

Abstract
Background
Anesthetic preconditioning (APC) of the myocardium is mediated in part by reversible alteration of mitochondrial function. Nitric oxide (NO) inhibits mitochondrial respiration and may mediate APC-induced cardioprotection. In this study, the effects of isoflurane on different states of mitochondrial respiration during the oxidation of complex I-linked substrates and the role of NO were investigated.
Methods
Mitochondria were isolated from Sprague-Dawley rat hearts. Respiration rates were measured polarographically at 28ºC with a computer-controlled Clark-type O2 electrode in the mitochondria (0.5 mg/mL) with complex I substrates glutamate/malate (5 mM). Isoflurane (0.25 mM) was administered before or after adenosine diphosphate (ADP)-initiated state 3 respiration. The NO synthase (NOS) inhibitor L-N5-(1-iminoethyl)-ornithine (L-NIO, 10 μM) and the NO donor S-nitroso-N-acetylpenicillamine (SNAP, 1 μM) were added before or after the addition of ADP.
Results
Isoflurane administered in state 2 increased state 2 respiration and decreased state 3 respiration. This attenuation of state 3 respiration by isoflurane was similar when it was given during state 3. L-NIO did not alter mitochondrial respiration or the effect of isoflurane. SNAP only, added in state 3, decreased state 3 respiration and enhanced the isoflurane-induced attenuation of state 3 respiration.
Conclusion
Isoflurane has clearly distinguishable effects on different states of mitochondrial respiration during the oxidation of complex I substrates. The uncoupling effect during state 2 respiration and the attenuation of state 3 respiration may contribute to the mechanism of APC-induced cardioprotection. These effects of isoflurane do not depend on endogenous mitochondrial NO, as the NOS inhibitor L-NIO did not alter the effects of isoflurane on mitochondrial respiration.
Keywords: Isoflurane, Mitochondria, Complex I, Respiration, Nitric oxide
Introduction
Anesthetic preconditioning (APC) is effective as ischemic preconditioning in protecting the heart against ischemia-reperfusion injury by decreasing myocardial infarct size and im proving postischemic functional recovery [1, 2, 3]. Although the precise mechanisms responsible for APC-induced cardioprotection remain incompletely characterized, the reversible alteration of mitochondrial function initiated by volatile anesthetics has been implicated in cardioprotection [4]. The effects of volatile anesthetics on mitochondrial respiration and the electron transport chain (ETC) has been demonstrated in recent studies [5, 6, 7]. Ljubkovic et al. [6] showed that APC-treated heart mitochondria exhibited a better preservation of respiration and adenosine triphosphate (ATP) synthesis after hypoxia stress than non-APC treated mitochondria did. Hanley et al. [8] found that halothane, isoflurane, and sevoflurane dose-dependently inhibited NADH:ubiquinone oxidoreductase (complex I) in pig heart submitochondrial particles. These studies showed that volatile anesthetics might directly affect mitochondria complexes and reactive oxygen species (ROS) production to modulate mitochondrial respiration and bioenergetics.
Volatile anesthetics may trigger preconditioning, in part, by inducing the formation of ROS and reactive nitrogen species and by the release of nitric oxide (NO) [9, 10]. NO exerts a number of actions that are beneficial during myocardial ischemia-reperfusion [10, 11]. For example, it may act as a trigger and mediator of isoflurane-induced delayed preconditioning in rabbit myocardium [12]. Furthermore, isoflurane potentiates ischemic postconditioning via an NO-dependent mechanism [13]. It has been suggested that mitochondria contain NO synthase (NOS) and produce NO to regulate respiration [14]. NO inhibits mitochondrial respiration by binding to respiratory chain enzymes, and modulates mitochondrial ATP-dependent K+ channels [15]. Riess et al. [5] reported that, in isolated heart mitochondrial preparation, sevoflurane administered in state 2 attenuated state 3 respiration. The attenuation was mediated by ROS but not by mitochondrial KATP channel opening. Therefore, the interaction between NO and the ETC may be important in APC-induced cardiac protection.
