

Abstract
The existence of multidrug-resistant influenza viruses, coupled with the continuously antigenic shift and antigenic drift of influenza viruses, necessitates the development of the next-generation of influenza antivirals. As the AM2-S31N mutant persists in more than 95 % of current circulating influenza A viruses, targeting the AM2-S31N proton channel appears to be a logical and valid approach to combating drug resistance. Starting from compound 1, an isoxazole compound with potent AM2-S31N channel blockage and antiviral activity, in this study we report an expeditious synthetic strategy that allows us to promptly explore the structure–activity relationships of isoxazole-containing AM2-S31N inhibitors. Propelled by the convenient synthesis, the lead optimization effort yielded a number of potent antivirals with submicromolar efficacy against several human clinical isolates of influenza A viruses, including both oseltamivir-sensitive and -resistant strains.
Keywords: Influenza A virus, AM2 proton channel, AM2-S31N inhibitor, antiviral
Graphical Abstract

INTRODUCTION
Influenza is a contagious respiratory illness that affects an estimated 10–15% of the population in the United States during the annual influenza season.1 For immune-competent person, infection with seasonal influenza virus is usually self-confined and non–life-threating. However, the symptoms and disease outcomes are exacerbated in seniors, children, and immunocompromised individuals.2 Furthermore, various influenza strains differ significantly in terms of their lethality for the general population.3 Mortality rates are disproportionally high for emerging and re-emerging pandemic influenza viruses as well as highly pathogenic avian influenza viruses (HPAIVs).4 For example, the 1918 Spanish H1N1 influenza pandemic claimed an estimated 50 million lives worldwide in a matter of months.5 The recent 2009 H1N1 influenza pandemic led to 284,000 deaths globally in the first 12 months of outbreak as estimated by the CDC.6 The mortality rate for HPAIVs such as H5N1 is more than 50%;7 therefore, effective vaccines and antiviral drugs are clearly needed to confine and limit the impact of influenza viruses.8
Vaccines are generally effective in preventing influenza infection with an overall effectiveness of approximately 60%.9 Due to the six–months delay between antigen identification and vaccine production, viruses might mutate during this period, which would render vaccines less effective.10, 11 Moreover, vaccines are not always available at the beginning of an influenza outbreak. Thus small–molecule antiviral drugs that target the conserved viral proteins such as the AM2-S31N are highly desired.2, 12, 13
The AM2-S31N mutant is one of the most conserved viral proteins among currently circulating influenza A viruses, and more than 95% of the viruses carry this mutation.14 Although this mutation confers amantadine resistance, this is not a result of the amantadine drug selection pressure.15 Instead the AM2-S31N mutant is a natural mutation that happens to maintain nearly identical channel function as the wild-type AM2 (AM2-WT) but is resistant to amantadine.16 Our goal is to design potent channel blockers of the AM2-S31N mutant and develop them as the next-generation of influenza antivirals. Propelled by mechanistic studies of AM2-S31N drug inhibition and proton conductance using X-ray crystallography,17–23 NMR,19–21 and molecular dynamics simulations,24 we were able to design inhibitors that target the AM2-S31N mutant,14 which was previously deemed undruggable as no progress had been made for decades.14 Briefly, at the initial stage of drug design, there was no AM2-S31N structure available so molecular dynamics simulations were applied to generate homology models of AM2-S31N.20, 24 In the drug-free form, AM2 adapts two conformations, Openout-Closedin and Closedout-Openin. In the Openout-Closedin conformation the AM2 channel opens to the viral exterior to allow protons to diffuse into the channel, while in the Closedout-Openin conformation, the AM2 channel opens to the viral interior such that the protonated histidines can release protons to the viral cytoplasm. We have designed inhibitors that intend to target either the Openout-Closedin or the Closedout-Openin conformation of AM2-S31N, and so far we were only able to successfully target the Openout-Closedin conformation.14, 25 With one of the AM2-S31N inhibitor M2WJ332 in hand, we were able to solve the solution NMR structure of AM2-S31N (PDB: 2LY0), and the structure indeed showed that the drug M2WJ332 bound to AM2-S31N channel when it was in the Openout-Closedin conformation.21 In terms of the biological activity, the designed AM2-S31N inhibitors were found to have potent antiviral activity against multidrug-resistant influenza A viruses.26, 27 More importantly, the AM2-S31N inhibitors have a higher genetic barrier to drug resistance than amantadine, and resistant mutants that emerge under AM2-S31N inhibitors drug selection pressure in cell culture are rare mutations.26 In light of these promising results, we are interested in continuously developing AM2-S31N inhibitors and providing lead compounds for in vivo mouse studies.
Initial structure–activity relationship (SAR) studies of the AM2-S31N inhibitors revealed that potent channel blockers contain the following structural features: a hydrophobic template such as adamantane or hydroxyl adamantane, an ammonium linker, and an aryl head group with a hydrophobic substitution (Figure 1A).21, 28, 29 It has been found that the aryl head group prefers to be isoxazole and a representative example is compound 1 (Figure 1B). As previous studies suggested that the hydrophobic substitution at the 5–position of isoxazole is critical for the channel blockage and antiviral activity,29 we are interested in exploring various aryls and heteroaryls at this substitution. To facilitate iterative cycles of design, synthesis, and biological characterization, we aim to develop an expeditious synthetic strategy that allows us to install diverse aryls and heteroaryls at the last step of the synthesis (Figure 1C). In this study, we report the optimized synthesis route as well as the channel blockage and antiviral activity of the synthesized compounds. These efforts resulted in several promising lead compounds with improved potency and a high selectivity index against multidrug-resistant influenza A viruses.
Figure 1.

AM2-S31N inhibitors. (A) Pharmacophore of AM2-S31N inhibitors. (B) Representative isoxazole-containing AM2-S31N inhibitor. (C) Synthesis of potent AM2-S31N inhibitors by microwave-assisted Suzuki–Miyaura cross-coupling.
RESULTS AND DISCUSSION
CHEMISTRY
Previous synthesis of isoxazole-containing AM2-S31N inhibitors involved a five-step synthesis procedure.29 Specifically, condensation of methyl ketone 2 and dimethyl oxalate 3 using potassium tert-butoxide as the base gave the intermediate 4, which was cyclized to isoxazole ester 5 upon treatment with hydroxylamine hydrochloride under mild heating at 50 °C in methanol. The ester 5 was subsequently reduced to alcohol 6 by sodium borohydride and was then converted to bromide 7 by triphenyl phosphine and carbon tetrabromide in dichloromethane. The last step alkylation of bromide 7 with the amantadine analog 8 furnished the isoxazole-containing AM2-S31N inhibitor 9. Although this is a highly reliable synthesis route with overall yields ranging from 26% to 62% (depending on substrates), one apparent limitation of this strategy from the medicinal chemistry perspective is that the diversity at the 5-position of isoxazole was carried from the first step to the last step, which means that it takes five steps to synthesize every single compound. This is less than ideal as it poses a rate-limiting step in the iterative cycles of design, synthesis, and biological characterization. In searching for an alternative synthesis route to speed up the lead optimization process, we devised a late-step diversification strategy (Scheme 1B). In the newly proposed synthesis strategy, the diversity was introduced in the last step using Suzuki–Miyaura cross-coupling. The success of the proposed route relies on not only the synthesis of the key intermediate 14, which has not been reported before, but also the last step Suzuki–Miyaura cross-coupling reaction. As an initial trial, a condensation–cyclization reaction between diethyl oxalacetate sodium 10 and hydroxylamine in the presence of sodium bicarbonate gave the intermediate 11 with a 40% yield, which was then converted to the key intermediate ethyl 5-bromoisoxazole-3-carboxylate 14 using phosphoryl bromide. Although we were able to obtain this intermediate 14, the yield for second step bromination was only 23%. We then switched to another strategy which started from 3+2 cycloaddition between a nitrile oxide generated from 2-chloro-2-(hydroxyimino)acetate 12 and ethynyltributylstannane to give the intermediate 13 in a 75% yield.30 Replacement of the tributylstannane group in 13 by bromide using bromine proceeded smoothly with a decent yield of 90%. With the key intermediate 14 in hand, we then proceeded to the reduction of 14 with diisobutylaluminium hydride at 0 °C to give the alcohol intermediate 15, which was then oxidized by Dess–Martin periodinane to give the aldehyde 16. The yields for these two steps were 85% and 64%, respectively. Reductive amination of the isoxazole aldehyde 16 with adamantane analogues 8a–b using previously optimized condition gave intermediate 17a–b in high yields: 79% for 17a and 82% for 17b.
