

Abstract
We combine the labeling of newly transcribed RNAs with 5-ethynyluridine with the characterization of bound proteins. This approach, named capture of the newly transcribed RNA interactome using click chemistry (RICK), systematically captures proteins bound to a wide range of RNAs, including nascent RNAs and traditionally neglected nonpolyadenylated RNAs. RICK has identified mitotic regulators amongst other novel RNA-binding proteins with preferential affinity for nonpolyadenylated RNAs, revealed a link between metabolic enzymes/factors and nascent RNAs, and expanded the known RNA-bound proteome of mouse embryonic stem cells. RICK will facilitate an in-depth interrogation of the total RNA-bound proteome in different cells and systems.
Current approaches for systematically characterizing the RNA-bound proteome (or RNA interactome) are mainly based on the capture of polyadenylated (polyA) RNAs, mostly mRNAs, with oligo(dT)-coated beads1,2. Yet, polyA tails are only added to newly transcribed RNAs during their processing to a mature form, whilst some mature mRNAs are either non-polyA or bimorphic3. Moreover, mature non-polyA RNA species comprise a substantial fraction of all transcribed sequences4,5. Methodologies allowing the capture of RNA-binding proteins (RBPs) interacting with all types of RNAs could not only expand our current view of the RNA interactome but also help to understand the function of non-polyA RNAs in physiology and disease.
We have developed a versatile method to capture the interactome of newly transcribed RNAs, based on linking 5- ethynyluridine (EU)-labeled RNAs and biotin using the click reaction6. We termed this method RICK and applied it to HeLa cells and mouse embryonic stem cells (mESCs) to identify numerous novel RBPs.
RESULTS
Capture of the newly transcribed RNA interactome using RICK
We labeled RNA in HeLa cells with EU, fixed the cells, and then biotinylated the EU using the click reaction6. Next, we extracted RNA–protein complexes using streptavidin-coated beads. We initially used a 16-h treatment with EU to ensure efficient isolation of all types of newly transcribed RNAs6 (Fig. 1 and Supplementary Fig. 1a). Measurement of streptavidin-conjugated horseradish peroxidase activity showed a strong signal that was diminished when cotreated with RNase (Supplementary Fig. 1b).
Figure 1.
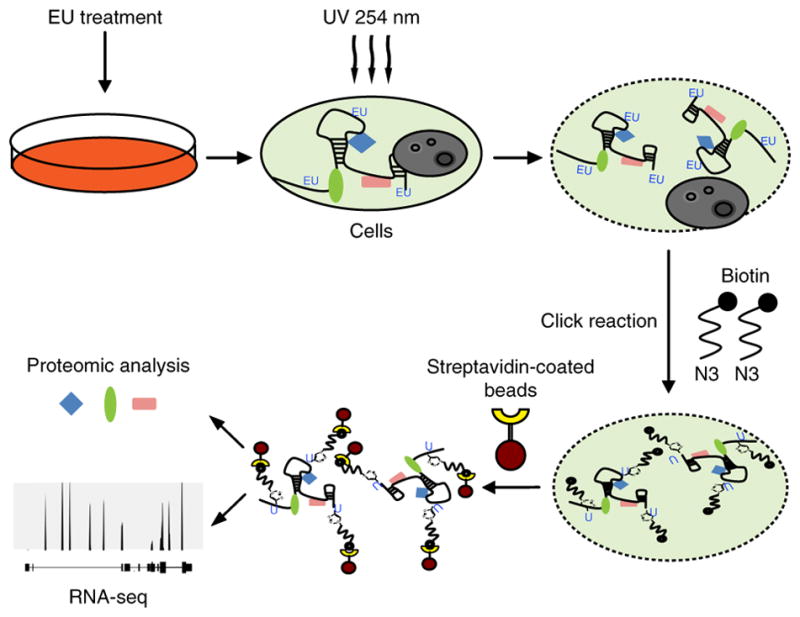
Establishment of a new technique to capture the newly transcribed RNA interactome. Schematic representation of the RICK procedure.
We then performed the same procedure, but used 254-nm UV to crosslink RNA with protein, and magnetic streptavidin- conjugated beads to capture the EU-labeled RNA-protein complexes (Fig. 1). Gel electrophoresis and silver staining confirmed that RICK successfully isolates proteins directly interacting with EU-labeled RNAs (Supplementary Fig. 1c). The specificity of the pull down was confirmed by loss of protein pull-down signal when the samples were not crosslinked or were cotreated with RNase (Supplementary Fig. 1c). We used western blotting to validate the capture of known RBPs by RICK, and β-ACTIN, β-TUBULIN were used as negative controls (Supplementary Fig. 1d). Thus, we have established a novel methodology to specifically isolate proteins interacting with newly transcribed RNAs.
Determination of RNA species captured by RICK
We performed RNA sequencing (RNA-seq) of a RICK pull-down sample to understand the nature of the captured RNAs. Our analysis (Fig. 2a, Supplementary Fig. 2a, and Supplementary Table 1) showed obvious differences compared to the oligo(dT) capture data set by Castello et al.1, in which ~80% of the captured RNA were mRNAs.
Figure 2.

Analysis of transcripts isolated by RICK. (a) Distribution of RNA species isolated in a representative RICK experiment. Values for the input sample are also shown. The same RICK data set was used for the analysis in b–d. (b,c) Metagene representation of Pol II15 (black) occupancy and RICK (green) or oligo(dT) capture (blue) RNA-seq signals at gene promoter or gene body of genes with TR > 4 (n = 5,889) (b) and TR < 4 (n = 6,838) (c). (d) Metagene representation of H3K27ac occupancy16 (black), H3K4me1 occupancy16 (magenta), and RICK (green) or oligo(dT) capture (blue) RNA-seq signals at potential enhancer sites; n = 18,272. (e) RT-qPCR analysis of different RNA species isolated by RICK or oligo(dT) capture. ‘RICK control’ indicates samples without EU treatment in RICK experiments; ‘Oligo(dT) control’ indicates samples isolated using beads without oligo(dT) probes in oligo(dT) capture; n = 3 biologically independent experiments and data are shown as the mean ± s.d. P value is shown (Student’s t-test, two tailed).
Next, we studied the enrichment in RICK samples versus oligo(dT) capture of three relevant non-polyA RNA species: circular RNAs (circRNAs)7, proximal promoter RNAs (ppR-NAs)8,9, and enhancer RNAs (eRNAs)10,11. We observed 6,199 backsplicing events, suggestive of circRNAs, in the RICK sample compared to only 57 in the oligo(dT) capture data set by Castello et al.1 (Supplementary Fig. 2b and Supplementary Table 2). Of the 6,199 back-splicing events, 828 overlapped with circBase data sets12, compared to only 2 in the oligo(dT) capture. The normalized number of junction reads13 was also higher in RICK (64.6 mapped back-splice junction reads per million reads) than oligo(dT) capture (8.2). Examples of candidate circRNAs enriched in RICK are shown in Supplementary Figure 2c; circ-6763 was validated by Sanger sequencing of a RICK sample (Supplementary Fig. 2d). For assessing ppRNAs, we divided genes in transcriptionally paused or not paused based on the RNA polymerase II (Pol II) traveling ratio (TR)9,14 (Supplementary Fig. 3a) using a previously reported data set15. Transcriptionally paused genes (TR > 4) showed higher RNA-seq signals accumulated around the TSS in RICK samples compared to oligo(dT) capture1, suggesting efficient isolation of ppRNAs only by RICK, whilst for nonpaused genes (TR < 4) the difference was small (Fig. 2b,c and Supplementary Fig. 3b). A transcriptionally paused transcript captured by RICK and not by the oligo(dT) capture method, the ACOX1 locus, is shown in Supplementary Figure 3c. We also observed robust enrichment of RNA-seq signals, suggestive of eRNAs, across putative enhancer regions16 in RICK samples (6.05%) but not in oligo(dT) capture (0.11%) (Fig. 2d). This was further confirmed by comparison with the FANTOM5 database for bidirectional eRNAs17 (Supplementary Fig. 3d). For example, a specific transcribed enhancer on chromosome 1 could only be detected by RICK (Supplementary Fig. 3e).