Because mitochondrial O2 consumption is different in each respiratory state, it is im portant to understand the effects of volatile anesthetics on the different states of mitochondrial respiration. The difference in O2 consumption in each state is tightly linked to the regulation of tissue O2 levels [16]. In addition, the respiration rate of mitochondria is one of the main factors for the regulation of mitochondrial ROS generation [17, 18]. To date, the role of volatile anesthetics on different states of mitochondrial respiration (i.e., states 2–4) has not been addressed in detail. It is also unclear whether NO, like mitochondrial ROS, is involved in the effect of isoflurane on mitochondrial respiration. This study was designed to explore: (1) the effect of isoflurane on different mitochondrial respiration states during the oxidation of complex I-linked substrates, and (2) whether mitochondrial NO is involved in the effect of isoflurane on different mitochondrial respiratory states.
Materials and Methods
Isolation of Mitochondria
Male SD rats at 8–10 weeks of age maintained on a standard diet were used. Animals used in this study were approved by the Suzhou Science and Technology Town Hospital Institutional Animal Care and Use Committee. They were anesthetized with pentobarbital sodium (50 mg/kg i.p.) and following decapitation, the hearts were quickly excised and plunged into an ice-cold isolation buffer containing (in mM) mannitol 200, sucrose 50, KH2PO4 5, EGTA 1, 3-(N-morpholino)propanesulfonic acid (MOPS) 5, and 0.1% bovine serum albumin (pH 7.3, adjusted with potassium hydroxide). Large vessels and auricles were discarded and remaining blood was removed. The ventricles were minced into 1-mm3 pieces. The isolation of mitochondria was performed at 4ºC as described previously [6]. The tissue was homogenized 4 times for 15 s each time with 30 s intervals. The homogenate was centrifuged at 800 g for 10 min and the obtained pellet was rehomogenized and recentrifuged. The resulting supernatants were centrifuged at 6,000 g for 10 min, the pellets resuspended in 25 mL isolation buffer, and the suspension was then centrifuged at 800 g for another 10 min. The supernatant containing the mitochondrial fraction was further centrifuged at 8,000 g for 10 min. This final pellet was resuspended in 1 mL isolation buffer without EGTA and kept on ice. The total protein concentration was determined by bicinchonicic acid protein assay with bovine serum albumin as a standard.
Measurement of Mitochondrial Oxygen Consumption
Mitochondrial respiration was measured polarographically at 28°C with a computer-controlled Clark-type O2 electrode (Hansatech Instruments Ltd, Norfolk, UK) in 0.5 mL respiratory buffer containing (in mM): KCl 130, K2PH4 5, MOPS 20, EGTA 2.5, Na4P2O7 0.001 EDTA, and 0.1% BSA (pH 7.2 adjusted with potassium hydroxide). Mitochondrial suspension (0.5 mg/mL) were added in respiratory buffer in the presence of complex I substrate glutamate (5 mM) plus malate (5 mM).
Experimental Protocols
To study the actions of isoflurane and NO on mitochondrial respiration, isoflurane (0.25 mM), the NO donor S-nitroso-N-acetylpenicillamine (SNAP, 1 μM), or the NOS inhibitor L-N5-(1-iminoethyl)-ornithine (L-NIO, 10 μM) were added according to the protocols in Figure 1. Glutamate (5 mM) and malate (5 mM) were added at 2 min and adenosine diphosphate (ADP, 250 μM) was added at 4 min. Isoflurane was added at 3 min or after ADP was administered (Fig. 1a). L-NIO or SNAP was added before substrates were administered (1 min, Fig. 1b) or after ADP-initiated state 3 respiration (4.5 min, Fig. 1c).
Fig. 1.
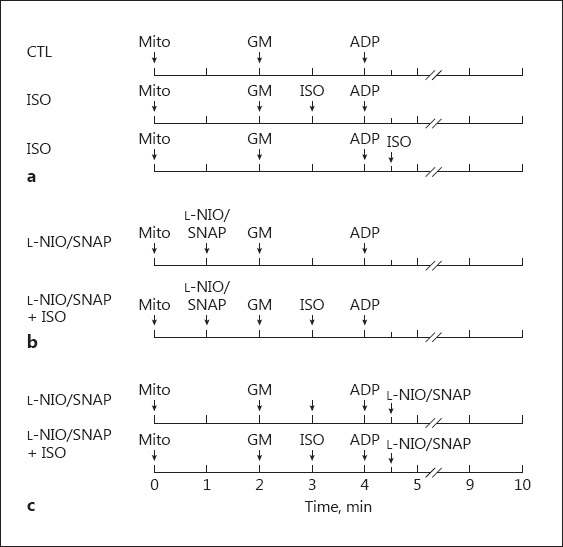
Experimental protocols used to measure mitochondrial respiration. State 2 respiration was initiated by the addition of 5 mM glutamate and malate; state 3 respiration was initiated by the addition of 250 μM ADP. a Isoflurane (0.25 mM) was added in state 2 or state 3 after applying the complex I substrates glutamate and malate. b The NOS inhibitor L-NIO or the NO donor SNAP was added in state 1, 2, or 3 with or without isoflurane. CTL, control group; ISO, isoflurane; GM, glutamate and malate; Mito, mitochondria; ADP, adenosine diphosphate.