Scheme 1.

Previously reported and newly discovered synthesis routes to isoxazole-containing AM2-S31N inhibitors.
The next critical step was to establish a cross-coupling condition to install a variety of aryls and heteroaryls at the 5-position of isoxazole, which has not been widely explored.31, 32 To this end, we chose N-[(5-bromo-1,2-oxazol-3-yl)methyl]adamantan-1-amine 17a and phenylboronic acid as a model reaction for reaction condition optimization (Table 1). Using microwave irradiation, a series of Suzuki coupling conditions with variations in catalyst, solvent, base, and reaction time were explored (Table 1). It was found that Pd(PPh3)4 was a preferred catalyst over Pd(OAc)2, Pd2dba3, and Pd(dppf)Cl2. Solvent and temperature also played important roles in this reaction and the highest yield (78%) was achieved when dioxane was used as the solvent and the reaction was heated at 150 °C (Table 1, entry 7). A reaction performed at a lower temperature, 120 °C, had a lower yield (Table 1, entry 11). Reduction of the reaction time from 60 minutes to 30 minutes had little to no effect on the yield (Table 1, entry 7 versus entry 10). Nevertheless, considering the yield might be substrate-dependent, a 60-minute reaction time was employed for the following cross-coupling reactions. In summary, the optimized reaction condition was 5% Pd(PPh3)4, 1.2 equivalents of boronic acid, 1.4 equivalents of sodium bicarbonate, and heating at 150 °C for 60 minutes in dioxane under microwave irradiation.
Table 1.
Optimization of the Suzuki–Miyaura cross-coupling reaction for isoxazole
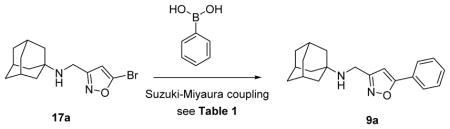
| ||||||
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
| Entry | Tempa °C | Catalyst | Solvent | Time | Base | Yieldb (%) |
|
|
|
|
|
|
|
|
| 1 | 120 | Pd(OAc)2c | Toluene/H2O | 30 min | K3PO4 | 15 |
| 2 | 100 | Pd2dba3c | DME | 60 min | K3PO4 | 47 |
| 3 | 100 | Pd2dba3c | Dioxane/H2O | 60 min | K3PO4 | 37 |
| 4 | 100 | Pd2dba3c | Dioxane | 60 min | K3PO4 | 51 |
| 5 | 90 | Pd(dppf)Cl2 | DME | 60 min | K2CO3 | 55 |
| 6 | 150 | Pd(PPh3)4 | Dioxane/H2O | 60 min | Na2CO3 | 70 |
| 7 | 150 | Pd(PPh3)4 | Dioxane | 60 min | Na2CO3 | 78 |
| 8 | 150 | Pd(PPh3)4 | Toluene | 60 min | Na2CO3 | 68 |
| 9 | 150 | Pd(PPh3)4 | DME | 60 min | Na2CO3 | 57 |
| 10 | 150 | Pd(PPh3)4 | Dioxane | 30 min | Na2CO3 | 77 |
| 11 | 120 | Pd(PPh3)4 | Dioxane | 60 min | Na2CO3 | 70 |
Microwave irradiation
Isolated Yield
Tricyclohexylphosphine was used as a ligand
With the optimized cross-coupling condition, we then synthesized a library of isoxazole-containing compounds (Scheme 2) with the substations at the 5-position of isoxazole being substituted benzene (9a, 9d–9n), pyridine (9b–9c), 1,1′-diphenyl (9o), naphthalene (9p), and substituted thiophene (9q–9v). It was found that this cross-coupling condition was not very sensitive to the substrate, and the reaction proceeded smoothly in the presence of functional groups such as amine, alcohol, and phenol, with yields ranging from 60% to 76%. Overall, this synthesis strategy proved to be robust and amenable for parallel synthesis.
Scheme 2.

Synthesis of a library of isoxazole-containing AM2-S31N inhibitors using the optimized Suzuki–Miyaura cross-coupling reaction condition.
CHANNEL INHIBITION, ANTIVIRAL ACTIVITY, AND CYTOTOXICITY OF ISOXAZOLE-CONTAINING AM2-S31N INHIBITORS
All synthesized compounds were tested for channel blockage against the AM2-S31N proton channel and antiviral activity against the A/California/07/2009 (H1N1) virus using the two-electrode voltage clamp (TEVC) assay and plaque assay, respectively. TEVC is one of the standard assays used to measure AM2 ion channel blockage by small molecules. Briefly, the AM2-S31N channel was expressed in the oocyte cell membrane and was activated by acidic pH (pH = 5.5), compound was added at 100 μM concentration and the channel conductance current at the two-minute time point was recorded. The activity of the compound was expressed as a percentage of current inhibition, which was calculated by dividing the current at the two-minute time point by the maximum conductance at pH 5.5. For AM2-S31N proton channel inhibition, it was found that benzene and thiophene are the preferred aryl substitutions at the 5-position of isoxazole (Table 2), while pyridines (9b and 9c) were not tolerated. The channel blockage was also sensitive to the substitutions on the benzene and thiophene rings. For example, when R equals benzene, compounds with chloride (9d) and thiomethyl (9f) substitutions at the ortho position had similar channel blockage as compound 9a, while compounds with methoxyl (9e) and hydroxyl (9h) had reduced channel blockage activity. When a mesyl group was present at the ortho position, compound 9g was nearly completely inactive. Compounds with substitutions at the meta position of benzene (9i and 9j) were also less potent in blocking the AM2-S31N channel than 9a, suggesting substitution at the meta position is not tolerated. For compounds with substitutions at the para position of benzene, dimethylamine (9k), cyano (9l), and methyl ester (9n) were tolerated, while amide (9m) was not compatible (47.4 ± 0.5%). Compounds with either 1,1′-diphenyl (9o) or naphthalene (9p) substitution had decreased channel blockage activity, probably due to steric hindrance. In contrast, when R equals thiophene, compounds 9q–9v all showed potent channel blockage (>78% at 100 μM). We also tested a few selected compounds (9a, 9d, 9f, 9h, 9q, 9s, 9t, and 9v) against the wild-type AM2 channel (AM2-WT). It was found that none of the compounds had inhibition greater than 30% at 100 μM, except compound 9v, which showed moderate inhibition (38.2 ± 1.6%). This result was not a surprising finding as this class of compounds were designed to specifically target the amantadine-resistant AM2-S31N mutant channel. In the next step, all compounds were tested for their antiviral activity in plaque assays at 10 μM against the amantadine-resistant influenza A virus A/California/07/2009 (H1N1), which contains the AM2-S31N mutation. Several compounds were able to reduce the plaque formation to less than 20 % compared to that of the DMSO control (9a, 9d, 9f, 9h, 9q, 9r, 9s, 9t, 9u, and 9v). All of these compounds (9a, 9d, 9f, 9h, 9q, 9r, 9s, 9t, 9u, and 9v) also had potent channel blockage against the AM2-S31N channel (> 70% at 100 μM), which is consistent with their potent antiviral activity. To further dissect the correlation between channel blockage efficacy and antiviral activity of all the synthesized AM2-S31N inhibitors, we plotted the percentage of channel blockage against the percentage of plaque formation and calculated the correlation coefficient (r). The correlation efficient r was calculated to be −0.81, suggesting that there was a strong correlation between the compound’s channel blockage and antiviral activity. Specifically, twelve out of the twenty-two compounds (9a, 9d, 9e, 9f, 9g, 9l, 9m, 9n, 9r, 9t, 9u, and 9v) fall on the curve, meaning that their channel blockage correlated well with their antiviral activity. However, there were compounds that fell both above and under the curve. Four compounds (9b, 9i, 9j, and 9k) fell to the northeast side of the curve (above the curve), meaning that their actual antiviral activity was less potent than that predicted by their AM2-S31N channel blockage efficacy. Likewise, six compounds (9c, 9h, 9o, 9p, 9q, and 9s) fell to the southwest side of the curve (below the curve), meaning that their actual antiviral activity was more potent than that predicted by their AM2-S31N channel blockage efficacy. The lack of a straight linear correlation between the results from the in vitro target-specific electrophysiological assay and the cellular antiviral assay might be due to many factors, such as the compound’s membrane permeability, off-target effects, binding kinetics, and etc. Specifically, in the antiviral plaque assay, compounds were incubated with the virus-infected cells for two days; thus the percentage of plaque formation represents the true potency of the compound. In contrast, the electrophysiological assay is under kinetic control: the percentage of current inhibition was recorded at the two-minute time point after applying the compounds in which the drug-binding equilibrium may or may not be achieved.26 Therefore, the electrophysiological percentage inhibition might underestimated the compound’s true potency if the compound showed slow binding kinetics. In summary, the electrophysiological assay results should be viewed as a means to confirm the compound’s mechanism of action but not a predictor for the compound’s cellular antiviral potency. Understanding this concept is critical for the lead optimization, as it suggests that the structure–activity relationship studies should not only focus on improving the channel blockage activity, but also on improving the cellular antiviral potency.