Quantitative RT-PCR (RT-qPCR) verified the isolation of selected non-polyA RNAs extracted using RICK, and the same RNAs were less enriched or absent in oligo(dT) capture samples (Fig. 2e and Supplementary Fig. 3f). Control mRNAs showed similar or higher levels using oligo(dT) capture (Fig. 2e). In conclusion, RICK successfully isolates a wide variety of RNA species not limited to polyA RNAs, suggesting that it can also enrich RBPs not isolated by oligo(dT) capture methods.
Characterization of proteins isolated by RICK
We analyzed the proteins isolated by RICK with liquid chromatography–tandem mass spectrometry (LC-MS/MS). This yielded 1,353 proteins divided into high- (720) or low-confidence (633) subsets (Supplementary Fig. 4a,b and Supplementary Table 3). We compared the 720 high-confidence proteins with the 860 proteins of the ‘HeLa mRNA interactome’ recovered by oligo(dT) capture1. 350 proteins were present in both data sets, whilst 370 were only in RICK (Supplementary Fig. 4c and Supplementary Table 3). Although 510 proteins of the ‘HeLa mRNA interactome’ were not included in the high-confidence RICK group, 159 of those were in the low-confidence RICK group (Supplementary Table 3d). Because RICK and oligo(dT) capture have different biases in the types of captured RNAs, it is no surprise that many proteins are unique to each method.
We also produced a new HeLa oligo(dT) capture data set and analyzed it using the same criteria as our RICK data set. We identified 554 high-confidence proteins, which, as expected, had a significant overlap (452 proteins, 81.6%) with the ‘HeLa mRNA interactome’ (Supplementary Fig. 4c and Supplementary Table 3e). Moreover, of the 370 high-confidence proteins identified by RICK and absent in the ‘HeLa mRNA interactome’, only 26 were present in our oligo(dT) capture (Supplementary Fig. 4c and Supplementary Table 3); we termed the remaining 344 candidate RBPs ‘RICK-exclusive RBPs’ (Supplementary Table 3).
We then performed oligo(dT) capture in the presence of EU to evaluate whether its incorporation affects RNA–protein interactions. Unsupervised clustering of the ion intensities showed that our EU+ and EU– oligo(dT) capture samples were highly correlated with each other but were poorly correlated with the RICK samples (Fig. 3a). We identified 469 high-confidence proteins in the oligo(dT) capture samples of HeLa cells treated with EU (Supplementary Table 3), of which only 53 were not present without EU (Supplementary Fig. 4d), and only two of those 53 proteins were amongst the 344 RICK-exclusive RBPs (Supplementary Table 3).
Figure 3.

Analysis of proteins isolated by RICK. (a) Hierarchical clustering of the ion intensities of three different LC-MS/MS experiments (n = 3 biologically independent experiments for both RICK and oligo(dT) capture (with or without EU)), colors in the heatmap indicate the pairwise Pearson correlation between the different data sets (n = 1,169). (b) Venn diagram comparing the 344 RICK-exclusive RBPs and different oligo(dT) capture studies using human cells2,18,19. The serial RNA interactome capture (serIC) study by Conrad et al.19 is a human RNA interactome using oligo(dT) capture of the nuclei of K562 cells.
In addition, comparison of the 344 RICK-exclusive RBPs with three reported human oligo(dT) capture studies2,18,19 showed that most of these proteins (295, 85.8%) are truly unique to RICK (Fig. 3b and Supplementary Table 3), so we termed them ‘RICK-unique RBPs’. Western blotting for a panel of candidates identified by RICK, including several RICK-unique RBPs such as the mitotic regulator CDK1 and the 7-methylguanosine (m7G) methyltransferase METTL120, validated that these proteins are precipitated by RICK but not by oligo(dT) capture (Supplementary Fig. 4e). Conversely, classical RBPs were present in samples from both methodologies.
Therefore, RICK isolates many proteins not identified in oligo(dT) capture studies, and the differences are neither caused by variations in cell culture, proteomic procedures, nor introduced by EU labeling.
Functional analysis of proteins isolated with RICK
We performed Gene Ontology (GO) analysis of the 295 RICK-unique RBPs, observing enrichment of biological processes related to mitosis (Fig. 4a and Supplementary Table 4). KEGG pathway analysis also included ‘cell cycle’ (P = 3.16 × 10−11) amongst the top ten most significantly enriched pathways (Fig. 4b and Supplementary Table 4). Conversely, GO and KEGG analysis of the 425 proteins identified by RICK and present in oligo(dT) capture data sets showed mostly RNA-related processes (Supplementary Fig. 5a,b and Supplementary Table 4). We also observed enrichment of other KEGG and GO terms, such as ‘proteasome’ and multiple terms related to transcriptional regulation, amongst the 295 RICK-unique RBPs (Supplementary Fig. 5c,d; Supplementary Table 4; and Supplementary Note). PAR-CLIP-biotin chemiluminescent nucleic acid detection21 validated the interaction of RNA with 23 of 28 tested proteins identified by RICK, including CDK1 and METTL1 (Supplementary Fig. 5e,f and Supplementary Table 4). Taken together, these data further reinforce the uniqueness of RICK in isolating proteins that were not identified using oligo(dT) capture methods and reveal the function of RNAs in unanticipated cellular processes.
Figure 4.

Functional characterization of novel candidate RBPs identified by RICK. (a) GO analysis of the 295 RICK-unique RBPs; the top ten GO terms with the smallest P value in the indicated categories are shown (Fisher’s exact test, one tailed). The number of proteins for each individual term is also shown. Biological processes related to mitosis are marked in red. (b) KEGG pathway analysis of the 295 RICK-unique RBPs. The number of proteins for each individual pathway is also shown; P value is also shown (Fisher’s exact test, one tailed).