These experiments used the conventional definitions for mitochondrial respiration states. State 2 is the state of O2 consumption in the presence of substrates without added ADP. State 3 is the state of maximal O2 consumption after the addition of ADP (ADP phosphorylation). State 4 is the state of minimal O2 consumption after the added ADP is depleted. The representative traces of respiration are shown in Figure 2. The traces indicate a high functional quality and structural integrity of mitochondria after the isolation procedure, as shown by the respiratory control ratio (RCR, state 3/state 4) of 9.8 ± 1.5 with the substrates glutamate and malate.
Fig. 2.
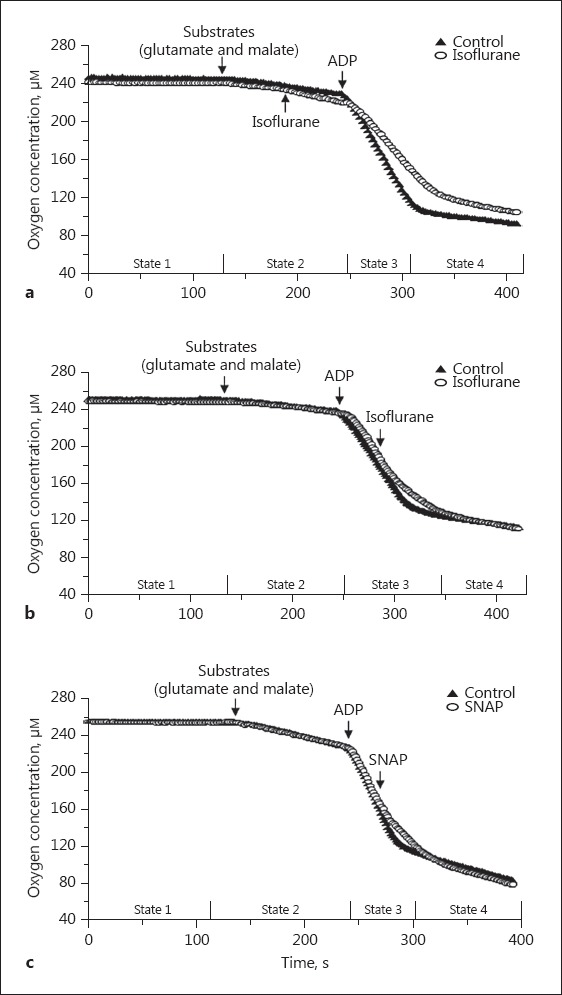
Representative recording traces of mitochondrial respiration. Isoflurane (0.25 mM) was added in state 2 (a) or state 3 (b). c The NO donor, SNAP (1 μM) was added in state 3. ADP, adenosine diphosphate.
Drugs and Chemicals
Isoflurane was purchased from Abbot Laboratories (Abbott Park, IL, USA). L-NIO was obtained from Calbiochem (San Diego, CA, USA). ADP, glutamate, malate, SNAP, and protease were obtained from Sigma (St. Louis, MO, USA). Isoflurane (10 μL) stock solution was made by putting 10 μL of anesthetic in 1 mL respiration buffer, and then sonicating for 10 min; 5 μL of this stock solution was added into the chamber containing 500 μL of mitochondrial suspension. The concentration of isoflurane was analyzed by gas chromatography (Schimadzu, Kyoto, Japan). The average concentration of isoflurane was 0.25 ± 0.02 mM. L-NIO was dissolved in respiration buffer at 1 mM and SNAP was dissolved in DMSO at 100 mM, and they were then diluted to 100 μM by respiration buffer as a stock solution before the experiment. The stock solution of L-NIO or SNAP was added to 500 μL mitochondrial suspension.
Statistical Analysis
All data are expressed as means ± SD. A two-way analysis of variance was used to assess the overall difference between groups. The Student Newman Keuls' multiple-comparison post hoc test was used to differentiate within-group differences. Differences between means were considered significant when p < 0.05 (two-tailed).
