

Abstract
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy in adults for which there is currently no treatment. The pathogenesis of this autosomal dominant disorder is associated with the expansion of CTG repeats in the 3′-UTR of the DMPK gene. DMPK transcripts with expanded CUG repeats (CUGexpDMPK) are retained in the nucleus forming multiple discrete foci, and their presence triggers a cascade of toxic events. Thus far, most research emphasis has been on interactions of CUGexpDMPK with the muscleblind-like (MBNL) family of splicing factors. These proteins are sequestered by the expanded CUG repeats of DMPK RNA leading to their functional depletion. As a consequence, abnormalities in many pathways of RNA metabolism, including alternative splicing, are detected in DM1. To date, in vitro and in vivo efforts to develop therapeutic strategies for DM1 have mostly been focused on targeting CUGexpDMPK via reducing their expression and/or preventing interactions with MBNL1. Antisense oligonucleotides targeted to the CUG repeats in the DMPK transcripts are of particular interest due to their potential capacity to discriminate between mutant and normal transcripts. However, a growing number of reports describe alternative strategies using small molecule chemicals acting independently of a direct interaction with CUGexpDMPK. In this review, we summarize current knowledge about these chemicals and we describe the beneficial effects they caused in different DM1 experimental models. We also present potential mechanisms of action of these compounds and pathways they affect which could be considered for future therapeutic interventions in DM1.
Keywords: myotonic dystrophy type 1, myotonic dystrophy type 1 pathogenesis, sequestration of muscleblind-like 1, antisense oligonucleotides, aberrant alternative splicing, small molecule compounds
Introduction
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy in adults leading to disability and shortened lifespan (1). There is currently no treatment for DM1. The symptoms of this disease include myotonia, muscle weakness and wasting, cardiac conduction defects, diabetes and insulin resistance, and cataracts. DM1 belongs to a larger group of microsatellite disorders associated with expansions of simple repetitive elements within specific genes (2). This autosomal dominant disease is caused by the expansion of CTG repeats in the 3′-UTR of the DMPK gene and its pathogenesis is mediated, at least in part, by a toxic RNA gain-of-function mechanism. Molecular hallmarks of DM1 cells expressing mutant DMPK transcripts (CUGexpDMPK) are nuclear RNA foci. Their presence has an adverse effect on host cells leading to a broad spectrum of abnormalities. Recent studies of the dynamics of CUG repeat foci have revealed that these are unstable, constantly aggregating, and disaggregating structures (3), and muscleblind-like (MBNL)1 protein is directly involved in the stochastic process of foci formation. Being associated with CUGexpDMPK, the MBNL1 protein has a role in stabilizing RNA aggregates and its downregulation resulted in the decrease of RNA foci accumulation (3). However, MBNL1 depletion does not completely eliminate CUG foci, which suggests involvement of other proteins in mutant transcripts retention. In fact, as demonstrated experimentally, proteins other than MBNL1 may be recruited to the CUG repeat inclusions. Such recruitment may involve limited colocalization, as shown for hnRNP H, hnRNP F, and DDX6 proteins, or it may represent only close association with the RNA foci as shown for SC35 protein (4–7). In addition to the depletion of proteins in CUG RNA foci, DM1 pathogenesis also involves aberrant protein synthesis and/or their altered stability as shown for CUGBP1 protein (8, 9).
Muscleblind-like 1 and CUGBP1 are antagonistic regulators of splicing. MBNL1 is a zinc finger protein which recognizes both RNA sequence (YGCY) and structural elements (hairpins) containing pyrimidine mismatches on either normal splicing substrates or pathogenic mutant repeat RNA (10). As shown in vitro, the protein binds selectively to the stem region of expanded CUG RNA, and such interaction is detected in DM1 cells as MBNL1 sequestration and colocalization with mutant CUG repeats (11, 12). On the other hand, CUGBP1 protein binds in vitro to single-stranded regions of GU-containing transcripts; however, the protein is not enriched in the RNA foci. In DM1 cells, CUGBP1 becomes hyperphosphorylated, stabilized, and consequently, overexpressed (9). Changes in cellular levels and activities of MBNL1 and CUGBP1 proteins result in the abnormal expression of embryonic splice variants in adult tissues which is one of the molecular hallmarks of DM1 pathogenesis. Besides regulating splicing, both proteins are also involved in mRNA translation, RNA stability, protein secretion, and localization of alternative 3′UTR isoforms (13, 14). Their altered activity in DM1 cells also correlate with changes in signaling pathways of various protein kinases including cyclin-dependent kinases (CDKs), glycogen synthase kinase 3β (GSK3β), AKT, and protein kinase C (PKC) (8, 15, 16).
In vitro and in vivo efforts to develop DM1 therapeutic strategies have been mostly aimed at destroying the toxic ribonucleoprotein complexes via targeting the mutant CUGexpRNA and/or inhibiting its pathogenic interactions with MBNL1 protein, leading to the generation of several strategies that proved to have a beneficial effect in DM models (17, 18). Thus far, antisense technology that utilizes synthetic siRNAs (19), modified CAG antisense oligonucleotides (20–22), viral vector-mediated expression of hU7-snRNA-(CAG) (23), or a hammerhead RNA (ribozyme) (24) designed to cut CUG repeats, appear effective in DM1 cells and mouse models of the disease. Moreover, morpholino CAG oligonucleotides (25, 26) and several bioactive small molecules (27–34), which are CUG repeat binders, have been reported as potential therapeutic agents for DM1, capable of inhibiting the interactions between expanded CUG RNA and MBNL1 protein.
As our understanding of the pathogenesis of DM1 has grown over the past years, the focus of the research encompassed more molecular events being important for the progression of the disease, e.g., aberrantly spliced genes, RAN translation (35), and deregulation of miRNAs (36). Considering that DM1 is a multisystem disorder, it seems reasonable to target multiple molecules and pathways of the misregulated DM1 apparatus with the aim of developing a beneficial strategy to combat DM1.
Over the past few years, experimental evidence has indicated that, indeed, small molecule chemicals affecting different cellular pathways independently of CUGexpDMPK can mitigate some of the molecular hallmarks of DM1 pathogenesis (18). However, it remains elusive, how the molecules alleviate DM1 features. Their efficiency, though, indicates that it is reasonable to search for novel candidate therapeutic targets that will provide new opportunities for future studies aiming to decipher the complex pathomechanism of DM1. In this review, we summarize current knowledge about such molecules, acting independently of direct binding to CUGexpDMPK, and we describe beneficial effects they caused in different DM1 experimental models. We also present potential mechanisms of action of these compounds and cellular pathways they affect which could be considered for future therapeutic interventions in DM1.
Inhibitors of Transcription
A few compounds that inhibit transcription and alleviate some of the molecular symptoms of DM1 have been identified in recent years. These include pentamidine and its analogs, as well as actinomycin D (ActD) (Figure 1) (37–40).
Figure 1.