RICK identifies proteins with preferential binding to non-polyA RNAs
Considering the striking differences in RNA species isolated by RICK and oligo(dT) capture, it seemed plausible that a considerable proportion of the 295 RICK-unique RBPs bind preferentially to non-polyA RNAs. This specificity could be due to stoichiometry, subcellular localization, or potentially intrinsic binding specificity. To clarify this, we added another step consisting of three consecutive rounds of incubation with oligo(dT)-coated beads to our standard RICK protocol (Supplementary Fig. 6a). We confirmed efficient removal of polyA RNAs by RT-qPCR, whilst the control 18S RNA remained unaffected (Supplementary Fig. 6b). LC-MS/MS identified 914 high-confidence proteins, which we termed ‘polyA-depleted RICK proteins’. Of these 914 proteins, 576 overlapped with the 720 high-confidence proteins of the standard RICK procedure (Fig. 5a and Supplementary Table 5). Further analysis showed that 204 (69.2%) of 295 RICK-unique RBPs overlap with the 914 polyA-depleted RICK proteins (Fig. 5b and Supplementary Table 5). Importantly, GO analysis of these 204 proteins showed enrichment of biological processes related to mitosis (Supplementary Table 5). We also noticed that 39 of the remaining 91 RICK-unique RBPs were amongst the low-confidence proteins identified in the polyA-depleted RICK (Supplementary Table 5). On the other hand, amongst the 710 polyA-depleted RICK proteins not overlapping with the RICK-unique RBPs, 563 were present in oligo(dT) capture data sets (Fig. 5b), suggesting that these proteins have a mixed tendency to bind polyA and non-polyA RNAs. We then used western blotting to confirm that a panel of selected RICK-identified RBPs, including CDK1 and METTL1, remain enriched after polyA RNA removal (Supplementary Fig. 6c). In conclusion, these results demonstrate that RICK can be used to identify proteins binding preferentially to non-polyA RNAs.
Figure 5.

Identification of METTL1-interacting RNAs. (a) Venn diagram comparing the 720 high-confidence proteins identified by RICK and the 914 high-confidence proteins identified by polyA-depleted RICK. (b) Venn diagram comparing the 295 RICK-unique RBPs and the 914 high-confidence proteins identified by polyA-depleted RICK. The column indicates distribution of the 710 polyA-depleted RICK proteins compared to high-confidence proteins of different oligo(dT) capture studies (green, proteins present in different oligo(dT) capture data sets1,2,18,19; purple, proteins not present in the same oligo(dT) capture data sets). (c) Distribution of RNA species identified by METTL1 PAR-CLIP sequencing. The average of n = 2 biologically independent experiments is shown. (d) RIP-qPCR analysis of different RNAs binding to METTL1 that were identified in the METTL1 PAR-CLIP sequencing. The enrichments were normalized to input; GFP was used as a negative control. Random hexamers or oligo(dT) primers were used for the reverse transcription as indicated. METTL1-interacting mRNAs are marked in gray as opposed to VTRNA1-3; n = 3 biologically independent experiments, and data are shown as the mean ± s.d.; P value is shown (Student’s t-test, two tailed). (e) The most enriched METTL1-binding motifs on all target RNAs (n = 26,311 and 20,816 for Rep1 and Rep2, respectively) identified in two independent PAR-CLIP sequencing experiments; P value is shown (Fisher’s exact test, one tailed).
Characterization of RNAs interacting with METTL1 and CDK1
We chose two RICK-unique RBPs, METTL1 and CDK1, to study their interacting RNAs using PAR-CLIP sequencing22. Two independent PAR-CLIP sequencing experiments for METTL1 showed extensive overlap in the captured RNAs (Supplementary Fig. 7a), a large proportion of which were captured with RICK too (Supplementary Fig. 7b). Further analysis confirmed prominent binding of METTL1 to tRNAs (31.8% of the PAR-CLIP tags). There was also binding to diverse—presumably—non-polyA RNAs such as intronic RNAs (5.9%) and RNAs transcribed from intergenic regions (not annotated as lincRNAs; 1.7%) (Fig. 5c). In addition, 37.8% of sequences mapped to mRNAs, but these mapped sequences could correspond to immature transcripts, circRNAs, or ppRNAs. Sequencing tracks for specific target RNAs are shown in Supplementary Figure 7c. We next performed RNA immunoprecipitation (RIP) followed by RT-qPCR (RIP-qPCR) using random hexamers or oligo(dT) primers to discern whether METTL1 target RNAs have polyA tails (Supplementary Fig. 7d). RIP-qPCR validated the interaction of METTL1 with the non-polyA RNA vault RNA1-3 (VTRNA1-3)3 (Fig. 5d). RIP-qPCR also showed enrichment for seven mRNAs using random hexamers, whilst only three of these could be comparably amplified using oligo(dT) primers, supporting the fact that METTL1 binds to non-polyA RNAs containing mRNA sequences (Fig. 5d). These results were validated using semiquantitative PCR (Supplementary Fig. 7e). In addition, motif discovery of our METTL1 PAR-CLIP sequencing data showed that ‘CUCUUCG’ is amongst the most enriched binding motifs in the two replicate experiments (Fig. 5e). Separate analysis of non-tRNAs and tRNAs showed that the two most enriched motifs in each group are not identical, but they all contain a ‘UUCG’ core sequence, which is commonly found in the T arm of many tRNAs (Supplementary Fig. 7f,g). The latter is consistent with reports showing that METTL1 recognizes the T arm of tRNAs to perform m7G modification23,24.
The analysis of CDK1 PAR-CLIP sequencing revealed binding to RNAs transcribed from intergenic regions (4.0%) and intronic RNAs (6.1%), amongst others (Supplementary Fig. 8a). There was also extensive overlap with the RNAs captured by RICK (Supplementary Fig. 8b). Sequencing tracks for two intergenic RNAs bound by CDK1 are shown in Supplementary Figure 8c. RIP-qPCR confirmed the capture of selected intergenic RNAs identified in the sequencing (Supplementary Fig. 8d). Moreover, of six selected mRNA sequences, only one showed enrichment with oligo(dT) primers using RIP-qPCR, potentially explaining why mRNAs comprise 58.6% of the CDK1 PAR-CLIP sequencing reads (Supplementary Fig. 8d).
Therefore, METTL1 and CDK1 bind extensively to non-polyA RNAs, hinting at overlooked RNA-related functions of these two proteins.
Capture of the nascent RNA interactome using RICK
We then performed short EU labeling (0.5, 1, and 2 h) of HeLa cells to study whether RICK can enrich the capture of proteins interacting with nascent (emerging or primary) RNAs6,25 (Supplementary Fig. 9a,b). We envisaged that this may allow studying RNA–protein interactions in the context of transcription. There was reduced capture of both RNAs and proteins compared to 16-h RICK on account of less EU incorporation (Supplementary Fig. 9c). Similarly, the percentage of sequences corresponding to 5′ UTR, CDS, and 3′ UTR was reduced with the short-labeling RICK, indicating that fewer mature mRNAs had accumulated (Supplementary Fig. 9d). Besides, rRNAs were more enriched, which might be caused by the higher concentration of EU (1 mM) compared to the 16-h RICK (0.25 mM).
Overall, we identified 208 distinct high-confidence proteins (149, 119, and 143 for 0.5, 1, and 2 h, respectively), which we termed ‘nascent enriched RBPs’ and 319 low-confidence proteins (Fig. 6a and Supplementary Table 6). We used western blotting to validate two nascent-enriched RBPs, RBM3 and TRIM28 (Supplementary Fig. 9e); conversely, HNRNPK and PTBP1 were present in both methodologies. As predicted, GO and KEGG pathway analyses of the 208 nascent-enriched RBPs showed terms enriched in transcription and RNA metabolism (Fig. 6b and Supplementary Table 6). Further study showed that only 43 of these 208 were not identified by oligo(dT) capture in HeLa cells (Supplementary Fig. 9f and Supplementary Table 6), consistent with the isolation of RBPs participating in transcription by oligo(dT) capture (e.g., HNRNPK)26. 22 of those 43 nascent-enriched RBPs were not amongst the 295 RICK-unique RBPs (Supplementary Fig. 9f and Supplementary Table 6). Notably, GO analysis of these 43 proteins showed significant (P = 2.02 × 10−7) enrichment of terms related to ‘coenzyme metabolic process’ or ‘cofactor metabolic process’ (P = 7.10 × 10−7) (Supplementary Fig. 9g and Supplementary Table 6). Eight of the 43 proteins were enzymes/factors related to these two GO terms (Supplementary Table 6d,e and Supplementary Note). Six of these eight proteins were present in 0.5-h RICK (Supplementary Fig. 9h and Supplementary Table 7), reinforcing the idea that they interact with nascent RNAs. These findings suggest a link between nascent RNA transcription and local metabolite production to regulate the epigenome27 and potentially the epitranscriptome28.
