

Abstract
We report the fabrication and performance of mechanically-responsive multilayer superhydrophobic composites. The application of tensile strain triggers the release of small molecules and proteins from these composites, with different tensile strain magnitudes and coating thickness influencing agent release. These mechanoresponsive composites consist of an absorbent drug core surrounded by an electrosprayed superhydrophobic protective coating that limits drug release in the absence of tensile strain. Coating thickness and applied tensile strain control release of chemotherapeutic cisplatin and enzyme β-galactosidase, as measured by atomic absorption and UV-Visible spectrophotometry, respectively, with preserved in vitro activity. Such mechanically-responsive drug delivery devices, when coupled to existing dynamic mechanical forces in the body or integrated with mechanical medical devices, such as stents, will provide local controlled dosing.
Keywords: Drug delivery, protein delivery, mechanoresponsive, superhydrophobic, biomaterials
Graphical Abstract
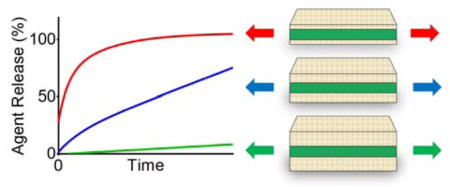
Stretching triggers the release of agents from multi-layered superhydrophobic composites Both the strain magnitude and superhydrophobic barrier coating thickness modulate release, resulting in the graded delivery of dye, chemotherapeutic cisplatin, and enzyme β-galactosidase with preserved in vitro activity.
Mechanically-responsive biomaterials are key components in drug delivery devices,[1,2] tissue engineering scaffolds,[3,4] and sensors.[5,6] The abundance of mechanical cues present within the body or applied externally to the body can serve as functional dynamic triggers.[2,7,8] For drug delivery applications, these mechanoresponsive devices provide spatiotemporal control over release of an active agent in response to compression,[9–11] tension,[12,13] or shear.[14–17]
In contrast, conventional polymeric drug delivery systems offer continuous release due to passive diffusion, hydration, or polymer degradation with control over the release profile based on both polymer and agent composition, structure, molecular weight, hydrophobicity, and/or charge as well as their interplay. Mechanoresponsive polymeric drug delivery systems provide an additional feature—active control via a mechanical stimulus. Of the agents delivered in the clinic, small molecules and proteins are the mainstay but each possesses specific delivery challenges. For example, water-soluble small molecules embedded in polymer systems typically have low encapsulation efficiency and require targeted or triggered delivery to effectively localize the drug payload, due to diffuse biodistribution and/or fast clearance.[18–20] Alternatively, the delivery of proteins suffers from similar obstacles as biologically-sensitive molecules are prone to denaturization and aggregation with low bioavailability.[21–23] Therefore, strategies to minimize the initial release, or trigger release on demand, as well as to modulate the release rate are of significant clinical interest.
To address these challenges and increase the portfolio of drug delivery strategies available, we are investigating multi-layered mechanoresponsive drug delivery systems, where the agent is loaded in a hydrophilic core and shielded by a protective superhydrophobic barrier from the surrounding milieu.[13] Release of the agent occurs upon the application of tensile strain through the formation of macroscopic cracks on the barrier layer that facilitate device wetting. Herein, we report the fabrication and performance of devices with varied superhydrophobic coating thicknesses (100, 200, and 350 μm) on a hydrophilic core substrate. The core substrate is amenable to solvent or aqueous loading of agents such that small molecules (dye, cisplatin) or proteins (β-galactosidase) are efficiently encapsulated. Application of tensile strains modulates agent release through strain magnitude, barrier coating thickness, and the nature of the entrapped agent. Biological function is preserved after release, as evident by the continued activity of β-galactosidase towards its substrate, and cytotoxicity of the released chemotherapeutic cisplatin to cancer cells in vitro.