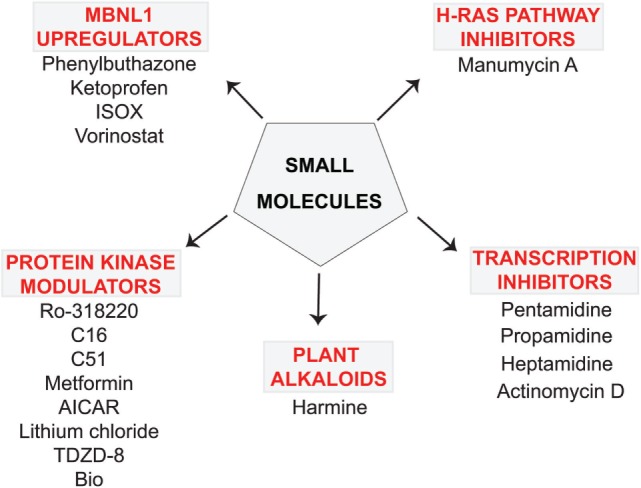
Small molecule compounds alleviating myotonic dystrophy type 1 (DM1) pathogenesis. Small molecule compounds which mitigated DM1 pathogenesis in different experimental systems are shown. The molecules are classified into a few categories depending on their presumable mechanisms of action. All the molecules are believed to act independently of direct interactions with expanded CUG repeats RNA.
Pentamidine
Pentamidine is an FDA-approved drug currently used to treat patients with Pneumocystis carinii infections (pneumonia) in acquired immunodeficiency syndrome, as well as patients with Trypanosomiasis and Leishmaniasis infections (41, 42). Pentamidine is a diamide composed of two phenyl amidine groups joined by a five-carbon methylene linker (Table 1). It has been speculated to inhibit the splicing of essential group I introns in P. carinii, preventing its growth (43, 44), and it was recently shown to inhibit translation by binding tRNA (45). Pentamidine may also block DNA replication (41), since its structure with DNA showed binding in the minor groove of the double-stranded molecule (46). Therefore, it is likely to interact with many different nucleic acid targets within cells. Interestingly, in DM1 patient’s cells and model organisms’ pentamidine disrupted CUG RNA foci, released MBNL1 protein, and reversed aberrant alterative splicing of some pre-mRNAs typically misspliced in DM1. Initially, these effects were thought to be caused by binding of the compound to the CUG repeats, because of its ability to bind DNA (40), however, subsequently it was found that its effect on the DM1 molecular phenotype was attributed to either decreased transcription of the CUG transcripts or increased degradation of the toxic RNA (38). So far, the beneficial effects of pentamidine have been shown in HeLa and HEK293 cells transfected with 960 CUG repeats and in model organisms, including the HSALR mice and a Drosophila model expressing 250 CTG repeats (37, 40) (Table S1 in Supplementary Material). Treatment of HeLa cells transiently expressing CUG repeats and transgenic HSALR mice showed, respectively, a decrease of CUG mRNA expression (and subsequent reduction of CUG RNA foci number), and a reduction of the HSA transcript. Correction of some misspliced pre-mRNAs, i.e., cTNT E5 and INSR E11 (in HeLa cells) and Clcn1 E7a and Serca1 E22 (in HSA mice) was attributed to the liberation of MBNL1 protein from diminished CUG foci and reduction of CUG transcript levels. This suggests that pentamidine does not directly block MBNL1 binding to the repeats and supports the hypothesis that it either inhibits transcription of the CTG repeats or increases the rate of CUGexpRNA degradation. However, in a DM1 Drosophila model, a behavioral and molecular improvement which included a minor rescue of cardiac defects reduced CUG foci, and Mbnl1 displacement was not correlated with diminished expression level of CUG repeats mRNA (37) (Table S1 in Supplementary Material). It was suggested that the beneficial effects induced by pentamidine are due to Mbnl1 diffusion and subsequent dispersion of toxic RNA in the nucleus, rather than by inhibition of transcription of the toxic RNA or its degradation.
Table 1.
Molecular formula and chemical structure of small molecule compounds which do not directly target expanded CUG repeats and alleviate pathogenesis of myotonic dystrophy type 1.
| Name | Molecular formula | Structure | Potential mechanism of action |
|---|---|---|---|
| Pentamidine | C19H24N4O2 | 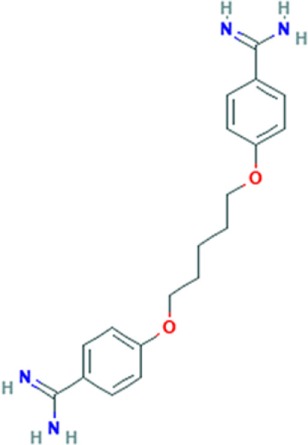 |
Inhibition of transcription |
| Propamidine | C17H20N4O2 | 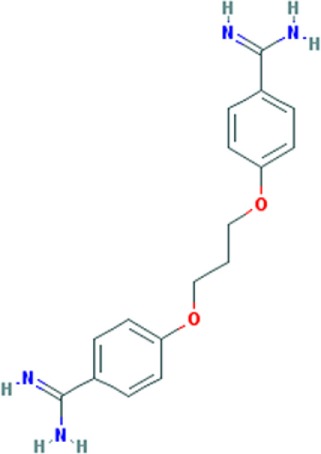 |
|
| Heptamidine | C21H28N4O2 | 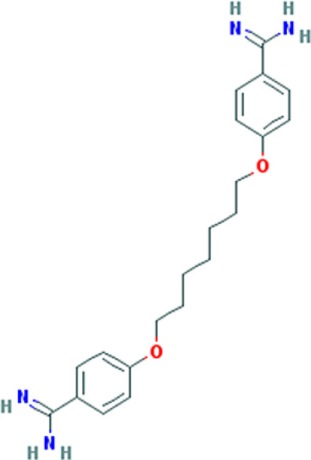 |
|
| Compound 13 | C18H16N4O | 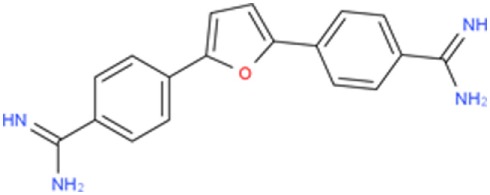 |
|
| Actinomycin D | C62H86N12O16 | 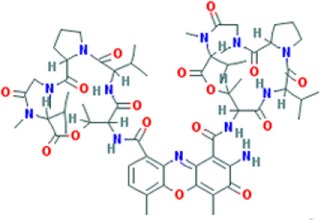 |
|
| Phenylbuthazone | C19H20N2O2 | 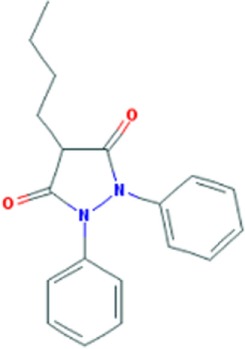 |
Upregultion of muscleblind-like 1 |
| Ketoprofen | C16H14O3 | 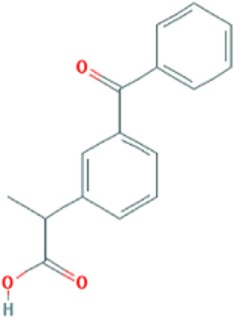 |
|
| ISOX | C22H30N4O6 | 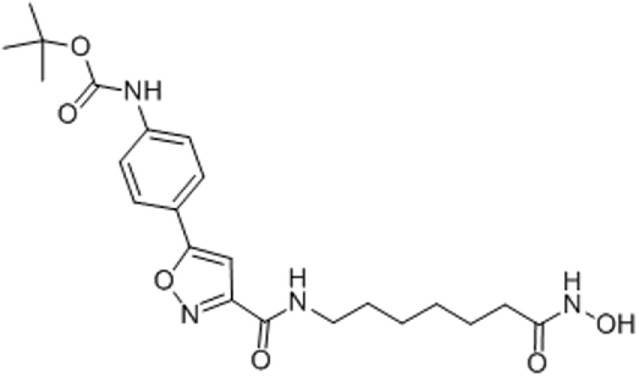 |
