Modular Valve Positioner (Hamilton Company, Reno, NV) and controlled by custom software (Easy Oocyte, Emory University, Atlanta, GA)to exchange solutions. Oocyte currents were recorded at a holding potential of −40 mV using a two-electrode voltage-clamp amplifier (OC-725B or OC-725C, Warner Instruments, Hamden, CT). For some recordings, 1 mM 2-(hydroxypropyl)-β-cyclodextrin was added to wash solutions to ensure modulators did not adhere to the tubing, valves, or recording chamber. In MTSEA experiments, fresh MTSEA solutions (0.2 mM) were prepared and utilized in less than 30 min to limit compound degradation.
Whole cell patch-clamp recordings
HEK293 cells (CRL-1573, ATCC, Manassas, VA) were maintained in a tissue culture room solely reserved for their culturing and transfected using a CaPO4 protocol to express the diheteromeric NMDARs as previously described (Hansen et al., 2013). Cell lines were obtained directly from the supplier, were expanded, frozen and kept in liquid nitrogen in a dewar. Mycoplasma testing was not performed. cDNA ratios for transfection were 1:1:1 (GluN2D) or 1:1:5 (GluN2A) for GluN1:GluN2:GFP. Cells were maintained in 5% CO2 at 37°C for 12–36 hr post-transfection. Prior to and during recording, cells were bathed in a solution containing (in mM) 150 NaCl, 10 HEPES, 3 KCl, 0.5 CaCl2, 0.01 EDTA (pH 7.4). Cells were patched with borosilicate glass micropipettes (3–4 MΩ) that were filled with internal recording solution that contained (in mM) 95 CsGluconate, 5 CsCl, 40 HEPES, 8 NaCl, 5 MgCl2, 10 BAPTA, 0.6 EGTA, 2 Na2ATP, and 0.3 NaGTP (pH 7.35). The whole cell recording conformation was achieved, and the cell lifted into the flow of solution from a two barreled theta glass or a triple barrel square glass perfusion system to perform rapid solution exchange experiments. The theta/triple barreled glass was translated using a piezoelectric manipulator to exchange the solution around the cell. Calibration of the perfusion manifold was performed each day to ensure the 10–90% rise time of the solution exchange (switching between 0/100% and 50/50% H2O/external solution) around an open tip was less than 1.5 ms for a theta tube and 4 ms for a transition into an outside lane and 2 ms for a transition between lanes for a triple barreled manifold. For some recordings, 1 mM 2-(hydroxypropyl)-β-cyclodextrin was added to wash solutions to ensure modulators did not adhere to the tubing, valves, or recording chamber.
Statistical analysis
Concentration-response curves for both positive and negative modulators were fitted by the Hill equation:
where I is the measured response of receptor activation, Extent is the maximal predicted modulation of the glutamate/glycine response, [M] is the concentration of the modulator being used, h is the Hill slope, and EC50 is the half-maximal effective concentration of the modulator. Modulator concentration response data were plotted as a percentage of unmodulated response and displayed fitted curves were obtained by fitting all data simultaneously.
Glutamate and glycine concentration-response curves were fit with the Hill equation as follows:
where I is the measured response of receptor activation, [A] is the concentration of the agonist, h is the Hill slope, and EC50 is the half-maximally effective concentration of the agonist. Agonist concentration-response data, unless otherwise stated, were plotted as a percentage of maximal response and displayed fitted curves were obtained by fitting all data simultaneously with the appropriate Hill equation.
The time course for the onset and offset of positive and negative modulation was fitted using a single exponential function:
where t is time, It is the difference in current at a given time point as compared to the response at t = 0, A is the amplitude of the exponential, τ is the time constant for the exponential and c is a constant. Additionally, some of the complex modulator time-courses were fitted with the sum of a single exponential equation and a linear component
where m is the slope.
Measurements are given as mean ± SEM unless otherwise indicated. The number of replicate experiments used was chosen to ensure a power level of at least 0.80 when α = 0.05 and when detecting appropriate effect sizes. EC50 and IC50 values and confidence intervals reported were calculated by averaging the log(EC50) or log(IC50) value, determining confidence intervals for mean log(EC50) or log(IC50), and converting back to units of molarity. Statistical significance evaluations (α set to 0.05) were performed using a one-way ANOVA with Dunnett’s multiple comparison test, a paired t–test, or other appropriate tests as described.
Modelling receptor function
Matlab (Mathworks, Natick, MA) scripts were written based on previously described approaches to modelling channel function (Colquhoun and Hawkes, 1977; Colquhoun and Hawkes, 1995). Briefly, a Q matrix was constructed for a model that accounts for macroscopic responses (Lester and Jahr, 1992). The glutamate association and dissociation rates were from (Erreger et al., 2005), and unidirectional rate constants for gating and modulator binding were chosen to allow the model to be used for modulator competition. The differential equations derived by the Q matrix were solved using an ordinary differential equation solver (ode23s, Matlab) and occupancy of each state was determined at equilibrium given the concentrations of the theoretical ligands.
Supplemental methods
Synthetic chemistry experimental details
All reagents were obtained from commercial suppliers and used without further purification unless specified otherwise, and were > 90% pure as determined by the manufacturer. Reaction progress was monitored by thin layer chromatography (TLC) on pre-coated glass plates (silica gel 60 F254, 0.25 mm) or liquid chromatography-mass spectrometry (LC-MS) using an analytical column (ZORBAX Eclipse XDB-C18, 4.6 x 50 mm, 3.5 µm, Agilent). When noted, flash chromatography was performed on a Teledyne (Lincoln, NE) ISCO Combiflash Companion with prepackaged Teledyne Redisep or Silicycle disposable normal phase silica columns. Proton and carbon NMR spectra were recorded on an INOVA-400 (400 MHz), VNMRS 400 (400 MHz), INOVA-600 (600 MHz), or Unity-600 (600 MHz). All chemical shifts are reported in parts per million and referenced to the residual solvent peak. Mass spectra were performed by the Emory University Mass Spectroscopy Center on either a VG 70-S Nier Johnson or JEOL instrument. The IR spectra were acquired with a Nicolet Avatar 370 DTGS. Final compound purity was established either through high-performance liquid chromatography (HPLC) under the conditions listed or by elemental analysis performed by Atlanta Microlab Inc. where C, H, N agreed with proposed structures within ±0.4% of theoretical values unless indicated otherwise.
Separation of EU1794-4 enantiomers
Semi-preparative separation of EU1794-4 enantiomers from racemic EU1794-4 (0.020 g) was accomplished using a ChiralPak OD-RH (21 mm × 250 mm, 5 μm) with the following conditions: 10.4 mL/min flow rate, 10 mL injection volume (2 mg/1 mL), 30% ACN/70% water with 0.1% formic acid over 120 min to afford (−)-EU1794-4, tR 62 min; (+)-EU1794-4, tR 65 min. The enantiomeric excess (ee) was determined using a ChiralPak OD-RH column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 5 μL injection volume, 30% ACN/70% water with 0.1% formic acid. (−)-EU1794-4: tR 29 min; 88% ee; [α]D20 = −185 (c 1.0, dry CHCl3). (+)-EU1794-4: tR 34 min, 73% ee; [α]D20 = +191 (c 1.0, dry CHCl3).
Separation of EU1794-27 enantiomers
Semi-preparative separation of EU1794-27 enantiomers from racemic EU1794-27 (0.020 g) was accomplished using a ChiralPak OD-RH (21 mm × 250 mm, 5 μm) with the following conditions: 10.4 mL/min flow rate, 10 mL injection volume (2 mg/1 mL), 60% ACN/40% water with 0.1% formic acid over 120 min to afford (−)-EU1794-27, tR 55 min; (+)-EU1794-27, tR 59 min. The enantiomeric excess (ee) was determined using a ChiralPak OD-RH column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 5 μL injection volume, 40% ACN/60% water with 0.1% formic acid. (−)-EU1794-27: tR 13 min; 88% ee; [α]D20 = −172 (c 1.0, dry CHCl3). (+)-EU1794-27: tR 15 min, 46% ee; [α]D20 = +178 (c 1.0, dry CHCl3).
General Procedure A: Preparation of aminothiophene compounds in alcohol solvent
To a solution of aldehyde/ketone (1.0 eq.) in alcohol (0.2-0.4 M, corresponding to the ester moiety for each reaction, see below) was added cyanoacetate (1.1 eq.), elemental sulfur (1.1 eq.), and morpholine (1.4 eq.). The reaction mixture was heated to 50 °C in an oil bath. When TLC or LC-MS indicated complete conversion (12-24 h.), the reaction mixture was concentrated in vacuo. The resulting residue was taken up in EtOAc and washed with water (2x) and brine (2x). The organic layer was dried over MgSO4 and filtered. The solvent was removed in vacuo. The resulting product was subjected to flash column chromatography.
General Procedure B: Preparation of aminothiophene compounds in DMF
To a solution of aldehyde/ketone (1.0 eq.) in DMF (1.0 M) was added cyanoacetate (1.1 eq.), elemental sulfur (1.1 eq.), and morpholine (1.4 eq.). The reaction mixture stirred at room temperature for 24 hours and then concentrated in vacuo. The resulting residue was taken up in EtOAc and washed with water (3x) and brine (3x). The organic layer was dried over MgSO4 and filtered. The solvent was removed in vacuo. The resulting product was subjected to flash column chromatography.