Superhydrophobic materials, defined as having an advancing water contact angle greater than 150°,[24–26] have been successfully incorporated in drug delivery systems.[27] These systems demonstrate entrapped air stability over weeks to months, and are thus robust barriers against release in the absence of mechanical stimuli. Utilizing this strategy, we fabricated mechanoresponsive systems (Figure 1) composed of an inner hydrophilic polyester cellulose drug core that wets immediately with water (Figure 1c, water contact angle = 0°) and outer superhydrophobic polymeric coatings of varying thicknesses. Both the low surface energy of the coating composition, consisting of biocompatible[28,29] and biodegradable polymers poly(caprolactone) [PCL, Mw = 45 kDa] and poly(glycerol monostearate carbonate-co-caprolactone)[30,31] [PGC-C18, Mw = 140 kDa], and the high surface roughness via electrospraying (Figure 1a),[32] results in superhydrophobic coatings. Coatings of 100 μm, 200 μm, and 350 μm were electrosprayed onto the cellulose polyester core (Figure 1b), rendering the polymeric multi-layer structure superhydrophobic (Figure 1c–d) by increasing electrospraying time (10 w/v% 1:1 PGC-C18:PCL blend in chloroform, 2 mL/hour at 20–25 kV). A representative SEM image of the 350 μm coating shows surfaces composed of 2–7 μm interconnected particles with an advancing water contact angle of 167° (Figure 1d). A cross-sectional SEM image of the composite is available in Figure S1c. 100 μm and 200 μm coatings exhibit similar morphology and measured contact angles. Based on previous mechanical analysis,[13] the mismatch under tension between the electrosprayed coating and the drug-loaded core propagates parallel crack patterns perpendicular to the direction of applied strain, resulting in strain-dependent release while maintaining a robust barrier in the absence of tension.
Figure 1.

Fabrication method and characterization of superhydrophobic mechanoresponsive systems. a) The release agent (green) is first loaded into the mesh core and then electrosprayed with a mixture of PCL and PGC-C18 (yellow box). b) Various coating thicknesses of 100 μm, 200 μm, and 350 μm were fabricated (cross-sectional views shown). Characterization of c) hydrophilic core and d) superhydrophobic coatings with SEM (left; scale bars = 100 μm and 10 μm, respectively) and advancing water contact angle measurements (right). Figure not drawn to scale.
As an initial proof of concept, green dye (Yellow 5/Blue 1 (Mw Blue 1 = 0.8 kDa, Mw Yellow 5 = 0.5 kDa)) was used as a model hydrophilic compound and loaded into the cellulose polyester core. We have previously reported that composites with hydrophobic, but not superhydrophobic, coatings composed of only PCL resulted in rapid wetting and release of agents in the absence of tensile strain.[13] In the absence of tensile strain, 100 μm superhydrophobic coatings minimized release of agents, with less than 10% release over 2 hours, while 50 μm superhydrophobic coatings did not provide a sufficient barrier to agent release (Figure S3).[13] With superhydrophobic coatings of 350 μm, the system displayed strain-dependent release at 10%, 30%, and 50% strains with significant differences after 5 minutes when submerged in simulated biological fluid (PBS with 10% fetal bovine serum) (Figure 2a). After 1 hour, ~87%, ~44%, and ~9% of the dye had released at 50%, 30%, and 10% strains, respectively, as detected by UV-Visible spectrophotometry at 630 nm.
Figure 2.

Release kinetics of model hydrophilic dye from superhydrophobic mechanoresponsive systems with a) 350 μm coating with various tensile strains, and varying coating thickness at tensile strains of b) 10% with c) corresponding photographs of coating cracks (top: 100 μm coating, bottom: 350 μm coating), d) 30%, and e) 50% with f) corresponding photographs of coating cracks (top: 100 μm coating, bottom: 350 μm coating). Error bars denote + SD, * denote p < 0.05, N = 3, scale bar = 0.5 cm.