|
| Vorinostat | C14H20N2O3 | 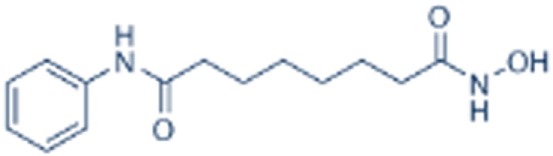 |
|
| Manumycin A | C31H38N2O7 | 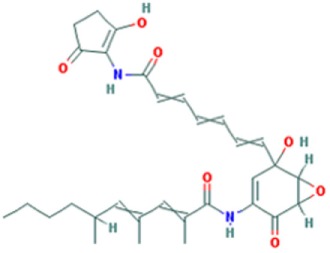 |
Inhibition of H-RAS pathway |
| Metformin | C4H11N5 | 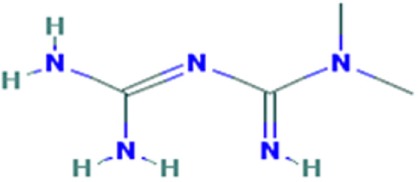 |
Modulation of protein kinases |
| AICAR | C9H14N4O5 | 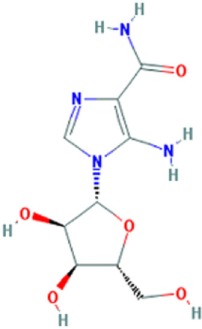 |
|
| Ro 31-8220 | C25H23N5O2S | 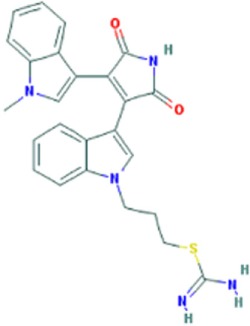 |
|
| C16 | C13H8N4OS | 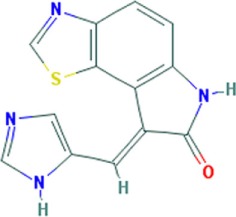 |
|
| C51 | C23H21N5 | 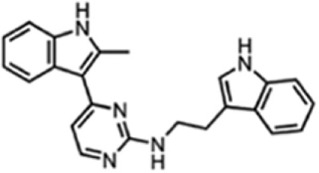 |
|
| Lithium chloride | LiCl |  |
|
| TDZD-8 | C10H10N2O2S | 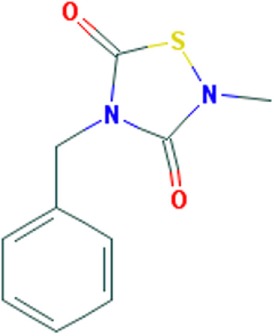 |
|
| 6-Bromoindirubin-3′-oxime | C16H10BrN3O2 | 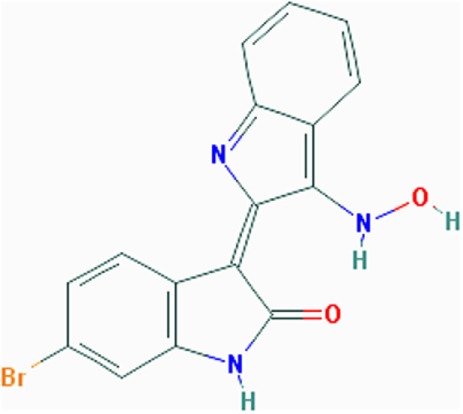 |
|
| Harmine | C13H12N2O | 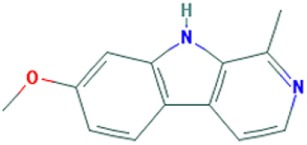 |
Undetermined |
Pentamidine Analogs
The results obtained with pentamidine as an anti-DM1 drug were promising; however, they showed a certain degree of toxicity and thus needed optimization, and the mechanism of action (MoA) needed clarification. To improve the physiochemical properties of pentamidine and to decrease its toxicity, a series of analogs of pentamidine, containing between three to nine methylene carbons, were analyzed for their ability to rescue splicing of transiently transfected minigenes in a HeLa cell model of DM1 and in HSALR mouse model (Table S1 in Supplementary Material). Two different minigenes, i.e., cTNT E5 and INSR E11, containing exons that are misspliced in DM1 were tested. Propamidine (methylene linker length of 3) and heptamidine (methylene linker length of 7) (Table 1) were identified as the most promising analogs in rescuing missplicing in both DM1 models (38). Although analogs with more methylenes rescued missplicing more efficiently, an increase of the methylene linker length also reduced solubility and increased toxicity. Interestingly, in the HeLa cell model only propamidine was able to reduce CUG repeat transcript levels in a dose-dependent manner (however, not as effectively as pentamidine), whereas no reduction was observed with heptamidine treatment before significant cell death occurred. The cTNT splicing was improved to varying degrees with different linker analogs, however, no correction was observed with propamidine at concentrations which were not toxic. Furthermore, the improvement was not dependent on inhibition of CUG transcript expression. Unlike the cTNT, all linker analogs partially or fully rescued the missplicing of INSR E11 after CUG repeats expression. Different MBNL targets may require different concentrations of the proteins to be properly regulated, which could explain the results found regarding correction of alternative splicing of different genes (47).
Treatment of HSALR DM1 mice with heptamidine caused reduction of both the mRNA and pre-mRNA levels of the transgene, while in HSASR mice expressing short CTG repeats, no such reduction was detected. This suggests that the effect was dependent on the presence of extended repeats, rather than the HSA promoter, transgene, or the short repeats. In the HSALR mouse model, heptamidine completely reversed the missplicing of Clcn1 E7a, while splicing of Atp2a1 E22 was rescued partially and at lower concentrations than pentamidine. In addition, heptamidine caused a rapid (after 1 week) and significant reduction of myotonia, as well as diminished levels of HSALR transcript (Table S1 in Supplementary Material). Nevertheless, it showed high toxicity in the mouse model. However, variation of the linker length of pentamidine resulted in significant improvements and is encouraging to develop a therapy for DM1. Reduction of the CUG transcript in in vitro transcription assays as well as in both DM1 models, i.e., HeLa cells and HSALR mice, as well as reduced formation of nuclear foci, suggest that pentamidine and its analogs most likely inhibit transcription of CTG*CAG repeat DNA. However, other possible mechanisms cannot be ruled out as pentamidine is a DNA-binding molecule and may affect expression of other genes, some of which may be regulators of alternative splicing.