Results
Effect of Isoflurane on State 2 and State 3 Respiration
In this protocol, isoflurane administered in state 2 increased the state 2 respiration rate and significantly attenuated state 3 respiration compared to the control group (Fig. 3a). Consequently, the RCR was lower in the isoflurane group than in the control group (Fig. 3b). To observe the direct effect of isoflurane on state 3 respiration, the anesthetic was applied shortly after ADP was added, and isoflurane also attenuated the state 3 respiration (Fig. 3a). In this case, the RCR was also lower than in the control group.
Fig. 3.
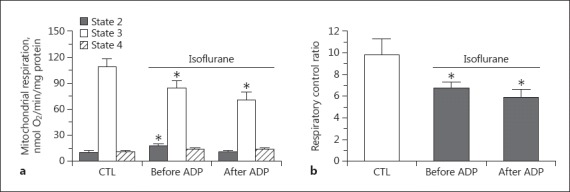
Summarized data of the effects of isoflurane (0.25 mM) on mitochondrial respiration (states 2–4; a) and respiratory control ratio (b) when isoflurane was added, before or after ADP initiated state 3 respiration in the presence of the complex I substrates glutamate and malate. * p < 0.05 compared to control group. # p < 0.05 compared to the group in which isoflurane was given in state 2. CTL, control group; ADP, adenosine diphosphate.
Role of NOS Inhibitor L-NIO in the Effect of Isoflurane
To study the role of NO in the effect of isoflurane on mitochondrial respiration, the NOS inhibitor L-NIO was added before the administration of substrates and isoflurane (Fig. 1b) or after ADP application in state 3 (Fig. 1c). L-NIO did not alter state 3 respiration and did not affect the isoflurane-induced increase in state 2 respiration or decrease in state 3 respiration (Fig. 4). L-NIO given in state 3 with or without isoflurane treatment did not have any significant effect on mitochondrial respiration when compared to the control group or the isoflurane-only group (data not shown).
Fig. 4.
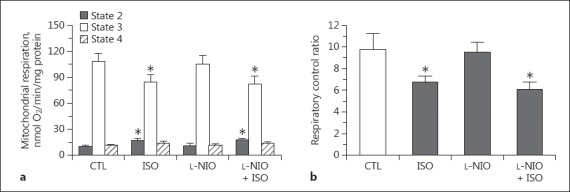
Summarized data of the effects of the NOS inhibitor L-NIO (10 μM, applied in state 1) on mitochondrial respiration (states 2–4; a) and respiratory control ratio (b) with or without isoflurane (applied in state 2) in the presence of the complex I substrates glutamate and malate. * p < 0.05 compared to control group. CTL, control group; ISO, isoflurane.
Role of Exogenous NO in the Effect of Isoflurane
To observe the effect of exogenous NO on mitochondrial respiration, the NO donor SNAP was added before (Fig. 1b) or after ADP-initiated state 3 respiration (Fig. 1c). SNAP alone added before substrate administration (state 2) did not alter mitochondrial respiration and did not affect isoflurane-induced respiration changes (data not shown). However, SNAP administered in state 3 attenuated state 3 respiration compared to in the control (Fig. 5a; 86.2 ± 6.3 vs. 108.8 ± 9.3 nmol O2/min/mg protein, p < 0.05). In the presence of isoflurane, SNAP further enhanced the attenuation of state 3 respiration (78 ± 11 vs. 61 ± 5%, nmol O2/min/mg protein, p < 0.05), but did not alter the increase in state 4 respiration by isoflurane. The decrease of RCR with SNAP in the presence of isoflurane was greater than in the isoflurane group or the SNAP-only group (Fig. 5b).
Fig. 5.
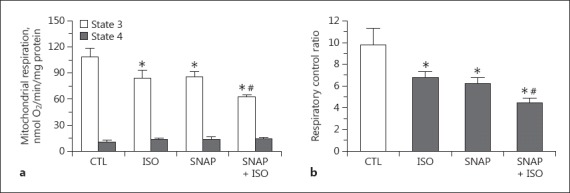
Summarized data of the effects of the NO donor SNAP (1 μM, applied in state 3) on mitochondrial respiration (a) and respiratory control ratio (b) with or without isoflurane (applied in state 2) in the presence of the complex I substrates glutamate and malate. * p < 0.05 compared to control group. # p < 0.05 compared to isoflurane group. CTL, control group; ISO, isoflurane.