Applying uniaxial tensile strain of 10% (stretched to a length 10% longer than the original length of the mesh), the mechanoresponsive systems exhibited greater release with decreasing coating thickness (Figure 2b). After 15 minutes at 10% strain, the devices with 100 μm coatings had significantly greater cumulative release than the devices with 200 μm and 350 μm coatings. Between 40 and 50 minutes, there were significant differences in release between the 200 μm and 350 μm coatings. After 2 hours, ~55%, ~22%, and ~14% of the dye cumulatively released with the 100 μm, 200 μm, and 350 μm coatings, respectively.
According to our recent study,[13] applying greater tensile strain results in the development of cracks, increasing in both area and number, for water to infiltrate through the coating and subsequently release loaded agents. In this current study, the increase in coating thickness yields fewer and smaller cracks at low strains (Figure 2c, 10% tensile strain). To overcome the hindrance of the thicker barrier, higher tensile strains must be applied (350 μm coating at 30% strain vs 100 μm coating at 10% strain) to achieve a similar release profile (no statistical difference, Figure S2a).
However, further increasing strain causes the cracks to enlarge (Figure 2f), providing rapid water infiltration, and resulting in greater release compared to lower strains but no significant differences in release among different coating thicknesses subjected to the same higher strain magnitudes. For example, at 30% strain, the release is slower from the thicker coating (350 μm) but there is no statistical difference in the release compared to the 100 μm coating (Figure 2d). Furthermore, at 50% (Figure 2e) and 100% strains (Figure S2b) the release is further accelerated with no statistical difference in the cumulative release.
Successful demonstration of hydrophilic dye release based on strain magnitude and coating thickness led to the determination of release kinetics and in vitro efficacy of cisplatin, a hydrophilic chemotherapeutic, at 10% strain. Owing to its low molecular weight (0.3 kDa), the release kinetics are hypothesized to be analogous to the dye loaded mechanoresponsive system described above. Cisplatin was encapsulated with an efficiency of 97.8 +/− 8.8% and its release from the device was determined by atomic absorption. As shown in Figure 3a, there are significant differences in cisplatin release starting at 5 minutes for the 100 μm coating, compared to the 200 μm and 350 μm coatings. However, the release only significantly differs between the 200 μm and 350 μm coatings at 40 minutes and 50 minutes (at which ~44%, ~14%, and ~4% are released from the 100 μm, 200 μm, and 350 μm coatings, respectively). Despite these minimal differences in cumulative release, the amount of cisplatin delivered affects the corresponding viability of OE33 cells, an esophageal cancer cell line, with significant differences after 2 minutes and until 60 minutes between the 200 μm and 350 μm coatings (Figure 3b). At 40 minutes, there is still ~92% viability with the 350 μm coating, compared to ~53% and ~2% viability for the 200 μm and 100 μm coatings, respectively, demonstrating control over dosing kinetically and by coating thickness in vitro.
Figure 3.

a) Cumulative cisplatin release at 10% strain with varying coating thicknesses and b) corresponding in vitro viability of OE33, an esophageal cancer cell line. Error bars denote +/− SD, * denote p < 0.05, and N = 3.
Based on the dye and cisplatin release results with varied strain magnitudes and coating thicknesses, we determined the release kinetics for a model protein, β-galactosidase. β-galactosidase is a 116 kDa enzyme that catalyzes the conversion of β-galactosides into monosaccharides. To determine whether the activity of β-galactosidase is retained with our processing fabrication conditions, the enzymatic conversion of the substrate ortho-nitrophenyl-β-galactoside, a colorless compound, into β-D-galactose and ortho-nitrophenol, a yellow compound, was quantified by UV-Visible spectrophotometry at 420 nm.
Proteins form highly organized structures from which they generate specific functions. Hence, the preservation of protein activity is aligned with its structural stability, which can be denatured through physical parameters, such as temperature, light, dehydration, and pH.[33] To overcome this challenge, excipients, including buffering agents, osmolytes, carbohydrates, polymers, salts, surfactants, chelators anti-oxidants, and preservatives, are used to stabilize proteins.[34] Specifically, trehalose is a disaccharide and osmolyte that has been shown to maintain protein activity especially during drying or lyophilization.[35] With trehalose as an excipient, 72 +/− 4.5% of the loaded β-galactosidase remains enzymatically active (Figure 4a), as solvent exposure is minimized from the electrospray coating process at ambient temperatures. Therefore, these gentle processing conditions may also be amenable for the encapsulation and delivery of other active therapeutic proteins.