Further modifications of pentamidine and heptamidine to modify their size, solubility, degrees of freedom, number of hydrogen bond donors, and hydrophobicity led to the generation of new promising compounds. These molecules were tested for their ability to correct splicing defects in cellular and transgenic mouse models of DM1, as well as their toxicity relative to the parental compounds (48). Three different series of molecules varying in amidine substitution, central linker length, and planarity were synthesized. Compound 8, a molecule with an un-substituted propyl chain, was able to rescue missplicing in the DM1 HeLa cell model with no toxicity at the range of concentrations used, but it was half as effective as pentamidine. Modification of the planarity produced a linear molecule, compound 12, and a molecule with a concave shape, compound 13 (Table 1). Both compounds rescued missplicing similarly to pentamidine and they were not toxic over the range of concentrations needed for the rescue, unlike pentamidine. Compound 12 affected splicing in a DM-independent manner in a model without CUG expanded repeats, suggesting a modulation of splicing through a mechanism beyond targeting the CUG repeats. On the contrary, compound 13 only rescued alternative splicing in the presence of CUG repeats. Furthermore, it reduced foci in the DM1 HeLa cell model and partially rescued misplicing of Clcn1 E7a and Atp2a1 E22 in the HSALR mouse model at a similar level observed with heptamidine, but without the associated toxicity (39).
The MoA of pentamidine has been previously proposed to be inhibition of transcription through binding to the CTG repeats or reduction of the stability of the transcript. Nonetheless, binding to the CUG repeats and therefore displacement of MBNL1 proteins, especially in the case of the analogs generated has not been completely ruled out, and further work is required to clarify the MoA of the diamines as anti-DM1 drugs.
Actinomycin D
Actinomycin D (Table 1) is a polypeptide antibiotic which forms a stable complex with double-stranded DNA, inhibiting DNA-primed RNA synthesis and causing single-stranded breaks in DNA, therefore stopping the proliferation of cells (49, 50). ActD is an FDA-approved anti-cancer drug with activity to inhibit global transcription. More recently, this antibiotic was shown to improve the DM1 molecular phenotype (39). ActD does not bind to CUG repeat RNA in vitro, however, low concentrations, insufficient to affect global transcription, triggered a significant reduction of expression of the expanded CUG repeat RNA in HeLa cells and in DM1 fibroblasts, by 50–70% and by 44–60%, respectively. At effective dosages, ActD was mildly toxic to HeLa cells causing reduction of nuclear CUGexpRNA foci by 50% and releasing MBNL1 from the foci. In HSALR mice, the molecule specifically reduced mRNA levels of the repeat-containing HSA transgene and completely rescued the aberrant splicing of Clcn1 E7a, whereas partially corrected splicing of a few other genes, i.e., Atp2a1 E22, Mbnl1 E7, Vps39 E3, Nfix E7, and Ldb3 E11. Importantly, at the range of dose used these changes were not correlated with global inhibition of transcription (Table S1 in Supplementary Material). The MoA of ActD in DM1 is yet to be determined. However, its effects produced in the disease models suggest inhibition of transcription of the CUGexpRNA by binding to the CTG repeats in DNA and blocking the RNA polymerase. Such mechanism is feasible since it is known that ActD intercalates into GC-rich sequences, stabilizing topoisomerase-I DNA complexes and preventing RNA polymerase progression (51).
Compounds Upregulating MBNL1
Overexpression of MBNL1 protein has been shown to alleviate pathogenesis of DM1 by reversal of aberrant alternative splicing and rescue of myotonia (52). Accordingly, small molecules which upregulate expression of the splicing factor would have a potential of becoming therapeutic molecules for DM1. Two compounds with such capacity have been identified by Chen et al. (53). Phenylbutazone (PBZ) and ketoprofen (Table 1) belong to nonsteroidal anti-inflammatory drugs (NSAIDs) which are used to reduce pain and fever, prevent blood clots, and in higher doses, decrease inflammation. NSAIDs inhibit the activity of cyclooxygenase enzymes (COX-1 and/or COX-2). These enzymes participate in the synthesis of key biological mediators in the cells, such as prostaglandins which are involved in inflammation, and thromboxanes which are involved in blood clotting (54, 55).
It was shown that PBZ enhances MBNL1 expression in proliferating and differentiating C2C12 cells in a dose-dependent manner (53) (Table S1 in Supplementary Material). Consistent with this result, when analyzed in HSALR DM1 mice PBZ elevated the expression of Mbnl1 mRNA and protein in tibialis anterior and in quadriceps muscles. Interestingly, the expression of CUGBP1 remained unchanged after the treatment with PBZ. Consequently, the treated mice showed partial rescue of aberrant splicing of MBNL1-dependent exons, such as Clcn1 E7a, Nfix E7, and Rpn2 E17, whereas no splicing correction was found for CUGBP1-regulated exons. Amelioration of the molecular features in HSALR mice was further confirmed in behavioral and histological tests and the mice had an increase of grip strength and decrease in the number of muscle fibers with central nuclei (53). Although PBZ elevated Mbnl1 expression levels in HSA mouse muscles, colocalization of the protein with CUG RNA foci was markedly attenuated by its treatment. This result suggests that PBZ inhibits the interaction between CUG RNA foci and MBNL1 and reduces the ratio of MBNL1 in the mutant transcript. The precise MoA of PBZ in DM1 remains unclear and it is speculated that it may not be limited to the liberation of MBNL1 proteins from CUGexpRNA. The observed upregulation of MBNL1 mRNA was attributed to demethylation of its intron 1, making this region to act as an enhancer of transcription (53).
Ketoprofen is another NSAID which was able to upregulate MBNL1 levels in C2C12 cells (53). In a Drosophila model of DM1 expressing 480 interrupted CUG repeats the compound suppressed CUG-mediated lethality (56). The MoA of ketoprofen in DM1 has not yet been elucidated.
More recently, a flow cytometry-based screen led to identification of small compounds that upregulated MBNL1 and partially rescued splicing defects in DM1 patient-derived fibroblasts (57). Using engineered HeLa cells containing a ZsGreen fluorescent tag in the N-terminus of the MBNL1 sequence, the HDAC inhibitors ISOX, and Vorinostat (Table 1) were found to produce a 2- and 1.8-fold increase of the ZsGreen-MBNL1 signal, respectively (57). Treatment of either normal or DM1 fibroblasts with ISOX and Vorinostat produced a significant increase of MBNL1 levels, and partially rescued the splicing of SERCA1 e22 and INSR e11 in both cell lines (Table S1 in Supplementary Material). Since ISOX and Vorinostat are HDAC inhibitors, they may affect expression of SERCA1, INSR, and DMPK mRNAs. However, as shown by Zhang and coauthors, treatment of normal and DM1 fibroblasts with these molecules caused no significant changes in the genes levels. The MoA of ISOX and Vorinostat is not fully clear, but inhibition of HDAC appears to have a role in modulating MBNL1 levels, and the effects might be caused by inhibition of several HDACs. ISOX inhibits HDAC6 at low concentrations, but also inhibits HDAC1 and other HDACs at higher concentrations. Vorinostat is an FDA-approved HDAC inhibitor for the treatment of cutaneous T cell lymphoma, and inhibits class I and class II HDACs, altering gene transcription and causing cell cycle arrest. Nonetheless, in the light of these recent results, modulation of MBNL1 through mechanisms other than inhibition of HDAC cannot be completely ruled out (57).