General Procedure C: Preparation of (Z)-4-oxo-4-(thiophen-2-ylamino)but-2-enoic acid compounds
A solution of the aminothiophene (1.0 eq.) in anhydrous ether was added dropwise to a solution of maleic anhydride (1.0 eq.) in anhydrous ether for a total concentration of 0.1-0.2 M. Reaction was allowed to stir at room temperature or refluxed until complete consumption of starting material was observed by TLC or LC-MS (16 h.-6 days). The resulting precipitate was filtered, washed with ether, and carried on without further purification.
General Procedure D: Preparation of 2-(2-imino-4-oxothiazolidin-5-yl)-N-(thiophen-2-yl)acetamide compounds from (Z)-4-oxo-4-(thiophen-2-ylamino)but-2-enoic acid using EDCI
To a solution of carboxylic acid (1 eq.) in dry DMF (0.15 M) was added HOBt (1.1 eq.) and EDCI (1.5 eq.). The reaction mixture was stirred at room temperature for 1 hour or until all materials were in solution. To the solution was added thiourea (1.1 eq.). The reaction mixture was allowed to stir at room temperature for an additional hour and then heated to 60 °C for 24 hours. The reaction mixture was poured into EtOAc and washed with water (2x) and brine (2x). The organic extracts were combined, dried over anhydrous MgSO4 and concentrated in vacuo. Crude material was dissolved in hot methanol or ethanol and then slowly cooled to room temperature. The resulting precipitate was filtered and washed with cold solvent.
General Procedure E: Preparation of aryl maleimide compounds
A mixture of carboxylic acid (1.0 eq.), acetic anhydride (3.3-7.0 eq.), and sodium acetate (0.2 eq.) was heated to 75 °C in an oil bath. After consumption of starting material, reaction mixture was cooled to room temperature where it stirred for 12-18 hours. The reaction was diluted with water and extracted with DCM (3x). The organic extracts were dried over MgSO4, filtered, and concentrated. The resulting product was subjected to flash column chromatography.
General Procedure F: Preparation of 2-(2-imino-4-oxothiazolidin-5-yl)-N-(thiophen-2-yl)acetamide compounds from aryl maleimides
A mixture of maleimide (1.0 eq.) and thiourea (1.0 eq.) in absolute EtOH (0.1 M) stirred at room temperature for 24-48 hours. Reaction mixture was filtered, and solid material was washed with EtOH and DCM.
Chemical structure 1. ethyl 2-amino-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11b).
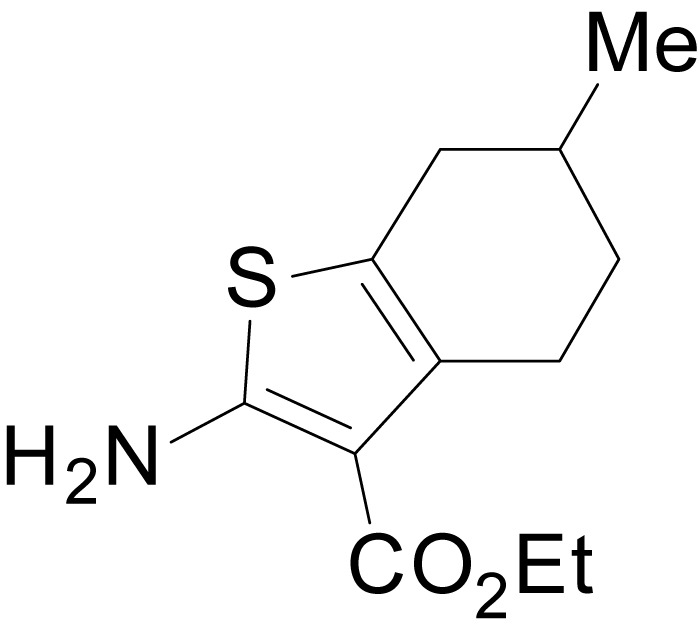
Compound 11b was prepared according to procedure A using 4-methylcyclohexanone (1.6 mL, 13.4 mmol) and ethyl 2-cyanoacetate (1.6 mL, 14.7 mmol) in absolute ethanol (0.2-0.4 M). The crude material was purified by flash column chromatography (ISCO, 120 g silica gel column, 0-10% EtOAc-Hexanes over 25 minutes) to afford the title compound as a white crystalline solid (3.14 g, 98%). 1H NMR [400 MHz, CDCl3] δ 5.92 (br s, 2H), 4.26 (q, J = 7.0 Hz, 2H), 2.85-2.90 (m, 1H), 2.53-2.63 (m, 2H), 2.11-2.17 (m, 1H), 1.81-1.88 (m, 2H), 1.29-1.37 (m, 4H), 1.05 (d, J = 6.4 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 166.3, 162.0, 132.3, 117.4, 105.8, 59.5, 32.8, 31.3, 29.7, 26.9, 21.7, 14.7; HRMS (ESI) [M+H]+, calc’d for C12H18NO2S 240.10528, found 240.10521.
Chemical structure 2. ethyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11c).
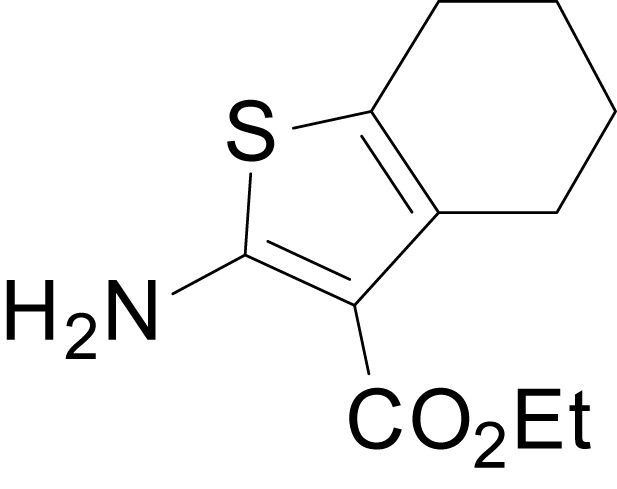
Compound 11c was prepared according to procedure A using cyclohexanone (5.3 mL, 50.9 mmol) and ethyl 2-cyanoacetate (6.0 mL, 56.0 mmol) in absolute ethanol (0.2-0.4 M). The crude material was recrystallized from absolute ethanol to afford the title compound as light yellow crystals (10.0 g, 87%). 1H NMR [400 MHz, CDCl3] δ 5.96 (br s, 2H), 4.26 (q, J = 7.1 Hz, 2H), 2.73-2.69 (m, 2H), 2.52- 2.48 (m, 2H), 1.81-1.71 (m, 4H), 1.34 (t, J = 7.2 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 166.3, 161.9, 132.6, 117.8, 105.9, 59.5, 27.1, 24.7, 23.4, 23.0, 14.7; HRMS (ESI) [M+H]+, calc’d for C11H16NO2S 226.08963, found 226.08966; IR (solid ATR): 3400, 3294, 2982, 2939, 2895, 1644, 1593, 1488, 1271 cm-1.
Chemical structure 3. isopropyl 2-amino-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11f).
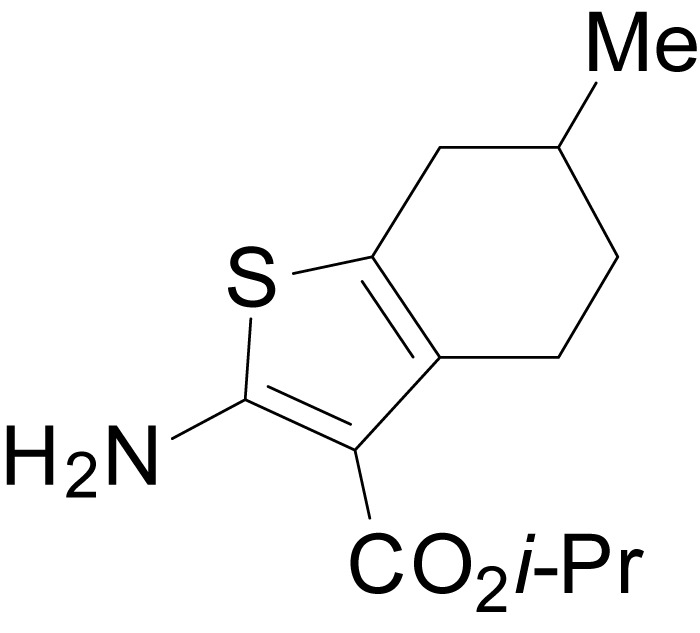
Compound 11f was prepared according to procedure A using 4-methylcyclohexanone (2.2 mL, 17.8 mmol) and isopropyl 2-cyanoacetate (2.5 mL, 19.6 mmol) in isopropanol (0.2-0.4 M). The crude material was purified by flash column chromatography (ISCO, 80 g silica gel column, 0-20% EtOAc-Hexanes over 25 minutes) to afford the title compound as an off-white solid (4.42 g, 98%). 1H NMR [400 MHz, CDCl3] δ 5.96 (br s, 2H), 5.16 (septet, J = 6.3 Hz, 1H), 2.84-2.91 (m, 1H), 2.52-2.65 (m, 2H), 2.10-2.18 (m, 1H), 1.80-1.90 (m, 2H), 1.25-1.35 (m, 1H), 1.32 (d, J = 6.4 Hz, 3H), 1.31 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.7 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 165.9, 161.9, 132.3, 117.4, 106.1, 66.8, 32.9, 31.3, 29.6, 27.0, 22.4, 21.7; HRMS (ESI) [M+H]+, calc’d for C13H20NO2S 254.12093, found 254.12052.
Chemical structure 4. isopropyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11h).