Figure 4.

a) Preservation of β-galactosidase activity with and without trehalose excipient before and after encapsulation, b) tensile strain-dependent release of enzyme β-galactosidase, and c) β-galactosidase release with varying superhydrophobic coating thickness. Error bars denote +/− SD, * denote p < 0.05, N = 3
With an intermediate coating thickness of 200 μm, the mechanoresponsive system exhibits strain-dependent release of β-galactosidase at 0%, 50%, and 100% strain with significant differences in release after 5 minutes until 30 minutes (p < 0.05) (Figure 4b). Half of the β-galactosidase releases within 5 minutes at 100% strain, while it requires 30 minutes to achieve the same cumulative release at 50% strain. However, due to its larger molecular weight compared to both the dye and cisplatin in the previous examples, the diffusion of β-galactosidase is slower and therefore requires a greater strain magnitude to trigger release (50% vs 10%). At the intermediate 50% strain, the effects of coating thickness are visible: half of the β-galactosidase releases within 10 minutes, 40 minutes, and 90 minutes with the 100 μm, 200 μm, and 350 μm coatings, respectively (Figure 4c). After 5 minutes, there are significant differences in release with the 350 μm coating compared to the 250 μm and 100 μm coatings, and after 25 minutes between the 200 μm and 100 μm coatings.
As proof-of-concept, we selected a hydrophilic non-elastic blend of polyester/cellulose for the core material. However, the electrospraying process is amenable to coating various substrates, ranging from aluminum foil, nitrile rubber, cotton fabric, and collagen.[36] While the coating affords barrier properties, the choice of substrate determines the bulk mechanical properties and agent loadability of the composite. The high absorbency of the polyester/cellulose core allows facile loading of hydrophilic small molecules and proteins, in addition to lipophilic agents, as we have previously reported.[13] However, this versatile absorbance is inelastic, thus limiting release to non-reversible tensile strain.
The concept of utilizing crack patterns under tensile strain has been previously explored for both drug delivery and tissue engineering, but our strategy differs from the published literature. For example Zhu et al[37] demonstrate precise control of periodic parallel crack patterns in polydimethylsiloxane from tensile strains of 5% to 50%, whereby the underlying material is exposed for template-free protein patterning. Both myoblasts and neuroblasts reversibly spread out and retract across fibronectin-coated cracks in vitro. Alternatively, Mertz et al[38] use polyelectrolyte multilayered systems that allow an increase in enzymatic activity of embedded alkaline phosphatase under 70% tensile strain. Too few polyelectrolyte layers result in permeable diffusion of enzyme substrate while thicker layers minimize the exposure of catalytic sites, reducing enzymatic activity. However, our approach differs as it releases agents rather than immobilizing them and exhibits a graded strain-dependent response compared to the switchable release from these examples. Nevertheless, the surface layers are critical to the architecture amongst all systems, as with oxidized PDMS coated with Pluronic in the former example, capping polyelectrolyte layers in the latter example, and superhydrophobic coatings in our mechanoresponsive system.
In summary, applying various tensile strains triggers the release of encapsulated agents from multi-layered superhydrophobic composites. These composites are composed of an inner inelastic hydrophilic core and an outer electrosprayed superhydrophobic coating. The superhydrophobic coating is a barrier against water infiltration, and limits the release of hydrophilic dyes, a chemotherapeutic, and a protein. Coating thickness and applied tensile strain magnitude control agent release. Thicker coatings afford slower release rates with greater overall cell viability and decreased enzymatic activity in vitro. However, increases in strain magnitude overcome this barrier dependence of the coating thickness with relatively rapid agent release. Nevertheless, the functionality of the agents is preserved after device loading, indicating that this mild fabrication approach is amenable to the encapsulation of a wide-range of agents for biomedical applications. For example, integration of these drug-loaded multi-layered superhydrophobic composites with existing medical devices that perform solely a mechanical function (i.e., mechanotherapy) would enhance their current capability to include both pharmacotherapy and mechanotherapy.