Inhibitors of H-Ras Pathway
The Ras family includes three members: H-Ras, K-Ras, and N-Ras. They play roles in a large number of biological processes including cell morphology, survival, apoptosis, gene expression, and alternative splicing regulation (58). Posttranslational modifications of Ras proteins lead to their activation and these modifications include farnesylation of H-Ras, as well as farnesylation and geranylgeranylation of K-Ras and N-Ras (59). Manumycin A (Table 1) is an FDA-approved antibiotic that acts as a potent and selective farnesyltransferase (FTase) inhibitor (60). By inhibiting FTase, manumycin A prevents activation of H-Ras protein but has no effect on K-Ras and N-Ras. It has been reported that some of DM1 features can be alleviated by exposure to manumycin A (Figure 2) (61). Treatment of HSALR DM1 mice with the compound led to correction of Clcn1 E7a splicing, what was linked to the inhibition of H-Ras activity since siRNA knockdown of endogenous H-Ras protein recapitulated improvement of the splicing. Such effect was not detected when the two other Ras proteins were downregulated. Although skeletal muscle injections with manumycin A corrected aberrant splicing of Clcn1 in DM1 mice, splicing of two other genes, Serca1 E22 and m-Titin Mex5, was not altered (61). Importantly, in this experimental model of DM1, manumycin A did not alter expression of Mbnl1 and Cugbp1, which are involved in splicing regulation of Clcn1 E7a, Serca1 E22, and m-Titin Mex5 (Table S1 in Supplementary Material). Thus, it was concluded that the effect of manumycin A on aberrant splicing was independent of these two splicing factors. Therefore, it is possible that manumycin A, which acts as a Ras FTase inhibitor, triggers alterations in H-Ras signaling. This may influence a trans-acting factor(s) other than MBNL1 and CUGBP1 involved in alternative splicing and may contribute to chloride channel splicing. However, a category of trans-acting factor(s) affected remains unknown.
Figure 2.

Therapeutic effects of small molecules on myotonic dystrophy type 1 (DM1) pathogenesis. DM1 is characterized by the presence of RNA foci which are aggregations of the mutant CUGexp transcript with muscleblind-like (MBNL)1 and other proteins. CUGBP1 is not sequestered by foci, but it is upregulated. The imbalance of these two alternative splicing proteins causes the aberrant alternative splicing of many pre-mRNAs. Treatment of DM1 cells and model organisms with small molecules that target the DNA and/or affect proteins involved in the DM1 pathogenesis can lead to beneficial effects, such as inhibition of transcription of the mutant transcript, or its degradation, release of MBNL1 protein from RNA foci, downregulation of CUGBP1 protein, and ultimately the correction of the aberrant splicing.
Modulators of Protein Kinases
Protein Kinase Inhibitors
Pathogenesis of DM1 has been linked to disrupted protein kinase signaling pathways. Altered expression and nonspecific activation have been shown for PKC, Src family kinases, CDKs, GSK3β, and protein kinase AKT (8, 15, 16). Current studies in DM1 with the use of small molecule inhibitors of protein kinases have shed light on a potential alternative strategy in DM1 therapeutic intervention. Screening of several libraries of small molecule compounds, including phosphatase and kinase inhibitors, using a medium throughput phenotypic assay by Ketley et al. led to the identification of such molecules (62). Based on the identification of nuclear foci in DM1 cells using in situ hybridization and high-content imaging, Ro 31-8220 (Table 1) was identified as a compound of potential therapeutic benefit in DM1. The compound eliminated nuclear CUGexpRNA foci, reduced MBNL1 protein in the nucleus, affected SERCA1 E22 alternative splicing, and decreased steady-state levels of CUGBP1 protein (Figure 2). Ro 31-8220 was previously identified as a PKC inhibitor and seen to affect the hyperphosphorylation of CUGBP1 and ameliorate the cardiac phenotype in a DM1 mouse model (8, 63). Nonetheless, Ketley et al. demonstrated that Ro 31-8220 acts independently of PKC on DM1 pathomechanism, suggesting the involvement of other kinases. Although the MoA of Ro-31-8220 still requires further investigation, the compound is likely to work independently of CUG repeat RNA binding.
Another study has identified two compounds, C16, an imidazolo-oxindole inhibitor and C51, a pyrimidine-based inhibitor (Table 1), of kinase inhibitory activity with the potential to alleviate DM1 molecular phenotype (64). Previous studies have described these two compounds as ATP-site directed PKR inhibitors (30, 65); however, activity of C16 against kinases other than PKR has also been reported (66). C16 has been successfully used in vitro and in vivo as an effective and versatile neuroprotective agent and shown to have potential value in the treatment of neurodegenerative diseases (66–69). Results by Wojciechowska et al. demonstrate that C16 and C51 may have therapeutic potential in patients with DM1 as well. These compounds produced a redistribution of the MBNL1 protein sequestered in CUGexpRNA foci and a reduction of the steady-state levels of CUGBP1. These actions were accompanied by correction of the aberrant alternative splicing of MBNL1-dependent (SERCA1 E22, DMD E78, MBNL1 E7, and LDB3 E7) and CUGBP1-dependent (ITGA6 E24, MTMR3 E16, and SORBS1 E6) pre-mRNA targets, shifting the patterns of spicing toward those observed in non-DM cells. However, despite causing the nuclear CUGexpRNA foci to become less abundant, as determined by in situ RNA hybridization and immunocytochemistry for MBNL1 protein, the compounds did not eliminate foci completely.
GSK3β Inhibitors
Glycogen synthase kinase 3β has been seen to have a role in DM1 pathogenesis (16). This kinase phosphorylates cyclin D3 at T283, which triggers degradation of the cyclin. Cyclin D3 associates with CDK4 and the resulting complex phosphorylates CUGBP1 at S302 and regulates the translational activity of the protein. CUGBP1 regulates translation of proteins important for skeletal muscle development, thus its normal translational activity is required for a proper myogenesis. In muscle biopsies of DM1 patients, GSK3β was found to be increased, causing a degradation of cyclin D3 and a reduced phosphorylation of CUGBP1 at S302, what was linked to delayed myogenesis. The presence of CUG expanded repeats appears to increase the stability of GSK3β by causing the autophosphorylation of the protein at Y216 (16, 70). Altogether, these observations point at GSK3β as a potential therapeutic target in DM1. Indeed, treatment of HSALR mice with two GSK3β inhibitors, lithium and TDZD-8 (thiadiazolidine) (Table 1), reduced the levels of GSK3β in skeletal muscle and normalized the levels of cyclin D3, restoring CUGBP1 translational function. These changes improved skeletal muscle strength in the mice and reduced myotonia, suggesting that correction of GSK3β may have a beneficial effect on myofiber regeneration (16).
Another GSK3β inhibitor, 6-bromoindirubin-3′-oxime (BIO) (Table 1), was used to treat young HSALR mice, resulting in recovery of the grip strength to near-normal levels and the effect was maintained for several months after completion of the treatment. In addition, the levels of GSK3β and cyclin D3 in 12-month-old mice previously treated with BIO were similar to those in control mice, and the translational activity of CUGBP1 was corrected. Results from this study suggest that inhibition of GSK3β in young mice is sufficient to maintain corrected levels of the GSK3β-cyclin D3-CUGBP1 pathway and nearly normal muscle health over a long period of time after completion of the treatment (71).