Discussion
A better understanding of how isoflurane and other volatile anesthetics mediate cardioprotection against ischemia and reperfusion injury could have clinical implications. The cardioprotective effects of volatile anesthetics, including isoflurane, have, in part, been at tributed to the direct inhibition of mitochondrial electron transport system complexes, and altered mitochondrial bioenergetics in hearts [5, 6, 7, 8]. Although an attenuation of mitochondrial state 3 respiration by volatile anesthetics and the role of ROS have been reported [5], the effect of isoflurane administered in different states of mitochondrial respiration and the role of NO in this process have not been investigated.
This study shows that (1) isoflurane administrated in state 2 increased state 2 respiration and decreased state 3 respiration, (2) the decrease in state 3 respiration was greater when isoflurane was applied in state 3 than in state 2, (3) the NOS inhibitor L-NIO did not alter the effect of isoflurane on respiration, and (4) the NO donor SNAP administered during state 3 respiration enhanced the isoflurane-induced attenuation of state 3 respiration.
We found that isoflurane induced an increase in state 2 respiration and a decrease in RCR compared to the control group. These results suggest that isoflurane may cause a small proton leak through the inner mitochondrial membrane, and partially uncouple respiration, resulting in increased respiration. It has been reported that isoflurane attenuated the magnitude and increased the duration of state 3 NADH oxidation. These effects are likely due to restricted proton pumping by complex I causing a slowed and prolonged proton entry into complex V after adding ADP [7].
Ischemic preconditioning also induces a mild proton leak that is involved in the mechanism of cardioprotection [19]. This uncoupling effect of isoflurane during state 2 or state 4 respiration may contribute to the mechanism of APC-induced cardioprotection. Uncoupling decreases proton driving force (ΔμH+) to facilitate the electron flow and reduce the toxic increase of superoxide production [20]. It has been confirmed that isoflurane preconditioning elicits partial mitochondrial uncoupling, with preserved respiration and ATP synthesis and a reduced mitochondrial Ca2+ uptake [6].
Isoflurane administered in state 2 attenuated state 3 respiration and decreased RCR during the oxidation of the complex I-linked substrates glutamate and malate. This effect of isoflurane is consistent with the findings of the studies by Agarwal et al. [7] and Riess et al. [5]. Riess et al. [5] found that sevoflurane reduced state 3 respiration when applied in state 2. To observe the direct effect of isoflurane on state 3 respiration independent of its effect on state 2 respiration, we added isoflurane in state 3. We observed that the attenuation of respiration by isoflurane was greater when it was added in state 3 than when it was added in state 2.
Although the mechanism causing this different effect of isoflurane on different respiratory states is not known, isoflurane may inhibit enzyme activity in the respiring state (state 3) more effectively than in the quiescent state (state 2). Cytochrome c oxidase (complex IV) is known as the terminal enzyme in the mitochondrial respiratory chain. It reduces dioxygen to water, using reducing equivalents supplied by ferrocytochrome c [16, 21]. It limits the respiration rate in the different respiring states. The control coefficient of cytochrome c oxidase over mitochondrial respiration is higher in state 3 than in state 4 [22]. Therefore, the inhibition of isoflurane on cytochrome c oxidase could be more significant when it is applied in state 3. Cytochrome c oxidase is more active during ADP-initiated phosphorylation. In addition, the increase of state 2 respiration by isoflurane may also contribute to the further decrease in state 3 respiration than when isoflurane was administered in state 3.
Recent studies have proposed that mitochondria contain NOS and can produce significant amounts of NO to regulate respiration [9, 19]. Therefore, NO may modulate the effects of volatile anesthetics on mitochondrial respiration. NO acts as a multisite inhibitor of the mitochondrial ETC. The most sensitive and widely studied target for NO in mitochondria is the terminal enzyme of the ETC, cytochrome c oxidase [9, 23, 24]. NO competes with O2 at the binuclear CuB/cytochrome a3 site. This competition with O2 results in the inhibition of the enzyme, suggesting that NO is an important physiological regulator of mitochondrial oxidative phosphorylation [9, 23, 24]. Reports [25, 26] that the presence of the mitochondrial NOS isoenzyme is a constitutive protein in the mitochondrial inner membrane support a potential physiological role for NO in mitochondrial respiration. Mitochondrial NOS activity has been shown to be susceptible to pharmacological regulation and metabolic states [27]. Volatile anesthetics may activate mitochondrial NOS to produce NO, which leads to the isoflurane-induced alteration of respiration. To test this hypothesis, the NOS inhibitor L-NIO was given before or after the administration of isoflurane. Our results showed that L-NIO did not affect mitochondrial respiration and did not alter the effect of isoflurane on mitochondrial respiration. This suggests that the effect of isoflurane on mitochondrial respiration is independent of endogenous mitochondrial NO generation.