Experimental Section
Polymer synthesis
Poly(glycerol carbonate-co-ε-caprolactone), the precursor to poly(glycerol monostearate carbonate-co-caprolactone), was prepared as previously described and characterized.[31] The Tin(II) catalyzed (0.002 eq) polymerization of monomers ε-caprolactone and 5-benzyloxy-1,3-dioxan-2-one 4:1 (mol) was conducted at 140°C for 18 hours over nitrogen. Further deprotection by hydrogenolysis with palladium at 50 psi and stearic acid grafting via DCC coupling yielded poly(glycerol monostearate carbonate-co-ε-caprolactone) (PGC-C18, 1:4 comonomer ratio, Mw = 140 kDa, PDI = 1.5) after repeated precipitations in cold methanol. This corresponds to approximately n = 71 and × = 395 repeating units for PGC-C18 and PCL, respectively as shown in Figure 1a.
Device fabrication
Solutions of dye (50 μL), β-galactosidase (100 μL of 2 mg mL−1 in 0.3 M trehalose), and cisplatin (50 μL of 25 mg mL−1 anhydrous dimethylformamide) were loaded onto the cellulose and polyester blended mesh, followed by air-drying for 24 hours. Loaded core substrates were affixed to a rotating and translating grounded aluminum drum for even coating coverage. PGC-C18 and PCL (1:1) was dissolved in chloroform at 10% (w v−1) and electrosprayed (20–25 kV) at a flow rate of 2 mL hr−1 and tip-to-collector distance of 15 cm at 30–40% relative humidity. Cisplatin was loaded at 2.4 wt% with 97.8 +/− 8.8% encapsulation efficiency, as previously reported.[13] β-galactosidase was loaded at 0.54 +/− 0.02 wt% with 72 +/− 4.5% encapsulation efficiency.
Tension-mediated release
Devices were placed between the grips of Instron 5848 tensile tester with 100 N load cell and submerged in a bath of PBS with 10% v v−1 FBS, or RPMI cell culture media (10% v v−1 FBS, cisplatin-loaded devices) with magnetic stir bar. All devices were stretched at 7% strain per second until the desired strain magnitude was reached. Dye concentrations were measured with HP 8453 UV-Visible spectrophotometer at 630 nm. Cisplatin concentrations in RPMI with 10% v v−1 FBS was assessed using Varian AA240Z atomic absorption spectrophotometer at 265.5 nm with pyrolytic carbon-coated graphite tubes and Zeeman background correction.
Cell culture assays
OE33 esophageal cancer cells were seeded into 12-well plates at a density of 2 × 104 cells per well for 24 hours before aspiration and subsequent incubation of release media from cisplatin-loaded devices. After 72 hours, in vitro cell viability was measured using the tetrazolium-based colorimetric MTS assay with absorbance at 492 nm with Beckman Coulter AD 340 Plate Reader.
β-galactosidase activity
β-galactosidase activity was measured based on the released enzyme’s ability to convert ortho-nitrophenyl-β-galactoside into ortho-nitrophenol, measured with HP 8453 UV-Visible spectrophotometer at 420 nm. Released β-galactosidase aliquots were incubated with 5.55 mM ortho-nitrophenyl-β-galactoside in 20 mM phosphate citrate buffer at pH 4.5. After 5 hours of incubation at 20°C the reaction was stopped with 200 mM borate buffer at pH 9.8.
Statistical analysis
Dye, drug, and protein release was normalized to the release area (excludes area of Instron mechanical tester grips). This data is presented as mean +/− standard deviation (or + standard deviation for ease of visualization) with a sample size of at least 3. Statistical significance was determined by one-way ANOVA at each time point with p < 0.05.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health T32 EB006359[39] (M.W.G, J.W.) and R01EB017722 (Y.L.C., M.W.G.), a National Science Foundation Graduate Research Fellowship DGE-1247312 (J.W.), and the Wallace H. Coulter Translational Partnership.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Julia Wang, Departments of Biomedical Engineering and Chemistry, Boston University, Boston, MA 02215 (USA).