Table 2.
Channel blockage, antiviral activity, and cytotoxicity of isoxazole-containing AM2-S31N inhibitors
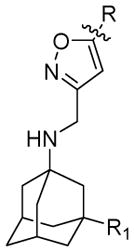
|
R | R1 | Compound ID | Electrophysiological TEVC assay*(% S31N channel inhibition) | Electrophysiological TEVC assay*(% WT channel inhibition) | % plaque formation at 10 μM drug concentration against A/California/07/2009 (H1N1) | CC50 (μM)MDCK cells | CC50 (μM) A549 cells |
|
H | 9a | 91.2 ± 1.5 | 19.7 ± 0.6 | 13.5 ± 2.3 | N.T. | N.T. | |
|
H | 9b | 53.0 ± 2.4 | N.T. | 105.5 ± 1.9 | N.T. | N.T. | |
|
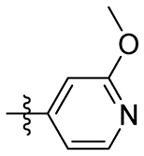
H | 9c | 58.0 ± 2.5 | N.T. | 21.7 ± 0.6 | N.T. | N.T. | |
|
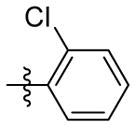
OH | 9d | 87.1 ± 0.3 | 24.6 ± 0.5 | 8.3 ± 0.8 | 64.1 ± 2.5 | 142.6 ± 11.4 | |
|
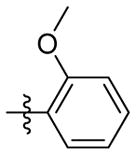
H | 9e | 51.8 ± 4.0 | N.T. | 62.1 ± 0.6 | N.T. | N.T. | |
|
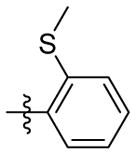
OH | 9f | 90.6 ± 0.6 | 28.8 ± 0.3 | 9.2 ± 0.3 | 59.3 ± 2.6 | 101.9 ± 4.5 | |
|
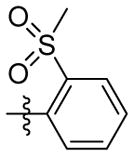
H | 9g | 9.8 ± 1.2 | N.T. | 110.0 ± 1.0 | N.T. | N.T. | |
|
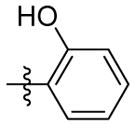
H | 9h | 71.5 ± 0.5 | 24.1 ± 1.9 | 3.4 ± 0.1 | 73.7 ± 1.0 | 78.2 ± 8.5 | |
|
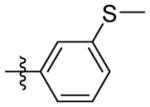
H | 9i | 69.6 ± 3.4 | N.T. | 56.3 ± 2.4 | N.T. | N.T. | |
|
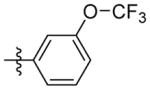
H | 9j | 56.7 ± 2.9 | N.T. | 72.7 ± 2.0 | N.T. | N.T. | |
|
H | 9k | 83.2 ± 2.2 | N.T. | 35.7 ± 0.6 | N.T. | N.T. | |
|
H | 9l | 72.2 ± 2.2 | N.T. | 28.0 ± 0.9 | N.T. | N.T. | |
|
H | 9m | 47.4 ± 0.5 | N.T. | 57.1 ± 2.4 | N.T. | N.T. | |
|
H | 9n | 73.6 ± 2.5 | N.T. | 24.1 ± 0.5 | N.T. | N.T. | |
|
|
H | 9o | 45.6 ± 0.6 | N.T. | 36.0 ± 2.1 | N.T. | N.T. | |
|
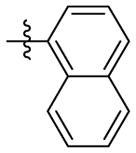
H | 9p | 56.8 ± 2.4 | N.T. | 36.1 ± 0.4 | N.T. | N.T. | |
|
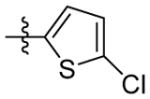
H | 9q | 78.8 ± 3.0 | 0 | 0 | 190.5 ± 14.2 | 208.6 ± 20.9 | |
|
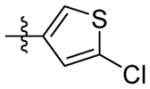
H | 9r | 87.9 ± 1.8 | N.T. | 10.9 ± 0.9 | 35.6 ± 1.9 | 52.6 ± 4.4 | |
|
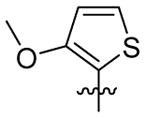
OH | 9s | 83.8 ± 3.3 | 14.5 ± 0.5 | 0.3 ± 0.1 | > 200 | > 200 | |
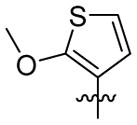
|
H | 9t | 94.2 ± 0.3 | 24.3 ± 1.3 | 7.2 ± 0.7 | 30.6 ± 5.0 | 35.5 ± 2.9 | |
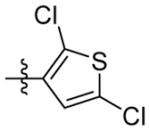
|
H | 9u | 87.7 ± 1.1 | N.T. | 10.1 ± 1.1 | 39.3 ± 2.2 | 61.4 ± 9.1 | |
| OH | 9v | 89.9 ± 1.3 | 38.2 ± 1.6 | 13.7 ± 0.1 | 87.6 ± 10.0 | N.T. |
Values represent the mean of three independent measurements.
The data are presented as percent inhibition at 100 μM at the two-minute time point. N.T. = not determined.
Another important factor that needs to be taken into consideration during lead optimization is the cellular toxicity. For this reason, the cytotoxicity of the potent lead compounds (9d, 9f, 9h, 9q, 9r, 9s, 9t, 9u, and 9v) was tested against both MDCK cells and human epithelial A549 cells using the neutral red method (Table 2).33 Compounds 9r, 9t, and 9u were found to have moderate cytotoxicity against both MDCK and A549 cells, with CC50 values less than 65 μM; thus these three compounds were excluded from further studies. Compounds 9d and 9f also showed moderate cytotoxicity against the MDCK cells (CC50 < 65 μM) but were less cytotoxic to the A549 cells (CC50 > 100 μM). Compounds 9h, 9q, 9s, and 9v appear to be well-tolerated for both MDCK and A549 cells and the CC50 values were greater than 70 μM.
Next, potent lead compounds with low cytotoxicity (9a, 9d, 9f, 9h, 9q, 9s, and 9v) were titrated against human clinical isolates of influenza A viruses in the plaque assay (Figure 3). According to the CC50 values (Table 2), the highest drug concentration applied in the plaque assay was 30 μM, which conferred minimal cellular cytotoxicity. It was found that all compounds had potent antiviral activity against the 2009 pandemic H1N1 virus A/California/07/2009 (H1N1) in a dose-dependent manner except compound 9v, which showed incomplete inhibition at the highest drug concentration of 30 μM (Figure 3A and 3B). Testing of compound 9v at a higher drug concentration was not attempted due to cellular cytotoxicity. The EC50 values for compounds 9a, 9d, 9f, 9h, 9q, and 9s range from 0.1 μM to 1.2 μM, with the most potent compound being 9q (EC50 = 0.1 ± 0.03 μM). Compound 9q was also non-cytotoxic, and the CC50 values for MDCK cells and A549 cells were 190.5 ± 14.2 μM and 208.6 ± 20.9 μM, respectively, resulting in selectivity indexes of 1905 and 2086 for MDCK and A549 cells, respectively. To further profile its antiviral spectrum, compound 9q was tested against three additional influenza A strains, A/Washington/29/2009 (H1N1), A/Denmark/528/2009 (H1N1), and A/Switzerland/9715293/2013 (H3N2). The A/Washington/29/2009 (H1N1) and A/Denmark/528/2009 (H1N1) viruses were chosen because they are representative examples of multidrug-resistant influenza A viruses and these two strains are resistant to both amantadine and oseltamivir due to AM2-S31N and H275Y mutations in their AM2 and neuraminidase genes, respectively. The A/Switzerland/9715293/2013 (H3N2) virus was chosen because it is an emerging predominant strain that was circulating in the 2014–2015 influenza season in North America.34 This strain is resistant to amantadine due to AM2-S31N mutation but is sensitive to oseltamivir. It was found that compound 9q had potent antiviral activity against all three drug-resistant influenza A strains, with EC50 values ranging from 0.1 μM to 0.2 μM. Overall, these results suggest that AM2-S31N inhibitors are potent antivirals against influenza A viruses with diverse genetic backgrounds.