Figure 6.

Capture of the nascent RNA interactome using RICK. (a) Venn diagram comparing the nascent-enriched RBPs identified with each of the short EU labeling times. (b) GO analysis for the 208 nascent-enriched RBPs; P value in the indicated categories is shown (Fisher’s exact test, one tailed).
Capture of the total RNA interactome of mESCs using RICK
Next, we aimed to demonstrate that RICK can be applied to other cell types. We confirmed efficient incorporation of EU in mESCs using streptavidin-conjugated horseradish peroxidase (Supplementary Fig. 10a), and performed LC-MS/MS, which identified 518 high-confidence and 304 low-confidence proteins (Supplementary Table 6). 160 of these 518 high-confidence proteins overlapped with the ‘mESC mRNA interactome’ reported by Kwon et al.29, whilst 358 were exclusive to RICK, and we thus termed them RICK-exclusive mESC RBPs (Supplementary Fig. 10b and Supplementary Table 6). GO analysis of these 358 candidate RBPs showed enrichment of RNA-binding or polyA-RNA-binding terms (Supplementary Fig. 10c and Supplementary Table 6). 95 of these 358 proteins were expressed at higher levels in mESCs than in differentiated cells30–33, pointing to a potential role in mESC self-renewal or pluripotency (Supplementary Fig. 10d and Supplementary Table 6). Consistently, we noticed that SON34, SMARCC1 (BAF155)35, and TRIM2836 were amongst the RICK-exclusive mESC RBPs. Thus, RICK can be applied to different cell types other than HeLa; further studies will be needed to ascertain the role in self-renewal/pluripotency of the newly identified candidate RBPs in mESCs.
DISCUSSION
We have developed a novel EU-based technology termed RICK to systematically characterize the total RNA interactome. Supporting our findings, Kanakkanthara et al.37 recently showed that CYCLIN A2 interacts with Mre11 mRNA in a CDK1- independent manner, whilst Brannan et al.38 reported computer-based prediction for novel RBPs that included several mitotic regulators. Deeper understanding of how mitotic regulators and RNAs are intertwined has implications for pathological states such as oncogenic transformation. As for the tRNA methyltransferase METTL123,24 we identified with RICK, one intriguing possibility is that, besides tRNA methylation, it induces m7G methylation of other RNA species to regulate their metabolism, though binding of METTL1 to those RNAs could have a different function.
Importantly, RICK can be easily modified to expand its potential applications, and we have shown proof of principle using short labeling times or incubation with oligo(dT) beads to remove polyA RNAs. Short-labeling RICK has revealed a potential link between metabolic enzymes/factors and nascent RNAs. Many metabolic enzymes/factors had been identified as bona fide RBPs in oligo(dT) capture studies1,39, but it was not known that some of them may interact with nascent RNAs. Short-labeling RICK could also be useful to differentiate RBPs interacting with newly transcribed RNAs from those interacting with steady-state RNAs. These differences could be used to map dynamic changes in the RNA interactome during acute cell stimulation or stem cell fate transitions. In addition, incorporation of EU into live animals might allow the characterization of RBPs in mammalian organs or during development.
Whilst our manuscript was under review, He et al.40 reported a methodology (RNA-binding region identification, RBR-ID) based on the incorporation of 4-thiouridine (4SU, another uridine analog) into newly transcribed RNAs in mESCs. Comparison of our 358 RICK-exclusive mESC RBPs with the 814 proteins identified using RBR-ID showed overlap of only 75 (20.9%) proteins (Supplementary Fig. 10e and Supplementary Table 6). This discrepancy might have been caused by the fact that the authors only isolated nuclear proteins. Future studies will be necessary to ascertain the advantages and disadvantages of RICK compared to other nucleoside-based technologies for RBP identification.
ONLINE METHODS
A step-by-step protocol is available as a Supplementary Protocol.
Cell lines and culture conditions
HeLa and HEK293T cells were cultured in DMEM (Dulbecco’s Modified Eagle’s medium, Hyclone) supplemented with 10% fetal bovine serum (FBS, Biowest), nonessential amino acids (Gibco), penicillin–streptomycin (HyClone), and GlutaMax (Gibco); E14 mESCs were cultured on 0.1% gelatin (Millipore)-coated plates in chemically defined N2B27-based medium41: DMEM/F12 (HyClone) and Neurobasal (Gibco) mixed 1:1, supplemented with N2 (Gibco), B27 (Gibco), nonessential amino acids, GlutaMAX, sodium-pyruvate (Cellgro), penicillin–streptomycin, 0.1 mM β-mercaptoethanol (Gibco), 1,000 U/mL leukemia inhibitory factor (LIF, Millipore), CHIR99021 (3 μM, Selleck), and PD0325901 (1 μM, Selleck).
Visualization of ethynyluridine incorporation
HeLa cells were cultured in medium with 0.25 mM (RiboBio) for 16 h, and mESCs were cultured with 0.5 mM EU. Cells were then fixed with 90% ethanol for 30 min, washed with phosphate-buffered saline (PBS) three times, and permeabilized with 0.5% Triton X-100 (in PBS) for 15 min. After three washes with PBS, permeabilized cells were incubated with 1 mL of click reaction buffer for each well of a 6-well plate (PBS containing 0.2 mM CuSO4, 250 μM biotinazide, and 2.5 mM sodium L-ascorbate) for 3 min. Cells were washed three or five times with 0.5% Triton X-100 (in PBS) supplemented with 2 mM EDTA and incubated with 1 mL horseradish-peroxidase-labeled streptavidin (0.5 μg/mL) for 1 h at room temperature. After three washes with PBS, DAB solution was added for 3–10 min to detect the EU-incorporation signal. Images were taken using a Zeiss Axiovert 40C inverted microscope.