In isolated mitochondria from cardiac myocytes, exogenous NO inhibited respiration and reduced the NAD(P)H redox state [28]. NO is able to inhibit the ETC, mainly at complex IV, regulating oxygen consumption and ATP generation [29]. The respiratory chain is inhibited by NO, either supplied exogenously or produced endogenously via NOS activation. The inhibition of respiration is reversible, although its dependence on concentration and time of exposure to NO is not clear [30]. We also tested whether exogenous NO modulates mitochondrial respiration in the presence of isoflurane. When the NO donor SNAP was added in state 3 respiration, we found that it attenuated state 3 respiration, consistent with other studies [14, 31]. The results also showed that the attenuation of state 3 respiration by isoflurane was enhanced by presence of SNAP. This suggests that extramitochondrial NO attenuates state 3 respiration and has a synergistic action with isoflurane on mitochondrial respiration. Borutaite and Brown [32] found that the reversible inactivation of mitochondrial enzymes by SNAP in isolated heart mitochondria resulted in a significant increase in H2O2 production. Therefore, SNAP may increase H2O2 production, which may further inhibit complex I, potentially contributing to the attenuation of state 3 respiration. It has been reported that H2O2 inhibits oxygen consumption in state 3, and further increases ROS production with complex I substrates. It does not associate with complex II substrate [33]. In the rat brain mitochondria, the NO substrate L-arginine was shown to decrease state 3 respiration when administered after ADP-initiated state 3 respiration [34]. It has been confirmed that, under physiological conditions, respiration is more sensitive to NO in state 3 than in state 4, due to there being greater control over respiration by cytochrome c oxidase in state 3 [16]. Taking previous studies into account, our finding of an enhancing effect of SNAP on the isoflurane-induced attenuation of state 3 respiration suggests that exogenous NO is involved in the modulation of mitochondrial respiration, and that isoflurane enhanced the inhibition of mitochondrial respiration in the presence of exogenous NO.
One limitation of our study is that the production of NO and ROS was not measured. The effects of endogenous NO on mitochondrial respiration with or without isoflurane need to be clarified. A study showed that mitochondrial KATP channel opening increased ROS production in isolated heart and liver mitochondria. The location of ROS generation is in complex I of the ETC [35]. Volatile anesthetics could increase the formation of ROS at complex I and/or complex III of the ETC by the reversible attenuation of mitochondrial electron transport [4]. The ROS generation then affects the mitochondrial respiration by acting on downstream effectors, including mitochondrial KATP channels that are normally closed at physiologic ATP levels [36]. We cannot exclude the relative contributions of KATP channel or other potassium channel opening in the isoflurane-induced effect on different mitochondrial respiration states. In addition, our in vitro experiments were generally carried out at atmospheric O2 concentrations. This gives an O2 concentration of about 150–200 μM in the chamber, which is above the concentrations that cells are exposed to in vivo. Whether O2 limitation of mitochondrial respiration occurs in vivo over the range of O2 (1–20 µM) remains to be determined [16]. Therefore, the role of exogenous or endogenous NO in the effect of isoflurane on mitochondrial respiration may depend on O2 tension.
Conclusion
Isoflurane has different effects on different states of mitochondrial respiration during the oxidation of the complex I substrates glutamate and malate. The uncoupling effect during state 2 respiration and the attenuation of state 3 respiration by isoflurane may contribute to the mechanism of APC-induced cardioprotection. Endogenous mitochondrial NO is not involved in the effect of isoflurane on mitochondrial respiration.
Disclosure Statement
The authors have no conflicts of interest to disclose.
Sources of Funding
This work was supported by grants No. SS201756 (to Dr. An) from the Science and Technology Development Plan of Suzhou City; No. BK20141187 (to Dr. Wang) from the Natural Science Foundation of Jiangsu Province, and No SYS201473 (to Dr. Qiao) from the Science and Technology Development Plan of Suzhou City, China. Dr. Wang also received support from the Project of Gusu Health Key Talent (SS201613).