Figure 3.
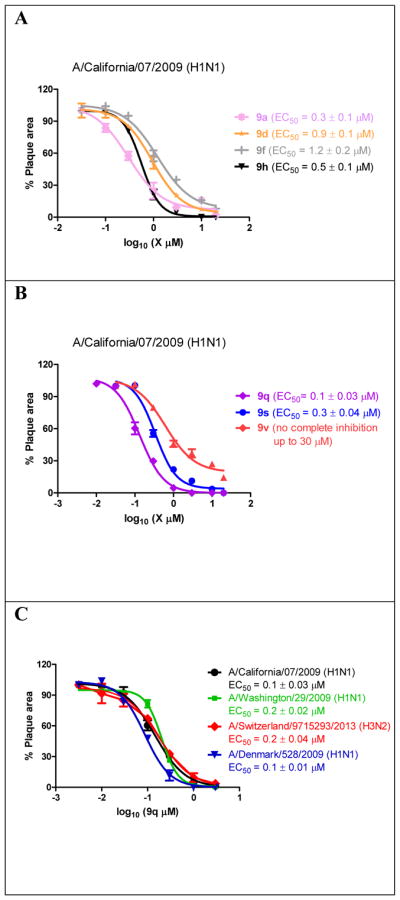
Antiviral efficacy of potent AM2-S31N inhibitors in inhibiting clinically relevant human influenza A viruses. The antiviral efficacy was determined using plaque reduction assays. (A) Dose response curves of compounds 9a, 9d, 9f, and 9h in inhibiting the A/California/07/2009 (H1N1) virus. (A) Dose response curves of compounds 9q, 9s and 9v in inhibiting the A/California/07/2009 (H1N1) virus. (B) Dose response curves of compound 9q in inhibiting the A/California/07/2009 (H1N1), A/Washington/29/2009 (H1N1), A/Switzerland/9715293/2013 (H3N2), and A/Denmark/528/2009 (H1N1) viruses. Each data point represents duplicate experiments.
To gain further insights how the most potent compound 9q binds to the AM2-S31N channel, molecular docking was performed (Figure. 4). In the drug-free form, AM2-S31N adapts the Closedout-Openin conformation as shown by the X-ray crystal structure (PDB: 3C02) in Figure 4A.22 The AM2-WT channel similarly adapts the Closedout-Openin conformation when it binds to amantadine.18 The overlay of the drug-free AM2-S31N structure (PDB: 3C02) with the amantadine-bound AM2-WT structure (PDB: 3C9J) was shown in Figure 4B. In contrast, when AM2-S31N inhibitors such as M2WJ332 binds to the AM2-S31N channel, the channel adapts the Openout-Closedin conformation as shown by the solution NMR structure (PDB: 2LY0).21 For this reason, the Openout-Closedin conformation of the AM2-S31N channel (PDB: 2LY0) was chosen for the docking of the most potent compound 9q. It was found that compound 9q binds to the AM2-S31N channel with its aryl group facing towards the N-terminus of the channel (Figure 4C). The adamantane ring of 9q is surrounded by the G34 residues, and the positively charged ammonium from 9q forms a hydrogen bond with one of the N31 side chain carbonyl group. The distal thiophene ring from 9q is surrounded by the V27 side chain methyl groups and this might be driven by hydrophobic interactions. It can be seen clearly from the overlay structures of the docking model and the drug-free AM2-S31N structure (Figure 4D) that when compound 9q binds to the AM2-S31N channel, the channel undergoes a conformation change, leading to the opening of the N-terminus of the channel to accommodate the bulky drug 9q. In summary, the docking model shows that compound 9q binds to the Openout-Closedin conformation of the AM2-S31N channel, which is consistent with previously solved solution NMR structure (PDB: 2LY0).21
Figure 4.

Molecular docking of compound 9q in the AM2-S31N channel. (A) X-ray crystal structure of drug-free AM2-S31N structure (PDB: 3C02).22 (B) Overlay of the drug-free AM2-S31N structure (orange) and the amantadine-bound AM2-WT structure (cyan) (PDB: 3C9J).18 (C) Docking model of compound 9q in the AM2-S31N channel. The solution NMR structure (PDB: 2LY0) of AM2-S31N was used for the docking.21 Docking was performed using Autodock Vina.35 (D) Overlay of the docking model (gray) and drug-free AM2-S31N structure (orange) (PDB: 3C02).22
CONCLUSIONS
Despite the urgent need of influenza antivirals to combat not only existing drug-resistant strains but also emerging or re-emerging strains that will undoubtedly appear in the near future, the drug discovery process, however, is unfortunately a lengthy process. With our continuous efforts in developing antivirals to target the predominant drug-resistant AM2-S31N mutant, we aimed to develop an expeditious synthesis strategy in order to speed up the iterative cycles of design, synthesis, and biological characterization. The synthesis route we devised allowed us to introduce a diverse set of substitutions at the 5-position of isoxazole at the last step, and the optimized Suzuki–Miyaura cross-coupling reaction condition was compatible with functional groups such as amine, hydroxyl, and phenol. All synthesized compounds were tested for their channel blockage in electrophysiological assays, and potent lead compounds were further validated in antiviral assays. Potent channel blockers were generally found to have greater antiviral efficacy; however, the correlation was not straightly linear (r = −0.81). It is worth highlighting that this was the first attempt to systematically correlate the channel blockage activity with antiviral activity for AM2-S31N inhibitors. The important message is that the TEVC assay results can be used to confirm the compound’s mechanism of action, but caution should be taken to use them to predict the antiviral activity. To corroborate with this conclusion, the most potent compound emerging from this study was compound 9q, which had single- to sub-micromolar efficacy against all four AM2-S31N-containing influenza A viruses (A/California/07/2009 (H1N1), A/Washington/29/2009 (H1N1), A/Denmark/528/2009 (H1N1), and A/Switzerland/9715293/2013 (H3N2)). However, this compound did not necessarily have the most potent channel blockage (78.8 ± 3.0% at 100 μM) when compared with other compounds such as 9a and 9f (>90% inhibition at 100 μM). Overall, these results suggest that lead optimization should not focus solely on improving the channel blockage potency, and optimizing antiviral activity should also be taken into consideration.
In summary, the potent antiviral efficacy, coupled with the high selectivity index of compound 9q, warrants further developing it as a next generation of antiviral drug. The major therapeutic value of AM2-S31N inhibitors is that they are active against both oseltamivir-sensitive and -resistant strains; therefore, they can be used either alone to combat oseltamivir-resistant strains or used in combination with oseltamivir to delay the evolution of resistant strains.
EXPERIMENTAL SECTION
Chemistry
All chemicals that were commercially available were used without further purification. Detailed synthesis procedures can be found below for reactions described in Scheme 1, Table 1, and Scheme 2. All final compounds were purified by flash column chromatography. 1H and 13C NMR spectra were recorded on a Bruker-400 NMR spectrometer. Chemical shifts are reported in parts per million referenced with respect to residual solvent (CD3OD) 3.31 ppm and (Chloroform-d) 7.24 ppm or from internal standard tetramethylsilane (TMS) 0.00 ppm. The following abbreviations were used in reporting spectra: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublets; ddd, doublet of doublet of doublets. All reactions were carried out under N2 atmosphere, unless otherwise stated. HPLC-grade solvents were used for all reactions. Flash column chromatography was performed using silica gel (230–400 mesh, Merck). Low-resolution mass spectra were obtained using an ESI technique on a 3200 Q Trap LC/MS/MS system (Applied Biosystems). The purity was assessed by using Shimadzu LC-MS with Waters XTerra MS C-18 column (part #186000538), 50 × 2.1 mm, at a flow rate of 0.3 mL/min; λ = 250 and 220 nm; mobile phase A, 0.1% formic acid in H2O, and mobile phase B′, 0.1% formic in 60% isopropanol, 30% CH3CN and 9.9% H2O. All compounds submitted for testing in TEVC assay and plaque reduction assay were confirmed to be > 95.0% purity by LC-MS traces. All compounds were characterized by proton and carbon NMR and MS.