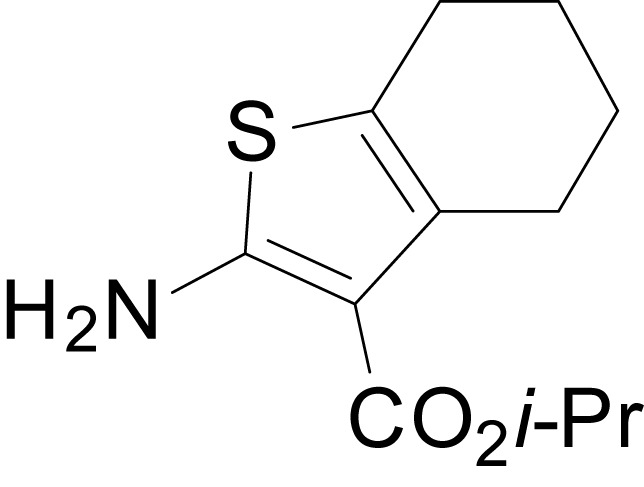
Compound 11h was prepared according to procedure A using cyclohexanone (2.1 mL, 20.4 mmol) and isopropyl 2-cyanoacetate (2.8 mL, 22.4 mmol) in isopropanol (0.2-0.4 M). The crude material was purified by flash column chromatography (ISCO, 120 g silica gel column, 0-10% EtOAc-Hexanes over 25 minutes) to afford the title compound as a white solid (4.12 g, 84%). 1H NMR [400 MHz, CDCl3] δ 6.12 (br s, 2H), 5.16 (septet, J = 6.2 Hz, 1H), 2.69-2.71 (m, 2H), 2.46-2.48 (m, 2H), 1.72-1.77 (m, 4H), 1.31 (d, J = 6.2 Hz, 6H); 13C NMR [100 MHz, CDCl3] δ 165.7, 162.0, 132.3, 117.4, 105.6, 66.6, 27.0, 24.5, 23.2, 22.9, 22.1; HRMS (ESI) [M+H]+, calc’d for C12H18NO2S 240.10528, found 240.10549; Elem. Anal. calc’d for C12H17NO2S, C, 60.22; H, 7.16; N, 5.85, found C, 60.49; H, 7.26; N, 5.79.
Chemical structure 5. tert-butyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11k).
Compound 11k was prepared according to procedure A using cyclohexanone (1.6 mL, 15.3 mmol) and tert-butyl 2-cyanoacetate (2.4 mL, 16.8 mmol) in absolute ethanol (40.0 mL). The crude material was purified by flash column chromatography (ISCO, 80 g silica gel, 0-20% EtOAc-Hexanes over 25 minutes) to afford the title compound as a yellow oil (2.45 g, 63%). 1H NMR [400 MHz, CDCl3] δ 5.83 (br s, 2H), 2.67-2.64 (m, 2H), 2.49-2.46 (m, 2H), 1.76-1.69 (m, 4H), 1.52 (s, 9H); 13C NMR [100 MHz, CDCl3] δ 165.8, 161.2, 132.7, 117.7, 107.4, 80.2, 28.8, 27.4, 24.8, 23.5, 23.1; HRMS (ESI) [M+H]+, calc’d for C13H20NO2S 254.12093, found 254.12087; IR (solid ATR): 3434, 3324, 2932, 1732, 1660, 1576, 1484, 1141 cm−1.
Chemical structure 6. butyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11l).
Compound 11l was prepared according to procedure B using cyclohexanone (1.2 mL, 11.6 mmol) and butyl 2-cyanoacetate (1.8 mL, 12.8 mmol). The crude material was purified by flash column chromatography (ISCO, 80 g silica gel, 0-10% EtOAc-Hexanes over 20 minutes) to afford the title compound as a yellow oil (2.35 g, 80%). 1H NMR [400 MHz, CDCl3] δ 5.96 (br s, 2H), 4.21 (t, J = 6.6 Hz, 2H), 2.72-2.69 (m, 2H), 2.52-2.49 (m, 2H), 1.82-1.66 (m, 6H), 1.45 (sextet, J = 7.4 Hz, 2H), 0.96 (t, J = 7.3 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 166.5, 161.9, 132.6, 117.8, 106.0, 63.5, 31.1, 27.2, 24.7, 23.4, 23.1, 19.6, 114.0; HRMS (ESI) [M+H]+, calc’d for C13H20NO2S 254.12093, found 254.12061; IR (solid ATR): 3433, 3325, 2932, 2857, 1664, 1576, 1485, 1265 cm-1.
Chemical structure 7. benzyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11m).
Compound 11m was prepared according to procedure B using cyclohexanone (0.7 mL, 6.5 mmol) and benzyl 2-cyanoacetate (1.1 mL, 7.1 mmol). The crude material was purified by flash column chromatography (ISCO, 40 g silica gel, 0-20% EtOAc-Hexanes over 15 minutes) to afford the title compound as a yellow tinted oil (1.38 g, 74%). 1H NMR [400 MHz, CDCl3] δ 7.43-7.33 (m, 5H), 6.00 (br s, 2H), 5.29 (s, 2H), 2.74-2.71 (m, 2H), 2.52-2.49 (m, 2H), 1.81-1.68 (m, 4H); 13C NMR [100 MHz, CDCl3] δ 166.0, 162.4, 136.9, 132.5, 128.7, 128.1, 128.1, 117.8, 105.5, 65.4, 27.2, 24.7, 23.4, 23.0; HRMS (ESI) [M+H]+, calc’d for C16H18NO2S 288.10528, found 288.10519; IR (solid ATR): 3442, 3325, 2980, 2934, 1659, 1575, 1483, 1261 cm-1.
Chemical structure 8. (Z)-4-((3-(ethoxycarbonyl)-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)amino)-4-oxobut-2-enoic acid (13b).
Compound 13b was prepared according to procedure C using compound 11b (2.6 g, 10.7 mmol). The crude yellow solid (3.54 g, 98%) was carried on without further purification. 1H NMR [400 MHz, CDCl3] δ 12.22 (s, 1H), 6.51 (d, J = 12.8 Hz, 1H), 6.45 (d, J = 12.8 Hz, 1H), 4.38 (q, J = 7.0 Hz, 2H), 2.95-2.99 (m, 1H), 2.78 (dd, J = 4.9, 15.9 Hz, 1H), 2.66-2.75 (m, 1H), 2.29-2.35 (m, 1H), 1.89-1.93 (m, 2H), 1.42 (t, J = 7.3 Hz, 4H), 1.09 (d, J = 6.7 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 166.9, 164.3, 162.1, 144.4, 137.8, 132.1, 130.8, 130.0, 115.0, 61.6, 32.8, 31.0, 29.3, 26.3, 21.5, 14.4; HRMS (ESI) [M+H]+, calc’d for C16H20NO5S, 338.10567, found 338.10550.
Chemical structure 9. (Z)-4-((3-(ethoxycarbonyl)-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)amino)-4-oxobut-2-enoic acid (13c).
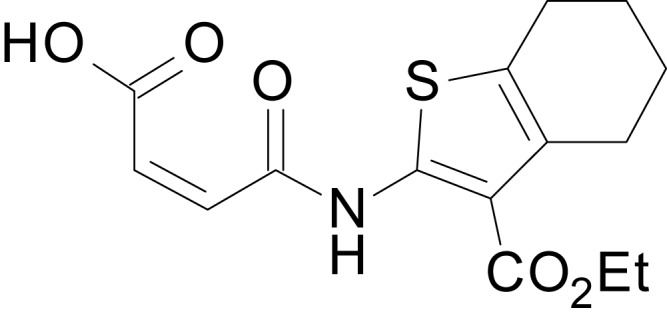
Compound 13c was prepared according to procedure C using compound 11c (3.0 g, 13.3 mmol). The crude yellow solid (4.02 g, 93%) was carried on without further purification. 1H NMR [400 MHz, CDCl3] δ 12.18 (br s, 1H), 6.49 (d, J = 12.8 Hz, 1H), 6.45 (d, J = 12.8 Hz, 1H,), 4.36 (q, J = 7.1 Hz, 2H), 2.78 (app. t, J = 5.2 Hz, 2H), 2.69 (app. t, J = 4.9 Hz, 2H), 1.78-1.83 (m, 4H), 1.40 (t, J = 7.3 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 166.8, 164.3, 162.0, 144.2, 137.7, 132.3, 131.0, 130.0, 115.1, 61.5, 26.4, 24.7, 22.9, 22.7, 14.4; HRMS (ESI) [M+H]+, calc’d for C15H18NO5S 324.09002, found 324.08965.
Chemical structure 10. (Z)-4-((3-(isopropoxycarbonyl)-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)amino)-4-oxobut-2-enoic acid (13f).