Activators of AMP-Activated Protein Kinase (AMPK)
Metformin (Table 1) is an FDA-approved antidiabetic drug and was earlier reported to improve hyperglycemia through increased insulin-independent glucose uptake in peripheral muscles of DM1 patients (72). Most recently, metformin was investigated in in vitro DM1 models of human embryonic stem cells and in primary myoblasts derived from patients (73). The drug appeared to modify the alternative splicing of a subset of genes associated with DM1 in these cell models, as it partially rescued the aberrant splicing of INSR E11, CLCN1 E7a, TNNT2 E5, ATP2A1 E22, and DMD E78. The effect on the modification of the alternative splicing has been linked to the inhibition of the complex I of the respiratory chain, which in turn raises the intracellular AMP/ATP ratio, which triggers the activation of AMPK. The role of AMPK activation in alternative RNA splicing was tested in DM1 myogenic progenitor cells and in myoblasts with the AMPK activator AICAR [5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside, Acadesine, N1-(β-d-ribofuranosyl)-5-aminoimidazole-4-carboxamide] (73). Treatment of DM1 cells promoted changes in similar subsets of pre-mRNAs as found for metformin. However, AICAR did not modulate the INSR E11 splicing in the cells used. Thus, these results suggest that activation of AMPK is partially involved in alternative splicing modulation, but metformin appears to trigger an additional molecular pathway. Interestingly, metformin decreases tyrosine kinase receptor signaling (73). The tyrosine kinase receptors include the epidermal growth factor receptor, the signaling pathway which controls INSR E11 inclusion via the inhibition of hnRNPA1 and hnRNPA2B1 expression (74, 75). This could explain the effect of metformin on the alternative splicing of INSR. The MoA behind other splicing events modulated by metformin but not by AMPK activation remains unclear.
Small Molecules of Natural Origin
Many small molecules have been synthesized as potential therapies for DM. However, only a few studies have reported utilization of molecules of natural origin (40, 76). Recently, a set of plant-derived alkaloids was identified as small molecules with an anti-DM1 effect (77). Using a novel CUG78–MBNL1 complex inhibition assay, a collection of isolated natural compounds and extracts from plants and fungal strains was screened. The bioactivity of the compounds was investigated in human DM1 cells and HSALR mice resulting in the identification of several alkaloids, including carboline harmine and isoquinoline berberine, which ameliorated certain aspects of the DM1 pathology in these models.
Aromatic alkaloids can interact with RNA, and indeed berberine and harmine (Table 1) have been reported to bind RNA structures (78, 79). In DM1 myoblasts, harmine reduced foci, nevertheless it did not improve the histology in gastrocnemius muscle of the HSALR mice, as the percentage of fibers with internalized nuclei was not altered by the treatment of the mice with the compound (77). However, although harmine increased the levels of MBNL1 in DM1 myoblasts and enhanced MBNL1-dependent alternative splicing of cTNT E5, INSR E11, and Clcn1 E7a (Table S1 in Supplementary Material), a similar effect was found in wild-type myoblasts, suggesting that inhibition of the CUG–MBNL1 complex is not the primary MoA of this alkaloid. Consequently, it was suggested that harmine acts through another mechanism that causes the increase of MBNL1 levels and the amelioration of the spliceopathy. Despite the side effects, harmine represents an interesting small molecule that could be optimized by chemical modifications to become a potential DM1 therapy.
Conclusion
Myotonic dystrophy type 1 is a life-shortening, debilitating disorder for which there is currently no treatment. Pathogenesis is associated with nuclear retention of mutant DMPK mRNA which attract or is attracted by various proteins. Experimental data suggest that the formation of riboprotein complexes is a necessary trigger for DM1 pathogenesis. Thus, compounds which reduce such inclusions would be therapeutically beneficial. Over the past few years, many efforts have been focused on the synthesis of small molecule chemicals specifically recognizing mutated CUG repeats and either cutting the toxic RNA or blocking their interactions with relevant proteins (19, 22, 26). In both cases, treatment improved DM1 molecular and behavioral features including fewer CUGexpRNA foci, liberation of MBNL1 protein, rescue of aberrant alternative splicing, and muscle pathology correction. Interestingly, similar improvements have also been observed with other small molecules affecting the DM1 mutation indirectly (38, 48, 62). Although there are many questions concerning the mode of action of these chemicals, in vitro and in vivo efficacy underline the notion that these molecules can have therapeutic benefit in DM1. Furthermore, several of the described molecules are FDA-approved drugs, potentially offering an opportunity of repositioning.
The development of therapeutic approaches based on small molecules has several advantages, including lower costs with ease of manufacturing, ease of management of the therapy, with the possibility to rapidly interrupting the treatment in case of toxicity, ease of administration and tissue delivery, opportunities of repositioning, and most notably, that the pharmaceutical industry has decades of experience in refining and improving potentially useful compounds via conventional medicinal chemistry-based approaches. Such efforts may prove fruitful for DM1.
Author Contributions
Conception and design and wrote the main manuscript text: ML-M, MW, and JDB. Prepared tables and figures: ML-M and MW. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. Work in the JDB’s lab is funded by grants from the Wellcome Trust (Seeding Drug Discovery grant number 107562/Z/15/Z; 2015), Muscular Dystrophy UK and the Myotonic Dystrophy Support Group. ML-M is supported by a studentship from the University of Nottingham. MW has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007–2013) under REA grants agreement No. PCOFUND-GA-2012-600181 and from the Polish National Science Center (2014/13/B/NZ5/03214).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fneur.2018.00349/full#supplementary-material.