References
- 1.An J, Varadarajan SG, Novalija E, Stowe DF. Ischemic and anesthetic preconditioning reduces cytosolic (Ca2+) and improves Ca(2+) responses in intact hearts. Am J Physiol Heart Circ Physiol. 2001;281:H1508–H1523. doi: 10.1152/ajpheart.2001.281.4.H1508. [DOI] [PubMed] [Google Scholar]
- 2.Wojtovich AP, Smith CO, Urciuoli WR, Wang YT, Xia XM, et al. Cardiac Slo2.1 is required for volatile anesthetic stimulation of K+ transport and anesthetic preconditioning. Anesthesiology. 2016;124:1065–1076. doi: 10.1097/ALN.0000000000001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian Y, Li H, Liu P, Xu JM, Irwin MG, et al. Captopril pretreatment produces an additive cardioprotection to isoflurane preconditioning in attenuating myocardial ischemia reperfusion injury in rabbits and in humans. Mediators Inflamm. 2015;2015:819232. doi: 10.1155/2015/819232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludwig LM, Tanaka K, Eells JT, Weihrauch D, Pagel PS, et al. Preconditioning by isoflurane is mediated by reactive oxygen species generated from mitochondrial electron transport chain complex III. Anesth Analg. 2004;99:1308–1315. doi: 10.1213/01.ANE.0000134804.09484.5D. [DOI] [PubMed] [Google Scholar]
- 5.Riess ML, Eells JT, Kevin LG, Camara AK, Henry MM, et al. Attenuation of mitochondrial respiration by sevoflurane in isolated cardiac mitochondria is mediated in part by reactive oxygen species. Anesthesiology. 2014;100:498–505. doi: 10.1097/00000542-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Ljubkovic M, Mio Y, Marinovic J, Stadnicka A, Warltier DC, et al. Isoflurane preconditioning uncouples mitochondria and protects against hypoxia-reoxygenation. Am J Physiol Cell Physiol. 2007;292:C1583–C1590. doi: 10.1152/ajpcell.00221.2006. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal B, Dash RK, Stowe DF, Bosnjak ZJ, Camara AK. Isoflurane modulates cardiac mitochondrial bioenergetics by selectively attenuating respiratory complexes. Biochim Biophys Acta. 2014;1837:354–365. doi: 10.1016/j.bbabio.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687–693. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Lang XE. Protein kinase C signaling pathway involvement in cardioprotection during isoflurane pre treatment. Mol Med Rep. 2015;11:2683–2688. doi: 10.3892/mmr.2014.3042. [DOI] [PubMed] [Google Scholar]
- 10.Swyers T, Redford D, Larson DF. Volatile anesthetic-induced preconditioning. Perfusion. 2014;29:10–15. doi: 10.1177/0267659113503975. [DOI] [PubMed] [Google Scholar]
- 11.Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- 12.Chiari PC, Bienengraeber MW, Weihrauch D, Krolikowski JG, Kersten JR, et al. Role of endothelial nitric oxide synthase as a trigger and mediator of isoflurane-induced delayed preconditioning in rabbit myocardium. Anesthesiology. 2005;103:74–83. doi: 10.1097/00000542-200507000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Ge ZD, Pravdic D, Bienengraeber M, Pratt PF, Jr, Auchampach JA, et al. Isoflurane postconditioning protects against reperfusion injury by preventing mitochondrial permeability transition by an endothelial nitric oxide synthase-dependent mechanism. Anesthesiology. 2010;112:73–85. doi: 10.1097/ALN.0b013e3181c4a607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 15.Shiva S, Oh JY, Landar AL, Ulasova E, Venkatraman A, et al. Nitroxia: the pathological consequence of dysfunction in the nitric oxide-cytochrome c oxidase signaling pathway. Free Radic Biol Med. 2005;38:297–306. doi: 10.1016/j.freeradbiomed.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 16.Brookes PS, Kraus DW, Shiva S, Doeller JE, Barone MC, et al. Control of mitochondrial respiration by NO*, effects of low oxygen and respiratory state. J Biol Chem. 2003;278:31603–31609. doi: 10.1074/jbc.M211784200. [DOI] [PubMed] [Google Scholar]
- 17.Saborido A, Soblechero L, Megias A. Isolated respiring heart mitochondria release reactive oxygen species in states 4 and 3. Free Radic Res. 2005;39:921–931. doi: 10.1080/10715760500188887. [DOI] [PubMed] [Google Scholar]