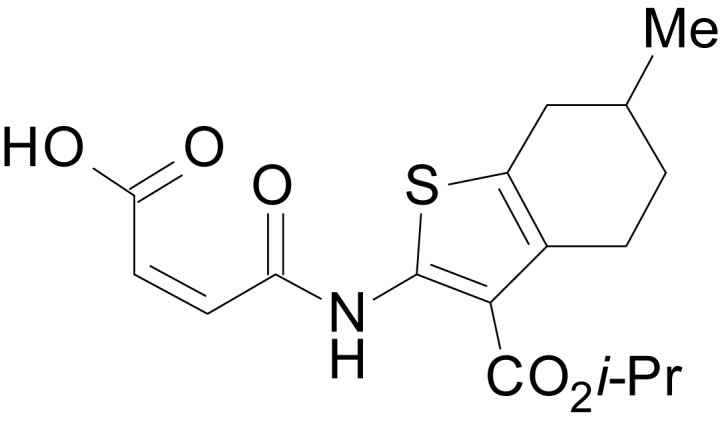
Compound 13f was prepared according to procedure C using compound 11f (2.0 g, 7.9 mmol). The crude yellow solid (2.69 g, 97%) was carried on without further purification. 1H NMR [400 MHz, CDCl3] δ 12.25 (br s, 1H), 6.49 (d, J = 12.8 Hz, 1H), 6.45 (d, J = 12.8 Hz, 1H), 5.22 (septet, J = 6.2 Hz, 1H), 2.93-2.97 (m, 1H), 2.64-2.79 (m, 2H), 2.26-2.33 (m, 1H), 1.84-1.92 (m, 2H), 1.29-1.43 (m, 7H), 1.08 (d, J = 6.1 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 166.4, 164.3, 162.0, 144.2, 137.8, 132.0, 130.7, 130.1, 115.3, 69.5, 32.7, 31.0, 29.3, 26.3, 22.1, 21.5; HRMS (ESI) [M–H]-, calc’d for C17H22NO5S 350.10677, found 350.10666.
Chemical structure 11. (Z)-4-((3-(isopropoxycarbonyl)-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)amino)-4-oxobut-2-enoic acid (13h).
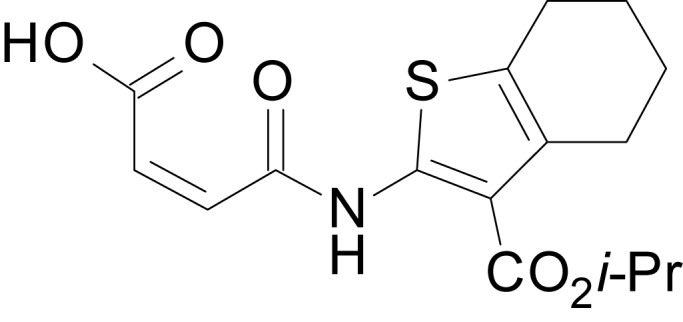
Compound 13h was prepared according to procedure C using compound 11h (1.5 g, 6.3 mmol). The crude yellow solid (2.01 g, 95%) was carried on without further purification. 1H NMR [600 MHz, CDCl3] δ 12.27 (br s, 1H), 6.50 (d, J = 12.8 Hz, 1H), 6.46 (d, J = 12.8 Hz, 1H), 5.23 (septet, J = 6.3 Hz, 1H), 2.79-2.80 (m, 2H), 2.69-2.70 (m, 2H), 1.81-1.82 (m, 4H), 1.39 (d, J = 6.0 Hz, 6H); 13C NMR [150 MHz, CDCl3] δ 166.5, 164.3, 162.1, 144.1, 137.8, 132.4, 131.1, 130.1, 115.5, 69.5, 26.6, 24.7, 22.9, 22.8, 22.2; HRMS (ESI) [M+H]+, calc’d for C16H20NO5S 338.10567, found 338.10578.
Chemical structure 12. (Z)-4-((3-(tert-butoxycarbonyl)-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)amino)-4-oxobut-2-enoic acid (13k).
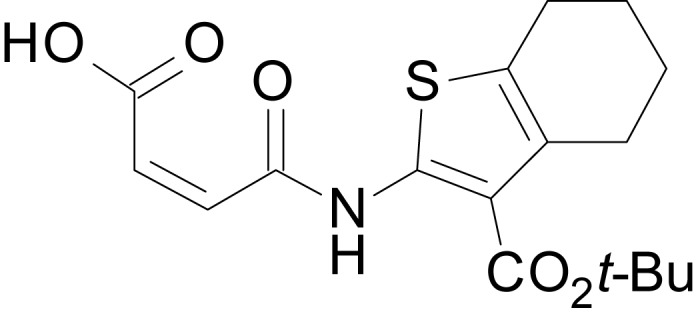
Compound 13k was prepared according to procedure C using compound 11k (2.4 g, 9.6 mmol). The crude yellow solid (2.88 g, 86%) was carried on without further purification. HRMS (ESI) [M+H]+, calc’d for C17H22NO5S 352.12132, found 352.12107.
Chemical structure 13. ethyl 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-6-methyl-4,5,6,7- tetrahydrobenzo[b]thiophene-3-carboxylate (18b).
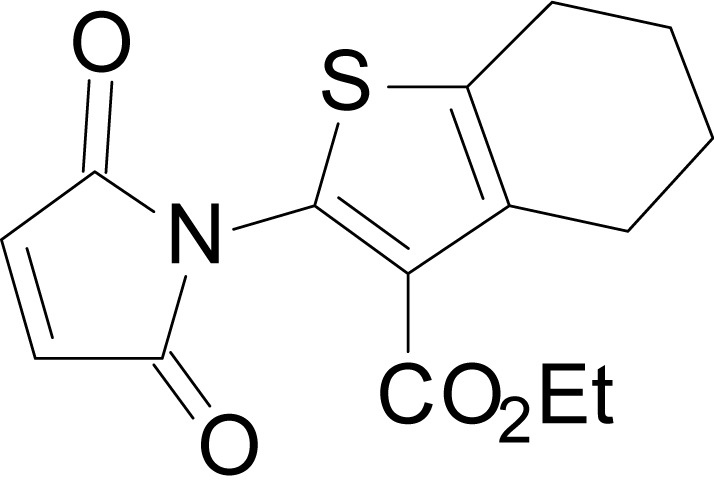
Compound 18b was prepared according to procedure E using compound 13b (1.0 g, 3.0 mmol). The crude material was purified by flash column chromatography (ISCO, 24 g silica gel, 0-18% EtOAc-Hexanes over 15 minutes) to afford the title compound as a yellow oil that solidified upon standing (0.769 g, 81%). HRMS (APCI) [M+H]+, calc’d for C16H18NO4S 320.09511, found 320.09520.
Chemical structure 14. isopropyl 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (18h).
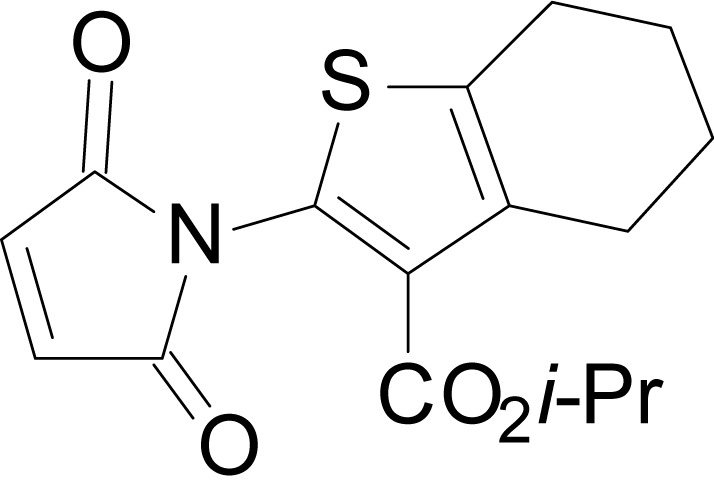
Compound 18h was prepared according to procedure E using compound 13h (0.8 g, 2.4 mmol). The crude material was purified by flash column chromatography (ISCO, 24 g silica gel, 0-25% EtOAc-Hexanes over 20 minutes) to afford the title compound as a golden brown oil (0.712 g, 94%). 1H NMR [400 MHz, CDCl3] δ 6.91 (s, 2H), 5.09 (septet, J = 5.3 Hz, 1H), 2.84 (app. t, J = 5.8 Hz, 2H), 2.75 (app. t, J = 5.5 Hz, 2H), 1.80-1.86 (m, 4H), 1.20 (d, J = 6.1Hz, 6H); HRMS (APCI) [M+H]+, calc’d for C16H18NO4S 320.09511, found 320.09527.
Chemical structure 15. tert-butyl 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (18k).
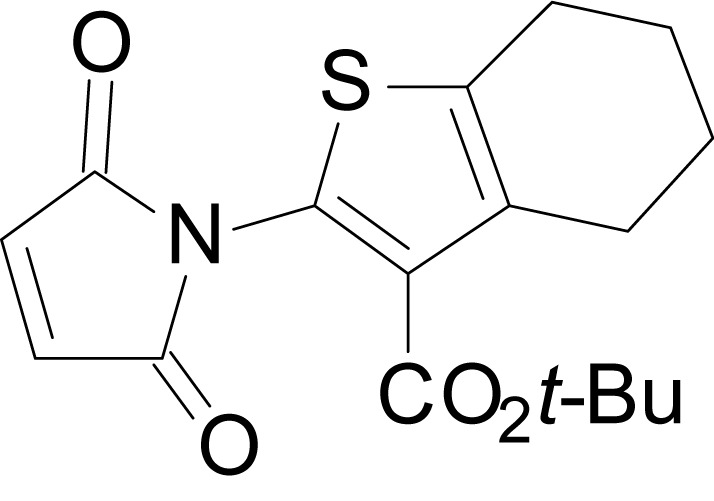
Compound 18k was prepared according to procedure E using compound 13k (2.0 g, 5.7 mmol). The crude material was purified by flash column chromatography (ISCO, 40 g silica gel, 0-18% EtOAc-Hexanes over 15 minutes) to afford the title compound as a light yellow solid (1.44 g, 76%). 1H NMR [400 MHz, CDCl3] δ 6.90 (s, 2H), 2.83-2.79 (m, 2H), 2.75-2.72 (m, 2H), 1.86-1.76 (m, 4H), 1.44 (s, 9H); 13C NMR [100 MHz, CDCl3] δ 169.1 (2C), 161.4, 136.6, 135.7, 135.1 (2C), 131.6, 130.4, 81.6, 28.4 (3C), 26.6, 25.4, 22.9, 22.6; HRMS (APCI) [M–Ot-Bu]+, calc’d for C13H10NO3S 260.03759, found 260.03737; IR (solid ATR): 2918, 1709, 1565, 1410, 1393, 1367, 1136, 691 cm−1.