Capture of the newly transcribed RNA interactome using click chemistry
For the 16-h RICK experiment using HeLa cells, three 10 cm-plates (Corning) of cells were grown to 80% confluence, and for mESCs three 10 cm-plates grow to 30% confluence. Then, cells were cultured with EU for 16 h. After washing three times with PBS, the plates were placed on ice and irradiated with 0.15 J/cm2 UV light at 254 nm. Cells were then fixed with 90% ethanol for 30 min, washed three times with PBS, and permeabilized with 0.5% Triton X-100 (in PBS) for 15 min. After three washes with PBS, permeabilized cells were incubated with 10 mL of click reaction buffer supplemented with 0.6 mM THPTA (tris(3-hydroxypropyltriazolylmethyl)amine) (Sigma-Aldrich, cat# 762342) and 1 mM aminoguanidine hydrochloride (Sigma-Aldrich, cat# 396494) for 3 min. The reaction was stopped by washing the cells with 0.5% Triton X-100 (in PBS) supplemented with 2 mM EDTA three times. Cells were then lysed in lysis buffer (20 mM Tris–HCl, pH 7.5, 500 mM LiCl, 1 mM EDTA pH 8.0, 0.5% lithium-dodecylsulfate (LiDS), and 5 mM DTT) supplemented with protease inhibitor cocktail (Roche, cat# 04693132001) and RNase inhibitor (TaKaRa, cat# 2313A), and they were harvested by scraping. Lysates were homogenized using a syringe with a 0.4 mm diameter-needle. 5% of the lysate was taken out as an input control. Complexes containing different RNA species and their associated proteins were isolated with streptavidin-conjugated magnetic beads (100 μl beads for each plate; Dynabeads MyOne Streptavidin C1, Thermo Fisher Scientific, cat# 65602). After incubation with the lysates for 2 h under continuous rotation, the beads were isolated on a magnetic stand and washed using lysis buffer, buffer 1 (20 mM Tris–HCl, pH 7.5, 500 mM LiCl, 1 mM EDTA pH 8.0, 0.1% LiDS, and 5 mM DTT), buffer 2 (20 mM Tris–HCl pH 7.5, 500 mM LiCl, 1 mM EDTA pH 8.0, and 5 mM DTT), and buffer 3 (20 mM Tris–HCl pH 7.5, 200 mM LiCl, 1 mM EDTA pH 8.0, and 5 mM DTT) for two times each under rotation (for 10 min at 4 °C). Afterwards, RNA or proteins were extracted from the captured complexes using proteinase K at 55 °C for 2 h (New England Biolabs, cat# P8107S) or RNase A at 37 °C for 1 h (Sigma-Aldrich, cat# R4875), respectively. All buffers were prepared with RNase-free H2O. For short-labeling (0.5, 1, 2 h) RICK in HeLa, we used EU at a final concentration of 1 mM; the rest of the procedure was similar to 16-h RICK. Additional information is available as a Supplementary Protocol.
Western blotting and silver staining
Proteins were loaded onto SDS-PAGE gel for separation. After transferring onto a PVDF membrane (Millipore), they were blocked for at least 1 h with 5% milk in TBST (0.1% Tween-20 in tris-buffered saline (TBS)) before incubating with the primary antibodies at 4 °C overnight. After three washes with TBST, the membranes were incubated for 2 h at room temperature with HRP- conjugated secondary antibodies and washed three times again with TBST. The signal was generated with ECL plus (Enhanced Chemiluminescence, Amersham, cat# RPN2232) and detected with FUSION SOLO 4M System (Vilber Lourmat). Detailed antibody information is provided in Supplementary Table 8. For silver staining, the gel was fixed with 50% methanol and 5% acetic acid for at least 40 min after SDS-PAGE gel separation and washed using 50% methanol followed by deionized H2O for 10 min each. The gel was then sensitized in 0.02% Na2S2O3 for 1 min and washed with deionized H2O twice for 1 min, followed by staining with 4 °C 0.1% AgNO3 for 20 min in the dark. Afterwards, the gel was washed twice with deionized H2O for 1 min. Then, a mixture of 2% Na2CO3 and 0.04% formaldehyde was added for developing the gel, and 5% acetic acid was used to stop the reaction. All the above solutions were prepared in deionized H2O.
RNA extraction, RT-qPCR, and high-throughput RNA sequencing
RNA complexes isolated by RICK or oligo(dT) capture were treated with proteinase K and then isolated using TRIzol. To facilitate the precipitation, 20 μg/mL glycogen (Thermo Fisher Scientific, cat# AM9510) in isopropanol was added. RNA was reconstituted in RNase-free H2O, and cDNA synthesis was performed using the SuperScipt III Kit (Thermo Fisher Scientific, cat# 18080093). RT-qPCR was performed using SYBR Premix Ex Taq kit (TaKaRa, cat# RR420A); detailed primer information is provided in Supplementary Table 8. For RICK RNA-seq, RNAs were fragmented with ZnCl2 at 94 °C for 3 min to approximately 200 nucleotides, subjected to first-strand and second-strand synthesis, adaptor ligation, and then low-cycle amplification using TruSeq RNA LT/HT Sample Prep Kit (Illumina). The purified library products were evaluated using the Agilent 2200 TapeStation and Qubit 2.0 (Life Technologies) and diluted to 10 pM for cluster generation followed by high-throughput sequencing (1 × 50 bp) on HiSeq 2500 (Illumina).
Bioinformatics analysis of high-throughput sequencing
For RICK RNA-seq, data were aligned to the hg19 human genome assembly using Bowtie2 (v2.2.5)42 with default settings. Distribution of the aligned reads was calculated according to GENCODE v19 with in-house scripts. Pol II data from HeLa cells were taken from Gene Expression Omnibus (GEO, GSE53008)15 and aligned to the hg19 genome. Genome-wide sequence read density maps were generated using the glbase track module43. Only a single read per base pair per strand was allowed in the generated tracks. For TR calculation, we analyzed the same ChIP-seq data set for Pol II in HeLa cells. The TR for all Ensembl transcripts (GENCODE v19) was generated by measuring the mean sequence tag density −30/+300 bp around the TSS and dividing this by the mean sequence tag density within the remaining transcript body.
Pseudogenes, antisense transcripts, and any transcript with a transcript body less than 2 kb long were not considered for the TR calculation. This left 12,727 valid transcripts, of which 5,889 had a TR > 4 and 6,838 transcripts a TR < 4. Sequence tag pileups were drawn using glbase. H3K4me1 and H3K27ac ChIP-seq data in HeLa cells were obtained from GSE29611 (ref. 16). Reads were aligned to the hg19 genome using Bowtie2 (v2.2.5) with default settings. Binding peaks were called using MACS with default settings, and the cobinding sites of H3K4me1 and H3K27ac were defined as potential enhancer regions. The densities of oligo(dT) capture RNA-seq1, ChIP-seq, and RICK RNA-seq were calculated at potential enhancer regions. FANTOM5 (ref. 17) enhancers were lifted over to hg38 human genome, and enhancers specific to HeLa cells were defined if they had >0.2 tags per million deepCAGE tags in any of the two HeLa RICK RNA-seq samples. This left 2,189 eRNAs expressed in HeLa cells. Read densities were combined, and the metagene plot was generated by R. For candidate circRNA identification, raw sequencing reads were first preprocessed to remove duplicated fragments, adapters, and low-quality bases. The remaining high-quality reads for each sample were further aligned to the hg19 human genome using a spliced read aligner TopHat44 with the following parameters: transcriptome-mismatches = 3; mate-std-dev = 50; genome-read-mismatches = 3 with the single-end alignment option as well as the annotated gene model file downloaded from UCSC (http://genome.ucsc.edu/). To maximize the removal of reads resulting from linear transcripts, the canonical junctions deduced from TopHat for all data were initially pooled together. Subsequently, the reads ignored by TopHat were aligned to the canonical junction pool and then to the genome for the second time using Bowtie2 with the settings ‘–very-sensitive–mm–score-min = C,-15,0’. Finally, the terminal parts of each unmapped read were extracted and aligned to the genome independently. Next, after extending the anchor alignments, we defined the original read as a circRNA candidate when paired chiastic anchor mapping and GT-AG splicing signals were identified. If the anchors aligned to multiple positions, all possible combinations of paired anchor alignments belonging to the same original read were analyzed. When multiple junction interpretations were possible for a read, the back-spliced junction was distinguished when it harboeds the minimum mismatch number, and hence the false discoveries resulting from repetitive sequences were largely avoided. To exclude the possibility that the number of identified circRNA candidates was due to the different depths of sequencing, we normalized the number of junction reads (indicative of circRNAs) to total sequencing reads. The original RNA-seq data of the oligo(dT) capture study by Castello et al.1 was obtained from the authors. For PAR-CLIP sequencing analysis, adapters and reads shorter than 15 nucleotides were removed. Identical reads were collapsed using FASTQ/A Collapser. Then, reads were first aligned to hg19 Repeat Masker sequences (downloaded from UCSC table browser with tRNA sequences removal); unaligned reads were further aligned to hg19 genome assembly using bowtie version 1.1.2 with the following configuration, -p 8 -v 2 -m 1 -f. Aligned reads were used for peak calling with PARalyzer (v1.5)45, requiring a minimum cluster size of 10 nucleotides, a minimum conversion count of 2 T-to-C conversion (recommended settings), and five minimum read counts for cluster inclusion. Peaks were categorized to two groups (with/without peaks annotated as tRNA) and further used for de novo motif finding using HOMER’s (v4.7) findMotifsGenome. pl program46 with the settings ‘-rna -S 10 -len 4,5,6,7’ and annotated according to GENCODE v19 for downstream distribution analysis. Distribution of the aligned reads was calculated according to GENCODE v19 with in-house scripts.