- 18.Paital B, Chainy GB. Effects of temperature on complexes I and II mediated respiration, ROS generation and oxidative stress status in isolated gill mitochondria of the mud crab Scylla serrata. J Therm Biol. 2014;41:104–111. doi: 10.1016/j.jtherbio.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395:611–618. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- 21.Zaslavsky D, Gennis RB. Proton pumping by cytochrome oxidase: progress, problems and postulates. Biochim Biophys Acta. 2000;1458:164–179. doi: 10.1016/s0005-2728(00)00066-9. [DOI] [PubMed] [Google Scholar]
- 22.Groen AK, Wanders RJ, Westerhoff HV, van der Meer R, Tager JM. Quantification of the contribution of various steps to the control of mitochondrial respiration. J Biol Chem. 1982;257:2754–2757. [PubMed] [Google Scholar]
- 23.Pannala VR, Camara AK, Dash RK. Modeling the detailed kinetics of mitochondrial cytochrome c oxidase: catalytic mechanism and nitric oxide inhibition. J Appl Physiol. 2016;121:1196–1207. doi: 10.1152/japplphysiol.00524.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarti P, Arese M, Forte E, Giuffre A, Mastronicola D. Mitochondria and nitric oxide: chemistry and pathophysiology. Adv Exp Med Biol. 2012;942:75–92. doi: 10.1007/978-94-007-2869-1_4. [DOI] [PubMed] [Google Scholar]
- 25.Zaobornyj T, Ghafourifar P. Strategic localization of heart mitochondrial NOS: a review of the evidence. Am J Physiol Heart Circ Physiol. 2012;303:H1283–H1293. doi: 10.1152/ajpheart.00674.2011. [DOI] [PubMed] [Google Scholar]
- 26.Kato K, Giulivi C. Critical overview of mitochondrial nitric-oxide synthase. Front Biosci. 2006;11:2725–2738. doi: 10.2741/2002. [DOI] [PubMed] [Google Scholar]
- 27.Zaobornyj T, Valdez LB. Heart mitochondrial nitric oxide synthase: a strategic enzyme in the regulation of cellular bioenergetics. Vitam Horm. 2014;96:29–58. doi: 10.1016/B978-0-12-800254-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 28.Kohlhaas M, Nickel AG, Bergem S, Casadei B, Laufs U, et al. Endogenous nitric oxide formation in cardiac myocytes does not control respiration during beta-adrenergic stimulation. J Physiol. 2017;595:3781–3798. doi: 10.1113/JP273750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tengan CH, Moraes CT. NO control of mitochondrial function in normal and transformed cells. Biochim Biophys Acta. 2017;1858:573–581. doi: 10.1016/j.bbabio.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarti P, Arese M, Bacchi A, Barone MC, Forte E, et al. Nitric oxide and mitochondrial complex IV. IUBMB Life. 2003;55:605–611. doi: 10.1080/15216540310001628726. [DOI] [PubMed] [Google Scholar]
- 31.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 32.Borutaite V, Brown GC. S-nitrosothiol inhibition of mitochondrial complex I causes a reversible increase in mitochondrial hydrogen peroxide production. Biochim Biophys Acta. 2006;1757:562–566. doi: 10.1016/j.bbabio.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 33.Sanz A, Caro P, Gomez J, Barja G. Testing the vicious cycle theory of mitochondrial ROS production: effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. J Bioenerg Biomembr. 2006;38:121–127. doi: 10.1007/s10863-006-9011-8. [DOI] [PubMed] [Google Scholar]
- 34.Lores-Arnaiz S, D'Amico G, Czerniczyniec A, Bustamante J, Boveris A. Brain mitochondrial nitric oxide synthase: in vitro and in vivo inhibition by chlorpromazine. Arch Biochem Biophys. 2004;430:170–177. doi: 10.1016/j.abb.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Andrukhiv A, Costa AD, West IC, Garlid KD. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol. 2006;291:H2067–H2074. doi: 10.1152/ajpheart.00272.2006. [DOI] [PubMed] [Google Scholar]
- 36.Zaugg M, Lucchinetti E, Spahn DR, Pasch T, Schaub MC. Volatile anesthetics mimic cardiac preconditioning by priming the activation of mitochondrial K(ATP) channels via multiple signaling pathways. Anesthesiology. 2002;97:4–14. doi: 10.1097/00000542-200207000-00003. [DOI] [PubMed] [Google Scholar]