Synthesis Procedures
Ethyl 5-oxo-4,5-dihydroisoxazole-3-carboxylate (11)
To a suspension of diethyl oxalacetate 10 (3 g, 15.94 mmol) in water (20 mL) was added sodium bicarbonate (1.2g, 17.1 mmol) and hydroxylamine hydrochloride (1.2g, 34.5 mmol) at room temperature. The mixture was stirred for 18 hours. Then HCl (1M, 20 mL) was added and the resulting solution was extracted with dichloromethane. The combined organic layers were then dried over MgSO4, filtered and concentrated under reduced pressure. The resulting mixture was then purified by silica gel flash column chromatography (0–10% EtOAc/Hexane) to give the final product as a white solid (1.01 g) 1H NMR (400 MHz, CDCl3) δ 4.45 (q, J = 7.1 Hz, 2H), 3.71 (s, 2H), 1.43 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.83, 158.11, 157.51, 63.24, 33.25, 14.00. C6H7NO4 EI-MS: m/z (M+H+): 158.1 (calculated), 158.0 (found). Yield: 40%.
Ethyl 5-(tributylstannyl)isoxazole-3-carboxylate (13)
To a solution of ethyl 2-chloro-2-(hydroxyimino)acetate (1.2 g, 7.9 mmol) in anhydrous dichloromethane (20 mL) was added ethynyltributylstannane (2.5g, 7.9 mmol) and potassium carbonate (1.2g, 8.7 mmol) at room temperature. The mixture was stirred for 24 hours and was then quenched with water. The resulting solution was extracted with dichloromethane. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The mixture was then purified by silica gel flash column chromatography (0–5% EtOAc/Hexane) to give the final product as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.82 (s, 1H), 4.47 (q, J = 7.1 Hz, 2H), 1.74 – 1.56 (m, 8H), 1.45 (t, J = 7.1 Hz, 3H), 1.40 – 1.20(m, 16H), 0.98 – 0.89 (m, 12H). C18H33NO3Sn EI-MS: m/z (M+H+): 432.1 (calculated), 432.0 (found). Yield: 75%.
Ethyl 5-bromoisoxazole-3-carboxylate (14)
To a mixture of 11 (550 mg, 3.5 mmol) in toluene (50 mL) was added phosphoryl bromide (5.5g, 17.5 mmol) and triethyl amine (2 mL) at 0 °C. The mixture was stirred at room temperature for one hour and then heated at 80 °C for 48 hours. The resulting residue was cooled to room temperature, quenched with water, and extracted with dichloromethane. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The mixture was then purified by silica gel flash column chromatography (0–10% EtOAc/Hexane) to give the final product as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.70 (s, 1H), 4.43 (q, J = 7.1 Hz, 2H), 1.47 – 1.35 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 158.75, 157.95, 143.16, 107.10, 62.58, 14.09. C6H7BrNO3 EI-MS: m/z (M+H+): 220.0 (calculated), 220.1 (found). Yield: 23%.
Ethyl 5-bromoisoxazole-3-carboxylate (14)
To a mixture of 13 (0.9 g, 2.1 mmol) and sodium carbonate (250 mg, 2.3 mmol) in dichloromethane (15 mL) was added bromine (82 uL, 3.1mmol) at room temperature. The mixture was stirred for 24 hours and then quenched with sodium thiosuflate solution (10%, 10 mL). The resulting residue was extracted with dichloromethane. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The mixture was then purified by silica gel flash column chromatography (0–10% EtOAc/Hexane) to give the final product as a colorless oil (415 mg). Yield: 90%.
(5-bromoisoxazol-3-yl)methanol (15)
To a solution of 14 (160 mg, 0.73 mmol) in tetrahydrofuran (10 mL) at 0 °C was added diisobutylaluminum hydride (4 mL, 1.1 M, 4.4 mmol) and the resulting solution was stirred for 2 hours. Then the reaction was quenched by adding 1M HCl (10 mL) and the solution was stirred at room temperature for 2 hours. The resulting solution was extracted with dichloromethane. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The mixture was clean on NMR and was used directly for the next step oxidation without further purification. 1H NMR (400 MHz, CDCl3) δ 6.37 (d, J = 1.1 Hz, 1H), 4.72 (d, J = 5.0 Hz, 2H), 3.21 (br, 1H). 13C NMR (101 MHz, CDCl3) δ 165.59, 141.80, 105.48, 56.81. C4H5BrNO2 EI-MS: m/z (M+H+): 178.0 (calculated), 178.1 (found). Yield: 85%.
5-bromoisoxazole-3-carbaldehyde (16)
To a mixture of alcohol 15 (200 mg, 1.12 mmol) in dichloromethane was added Dess-Martin periodinane (712 mg, 1.68 mmol) and the solution was stirred for 1 hour. The reaction was quenched with sodium thiosulfate (10%, 5 mL) and saturated sodium bicarbonate (5 mL) solution. The resulting mixture was extracted with ethyl ether (2 × 10 mL) and ethyl acetate (2 × 10 mL). The organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The mixture was then purified by silica gel flash column chromatography (0–20 % EtOAc/Hexane) to give 16 as pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 10.07 (s, 1H), 6.68 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 183.28, 163.12, 143.97, 104.07. C4H3BrNO2 EI-MS: m/z (M+H+): 176.0 (calculated), 176.1 (found). Yield: 64%.
N-[(5-bromo-1,2-oxazol-3-yl)methyl]adamantan-1-amine (17a)
Adamantane 8a (147 mg, 0.97 mmol) and 16 (170 mg, 0.97 mmol) were mixed with 1.5 mL of titanium (IV) isopropoxide. The resulting slurry was heated in microwave at 120 °C for 30 minutes. Then the solution was cooled down to room temperature and CH3OH (10 mL) was added. The resulting solution was cooled to 0 °C and NaBH4 (147 mg, 3.88 mmol) was added portionwise in 10 minutes. The solution was then warmed to room temperature and stirred for another hour. The reaction was quenched with 1M NaOH and filtered through celite. The filtrate was extracted with dichloromethane and was purified by silica gel flash column chromatography (5–10 % CH3OH/CH2Cl2) to give the final product as a white solid. 1H NMR (400 MHz, CDCl3) δ 6.25 (t, J = 0.9 Hz, 1H), 3.90 (s, 2H), 2.07 (s, 3H), 1.74 – 1.53 (m, 12H). C14H20BrN2O EI-MS: m/z (M+H+): 311.0 (calculated), 311.1 (found). Yield: 79%.
3-{[(5-bromo-1,2-oxazol-3-yl)methyl]amino}adamantan-1-ol (17b)
Compound 17b was synthesized using the same procedure as described for 17a. 1H NMR (400 MHz, CDCl3) δ 7.23 (s, 1H), 6.24 (s, 1H), 3.89 (s, 2H), 2.26 (br, 3H), 1.75 – 1.40 (m, 14H). 13C NMR (101 MHz, CDCl3) δ 174.88, 140.35, 105.30, 69.59, 54.12, 50.09, 44.31, 41.29, 37.56, 37.35, 37.15, 35.00, 30.85, 30.83. C14H20BrN2O2 EI-MS: m/z (M+H+): 327.1 (calculated), 327.0 (found). Yield: 82%.
General procedure of Suzuki coupling reaction (Table 1)
To a solution of admantane isoxazole bromide (17a–b) (1 eq) in dioxane (2 mL) was added boronic acid or boronic ester (1.2 eq) and sodium carbonate (1.4 eq). The mixture was purged with dry nitrogen for 5 minutes. Then tetrakis(triphenylphosphine)palladium(0) (0.05 eq) was added and the solution was heated in microwave at 150 °C for 30 minutes. The solvent was removed under reduced pressure, and the resulting residue was extracted with dichloromethane and water. The organic layer was separated, dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The mixture was then purified by silica gel flash column chromatography (0–10% CH3OH/CH2Cl2) to give the final product.
N-[(5-phenyl-1,2-oxazol-3-yl)methyl]adamantan-1-amine (9a)
Characterization of 9a was reported before.21
N-((5-(pyridin-3-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9b)
1H NMR (400 MHz, CDCl3) δ 9.02 (dd, J = 2.3, 0.9 Hz, 1H), 8.69 (dd, J = 4.8, 1.7 Hz, 1H), 8.15 (ddd, J = 7.9, 2.3, 1.7 Hz, 1H), 7.41 (ddd, J = 7.9, 4.8, 0.9 Hz, 1H), 6.58 (t, J = 0.8 Hz, 1H), 4.01 (d, J = 0.9 Hz, 2H), 2.14 (s, 3H), 1.84 – 1.59 (m, 12H). C19H24N3O EI-MS: m/z (M+H+): 310.4 (calculated), 310.0 (found). Yield: 61%.