Sample preparation for LC-MS/MS
For LC-MS/MS analysis, isolated protein samples from different experiments (using the same initial amount of cells for RICK, polyA-depleted RICK, and our oligo(dT) capture) were reduced with 50 mM tris(2-carboxyethyl) phosphine hydrochloride (Sigma-Aldrich) and 200 mM S-methyl methanethiosulfonate (Sigma-Aldrich) separately, followed by washing with 70% ethanol, 8 M urea, and 0.25 M triethylammonium bicarbonate (Sigma-Aldrich). Proteins were digested into peptides with 1 μg/μl trypsin in 5 mM triethylammonium bicarbonate at 37 °C overnight. An Ultimate 3000 HPLC system (Dionex) equipped with a 2 mm inner diameter and 100 mm long, Gemini 3 μm NX-C18 110 Å, LC column (Phenomenex, cat# 00D-4453-B0), was used for fractionation. Peptides were loaded into the column and washed with 5% acetonitrile (ACN, pH 10.0). Peptide fractionation was performed using ACN with a linear binary gradient from 15 to 50% at 0.2 mL/min over 45 min. Finally, the column was washed with 90% ACN for 10 min and returned to 5% ACN for 10 min. The UV detector was set at 214/280 nm and fractions were collected every minute. In total, 10 fractions were separated for each sample and dried by vacuum centrifuge for subsequent nano-reversed phase liquid chromatography fractionation.
LC-MS/MS
Each fraction was resuspended in loading buffer (containing 0.1% formic acid and 2% ACN) and separated using an Ultimate 3000 nano-LC system equipped with a C18 reverse phase column (100 μm inner diameter, 10 cm long, 3 μm resin). Peptides were separated using mobile phase A solution (containing 0.1% FA and 5% ACN) and mobile phase B solution (containing 0.1% FA and 95% ACN). Specifically, peptides were separated with a gradient of 5% to 40% phase B solution in phase A solution at a flow rate of 300 nl/min over 70 min. Then, the LC eluate was subjected to analysis with Q Exactive Hybrid Quadrupole-Orbitrap MS system (Thermo Fisher Scientific) in an information-dependent acquisition mode. MS spectra were acquired across the mass range of 350–1,800 m/z in high-resolution mode (70,000) using 250 ms accumulation time per spectrum. A maximum of 20 precursors per cycle were chosen for fragmentation from each MS spectrum with 100 ms minimum accumulation time for each precursor and dynamic exclusion for 20 s. Tandem mass spectra were recorded in high-resolution mode (>17,500) with rolling collision energy on. Peptides were selected for MS/MS using high-energy collision dissociation (HCD) operating mode with a normalized collision energy setting of 30.0. All the above solutions for LC-MS/MS were prepared in deionized H2O.
Bioinformatics analysis of LC-MS/MS data
First, MaxQuant was used to extract the intensities and unique peptide counts from LC-MS/MS data. For pairwise correlation analysis of the identified proteins, we used ggpairs to generate scatter plots and calculate pairwise correlation coefficient based on the ion intensities after normalization (http://CRAN.R-project.org/package=GGally). To define the pool of RNA-interacting proteins obtained with RICK, the modified RICK, and oligo(dT) capture with or without EU, we applied the following criteria: first, we included ion signals that were identified in at least two out of three experiments in experimental groups but not in controls. Second, for signals identified in both experimental and control groups, we selected the proteins with a ratio bigger than three compared to controls. We further filtered them by selecting only those proteins with a unique peptide count higher than two identified in at least two out of three independent experiments. GO analyses were performed using Fisher’s exact test using the latest version of GO database (http://www.ebi.ac.uk/QuickGO/) implemented in omicsbean proteomics toolkit (www.omicsbean.com:88). Adjusted P value < 0.05 was considered as statistically significant. KEGG analyses were performed using KEGG Mapper v2.8 (http://www.kegg.jp/kegg/mapper.html). Adjusted P value < 0.05 was considered as statistically significant. The 358 RICK-exclusive mESC RBPs was compared to the 814 RBR-ID40 identified mouse protein by using venny 2.1.0.
Plasmids
cDNAs were amplified from HEK293T or HeLa cells. They were cloned into pMXs vectors (purchased from Addgene) with FLAG tags. All plasmid sequences were confirmed by Sanger sequencing. Primers used for cloning are listed in Supplementary Table 8; other plasmids used in this study were purchased from Vigene and are also listed in Supplementary Table 8.