Dr. Yolonda L. Colson, Division of Thoracic Surgery, Department of Surgery, Brigham and Women’s Hospital, Boston, MA 02115 (USA)
Prof. Mark W. Grinstaff, Departments of Biomedical Engineering and Chemistry, Boston University, Boston, MA 02215 (USA)
References
- 1.Zhang Y, Yu J, Bomba HN, Zhu Y, Gu Z. Chem Rev. 2016;116:12536. doi: 10.1021/acs.chemrev.6b00369. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Kaplan JA, Colson YL, Grinstaff MW. Adv Drug Deliv Rev. 2017;108:68. doi: 10.1016/j.addr.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim BC, Moraes C, Huang J, Thouless MD, Takayama S. Biomater Sci. 2014;2:288. doi: 10.1039/C3BM60276A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riehl BD, Park JH, Kwon IK, Lim JY. Tissue Eng Part B Rev. 2012;18:288. doi: 10.1089/ten.teb.2011.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Q, Gossweiler GR, Craig SL, Zhao X. J Mech Phys Solids. 2015;82:320. [Google Scholar]
- 6.Rogers JA, Someya T, Huang Y. Science. 2010;327:1603. doi: 10.1126/science.1182383. [DOI] [PubMed] [Google Scholar]
- 7.Nemir S, West JL. Ann Biomed Eng. 2010;38:2. doi: 10.1007/s10439-009-9811-1. [DOI] [PubMed] [Google Scholar]
- 8.Ingber D. Ann Med. 2003;35:564. doi: 10.1080/07853890310016333. [DOI] [PubMed] [Google Scholar]
- 9.Wang PY. Biomaterials. 1989;10:197. doi: 10.1016/0142-9612(89)90024-0. [DOI] [PubMed] [Google Scholar]
- 10.Lee KY, Peters MC, Anderson KW, Mooney DJ. Nature. 2000;408:998. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]
- 11.Lee KY, Peters MC, Mooney DJ. Adv Mater. 2001;13:837. [Google Scholar]
- 12.Di J, Yao S, Ye Y, Cui Z, Yu J, Ghosh TK, Zhu Y, Gu Z. ACS Nano. 2015;9:9407. doi: 10.1021/acsnano.5b03975. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Kaplan JA, Colson YL, Grinstaff MW. Angew Chemie Int Ed. 2016;128:2846. doi: 10.1002/anie.201511052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marosfoi MG, Korin N, Gounis MJ, Uzun O, Vedantham S, Langan ET, Papa A, Brooks OW, Johnson C, Puri AS, Bhatta D, Kanapathipillai M, Bronstein BR, Chueh J, Ingber DE, Wakhloo AK. 2015 doi: 10.1161/STROKEAHA.115.011063. [DOI] [Google Scholar]
- 15.Kaplan JA, Barthélémy P, Grinstaff MW. Chem Commun. 2016;52:5860. doi: 10.1039/c6cc02221a. [DOI] [PubMed] [Google Scholar]
- 16.Holme MN, Fedotenko Ia, Abegg D, Althaus J, Babel L, Favarger F, Reiter R, Tanasescu R, Zaffalon P-L, Ziegler A, Müller B, Saxer T, Zumbuehl A. Nat Nanotechnol. 2012;7:536. doi: 10.1038/nnano.2012.84. [DOI] [PubMed] [Google Scholar]
- 17.Buscema M, Matviykiv S, Mészáros T, Gerganova G, Weinberger A, Mettal U, Mueller D, Neuhaus F, Stalder E, Ishikawa T, Urbanics R, Saxer T, Pfohl T, Szebeni J, Zumbuehl A, Müller B. J Control Release. 2017 doi: 10.1016/j.jconrel.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Vrignaud S, Benoit JP, Saulnier P. Biomaterials. 2011;32:8593. doi: 10.1016/j.biomaterials.2011.07.057. [DOI] [PubMed] [Google Scholar]