Chemical structure 16. butyl 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-4,5,6,7-tetrahydrobenzo[b]thiophene-3- carboxylate (18l).
Compound 18l was prepared according to procedure E using compound 13l (1.2 g, 3.4 mmol). The crude material was purified by flash column chromatography (ISCO, 40 g silica gel, 0-18% EtOAc-Hexanes over 15 minutes) to afford the title compound as a light yellow solid (1.04 g, 91%). 1H NMR [400 MHz, CDCl3] δ 6.91 (s, 2H), 4.13 (t, J = 6.7 Hz, 2H), 2.84 (app. t, J = 5.8 Hz, 2H), 2.75 (app. t, J = 5.8 Hz, 2H), 1.87-1.78 (m, 4H), 1.57 (p, J = 7.2 Hz, 2H), 1.34 (sextet, J = 7.5 Hz, 2H), 0.91 (t, J = 7.5 Hz, 3H); 13C NMR [100 MHz, CDCl3] δ 169.2 (2C), 162.21, 136.8, 135.9, 135.1 (2C), 133.0, 128.7, 64.7, 30.8, 26.6, 25.4, 22.8, 22.6, 19.3, 13.9; 62 HRMS (APCI) [M+H]+, calc’d for C17H20NO4S 334.11076, found 334.11128; IR (solid ATR): 3097, 2944, 1710, 1414, 1364, 1175, 836, 693 cm−1.
Chemical structure 17. benzyl 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-4,5,6,7-tetrahydrobenzo[b]thiophene-3- carboxylate (18m).
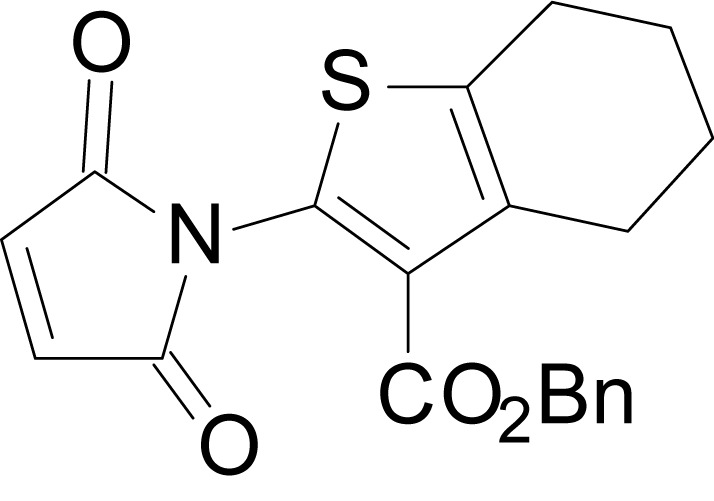
Compound 18m was prepared according to procedure E using compound 13m (0.4 g, 1.0 mmol). The crude material was purified by flash column chromatography (ISCO, 24 g silica gel, 0-25% EtOAc-Hexanes over 15 minutes) to afford the title compound as a light yellow solid (0.299 g, 78%). 1H NMR [400 MHz, CDCl3] δ 7.40-7.21 (m, 5H), 6.44 (s, 2H), 5.07 (s, 2H), 2.91-2.78 (m, 2H), 2.75-2.64 (m, 2H), 1.88-1.71 (m, 4H); 13C NMR [100 MHz, CDCl3] δ 169.2 (2C), 161.6, 137.0, 136.3, 135.4, 134.2 (2C), 133.3, 129.4 (2C), 128.8 (2C), 128.7, 128.3, 67.0, 26.4, 25.4, 22.8, 22.5; HRMS (APCI) [M+H]+, calc’d for C20H18NO4S 368.09511, found 368.09512; IR (solid ATR): 2942, 2850, 1763, 1672, 1638, 1537, 1176, 1137, 691 cm−1.
Chemical structure 18. (Z)-4-((3-((benzyloxy)carbonyl)-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)amino)-4-oxobut-2-enoic acid (13m).
Compound 13m was prepared according to procedure C using compound 11m (0.6 g, 2.1 mmol). The crude yellow solid (0.573 g, 70%) was carried on without further purification. HRMS (ESI) [M+H]+, calc’d for C20H20NO5S 386.10567, found 386.10587.
Chemical structure 19. ethyl 2-(2-(2-imino-4-oxothiazolidin-5-yl)acetamido)-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (EU1794-2).
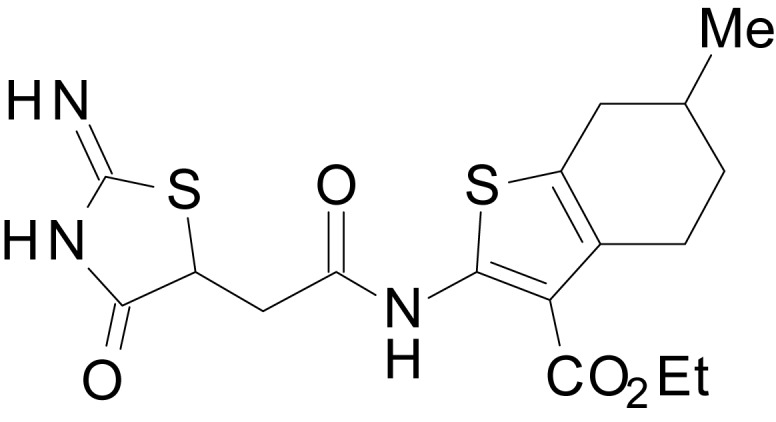
Compound EU1794-2 was prepared according to procedure D using compound 13b (0.3 g, 0.7 mmol). The crude material was precipitated from hot ethanol to afford the title compound (inseparable mixture of diastereomers) as a light yellow solid (0.0779 g, 27%). Compound EU1794-2 was also prepared according to procedure F using compound 18b (0.4 g, 1.3 mmol). The precipitate was rinsed with DCM to afford the title compound (inseparable mixture of diastereomers) as an off-white solid (0.406 g, 82%). 1H NMR [400 MHz, DMSO-d6] δ 10.98 (s, 1H), 9.03 (s, 1H), 8.80 (s, 1H), 4.42 (dd, J = 3.7, 10.4 Hz, 1H), 4.27 (q, J = 6.9 Hz, 2H), 2.99 (dd, J = 10.4, 15.3 Hz, 1H), 2.83-2.88 (m, 1H), 2.59-2.70 (m, 2H), 2.16-2.22 (m, 1H), 1.80 (m, 2H), 1.30 (t, J = 7.0 Hz, 4H), 1.01 (d, J = 6.7 Hz, 4H); 13C NMR [100 MHz, DMSO-d6] (13C resonance obstructed by solvent peak) δ 188.5, 182.0, 167.8, 164.7, 145.4, 130.2, 125.9, 111.7, 60.4, 51.4, 31.7, 30.5, 28.7, 25.6, 21.2, 14.1; HRMS (ESI) [M+H]+, calc’d for C13H16N3O4S2, 342.05768, found 342.05759; IR (solid ATR): 3224, 2927, 1673, 1562, 1534, 1496, 1460, 1435, 1376, 1284, 1257, 1231, 1166, 1142, 1081, 1027, 960, 843, 775, 741, 698, 636 cm-1; m.p. = 190-204 °C (dec.); Elem. Anal. calc’d for C13H15N3O4S2, C, 45.73; H, 4.43; N, 12.31, found C, 45.88; H, 4.46; N, 11.93.
Chemical structure 20. ethyl 2-(2-(2-imino-4-oxothiazolidin-5-yl)acetamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (EU1794-4).
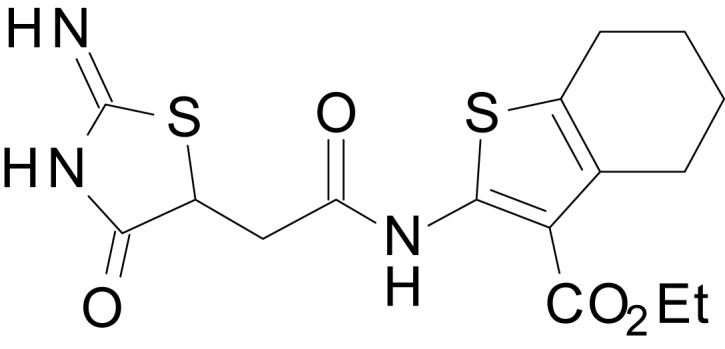
Compound EU1794-4 was prepared according to procedure D using compound 13c (0.4 g, 1.2 mmol). The crude material was precipitated from hot ethanol to afford the title compound as a light yellow solid (0.281 g, 60%). 1H NMR [400 MHz, DMSO-d6] δ 11.00 (s, 1H), 9.06 (s, 1H), 8.82 (s, 1H), 4.43 (dd, J = 3.9, 10.2 Hz, 1H), 4.28 (q, J = 7.0 Hz, 2H), 3.36-3.42 (m, 1H), 3.01 (dd, J = 10.2, 16.8, 1H), 2.69 (s, 2H), 2.59 (s, 2H), 1.72 (s, 4H), 1.31 (t, J = 7.0 Hz, 3H); 13C NMR [100 MHz, DMSO-d6] (13C resonance obstructed by solvent peak) δ 188.5, 182.0, 167.8, 164.7, 145.3, 130.5, 126.3, 111.8, 60.3, 51.4, 25.8, 23.7, 22.5, 22.3, 14.1; HRMS (ESI) [M+H]+, calc’d for C16H20N3O4S2, 382.08952, found 382.08902; IR (solid ATR): 3661, 3269, 3195, 2933, 1655, 1563, 1527, 1439, 1408, 1366, 1326, 1231, 1134, 1032, 996, 969, 784, 703, 629 cm−1; m.p. = 163 °C (dec.); Elem. Anal. calc’d for C16H19N3O4S2, C, 50.38; H, 5.02; N, 11.02, found C, 49.31; H, 5.00; N, 10.73 (Value did not fall within ± 0.4).