PAR-CLIP-biotin chemiluminescent nucleic acid detection and PAR-CLIP sequencing
HEK293T cells were transfected with plasmids expressing FLAG-tagged RBP candidates with 1 μg/μl Polyethylenimine (Polyscience). 24 h after transfection the cells were treated with 0.2 mM 4SU for another 16 h. Cells were then cross-linked with 0.4 J/cm2 of 365 nm UV light, scraped off with a rubber scraper in 2 mL PBS per plate, and collected by centrifugation at 500 g for 5 min at 4 °C. Crosslinked cell pellets were resuspended in 300 μl (for each plate) NP40 lysis buffer (50 mM HEPES pH 7.5,150 mM KCl, 2 mM EDTA, 1 mM NaF, 0.5% NP40, 0.5 mM DTT, RNase inhibitor, and protease inhibitor cocktail), and incubated on ice for 10 min. Lysates were homogenized by 0.4 mm syringe needle and centrifuged at 13,000 g for 15 min at 4 °C. The supernatants were digested with RNase T1 (Sigma-Aldrich, cat# R1003) at a final concentration of 1 U/mL at 22 °C for 15 min and cooled for 5 min on ice. Magnetic beads conjugated with FLAG antibodies (10 μl for each plate; Sigma-Aldrich, cat# M8823) were then added to the cell lysates and incubated for 2 h at 4 °C. After that, the beads were collected with a magnetic particle collector (Invitrogen). Beads were washed three times with 1 mL of washing buffer (50 mM HEPES-KOH pH 7.5, 300 mM KCl, 0.05% NP40, 0.5 mM DTT, and protease inhibitor cocktail) at 4 °C, and resuspended in washing buffer (10 μl for each plate) with RNase T1 at a final concentration of 10 U/mL for 15 min at 22 °C, followed by immediate cooling for 5 min on ice. Beads were washed three times with 1 mL of high-salt washing buffer (50 mM HEPES-KOH pH 7.5, 500 mM KCl, 0.05% NP40, 0.5 mM DTT, and protease inhibitor cocktail) at 4 °C. Beads were resuspended in dephosphorylation buffer (50 mM Tris-HCl, pH 7.9, 100 mM NaCl, 10 mM MgCl2, and 1 mM DTT supplemented with calf intestinal alkaline phosphatase (New England Biolabs, cat# M0290) at a final concentration of 0.5 U/mL), and the suspension was incubated for 10 min at 37 °C to catalyze the dephosphorylation of both 5′ and 3′ ends of the precipitated RNA. After incubation, the beads were washed twice with 1 mL of phosphatase washing buffer (50 mM Tris–HCl, pH 7.5, 20 mM EGTA, and 0.5% NP40) and twice with 1 mL of T4 poly-nucleotide kinase (PNK) buffer without DTT (50 mM Tris–HCl pH 7.5, 50 mM NaCl, and 10 mM MgCl2). For PAR-CLIP-biotin chemiluminescent nucleic acid detection, HEK293T cells (5 × 10 cm plates) were transfected with plasmids expressing FLAG-tagged RBP candidates. RNAs coimmunoprecipitated with the candidate RBPs were labeled with biotin according to the instructions of the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher Scientific, cat# 89880). After the labeling, the beads were washed three times with washing buffer (containing 50 mM HEPES-KOH pH 7.5, 300 mM KCl, 0.05% NP40, 0.5 mM DTT, RNase inhibitor, and protease inhibitor cocktail) for 5 min at 4 °C and then incubated with SDS-PAGE loading buffer. The beads were then heated at 95 °C for 5 min to release the RNA–protein complexes. The RNA–protein complexes were loaded and run with Novex Bis-Tris 4–12% PAGE gel in MOPS buffer (Invitrogen). The complexes were transferred from the gel onto the nitrocellulose membrane on ice for western blotting or chemiluminescence nucleic acid detection. For PAR-CLIP sequencing (25 plates, 10 cm), the RNAs coimmunoprecipitated with CDK1 or METTL1 were treated with 1 U/μl T4 PNK (New England Biolabs, cat# M0201) for 60 min at 37 °C to transfer a phosphate to 5′-OH group of the precipitated RNA. After that, the samples were washed, eluted, and run with Novex Bis-Tris 4–12% PAGE gel in MOPS buffer. RNA–protein complexes were then transferred onto a nitrocellulose membrane (in parallel, another membrane was used for PAR-CLIP-biotin chemiluminescent nucleic acid detection by using a FUSION SOLO 4M System to identify the position of the captured RNAs). The first membrane was cut accordingly into small slices, and 200 μl proteinase K buffer (100 mM Tris–HCl, pH 7.4, 50 mM NaCl, and 10 mM EDTA) supplemented with proteinase K (Sigma-Aldrich) at a final concentration of 4 μg/μl were added. The slices were then incubated at 37 °C for 20 min. After that, 200 μl PK/urea buffer (100 mM Tris–HCl pH 7.4, 50 mM NaCl, 10 mM EDTA, and 7 M urea) were added and incubated for another 20 min at 37 °C. Afterwards, the RNA was extracted by adding 400 μl acid phenol/CHCl3 (pH 4.3–4.7). Then, 1 μl of glycogen (20 μg/μl), 40 μl NaOAc (3 M, pH 5.5), and 1 mL 75% ethanol were added to assist the RNA precipitation at −20 °C overnight. Extracted RNAs were dissolved in 25 μl RNase-free water. The resultant RNAs were first ligated with 3′ RNA adaptor and then 5′ RNA adaptor. cDNAs were synthesized and amplified with a low-cycle PCR. The PCR products were selected based on size using PAGE gel according to instructions of TruSeq Small RNA Sample Prep Kit (Illumina). The purified library products were evaluated using the Agilent 2200 TapeStation and diluted to 10 pM for cluster generation and high-throughput sequencing (1 × 50 bp) on HiSeq 2500 (Illumina).
RNA immunoprecipitation
RIP experiments were performed as reported37. Briefly, 2 × 107 cells were washed with chilled PBS, harvested by scraping, centrifuged, and resuspended in an approximately equal pellet volume of polysome lysis buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES pH 7.0, 0.5% NP-40, and 1 mM DTT) supplemented with RNase inhibitors (RNaseOUT, Thermo Fisher Scientific) and protease inhibitor cocktail. Samples were homogenized using a syringe with a 0.4 mm diameter needle and spun down in a microcentrifuge at maximum speed for 10 min at 4 °C. Supernatants were precleared with 30 μl protein G beads (Invitrogen) at 4 °C. Then, the lysates were diluted with NT2 buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM MgCl2, and 0.05% NP-40) supplemented with 100 mM DTT and 20 mM EDTA. One-twentieth of the supernatant was saved as RNA input, and the remaining lysates were used for RIP with 30 μl anti-FLAG magnetic beads (Sigma-Aldrich) at 4 °C overnight. Beads were washed five times with NT2 buffer. Afterwards, the RNA–protein complexes were digested with pro-teinase K at 55 °C for 1 h. RNA was purified using TRIzol and analyzed by RT-qPCR or semiquantitative PCR. Detailed primer information is provided in Supplementary Table 8.
PolyA-depleted RICK
Cells (5 × 10 cm plates) were harvested by scraping and lysed in lysis buffer (100 mM Tris–HCl, pH 7.5, 500 mM LiCl, 10 mM EDTA pH 8.0, 1% LiDS, 5 mM DTT, and protease inhibitor cocktail) after click reaction, as described above. Lysates were homogenized using a syringe with a 0.4 mm diameter needle. 100 μl oligo(dT) beads (New England Biolabs, cat# S1419) were washed twice using lysis buffer and added to the cell lysate to incubate for 1 h at 4 °C. Beads bound to RNA–protein complexes were collected, and the remaining lysates were subjected to another two rounds of oligo(dT) capture. After that, the supernatants were incubated with Dynabeads MyOne Streptavidin C1 beads and then processed for RICK isolation. Then, the RNA–protein complexes bound to the Dynabeads MyOne Streptavidin C1 beads were extracted. The extracted proteins were digested and processed for LC-MS/MS analysis with Q Exactive Hybrid Quadrupole-Orbitrap MS system (Thermo Fisher Scientific) in an information-dependent acquisition mode, as described above.
Statistical analysis
All statistical calculations are included in the relevant figure legends. P value less than 0.05 was accepted as significant; *P < 0.05, **P < 0.01, ***P < 0.001.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
RICK RNA-seq and PAR-CLIP sequencing data shown in this paper have been deposited in GEO under accession code GSE100756. Source data files for Figures 2e, 5d, and Supplementary Figures 3f, 6b, 7d, 8d, and 9c are included in Supplementary Tables 9 and 10. All unprocessed scans of blots are available online as Supplementary Figures 11 and 12. Code to generate distribution of the aligned reads is available from the corresponding author upon request.