- 19.Eloy JO, Claro de Souza M, Petrilli R, Barcellos JPA, Lee RJ, Marchetti JM. Colloids Surfaces B Biointerfaces. 2014;123:345. doi: 10.1016/j.colsurfb.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 20.Ramazani F, Chen W, van Nostrum CF, Storm G, Kiessling F, Lammers T, Hennink WE, Kok RJ. Int J Pharm. 2016;499:358. doi: 10.1016/j.ijpharm.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Couvreur P, Puisieux F. Adv Drug Deliv Rev. 1993;10:141. doi: 10.1016/s0169-409x(97)00052-5. [DOI] [PubMed] [Google Scholar]
- 22.Sinha VR, Trehan A. J Control Release. 2003;90:261. doi: 10.1016/s0168-3659(03)00194-9. [DOI] [PubMed] [Google Scholar]
- 23.Fu K, Klibanov AM, Langer R. Nat Biotechnol. 2000;18:24. doi: 10.1038/71875. [DOI] [PubMed] [Google Scholar]
- 24.Wen G, Guo Z, Liu W. Nanoscale. 2017;9:3338. doi: 10.1039/c7nr00096k. [DOI] [PubMed] [Google Scholar]
- 25.Wolfs M, Darmanin T, Guittard F. Polym Rev. 2013;53:460. [Google Scholar]
- 26.Liu M, Wang S, Jiang L. Nat Rev Mater. 2017;2:17036. [Google Scholar]
- 27.Falde EJ, Yohe ST, Colson YL, Grinstaff MW. Biomaterials. 2016;104:87. doi: 10.1016/j.biomaterials.2016.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan JA, Liu R, Freedman JD, Padera R, Schwartz J, Colson YL, Grinstaff MW. Biomaterials. 2016;76:273. doi: 10.1016/j.biomaterials.2015.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Damodaran VB, Bhatnagar D, Murthy NS. Springer; Cham: 2016. pp. 1–22. [Google Scholar]
- 30.Wolinsky JB, Yohe ST, Colson YL, Grinstaff MW. Biomacromolecules. 2012;13:406. doi: 10.1021/bm201443m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolinsky JB, Ray WC, Colson YL, Grinstaff MW. Macromolecules. 2007;40:7065. [Google Scholar]
- 32.Yohe ST, Grinstaff MW. Chem Commun. 2013;49:804. doi: 10.1039/c2cc38012a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohtake S, Kita Y, Arakawa T. Adv Drug Deliv Rev. 2011;63:1053. doi: 10.1016/j.addr.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 34.Kamerzell TJ, Esfandiary R, Joshi SB, Middaugh CR, Volkin DB. Adv Drug Deliv Rev. 2011;63:1118. doi: 10.1016/j.addr.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 35.Ohtake S, Wang YJ. J Pharm Sci. 2011;100:2020. doi: 10.1002/jps.22458. [DOI] [PubMed] [Google Scholar]
- 36.Yohe ST, Grinstaff MW. Chem Commun. 2013;49:804. doi: 10.1039/c2cc38012a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu X, Mills KL, Peters PR, Bahng JH, Liu EH, Shim J, Naruse K, Csete ME, Thouless MD, Takayama S. Nat Mater. 2005;4:403. doi: 10.1038/nmat1365. [DOI] [PubMed] [Google Scholar]
- 38.Mertz D, Vogt C, Hemmerlé J, Mutterer J, Ball V, Voegel JC, Schaaf P, Lavalle P. Nat Mater. 2009;8:731. doi: 10.1038/nmat2504. [DOI] [PubMed] [Google Scholar]
- 39.Grinstaff MW, Kaplan HM, Kohn J. ACS Biomater Sci Eng. 2017 doi: 10.1021/acsbiomaterials.7b00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.