Chemical structure 21. isopropyl 2-(2-(2-imino-4-oxothiazolidin-5-yl)acetamido)-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (EU1794-19).
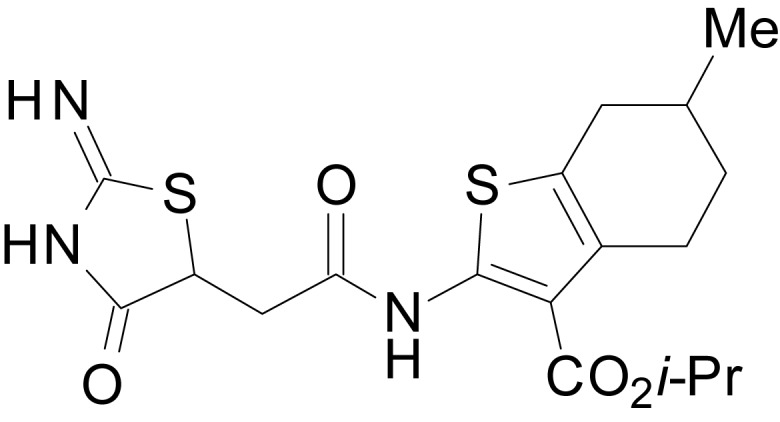
Compound EU1794-19 was prepared according to procedure D using compound 13f (0.7 g, 2.0 mmol). The crude material was precipitated from hot ethanol to afford the title compound as a white solid (0.134 g, 16%). 1H NMR [400 MHz, DMSO-d6] δ 11.00 (s, 1H), 9.04 (s, 1H), 8.81 (s, 1H), 4.43 (dd, J = 3.8, 10.2 Hz, 1H), 4.29 (q, J = 7.1 Hz, 2H), 3.39 (dd, J = 3.8, 16.9 Hz, 1H), 3.00 (dd, J = 10.4, 16.8 Hz, 1H), 2.71 (app. t, J = 5.9 Hz, 2H), 2.38 (app. s, 2H), 1.48 (app. t, J = 6.3 Hz, 2H), 1.32 (t, J = 7.0 Hz, 3H), 0.95 (s, 6H).; 13C NMR [100 MHz, DMSO-d6] δ 188.5, 181.9, 167.7, 164.7, 145.5, 129.1, 125.6, 111.5, 60.4, 51.4, 39.2, 37.1, 35.0, 29.7, 27.4, 27.3, 23.5, 14.0; HRMS (ESI) [M+H]+, calc’d for C16H24N3O4S2 410.12028, found 410.12066; IR (solid ATR): 3205, 2923, 1655, 1561, 1531, 1492, 1386, 1293, 1233, 1148, 1106, 1074, 1011, 979, 960, 912, 835, 783, 761, 736, 698, 645 cm-1; m.p. = 229 °C (dec.); HPLC purity: >95% tR = 1.300 min. (85% MeOH/Water with 0.1% formic acid, isocratic, 1.0 mL/min.); tR = 0.995 min. (75% MeCN/Water with 0.1% formic acid, isocratic, 1.0 mL/min.).
Chemical structure 22. isopropyl 2-(2-(2-imino-4-oxothiazolidin-5-yl)acetamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (EU1794-5).
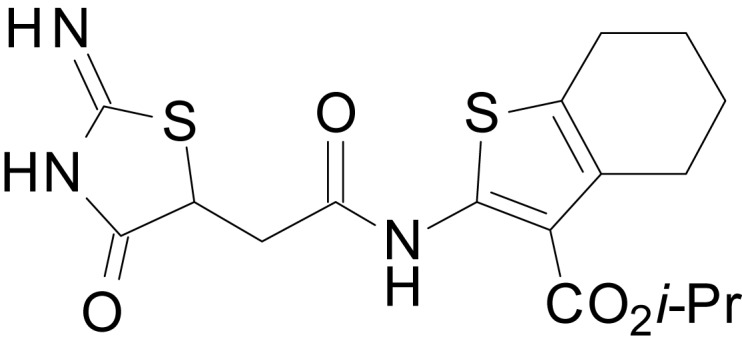
To a solution of 13h (0.2 g, 0.6 mmol) dissolved in dry DMF (5.0 mL) was added HATU (0.2 g, 0.6 mmol). The reaction mixture was allowed to stir at room temperature for 10 minutes. To the stirring mixture was added thiourea (0.1 g, 1.5 mmol) and DIPEA (0.5 mL, 3.0 mmol). The reaction was allowed to stir an additional 30 minutes before heating to 60 °C for 72 hours. The solvent was removed in vacuo. The crude material was purified by flash column chromatography (ISCO, 12 g silica gel column, 3-11% MeOH-DCM over 15 minutes). The resulting fractions were found to contain impurities. Material was re-purified by flash column chromatography (12 g silica gel column, 0-8% MeOH-DCM over 15 minutes) to afford the title compound as a light brown solid (0.0551 g, 23%). Compound EU1794-5 was also prepared according to procedure F using 18h (0.2 g, 0.7 mmol). The precipitate was rinsed with DCM to afford the title compound as a white solid (0.119 g, 42%). 1H NMR [400 MHz, DMSO-d6] δ 11.04 (s, 1H), 9.04 (s, 1H), 8.81 (s, 1H), 5.10 (septet, J = 6.3 Hz, 1H), 4.42 (dd, J = 3.9, 10.2 Hz, 1H), 3.40 (d, J = 3.5 Hz, 1H), 3.00 (dd, J = 10.2 16.8, 1H), 2.69 (br s, 2H), 2.58 (br s, 2H), 1.71 (br s, 4H), 1.31 (d, J = 6.3 Hz, 6H); 13C NMR [100 MHz, DMSO-d6] (13C resonance obstructed by solvent peak) δ 188.5, 181.9, 167.7, 164.4, 145.4, 130.4, 126.3, 112.0, 68.0, 51.4, 25.9, 23.7, 22.5, 22.3, 21.7 (2C); HRMS (ESI) [M+H]+, calc’d for C17H22N3O4S2 396.10463, found 396.10409; m.p. = 152-154 °C; HPLC purity: >95% tR = 2.082 min. (75-95% MeOH/Water with 0.1% formic acid, gradient over 3 minutes, 1.0 mL/min.); tR = 0.698 min. (85% MeCN/Water with 0.1% formic acid, isocratic, 1.0 mL/min.).
Chemical structure 23. tert-butyl 2-(2-(2-imino-4-oxothiazolidin-5-yl)acetamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (EU1794-27).
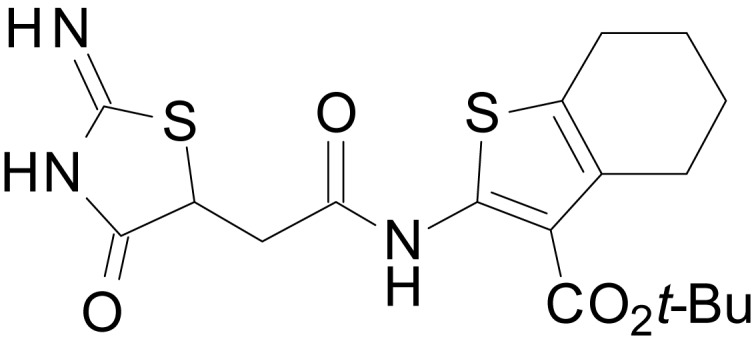
Compound EU1794-27 was prepared according to procedure F using compound 18k (0.5 g, 1.5 mmol). The precipitate was rinsed with EtOH and EtOAc to afford the title compound as a light yellow solid (0.403 g, 66%). 1H NMR [400 MHz, DMSO-d6] δ 11.03 (s, 1H), 9.03 (s, 1H), 8.80 (s, 1H), 4.43 (dd, J = 4.0, 10.2 Hz, 1H), 3.38 (dd, J = 3.7, 16.8 Hz, 1H), 3.00 (dd, J = 10.4, 16.8 Hz, 1H), 2.68 (app. br s, 2H), 2.58 (app. br s, 2H), 1.70 (app. br s, 4H), 1.54 (s, 9H); 13C NMR [100 MHz, DMSO-d6] (13C resonance obstructed by solvent peak) δ 188.5, 182.0, 167.7, 164.3, 145.2, 130.5, 126.1, 112.8, 81.5, 51.4, 28.0 (3C), 26.1, 23.8, 22.5, 22.4; HRMS (ESI) [M+H]+, calc’d for C18H24N3O4S2 410.12027, found 410.12011; IR (solid ATR): 3372, 2919, 1705, 1565, 1524, 1367, 1240, 1138 cm−1; HPLC purity: >95% tR = 0.765 min. (95% MeOH/Water with 0.1% formic acid, isocratic, 1.0 mL/min.).