Supplementary Material
Acknowledgments
We thank all other members of the Laboratory of RNA, Chromatin, and Human Disease for their support, and we also thank R. Johnson (University of Bern, Switzerland), X. Zhang (Institute of Biophysics, Chinese Academy of Sciences, Beijing), L.-L. Chen (Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai), Y. Zhou (Wuhan University, Wuhan), M. Tortorella, and Q. Zheng (Guangzhou Institutes of Biomedicine and Health, Guangzhou), Y. Xu (Sun Yat-sen University, Guangzhou), and P. Liu (Anhui University, Hefei) for their expert comments and technical support. C.W. is supported by Zhujiang Talent-Overseas Postdoc Funding Grant, M.J.K. and S.K. are supported by CAS President’s International Fellowships, and M.T. is supported by CAS-TWAS President’s PhD Fellowship. This work was supported by the National Key Research and Development Program of China (2016YFA0100701 to X.B., 2016YFA0100102 to M.A.E., 2016YFA0100301 to B.Q., and 2017YFA0504400 to J.Y.), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16030502 to M.A.E.), the Pearl River Science and Technology Nova Program of Guangzhou (201610010107 to X.B., 201610010053 to W.W.), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2015294) to X.B.; the Cooperation Program of the Research Grants Council (RGC) of Hong Kong and the National Natural Science Foundation of China (NSFC; 81261160506 to M.A.E.); the NSFC (31371513 and 31671537 to M.A.E., 91440115 to B.Z., 81401909 to M.Y.), and 91440110 to J.Y., the General Research Funds from the RGC of Hong Kong (14116014 to H.W. 14113514 to H.S.), the Focused Innovations Scheme (Scheme B: 1907307 to H.S.), the RGC Collaborative Research Fund from RGC (C6015-14G to H.S. and H.W.), the Bureau of Science, Technology and Information of Guangzhou Municipality (2012J5100040 and 201508030027 to M.A.E.); the Natural Science Foundation of Guangdong Province (2014A030312001 to M.A.E., 2016A050503037 and 2015A030308007 to B.Q.); the Strategic Emerging Industry Key Technology Project of Guangdong Province (2012A080800006 to B.Z.), the Shenzhen Peacock Team Project (KQTD2015033117210153 to Nan L.); the Introduced Innovative R&D Team Program of Guangdong Province (201001Y0104789252 to B.Z.), the NIH Grant (R00CA175290) and the Cancer Prevention Research Institute of Texas First-Time Tenure-Track Faculty Recruitment Grant (RR140071) to Y.C. The Guangdong Provincial Key Laboratory of Stem Cell and Regenerative Medicine is supported by the Science and Technology Planning Project of Guangdong Province, China (2017B030314056).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
X.B. and M.A.E. conceived the idea, and X.G. contributed to the idea; X.B. and X.G. performed most of the experiments; M.Y. contributed to the experiments; M.T., Yiwei L., S.K., J.Z., Na L., Yuan L., C.P.-Q., X.W., L.J., M.J.K., X.Z., Z.L., C.S., D.-H.L., X.L., W.W., M.H., Y.-L.L., C.W., T.W., G.Z., D.W., J.Y., Y.C., C.Z., R.J., Y.-G.Y., Y.W., B.Q., M.-L.A., A.P.H., H.S., and X.-D.F. provided technical support, relevant advice, or performed computational analyses; Nan L., B.Q., H.W., and B.Z. provided infrastructural support; X.B., B.Z., and M.A.E. provided most of the financial support and supervised the project; X.B., X.G., and M.A.E. wrote the manuscript. X.B., B.Z., and M.A.E. approved the final version of the manuscript.
References
- 1.Castello A, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 2.Baltz AG, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 3.Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011;12:R16. doi: 10.1186/gb-2011-12-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Q, et al. Poly A- transcripts expressed in HeLa cells. PLoS One. 2008;3:e2803. doi: 10.1371/journal.pone.0002803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng J, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 6.Jao CY, Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci USA. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang XO, et al. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–147. doi: 10.1016/j.cell.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai F, Gardini A, Zhang A, Shiekhattar R. Integrator mediates the biogenesis of enhancer RNAs. Nature. 2015;525:399–403. doi: 10.1038/nature14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glažar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–1670. doi: 10.1261/rna.043687.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Memczak S, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 14.Liu L, et al. Transcriptional pause release is a rate-limiting step for somatic cell reprogramming. Cell Stem Cell. 2014;15:574–588. doi: 10.1016/j.stem.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 15.Liu P, et al. Release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein (snRNP) activates hexamethylene bisacetamide-inducible protein (HEXIM1) transcription. J Biol Chem. 2014;289:9918–9925. doi: 10.1074/jbc.M113.539015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckmann BM, et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat Commun. 2015;6:10127. doi: 10.1038/ncomms10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conrad T, et al. Serial interactome capture of the human cell nucleus. Nat Commun. 2016;7:11212. doi: 10.1038/ncomms11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexandrov A, Martzen MR, Phizicky EM. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA. 2002;8:1253–1266. doi: 10.1017/s1355838202024019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao W, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okamoto H, et al. Substrate tRNA recognition mechanism of tRNA (m7G46) methyltransferase from Aquifex aeolicus. J Biol Chem. 2004;279:49151–49159. doi: 10.1074/jbc.M408209200. [DOI] [PubMed] [Google Scholar]
- 24.Leulliot N, et al. Structure of the yeast tRNA m7G methylation complex. Structure. 2008;16:52–61. doi: 10.1016/j.str.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Yang E, et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003;13:1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bao X, et al. The p53-induced lincRNA-p21 derails somatic cell reprogramming by sustaining H3K9me3 and CpG methylation at pluripotency gene promoters. Cell Res. 2015;25:80–92. doi: 10.1038/cr.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mews P, et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017;546:381–386. doi: 10.1038/nature22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roundtree IA, Evans ME, Pan T, He C. Dynamic rna modifications in gene expression regulation. Cell. 2017;169:1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon SC, et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013;20:1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- 30.Wong DJ, et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben-Porath I, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhattacharya B, et al. Gene expression in human embryonic stem cell lines: unique molecular signature. Blood. 2004;103:2956–2964. doi: 10.1182/blood-2003-09-3314. [DOI] [PubMed] [Google Scholar]
- 33.Ivanova N, et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 34.Lu X, et al. SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nat Cell Biol. 2013;15:1141–1152. doi: 10.1038/ncb2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho L, et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci USA. 2009;106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu G, et al. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanakkanthara A, et al. Cyclin A2 is an RNA binding protein that controls Mre11 mRNA translation. Science. 2016;353:1549–1552. doi: 10.1126/science.aaf7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brannan KW, et al. SONAR discovers RNA-binding proteins from analysis of large-scale protein-protein interactomes. Mol Cell. 2016;64:282–293. doi: 10.1016/j.molcel.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao Y, et al. ThecCardiomyocyte RNA-binding proteome: links to intermediary metabolism and heart disease. Cell Rep. 2016;16:1456–1469. doi: 10.1016/j.celrep.2016.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He C, et al. High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol Cell. 2016;64:416–430. doi: 10.1016/j.molcel.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ying QL, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutchins AP, Jauch R, Dyla M, Miranda-Saavedra D. glbase: a framework for combining, analyzing and displaying heterogeneous genomic and high-throughput sequencing data. Cell Regen (Lond) 2014;3:1. doi: 10.1186/2045-9769-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corcoran DL, et al. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011;12:R79. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.