References
- 1.Harper PS. Myotonic Dystrophy. 3rd ed London: W.B. Saunders; (2001). [Google Scholar]
- 2.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci (2007) 30:575–621. 10.1146/annurev.neuro.29.051605.113042 [DOI] [PubMed] [Google Scholar]
- 3.Querido E, Gallardo F, Beaudoin M, Menard C, Chartrand P. Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J Cell Sci (2011) 124:1703–14. 10.1242/jcs.073270 [DOI] [PubMed] [Google Scholar]
- 4.Holt I, Mittal S, Furling D, Butler-Browne GS, Brook JD, Morris GE. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells (2007) 12:1035–48. 10.1111/j.1365-2443.2007.01112.x [DOI] [PubMed] [Google Scholar]
- 5.Kim DH, Langlois MA, Lee KB, Riggs AD, Puymirat J, Rossi JJ. HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucleic Acids Res (2005) 33:3866–74. 10.1093/nar/gki698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paul S, Dansithong W, Jog SP, Holt I, Mittal S, Brook JD, et al. Expanded CUG repeats dysregulate RNA splicing by altering the stoichiometry of the muscleblind 1 complex. J Biol Chem (2011) 286:38427–38. 10.1074/jbc.M111.255224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pettersson OJ, Aagaard L, Andrejeva D, Thomsen R, Jensen TG, Damgaard CK. DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Res (2014) 42:7186–200. 10.1093/nar/gku352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell (2007) 28:68–78. 10.1016/j.molcel.2007.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem (2001) 276:7820–6. 10.1074/jbc.M005960200 [DOI] [PubMed] [Google Scholar]
- 10.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, et al. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res (2007) 35:5474–86. 10.1093/nar/gkm601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mankodi A, Teng-Umnuay P, Krym M, Henderson D, Swanson M, Thornton CA. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol (2003) 54:760–8. 10.1002/ana.10763 [DOI] [PubMed] [Google Scholar]
- 12.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet (2001) 10:2165–70. 10.1093/hmg/10.19.2165 [DOI] [PubMed] [Google Scholar]
- 13.Timchenko NA, Wang GL, Timchenko LT. RNA CUG-binding protein 1 increases translation of 20-kDa isoform of CCAAT/enhancer-binding protein beta by interacting with the alpha and beta subunits of eukaryotic initiation translation factor 2. J Biol Chem (2005) 280:20549–57. 10.1074/jbc.M409563200 [DOI] [PubMed] [Google Scholar]
- 14.Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell (2012) 150:710–24. 10.1016/j.cell.2012.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Botta A, Malena A, Tibaldi E, Rocchi L, Loro E, Pena E, et al. MBNL142 and MBNL143 gene isoforms, overexpressed in DM1-patient muscle, encode for nuclear proteins interacting with Src family kinases. Cell Death Dis (2013) 4:e770. 10.1038/cddis.2013.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones K, Wei C, Iakova P, Bugiardini E, Schneider-Gold C, Meola G, et al. GSK3beta mediates muscle pathology in myotonic dystrophy. J Clin Invest (2012) 122:4461–72. 10.1172/JCI64081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chau A, Kalsotra A. Developmental insights into the pathology of and therapeutic strategies for DM1: back to the basics. Dev Dyn (2015) 244:377–90. 10.1002/dvdy.24240 [DOI] [PubMed] [Google Scholar]
- 18.Konieczny P, Selma-Soriano E, Rapisarda AS, Fernandez-Costa JM, Perez-Alonso M, Artero R. Myotonic dystrophy: candidate small molecule therapeutics. Drug Discov Today (2017) 22:1740–8. 10.1016/j.drudis.2017.07.011 [DOI] [PubMed] [Google Scholar]
- 19.Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther (2013) 21:380–7. 10.1038/mt.2012.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Barriga A, Mulders SA, van de Giessen J, Hooijer JD, Bijl S, van Kessel ID, et al. Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol Ther Nucleic Acids (2013) 2:e81. 10.1038/mtna.2013.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JE, Bennett CF, Cooper TA. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc Natl Acad Sci U S A (2012) 109:4221–6. 10.1073/pnas.1117019109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci U S A (2009) 106:13915–20. 10.1073/pnas.0905780106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francois V, Klein AF, Beley C, Jollet A, Lemercier C, Garcia L, et al. Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat Struct Mol Biol (2011) 18:85–7. 10.1038/nsmb.1958 [DOI] [PubMed] [Google Scholar]
- 24.Langlois MA, Lee NS, Rossi JJ, Puymirat J. Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol Ther (2003) 7:670–80. 10.1016/S1525-0016(03)00068-6 [DOI] [PubMed] [Google Scholar]
- 25.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science (2009) 325:336–9. 10.1126/science.1173110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature (2012) 488:111–5. 10.1038/nature11362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc Natl Acad Sci U S A (2009) 106:16068–73. 10.1073/pnas.0901824106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol (2012) 7:856–62. 10.1021/cb200408a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jahromi AH, Nguyen L, Fu Y, Miller KA, Baranger AM, Zimmerman SC. A novel CUG(exp).MBNL1 inhibitor with therapeutic potential for myotonic dystrophy type 1. ACS Chem Biol (2013) 8:1037–43. 10.1021/cb400046u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jammi NV, Whitby LR, Beal PA. Small molecule inhibitors of the RNA-dependent protein kinase. Biochem Biophys Res Commun (2003) 308:50–7. 10.1016/S0006-291X(03)01318-4 [DOI] [PubMed] [Google Scholar]
- 31.Nakamori M, Taylor K, Mochizuki H, Sobczak K, Takahashi MP. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann Clin Transl Neurol (2016) 3:42–54. 10.1002/acn3.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, et al. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J Am Chem Soc (2012) 134:4731–42. 10.1021/ja210088v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, et al. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J Am Chem Soc (2009) 131:9767–79. 10.1021/ja9020149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong CH, Nguyen L, Peh J, Luu LM, Sanchez JS, Richardson SL, et al. Targeting toxic RNAs that cause myotonic dystrophy type 1 (DM1) with a bisamidinium inhibitor. J Am Chem Soc (2014) 136:6355–61. 10.1021/ja5012146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A (2011) 108:260–5. 10.1073/pnas.1013343108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perbellini R, Greco S, Sarra-Ferraris G, Cardani R, Capogrossi MC, Meola G, et al. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord (2011) 21:81–8. 10.1016/j.nmd.2010.11.012 [DOI] [PubMed] [Google Scholar]
- 37.Chakraborty M, Selma-Soriano E, Magny E, Couso JP, Perez-Alonso M, Charlet-Berguerand N, et al. Pentamidine rescues contractility and rhythmicity in a Drosophila model of myotonic dystrophy heart dysfunction. Dis Model Mech (2015) 8:1569–78. 10.1242/dmm.021428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coonrod LA, Nakamori M, Wang W, Carrell S, Hilton CL, Bodner MJ, et al. Reducing levels of toxic RNA with small molecules. ACS Chem Biol (2013) 8:2528–37. 10.1021/cb400431f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siboni RB, Nakamori M, Wagner SD, Struck AJ, Coonrod LA, Harriott SA, et al. Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep (2015) 13:2386–94. 10.1016/j.celrep.2015.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci U S A (2009) 106:18551–6. 10.1073/pnas.0903234106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frayha GJ, Smyth JD, Gobert JG, Savel J. The mechanisms of action of antiprotozoal and anthelmintic drugs in man. Gen Pharmacol (1997) 28:273–99. 10.1016/S0306-3623(96)00149-8 [DOI] [PubMed] [Google Scholar]
- 42.Paredes R, Laguna F, Clotet B. Leishmaniasis in HIV-infected persons: a review. J Int Assoc Physicians AIDS Care (1997) 3:22–39. [PubMed] [Google Scholar]