N-((5-(6-methoxypyridin-3-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9c)
1H NMR (400 MHz, CD3OD) δ 8.19 (dd, J = 5.2, 2.3 Hz, 1H), 8.05 (ddd, J = 7.2, 5.1, 2.1 Hz, 1H), 7.14 – 7.00 (m, 1H), 6.84 (d, J = 1.7 Hz, 1H), 4.53 (s, 2H), 3.31 (s, 3H), 2.31 – 2.18 (m, 3H), 2.01-1.96 (m, 6H), 1.86-1.72(m, 6H). C20H26N3O2 EI-MS: m/z (M+H+): 340.4 (calculated), 340.0 (found). Yield: 70%.
3-({[5-(2-chlorophenyl)-1,2-oxazol-3-yl]methyl}amino)adamantan-1-ol (9d)
1HNMR (400 MHz, CDCl3 + CD3OD): δ 7.80-7.78 (m, 1H), 7.51-7.48 (m, 1H), 7.44-7.41 (m, 1H), 7.34-7.30 (m, 2H), 4.21 (s, 2H), 2.31 (s, 2H), 1.96 (s, 2H), 1.91-1.85 (m, 4H), 1.65-1.62 (m, 4H), 1.51 (s, 2H). 13CNMR (100 MHz, CDCl3 + CD3OD) δ 171.97, 160.83, 135.98, 135.40, 134.86, 133.31, 131.17, 129.43, 108.82, 72.22, 64.48, 49.42, 46.66, 40.79, 39.29, 38.02, 34.10. C20H23ClN2O2 EI-MS: m/z (M+H+): 359.9 (calculated), 360.0 (found). Yield: 72%.
N-((5-(2-methoxyphenyl)isoxazol-3-yl)methyl)adamantan-1-amine (9e)
1H NMR (400 MHz, CDCl3) δ 7.76 (ddd, J = 7.8, 1.6, 0.5 Hz, 1H), 7.37 (ddd, J = 8.2, 7.3, 1.5 Hz, 1H), 7.30 (dd, J = 8.1, 1.3 Hz, 1H), 7.24 – 7.20 (m, 1H), 6.82 (s, 1H), 3.92 (s, 2H), 2.49 (s, 3H), 2.08 (s, 3H), 1.72 – 1.59 (m, 12H). C21H26N2O2 EI-MS: m/z (M+H+): 339.5 (calculated), 339.0 (found). Yield: 74%.
3-[({5-[2-(methylsulfanyl)phenyl]-1,2-oxazol-3-yl}methyl)amino]adamantan-1-ol (9f)
1H NMR (400 MHz, CDCl3 + CD3OD): δ 7.68-7.65 (m, 1H), 7.38 (s, 1H), 7.37-7.34 (m, 1H), 7.28-7.26 (m, 1H), 7.20-7.16 (m, 1H), 4.23 (s, 2H), 3.64 (s, 1H), 2.45 (s, 3H), 2.34 (s, 2H), 2.03 (s, 2H), 1.95-1.87 (m, 4H), 1.68 (m, 4H), 1.58 (s, 2H). 13C NMR (100 MHz, CDCl3 + CD3OD) δ 169.23, 156.74, 137.92, 130.33, 128.63, 126.06, 124.35, 104.26, 68.30, 60.35, 45.81, 42.86, 36.60, 35.61, 33.58, 30.01, 15.83. C21H26N2O2S EI-MS: m/z (M+H+): 371.5 (calculated), 371.0(found). Yield: 75%.
N-((5-(2-(methylsulfonyl)phenyl)isoxazol-3-yl)methyl)adamantan-1-amine (9g)
1H NMR (400 MHz, CD3OD) δ 7.70 – 7.60 (m, 1H), 7.40 (td, J = 7.4, 1.3 Hz, 1H), 7.35 – 7.29 (m, 1H), 7.23 (d, J = 7.3 Hz, 1H), 6.52 (s, 1H), 4.18 (s, 2H), 2.83 (s, 3H), 1.93 (q, J = 3.0 Hz, 4H), 1.68 (d, J = 2.9 Hz, 7H), 1.55 – 1.40 (m, 5H). C21H27N2O3S EI-MS: m/z (M+H+): 387.5 (calculated), 387.0 (found). Yield: 69%.
2-(3-(((adamantan-1-yl)amino)methyl)isoxazol-5-yl)phenol (9h)
1H NMR (400 MHz, CDCl3) δ 7.96 – 7.90 (m, 1H), 7.53 – 7.47 (m, 1H), 7.46 – 7.36 (m, 2H), 6.98 (s, 1H), 3.96 (s, 2H), 2.13 (s, 3H), 1.75 – 1.63 (m, 12H). C20H24N2O2 EI-MS: m/z (M+H+): 325.4 (calculated), 325.0 (found). Yield: 68%.
N-((5-(3-(methylthio)phenyl)isoxazol-3-yl)methyl)adamantan-1-amine (9i)
1H NMR (400 MHz, CD3OD) δ 7.56-7.35 (m, 2H), 7.32 – 7.24 (m, 2H), 6.82 (d, J = 0.7 Hz, 1H), 4.49 (d, J = 0.6 Hz, 2H), 2.46 (s, 3H), 2.28-2.21 (m, 3H), 1.98 (m, 6H), 1.88 – 1.66 (m, 6H). C21H27N2OS EI-MS: m/z (M+H+): 355.5 (calculated), 355.1 (found). Yield: 77%.
N-((5-(3-(trifluoromethoxy)phenyl)isoxazol-3-yl)methyl)adamantan-1-amine (9j)
1H NMR (400 MHz, CD3OD) δ 7.64 (d, J = 7.4 Hz, 1H), 7.51 (s, 1H), 7.46 – 7.41 (m, 1H), 7.28 (ddt, J = 8.2, 2.4, 1.1 Hz, 1H), 6.81 (s, 1H), 4.49 (d, J = 0.6 Hz, 2H), 2.25 (s, 3H), 1.98 (d, J = 3.0 Hz, 6H), 1.90 – 1.59 (m, 6H). C21H24F3N2O2 EI-MS: m/z (M+H+): 393.4 (calculated), 393.1 (found). Yield: 70%.
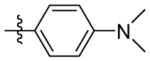
N-((5-(4-(dimethylamino)phenyl)isoxazol-3-yl)methyl)adamantan-1-amine (9k)
1H NMR (400 MHz, CDCl3) δ 7.96 – 7.90 (m, 1H), 7.53 – 7.47 (m, 1H), 7.46 – 7.36 (m, 2H), 6.98 (s, 1H), 3.96 (s, 2H), 2.13 (s, 3H), 1.75 – 1.63 (m, 12H). C22H29N3O EI-MS: m/z (M+H+): 352.5 (calculated), 352.0 (found). Yield: 76%.
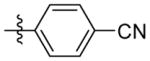
4-(3-(((adamantan-1-yl)amino)methyl)isoxazol-5-yl)benzonitrile (9l)
1H NMR (400 MHz, CDCl3) δ 7.88 – 7.71 (m, 2H), 7.28 (dt, J = 7.9, 1.0 Hz, 2H), 6.56 (s, 1H), 3.89 (s, 2H), 2.09 (s, 3H), 1.72 – 1.47 (m, 12H). C21H23N3O EI-MS: m/z (M+H+): 334.4 (calculated), 334.0 (found). Yield: 71%.
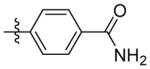
N-((5-(2-(methylsulfonyl)phenyl)isoxazol-3-yl)methyl)adamantan-1-amine (9m)
1H NMR (400 MHz, CDCl3) δ 9.63 (br, 2H), 7.64 (ddd, J = 6.8, 3.5, 1.7 Hz, 2H), 7.07 (dd, J = 5.6, 2.9 Hz, 2H), 6.87 – 6.74 (m, 1H), 4.19 (d, J = 4.5 Hz, 2H), 2.09 (s, 3H), 1.91 (d, J = 4.1 Hz, 6H), 1.75 – 1.49 (m, 6H). C21H26N3O2 EI-MS: m/z (M+H+): 352.5 (calculated), 352.1 (found). Yield: 73%.