Chemical structure 24. butyl 2-(2-(2-imino-4-oxothiazolidin-5-yl)acetamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (EU1794-29).
Compound EU1794-29 was prepared according to procedure F using compound 18l (0.5 g, 1.5 mmol). The precipitate was rinsed with EtOH to afford the title compound as a light yellow solid (0.325 g, 53%). 1H NMR [400 MHz, DMSO-d6] δ 11.38 (s, 1H), 10.05 (br s, 1H), 7.53 (s, 1H), 4.55 (dd, J = 3.2, 11.1 Hz, 1H), 4.24 (t, J = 6.6 Hz, 2H), 3.54 (dd, J = 3.1, 16.8 Hz, 1H), 2.96 (dd, J = 11.3, 16.8 Hz, 1H), 2.72 (app. br s, 2H), 2.57 (app. br s, 2H), 1.76 (app. br s, 4H), 1.70 (p, J = 7.2 Hz, 2H), 1.44 (sextet, J = 7.4 Hz, 2H), 0.96 (t, J = 7.3 Hz, 3H); 13C NMR [100 MHz, DMSO-d6] δ 189.1, 184.3, 167.0, 166.7, 146.9, 131.0, 127.2, 112.2, 64.7, 52.0, 40.2, 30.9, 26.5, 24.5, 23.1, 23.0, 19.6, 13.9; HRMS (ESI) [M+H]+, calc’d for C18H24N3O4S2 410.12027, found 410.11979; IR (solid ATR): 3307, 2939, 1650, 1567, 1521, 1414, 1245, 1115 cm-1; HPLC purity: >95% tR = 1.479 min. (85% MeOH/Water with 0.1% formic acid, isocratic, 1.0 mL/min.).
Chemical structure 25. benzyl 2-(2-(2-imino-4-oxothiazolidin-5-yl)acetamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (EU1794-25).
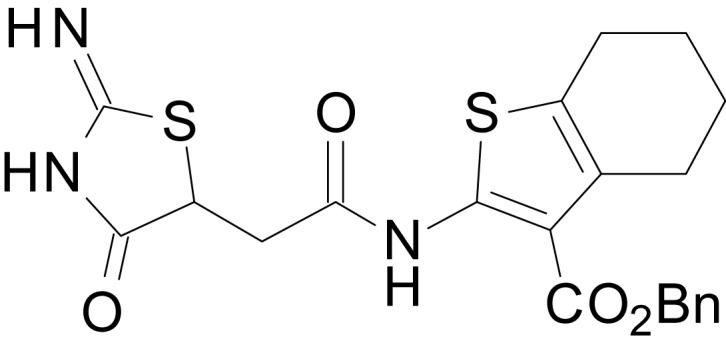
Compound EU1794-25 was prepared according to procedure F using compound 18m (0.3 g, 0.7 mmol). The precipitate was rinsed with DCM to afford the title compound as an off-white solid (0.124 g, 41%). 1H NMR [400 MHz, DMSO-d6] δ 10.98 (s, 1H), 9.04 (s, 1H), 8.80 (s, 1H), 7.49-7.29 (m, 5H), 5.33 (s, 2H), 4.42 (dd, J = 3.8, 10.2 Hz, 1H), 3.37 (dd, J = 4.0, 16.8 Hz, 1H), 2.96 (dd, 1H, J = 10.2, 16.9 Hz, 1H), 2.72-2.64 (m, 2H), 2.62-2.54 (m, 2H), 1.77-1.61 (m, 4H); 13C NMR [100 MHz, DMSO-d6] (13C resonance obstructed by solvent peak) δ 188.5, 181.97, 167.9, 164.4, 145.7, 136.1, 130.5, 128.6 (2C), 128.2, 128.1 (2C), 126.4, 111.6, 65.9, 51.4, 25.9, 23.7, 22.5, 22.3; HRMS (ESI) [M+H]+, calc’d for C21H22N3O4S2 444.10463, found 444.10497; IR (solid ATR): 2936, 1649, 1564, 1517, 1237, 1191, 1020 cm-1; HPLC purity: >95% tR = 2.470 min. (75-95% MeOH/Water with 0.1% formic acid, gradient over 3 minutes, 1.0 mL/min.).
Acknowledgements
We thank Phuong Le, Jing Zhang, and Anel Tankovic for excellent technical assistance. GNE-6901 and GNE-0723 were generously provided by Jesse Hanson and Genentech. This work was supported by the NINDS (NS036654, NS065371, SFT) and NIGMS (T32-GM008602). We also thank the Emory Chemical Biology Discovery Center for their assistance.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Stephen F Traynelis, Email: strayne@emory.edu.
Kenton J Swartz, National Institutes of Health, United States.
Funding Information
This paper was supported by the following grants:
National Institute of Neurological Disorders and Stroke NS036654,NS065371 to Stephen F Traynelis.
National Institute of General Medical Sciences GM008602 to Riley Perszyk.
Additional information
Competing interests
No competing interests declared.
BMK is an inventor of Emory-owned IP.
MPE is an inventor of Emory-owned IP.
DM is an inventor of Emory-owned IP.
DCL is a member of the Board of Directors at NeurOp Inc. and is an inventor of Emory-owned IP.
SFT is a co-founder of NeurOp Inc., a paid consultant for Janssen, a PI on a research grant from Janssen to Emory University, a member of the Scientific Advisory Board for Sage Therapeutics, and is an inventor of Emory-owned IP.
Author contributions
Conceptualization, Data curation, Software, Formal analysis, Validation, Investigation, Visualization, Methodology, Writing—original draft, Writing—review and editing.
Conceptualization, Resources, Supervision, Funding acquisition, Writing—original draft, Writing—review and editing.
Conceptualization, Resources, Data curation, Software, Formal analysis, Validation, Investigation, Visualization, Methodology, Writing—original draft, Writing—review and editing.
Conceptualization, Resources, Writing—original draft, Writing—review and editing.
Conceptualization, Resources, Writing—review and editing.
Resources, Investigation, Writing—review and editing.
Conceptualization, Resources, Writing—review and editing.
Conceptualization, Resources, Writing—review and editing.
Conceptualization, Resources, Writing—review and editing.
Conceptualization, Resources, Supervision, Funding acquisition, Writing—review and editing.
Conceptualization, Resources, Supervision, Funding acquisition, Writing—original draft, Writing—review and editing.
Additional files
References
- Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, Traynelis SF. Mechanism for noncompetitive inhibition by novel GluN2C/D N-methyl-D-aspartate receptor subunit-selective modulators. Molecular Pharmacology. 2011;80:782–795. doi: 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaloum M, Kazi R, Gan Q, Amin J, Wollmuth LP. A molecular determinant of subtype-specific desensitization in ionotropic glutamate receptors. The Journal of Neuroscience. 2016;36:2617–2622. doi: 10.1523/JNEUROSCI.2667-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. British Journal of Pharmacology and Chemotherapy. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard EA, Skolnick P, Olsen RW, Mohler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, Langer SZ. International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacological Reviews. 1998;50:291–314. [PubMed] [Google Scholar]
- Borovska J, Vyklicky V, Stastna E, Kapras V, Slavikova B, Horak M, Chodounska H, Vyklicky L. Access of inhibitory neurosteroids to the NMDA receptor. British Journal of Pharmacology. 2012;166:1069–1083. doi: 10.1111/j.1476-5381.2011.01816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Shieh C, Swanger SA, Tankovic A, Au M, McGuire M, Tagliati M, Graham JM, Madan-Khetarpal S, Traynelis SF, Yuan H, Pierson TM. GRIN1 mutation associated with intellectual disability alters NMDA receptor trafficking and function. Journal of Human Genetics. 2017;62:589–597. doi: 10.1038/jhg.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacological Reviews. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. Relaxation and fluctuations of membrane currents that flow through drug-operated channels. Proceedings of the Royal Society B: Biological Sciences. 1977;199:231–262. doi: 10.1098/rspb.1977.0137. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. A Q-matrix cookbook: How to write only one program to calculate the single-channel and macroscopic predictions for any kinetic mechanism. In: Sakmann B, Neher E, editors. Single-Channel Recording. Boston: Springer; 1995. [Google Scholar]
- Erreger K, Geballe MT, Dravid SM, Snyder JP, Wyllie DJ, Traynelis SF. Mechanism of partial agonism at NMDA receptors for a conformationally restricted glutamate analog. Journal of Neuroscience. 2005;25:7858–7866. doi: 10.1523/JNEUROSCI.1613-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Molecular and Cellular Neuroscience. 2011;48:308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A-2B subunits. PNAS. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos DH, Lupardus PJ, Grand T, Chen Y, Wang TM, Reynen P, Gustafson A, Wallweber HJ, Volgraf M, Sellers BD, Schwarz JB, Paoletti P, Sheng M, Zhou Q, Hanson JE. Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron. 2016;89:983–999. doi: 10.1016/j.neuron.2016.01.016. [DOI] [PubMed] [Google Scholar]
- Hackos DH, Hanson JE. Diverse modes of NMDA receptor positive allosteric modulation: Mechanisms and consequences. Neuropharmacology. 2017;112:34–45. doi: 10.1016/j.neuropharm.2016.07.037. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Traynelis SF. Structural and mechanistic determinants of a novel site for noncompetitive inhibition of GluN2D-containing NMDA receptors. Journal of Neuroscience. 2011;31:3650–3661. doi: 10.1523/JNEUROSCI.5565-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Traynelis SF. Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. Journal of Neuroscience. 2012;32:6197–6208. doi: 10.1523/JNEUROSCI.5757-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jørgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF. Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-D-aspartate receptors. Molecular Pharmacology. 2013;84:114–127. doi: 10.1124/mol.113.085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nature Reviews Neuroscience. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Recruiting extrasynaptic NMDA receptors augments synaptic signaling. Journal of Neurophysiology. 2008;99:524–533. doi: 10.1152/jn.01169.2007. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiological Reviews. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Petrovic M, Chodounska H, Vyklicky L. Molecular mechanism of pregnenolone sulfate action at NR1/NR2B receptors. Journal of Neuroscience. 2004;24:10318–10325. doi: 10.1523/JNEUROSCI.2099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Chodounska H, Vyklicky L. Subtype-dependence of N-methyl-D-aspartate receptor modulation by pregnenolone sulfate. Neuroscience. 2006;137:93–102. doi: 10.1016/j.neuroscience.2005.08.058. [DOI] [PubMed] [Google Scholar]