- 43.Zhang Y, Bell A, Perlman PS, Leibowitz MJ. Pentamidine inhibits mitochondrial intron splicing and translation in Saccharomyces cerevisiae. RNA (2000) 6:937–51. 10.1017/S1355838200991726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Li Z, Pilch DS, Leibowitz MJ. Pentamidine inhibits catalytic activity of group I intron Ca.LSU by altering RNA folding. Nucleic Acids Res (2002) 30:2961–71. 10.1093/nar/gkf394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun T, Zhang Y. Pentamidine binds to tRNA through non-specific hydrophobic interactions and inhibits aminoacylation and translation. Nucleic Acids Res (2008) 36:1654–64. 10.1093/nar/gkm1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edwards KJ, Jenkins TC, Neidle S. Crystal structure of a pentamidine-oligonucleotide complex: implications for DNA-binding properties. Biochemistry (1992) 31:7104–9. 10.1021/bi00146a011 [DOI] [PubMed] [Google Scholar]
- 47.Wagner SD, Struck AJ, Gupta R, Farnsworth DR, Mahady AE, Eichinger K, et al. Dose-dependent regulation of alternative splicing by MBNL proteins reveals biomarkers for myotonic dystrophy. PLoS Genet (2016) 12:e1006316. 10.1371/journal.pgen.1006316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siboni RB, Bodner MJ, Khalifa MM, Docter AG, Choi JY, Nakamori M, et al. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J Med Chem (2015) 58:5770–80. 10.1021/acs.jmedchem.5b00356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamitori S, Takusagawa F. Crystal structure of the 2:1 complex between d(GAAGCTTC) and the anticancer drug actinomycin D. J Mol Biol (1992) 225:445–56. 10.1016/0022-2836(92)90931-9 [DOI] [PubMed] [Google Scholar]
- 50.Muller W, Crothers DM. Studies of the binding of actinomycin and related compounds to DNA. J Mol Biol (1968) 35:251–90. 10.1016/S0022-2836(68)80024-5 [DOI] [PubMed] [Google Scholar]
- 51.Trask DK, Muller MT. Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D. Proc Natl Acad Sci U S A (1988) 85:1417–21. 10.1073/pnas.85.5.1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet (2006) 15:2087–97. 10.1093/hmg/ddl132 [DOI] [PubMed] [Google Scholar]
- 53.Chen G, Masuda A, Konishi H, Ohkawara B, Ito M, Kinoshita M, et al. Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci Rep (2016) 6:25317. 10.1038/srep25317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hemler M, Lands WE. Purification of the cyclooxygenase that forms prostaglandins. Demonstration of two forms of iron in the holoenzyme. J Biol Chem (1976) 251:5575–9. [PubMed] [Google Scholar]
- 55.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol (1971) 231:232–5. 10.1038/newbio231232a0 [DOI] [PubMed] [Google Scholar]
- 56.Garcia-Lopez A, Monferrer L, Garcia-Alcover I, Vicente-Crespo M, Alvarez-Abril MC, Artero RD. Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS One (2008) 3:e1595. 10.1371/journal.pone.0001595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang F, Bodycombe NE, Haskell KM, Sun YL, Wang ET, Morris CA, et al. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum Mol Genet (2017) 26:3056–68. 10.1093/hmg/ddx190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer (2003) 3:459–65. 10.1038/nrc1097 [DOI] [PubMed] [Google Scholar]
- 59.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science (1997) 277:333–8. 10.1126/science.277.5324.333 [DOI] [PubMed] [Google Scholar]
- 60.Hara M, Akasaka K, Akinaga S, Okabe M, Nakano H, Gomez R, et al. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc Natl Acad Sci U S A (1993) 90:2281–5. 10.1073/pnas.90.6.2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oana K, Oma Y, Suo S, Takahashi MP, Nishino I, Takeda S, et al. Manumycin A corrects aberrant splicing of Clcn1 in myotonic dystrophy type 1 (DM1) mice. Sci Rep (2013) 3:2142. 10.1038/srep02142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ketley A, Chen CZ, Li X, Arya S, Robinson TE, Granados-Riveron J, et al. High content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC independent pathway in myotonic dystrophy cell lines. Hum Mol Genet (2013) 23:1551–62. 10.1093/hmg/ddt542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest (2009) 119:3797–806. 10.1172/JCI37976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wojciechowska M, Taylor K, Sobczak K, Napierala M, Krzyzosiak WJ. Small molecule kinase inhibitors alleviate different molecular features of myotonic dystrophy type 1. RNA Biol (2014) 11:742–54. 10.4161/rna.28799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bryk R, Wu K, Raimundo BC, Boardman PE, Chao P, Conn GL, et al. Identification of new inhibitors of protein kinase R guided by statistical modeling. Bioorg Med Chem Lett (2011) 21:4108–14. 10.1016/j.bmcl.2011.04.149 [DOI] [PubMed] [Google Scholar]
- 66.Chen HM, Wang L, D’Mello SR. A chemical compound commonly used to inhibit PKR, {8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one}, protects neurons by inhibiting cyclin-dependent kinase. Eur J Neurosci (2008) 28:2003–16. 10.1111/j.1460-9568.2008.06491.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimazawa M, Hara H. Inhibitor of double stranded RNA-dependent protein kinase protects against cell damage induced by ER stress. Neurosci Lett (2006) 409:192–5. 10.1016/j.neulet.2006.09.074 [DOI] [PubMed] [Google Scholar]
- 68.Shimazawa M, Ito Y, Inokuchi Y, Hara H. Involvement of double-stranded RNA-dependent protein kinase in ER stress-induced retinal neuron damage. Invest Ophthalmol Vis Sci (2007) 48:3729–36. 10.1167/iovs.06-1122 [DOI] [PubMed] [Google Scholar]
- 69.Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-gamma-mediated disinhibition. Cell (2011) 147:1384–96. 10.1016/j.cell.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cole A, Frame S, Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J (2004) 377:249–55. 10.1042/BJ20031259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei C, Stock L, Valanejad L, Zalewski ZA, Karns R, Puymirat J, et al. Correction of GSK3beta at young age prevents muscle pathology in mice with myotonic dystrophy type 1. FASEB J (2018) 32:2073–85. 10.1096/fj.201700700R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kouki T, Takasu N, Nakachi A, Tamanaha T, Komiya I, Tawata M. Low-dose metformin improves hyperglycaemia related to myotonic dystrophy. Diabet Med (2005) 22:346–7. 10.1111/j.1464-5491.2005.01432.x [DOI] [PubMed] [Google Scholar]
- 73.Laustriat D, Gide J, Barrault L, Chautard E, Benoit C, Auboeuf D, et al. In vitro and in vivo modulation of alternative splicing by the biguanide metformin. Mol Ther Nucleic Acids (2015) 4:e262. 10.1038/mtna.2015.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Claperon A, Chretien Y, et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res (2013) 73:3974–86. 10.1158/0008-5472.CAN-12-3824 [DOI] [PubMed] [Google Scholar]
- 75.Quinn BJ, Dallos M, Kitagawa H, Kunnumakkara AB, Memmott RM, Hollander MC, et al. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma IGF-I and diminished receptor tyrosine kinase signaling. Cancer Prev Res (Phila) (2013) 6:801–10. 10.1158/1940-6207.CAPR-13-0058-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoskins JW, Ofori LO, Chen CZ, Kumar A, Sobczak K, Nakamori M, et al. Lomofungin and dilomofungin: inhibitors of MBNL1-CUG RNA binding with distinct cellular effects. Nucleic Acids Res (2014) 42:6591–602. 10.1093/nar/gku275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herrendorff R, Faleschini MT, Stiefvater A, Erne B, Wiktorowicz T, Kern F, et al. Identification of plant-derived alkaloids with therapeutic potential for myotonic dystrophy type I. J Biol Chem (2016) 291:17165–77. 10.1074/jbc.M115.710616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kumar GS. RNA targeting by small molecules: binding of protoberberine, benzophenanthridine and aristolochia alkaloids to various RNA structures. J Biosci (2012) 37:539–52. 10.1007/s12038-012-9217-3 [DOI] [PubMed] [Google Scholar]
- 79.Nafisi S, Malekabady ZM, Khalilzadeh MA. Interaction of beta-carboline alkaloids with RNA. DNA Cell Biol (2010) 29:753–61. 10.1089/dna.2010.1087 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.