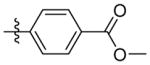
Methyl 4-(3-(((adamantan-1-yl)amino)methyl)isoxazol-5-yl)benzoate (9n)
1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 9.0 Hz, 2H), 7.06 – 6.96 (m, 2H), 6.46 (s, 1H), 3.91 (d, J = 0.3 Hz, 2H), 3.88 (s, 3H), 2.13 (s, 3H), 1.84 – 1.60 (m, 12H). C22H27N2O3 EI-MS: m/z (M+H+): 367.5 (calculated), 367.0 (found). Yield: 72%.
N-((5-([1,1′-biphenyl]-4-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9o)
1H NMR (400 MHz, CDCl3) δ 7.57 – 7.52 (m, 5H), 7.48 – 7.40 (m, 4H), 7.38 – 7.32 (m, 1H), 6.25 (t, J = 0.9 Hz, 1H), 3.90 (d, J = 0.8 Hz, 2H), 2.08 (s, 3H), 1.78 – 1.50 (m, 12H). C26H28N2O EI-MS: m/z (M+H+): 385.5 (calculated), 385.0 (found). Yield: 76%.
N-((5-(naphthalen-1-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9p)
1H NMR (400 MHz, CD3OD) δ 7.85 (dt, J = 7.8, 1.0 Hz, 2H), 7.76 (d, J = 7.6 Hz, 1H), 7.58 – 7.38 (m, 4H), 6.80 (d, J = 0.6 Hz, 1H), 4.47 (d, J = 0.5 Hz, 2H), 2.22 (s, 3H), 1.95 (d, J = 3.0 Hz, 6H), 1.84 – 1.69 (m, 6H). C24H27N2O EI-MS: m/z (M+H+): 359.5 (calculated), 359.0 (found). Yield: 70%.
N-((5-(5-chlorothiophen-2-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9q)
1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 6.0 Hz, 1H), 7.11 (d, J = 5.9 Hz, 1H), 6.22 (s, 1H), 3.89 (s, 2H), 2.07 (s, 3H), 1.74 – 1.55 (m, 13H). C18H22ClN2OS EI-MS: m/z (M+H+): 349.9 (calculated), 350.0 (found). Yield: 65%.
N-((5-(5-chlorothiophen-3-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9r)
1H NMR (400 MHz, CD3OD) δ 7.54 (s, 1H), 6.93 (s, 1H), 6.61 – 6.46 (s, 1H), 3.95 (s, 2H), 2.12 – 2.01 (m, 3H), 1.74 – 1.59 (m, 12H). C18H22ClN2OS EI-MS: m/z (M+H+): 349.9 (calculated), 350.0 (found). Yield: 60%.
3-({[5-(3-methoxythiophen-2-yl)-1,2-oxazol-3-yl]methyl}amino)adamantan-1-ol (9s)
Characterization of compound 9s was reported before.29
N-((5-(2-methoxythiophen-3-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9t)
1H NMR (400 MHz, CDCl3) δ 7.39 – 7.22 (m, 1H), 6.54 (d, J = 5.3 Hz, 1H), 6.28 (d, J = 1.0 Hz, 1H), 4.77 (br, 1H), 3.90 (d, J = 0.9 Hz, 2H), 3.83 (s, 3H), 2.09 (d, J = 3.8 Hz, 3H), 1.79 – 1.56 (m, 12H). C19H25N2O2S EI-MS: m/z (M+H+): 345.5 (calculated), 345.0 (found). Yield: 68%.
N-((5-(2,5-dichlorothiophen-3-yl)isoxazol-3-yl)methyl)adamantan-1-amine (9u)
1H NMR (400 MHz, CDCl3) δ 7.05 (s, 1H), 6.82 (s, 1H), 3.91 (d, J = 1.0 Hz, 2H), 2.07 (s, 3H), 1.74 – 1.57 (m, 12H). C18H21Cl2N2OS EI-MS: m/z (M+H+): 384.3 (calculated), 384.3 (found). Yield: 67%.
3-({[5-(2,5-dichlorothiophen-3-yl)-1,2-oxazol-3-yl]methyl}amino)adamantan-1-ol (9v)
1H NMR (400 MHz, CDCl3) δ 7.24 (s, 1H), 6.25 (s, 1H), 3.90 (d, J = 0.9 Hz, 2H), 2.32 – 2.22 (m, 2H), 1.70 – 1.42 (m, 13H). C18H21Cl2N2O2S EI-MS: m/z (M+H+): 400.3 (calculated), 400.0 (found). Yield: 60 %.
Two-electrode voltage clamp (TEVC) assay
The compounds were tested in a two-electrode voltage clamp assay using Xenopus laevis frog oocytes microinjected with RNA expressing either the AM2-WT or the AM2-S31N mutant of the AM2 protein, as previously reported.36 The potency of the inhibitors was expressed as percentage inhibition of A/M2 current observed after two minutes of incubation with 100 μM of compounds at pH 5.5. All measurements were repeated three times with different oocytes.
Plaque reduction assay
The plaque reduction assay was performed as previously reported,21 except MDCK cells expressing ST6Gal I were used instead of regular MDCK cells.37 Briefly, confluent monolayer of ST6Gal MDCK cells were incubated with ~100 pfu virus samples in DMEM with 0.5% BSA for 1 hour at 4 °C, then 37 °C for 1 hour. The inoculums were removed, and the cells were washed with phosphate buffered saline (PBS). The cells were then overlaid with DMEM containing 1.2% Avicel microcrystalline cellulose (FMC BioPolymer, Philadelphia, PA) and NAT (2.0 μg/mL). To examine the effect of the compounds on plaque formation, the overlay media was supplemented compounds at testing concentrations. At two days after infection, the monolayers were fixed and stained with crystal violet dye solution (0.2% crystal violet, 20% methanol). The viruses used for this assay were A/California/07/2009 (H1N1), A/Washington/29/2009 (H1N1), A/Denmark/528/2009 (H1N1) and A/Switzerland/9715293/2013 (H3N2), all of which contain the AM2-S31N mutant in their AM2 genes. Influenza A viruses A/Switzerland/9715293/2013 X-247 (H3N2), FR-1366, A/Washington/29/2009 (H1N1), FR-460, and A/California/07/2009 (H1N1), FR-201, were obtained through the Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA, USA. Influenza virus A/Denmark/528/2009 (H1N1) was obtained from Dr. Elena Govorkova at St. Jude Children’s Research Hospital.
Correlation coefficient
The correlation coefficient r between the compound’s channel blockage and antiviral activity was calculated using GraphPad Prism version 5.
Cytotoxicity assay and cytopathic effect assay
Evaluation of the cytotoxicity of compounds and the efficacy of compounds against influenza-induced cytopathic effect was carried out using the neutral red uptake assay.33 Briefly, 80,000 cells/mL of MDCK or A549 cells in DMEM medium supplemented with 10% FBS and 100 U/mL Penicillin-Streptomycin were dispensed into 96-well cell culture plates at 100 μL/well. Twenty-four hours later, the growth medium was removed and washed with 100 μL PBS buffer; then for the cytotoxicity assay, 200 μL fresh DMEM (No FBS) medium containing serial diluted compounds was added to each well. After incubating for 48 hours at 37 °C with 5% CO2 in a CO2 incubator, the medium was removed and replaced with 100 μL DMEM medium containing 40 μg/mL neutral red for four hours at 37 °C. The amount of neutral red uptake was determined at absorbance 540 nm using a Multiskan FC Microplate Photometer (Fisher Scientific). The CC50 values were calculated from best-fit dose response curves with variable slope in GraphPad Prism version 5.
Supplementary Material
Figure 2.
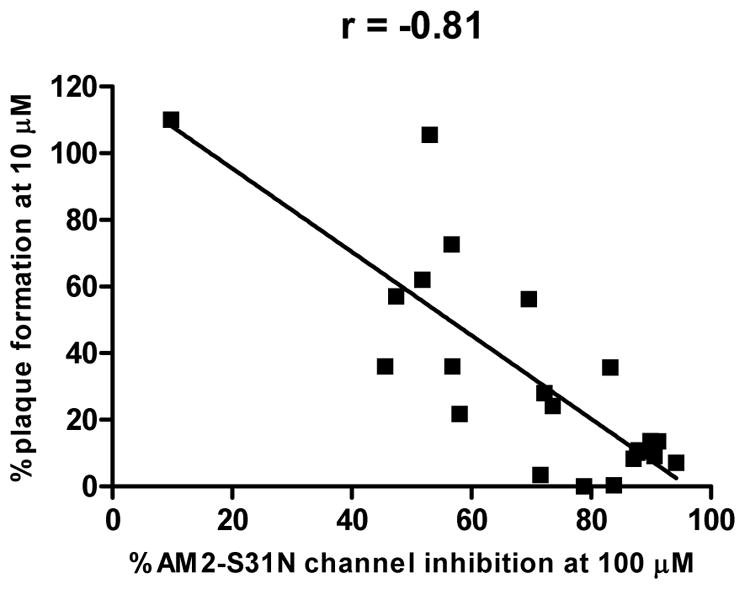
Correlation of channel blockage and antiviral activity for isoxazole-containing AM2-S31N inhibitors. P < 0.0001.