- Jones KS, VanDongen HM, VanDongen AM. The NMDA receptor M3 segment is a conserved transduction element coupling ligand binding to channel opening. Journal of Neuroscience. 2002;22:2044–2053. doi: 10.1523/JNEUROSCI.22-06-02044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. The Lancet Neurology. 2008;7:742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 2011;475:249–253. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman BM, Perszyk RE, Yuan H, Tahirovic YA, Sotimehin AE, Traynelis SF, Liotta DC. A novel class of negative allosteric modulators of NMDA receptor function. Bioorganic & Medicinal Chemistry Letters. 2015;25:5583–5588. doi: 10.1016/j.bmcl.2015.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. The Journal of Physiology. 1996;497 (Pt 3):761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri A, Burger PB, Swanger SA, Hansen KB, Zimmerman S, Karakas E, Liotta DC, Furukawa H, Snyder JP, Traynelis SF. Structural determinants and mechanism of action of a GluN2C-selective NMDA receptor positive allosteric modulator. Molecular Pharmacology. 2014;86:548–560. doi: 10.1124/mol.114.094516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RA, Jahr CE. NMDA channel behavior depends on agonist affinity. The Journal of Neuroscience. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malayev A, Gibbs TT, Farb DH. Inhibition of the NMDA response by pregnenolone sulphate reveals subtype selective modulation of NMDA receptors by sulphated steroids. British Journal of Pharmacology. 2002;135:901–909. doi: 10.1038/sj.bjp.0704543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan DT, Irvine MW, Costa BM, Fang G, Jane DE. Pharmacological modulation of NMDA receptor activity and the advent of negative and positive allosteric modulators. Neurochemistry International. 2012;61:581–592. doi: 10.1016/j.neuint.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, Liotta DC, Traynelis SF. A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nature Communications. 2010;1:90–8. doi: 10.1038/ncomms1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF. New advances in NMDA receptor pharmacology. Trends in Pharmacological Sciences. 2011;32:726–733. doi: 10.1016/j.tips.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF. Contribution of the M1 transmembrane helix and pre-M1 region to positive allosteric modulation and gating of N-methyl-D-aspartate receptors. Molecular Pharmacology. 2013;83:1045–1056. doi: 10.1124/mol.113.085209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Khatri A, Traynelis SF, Heldt SA. Potentiation of GluN2C/D NMDA receptor subtypes in the amygdala facilitates the retention of fear and extinction learning in mice. Neuropsychopharmacology. 2014;39:625–637. doi: 10.1038/npp.2013.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Chen W, Swanger SA, McDaniel MJ, Fan LZ, Hu C, Tankovic A, Kusumoto H, Kosobucki GJ, Schulien AJ, Su Z, Pecha J, Bhattacharya S, Petrovski S, Cohen AE, Aizenman E, Traynelis SF, Yuan H. Molecular mechanism of disease-associated mutations in the Pre-M1 helix of NMDA receptors and potential rescue pharmacology. PLoS Genetics. 2017;13:e1006536. doi: 10.1371/journal.pgen.1006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature Reviews Neuroscience. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Papouin T, Oliet SH. Organization, control and function of extrasynaptic NMDA receptors. Philosophical Transactions of the Royal Society B: Biological Sciences. 2014;369:20130601. doi: 10.1098/rstb.2013.0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic M, Sedlacek M, Horak M, Chodounska H, Vyklický L. 20-oxo-5beta-pregnan-3alpha-yl sulfate is a use-dependent NMDA receptor inhibitor. Journal of Neuroscience. 2005;25:8439–8450. doi: 10.1523/JNEUROSCI.1407-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nature Reviews Drug Discovery. 2011;10:685–697. doi: 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. Journal of Neuroscience. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlender DA, Savtchouk I, Volterra A. What do we know about gliotransmitter release from astrocytes? Philosophical Transactions of the Royal Society B: Biological Sciences. 2014;369:20130592. doi: 10.1098/rstb.2013.0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. Journal of Neuroscience. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, Traynelis SF, Liotta DC. Novel NMDA receptor modulators: an update. Expert Opinion on Therapeutic Patents. 2012;22:1337–1352. doi: 10.1517/13543776.2012.728587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GG, Williams KM, Wonnacott DM. Factors affecting the rate of racemization of amino acids and their significance to geochronology. The Journal of Organic Chemistry. 1978;43:1–5. doi: 10.1021/jo00395a001. [DOI] [Google Scholar]
- Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Köhr G. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. Journal of Neuroscience. 2000;20:4573–4581. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong KL, Jing Y, Prosser AR, Traynelis SF, Liotta DC. NMDA receptor modulators: an updated patent review (2013-2014) Expert Opinion on Therapeutic Patents. 2014;24:1349–1366. doi: 10.1517/13543776.2014.972938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Vance KM, Acker TM, Zimmerman SS, DiRaddo JO, Myers SJ, Bundgaard C, Mosley CA, Summer SL, Menaldino DS, Jensen HS, Liotta DC, Traynelis SF. A novel negative allosteric modulator selective for GluN2C/2D-containing NMDA receptors inhibits synaptic transmission in hippocampal interneurons. ACS Chemical Neuroscience. 2018;9:306–319. doi: 10.1021/acschemneuro.7b00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima N, Karakas E, Grant T, Simorowski N, Diaz-Avalos R, Grigorieff N, Furukawa H. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature. 2016;534:63–68. doi: 10.1038/nature17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Reviews. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, Sobolevsky AI. Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature. 2017a;549:60–65. doi: 10.1038/nature23479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, Sobolevsky AI. Structural bases of desensitization in AMPA receptor-auxiliary subunit complexes. Neuron. 2017b;94:569–580. doi: 10.1016/j.neuron.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Sobolevsky AI. Structural mechanisms of gating in ionotropic glutamate receptors. Biochemistry. 2018;57:267–276. doi: 10.1021/acs.biochem.7b00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KM, Hansen KB, Traynelis SF. GluN1 splice variant control of GluN1/GluN2D NMDA receptors. The Journal of Physiology. 2012;590:3857–3875. doi: 10.1113/jphysiol.2012.234062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. Journal of Neurophysiology. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Vyklicky V, Krausova B, Cerny J, Balik A, Zapotocky M, Novotny M, Lichnerova K, Smejkalova T, Kaniakova M, Korinek M, Petrovic M, Kacer P, Horak M, Chodounska H, Vyklicky L. Block of NMDA receptor channels by endogenous neurosteroids: implications for the agonist induced conformational states of the channel vestibule. Scientific Reports. 2015;5:10935. doi: 10.1038/srep10935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TM, Brown BM, Deng L, Sellers BD, Lupardus PJ, Wallweber HJA, Gustafson A, Wong E, Volgraf M, Schwarz JB, Hackos DH, Hanson JE. A novel NMDA receptor positive allosteric modulator that acts via the transmembrane domain. Neuropharmacology. 2017;121:204–218. doi: 10.1016/j.neuropharm.2017.04.041. [DOI] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nature Reviews Drug Discovery. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- Yelshanskaya MV, Singh AK, Sampson JM, Narangoda C, Kurnikova M, Sobolevsky AI. Structural bases of noncompetitive inhibition of AMPA-subtype ionotropic glutamate receptors by antiepileptic drugs. Neuron. 2016;91:1305–1315. doi: 10.1016/j.neuron.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshanskaya MV, Mesbahi-Vasey S, Kurnikova MG, Sobolevsky AI. Role of the ion channel extracellular collar in AMPA receptor gating. Scientific Reports. 2017;7:1050. doi: 10.1038/s41598-017-01146-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Mou TC, Dorsett KN, Volkmann RA, Menniti FS, Sprang SR, Hansen KB. Structural basis for negative allosteric modulation of GluN2A-containing NMDA receptors. Neuron. 2016;91:1316–1329. doi: 10.1016/j.neuron.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Erreger K, Dravid SM, Traynelis SF. Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. Journal of Biological Chemistry. 2005;280:29708–29716. doi: 10.1074/jbc.M414215200. [DOI] [PubMed] [Google Scholar]
- Zhu S, Paoletti P. Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Current Opinion in Pharmacology. 2015;20:14–23. doi: 10.1016/j.coph.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stein RA, Yoshioka C, Lee CH, Goehring A, Mchaourab HS, Gouaux E. Mechanism of NMDA receptor inhibition and activation. Cell. 2016;165:704–714. doi: 10.1016/j.cell.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]