Acknowledgments
This research is supported by startup funding from the University of Arizona, the 2015 PhRMA Foundation Research Starter Grant in Pharmacology and Toxicology, and NIH grant AI119187 to J.W. We thank Dr. David Bishop for proofreading and editing the manuscript.
ABBREVIATIONS USED
- WT
wild type
- DMEM
Dulbecco’s modified eagle medium
- MDCK
Madin–Darby Canine Kidney
- TEVC
two-electrode voltage clamps
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Cox N, Subbarao K. Global epidemiology of influenza: past and present. Ann Rev Med. 2000;51:407–421. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- 2.Webster RG, Govorkova EA. Continuing challenges in influenza. Ann N Y Acad Sci. 2014;1323:115–139. doi: 10.1111/nyas.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson MG, Shay DK, Zhou H, Bridges CB, Cheng PY, Burns E, Bresee JS, Cox NJ. Estimates of deaths associated with seasonal influenza-United States, 1976–2007 (Reprinted from MMWR, vol 59, pg 1057–1062, 2010) JAMA. 2010;304:1778–1780. [Google Scholar]
- 4.Monto AS, Webster RG. Textbook of Influenza. John Wiley & Sons, Ltd; West Sussex: 2013. Influenza pandemics: History and lessons learned; pp. 20–34. [Google Scholar]
- 5.Taubenberger JK, Morens DM. 1918 influenza: the mother of all pandemics. Emerg Infect Dis. 2006;12:15–22. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, Bandaranayake D, Breiman RF, Brooks WA, Buchy P, Feikin DR, Fowler KB, Gordon A, Hien NT, Horby P, Huang QS, Katz MA, Krishnan A, Lal R, Montgomery JM, Molbak K, Pebody R, Presanis AM, Razuri H, Steens A, Tinoco YO, Wallinga J, Yu H, Vong S, Bresee J, Widdowson MA. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2012;12:687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- 7.Horimoto T, Kawaoka Y. Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol. 2005;3:591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- 8.Loregian A, Mercorelli B, Nannetti G, Compagnin C, Palù G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci. 2014:1–25. doi: 10.1007/s00018-014-1615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov. 2015;14:167–182. doi: 10.1038/nrd4529. [DOI] [PubMed] [Google Scholar]
- 10.Lambert L, Fauci A. Influenza vaccines for the future. N Engl J Med. 2010;363:2036–2044. doi: 10.1056/NEJMra1002842. [DOI] [PubMed] [Google Scholar]
- 11.Wong S, Webby R. Traditional and new influenza vaccines. Clin Microbiol Rev. 2013;26:476–492. doi: 10.1128/CMR.00097-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preziosi P. Influenza pharmacotherapy: present situation, strategies and hopes. Expert Opin Pharmacother. 2011;12:1523–1549. doi: 10.1517/14656566.2011.566557. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Qiu JX, Soto C, DeGrado WF. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr Opin Struct Biol. 2011;21:68–80. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Li F, Ma C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Biopolymers. 2015;104:291–309. doi: 10.1002/bip.22623. [DOI] [PubMed] [Google Scholar]
- 15.Furuse Y, Suzuki A, Oshitani H. Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for emergence and spread of amantadine resistance. Antimicrob Agents Chemother. 2009;53:4457–4463. doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balannik V, Carnevale V, Fiorin G, Levine B, Lamb R, Klein M, DeGrado W, Pinto L. Functional studies and modeling of pore-lining residue mutants of the influenza A virus M2 ion channel. Biochemistry. 2010;49:696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci U S A. 2010;107:15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Cady SD, Balannik V, Pinto LH, DeGrado WF, Hong M. Discovery of spiro-piperidine inhibitors and their modulation of the dynamics of the M2 proton channel from influenza A virus. J Am Chem Soc. 2009;131:8066–8076. doi: 10.1021/ja900063s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Kein ML, DeGrado WF. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J Am Chem Soc. 2011;133:12834–12841. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc Natl Acad Sci U S A. 2013;110:1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomaston JL, DeGrado WF. Crystal structure of the drug-resistant S31N influenza M2 proton channel. Protein Sci. 2016;25:1551–1554. doi: 10.1002/pro.2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomaston JL, Alfonso-Prieto M, Woldeyes RA, Fraser JS, Klein ML, Fiorin G, DeGrado WF. High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proc Natl Acad Sci U S A. 2015;112:14260–14265. doi: 10.1073/pnas.1518493112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khurana E, Dal Peraro M, DeVane R, Vemparala S, DeGrado W, Klein M. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc Natl Acad Sci U S A. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J. M2 as a target to combat influenza drug resistance: what does the evidence say? Future Virology. 2016;11:1–4. [Google Scholar]
- 26.Ma C, Zhang J, Wang J. Pharmacological characterization of the spectrum of antiviral activity and genetic barrier to drug resistance of M2-S31N channel blockers. Mol Pharmacol. 2016;90:188–198. doi: 10.1124/mol.116.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li F, Ma C, Hu Y, Wang Y, Wang J. Discovery of potent antivirals against amantadine-resistant influenza A viruses by targeting the M2-S31N proton channel. ACS Infect Dis. 2016;2:726–733. doi: 10.1021/acsinfecdis.6b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Ma C, Wang J, Jo H, Canturk B, Fiorin G, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Discovery of novel dual inhibitors of the wild-type and the most prevalent drug-resistant mutant, S31N, of the M2 proton channel from influenza A virus. J Med Chem. 2013;56:2804–2812. doi: 10.1021/jm301538e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F, Ma C, DeGrado WF, Wang J. Discovery of highly potent inhibitors targeting the predominant drug-resistant S31N mutant of the influenza A virus M2 proton channel. J Med Chem. 2016;59:1207–1216. doi: 10.1021/acs.jmedchem.5b01910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JS, Cho YS, Chang MH, Koh HY, Chung BY, Pae AN. Synthesis and in vitro activity of novel isoxazolyl tetrahydropyridinyl oxazolidinone antibacterial agents. Bioorg Med Chem Lett. 2003;13:4117–4120. doi: 10.1016/j.bmcl.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 31.Grob JE, Nunez J, Dechantsreiter MA, Hamann LG. Regioselective synthesis and slow-release Suzuki–Miyaura cross-coupling of MIDA boronate-functionalized isoxazoles and triazoles. J Org Chem. 2011;76:10241–10248. doi: 10.1021/jo201973t. [DOI] [PubMed] [Google Scholar]
- 32.Ku YY, Grieme T, Sharma P, Pu YM, Raje P, Morton H, King S. Use of iodoacetylene as a dipolarphile in the synthesis of 5-iodoisoxazole derivatives. Org Lett. 2001;3:4185–4187. doi: 10.1021/ol0168162. [DOI] [PubMed] [Google Scholar]
- 33.Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3:1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- 34.Grohskopf LA, Sokolow LZ, Olsen SJ, Bresee JS, Broder KR, Karron RA. Prevention and control of influenza with vaccines: recommendations of the advisory committee on immunization practices, United States, 2015–16 influenza season. MMWR Morb Mortal Wkly Rep. 2015;64:818–825. doi: 10.15585/mmwr.mm6430a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, DeGrado WF, Pinto LH. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry. 2009;48:11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hatakeyama S, Sakai-Tagawa Y, Kiso M, Goto H, Kawakami C, Mitamura K, Sugaya N, Suzuki Y, Kawaoka Y. Enhanced expression of an α2,6-linked sialic acid on MDCK cells improves isolation of human influenza viruses and evaluation of their sensitivity to a neuraminidase inhibitor. J Clin Microbiol. 2005;43:4139–4146. doi: 10.1128/JCM.43.8.4139-4146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.