

Abstract
Cardiac hypertrophy is closely associated with a series of cardiovascular diseases, including heart failure and sudden death in particular. An in-depth comprehension of the pathogenesis of cardiac hypertrophy will improve the diagnosis and therapy of cardiac hypertrophy. It has been acknowledged that long noncoding RNAs/microRNAs (lncRNAs/miRNAs) are crucial regulators in diverse biological processes, including various cardiovascular diseases, in multiple manners. Nevertheless, the biological roles of lncRNA UCA1 and miR-184 in cardiac hypertrophy are scarcely reported. In this paper, qRT-PCR analysis exhibited that lncRNA UCA1 was highly expressed in mice heart treated with transverse aortic constriction (TAC) and the cardiomyocytes treated with phenylephrine (PE). On the contrary, miR-184 was downregulated under the same conditions. In addition, it was deduced that lncRNA UCA1 was reversely related with miR-184 in PE-triggered hypertrophic cardiomyocytes, confirmed by the Spearman correlation analysis. The knockdown of UCA1 or the overexpression of miR-184 lessened the enlarged surface area of cardiomyocytes and the elevated expressions of fetal genes (ANP and BNP) induced by PE. Later, it was determined that miR-184 was a direct target of UCA1, whereas the mRNA HOXA9 was a target of miR-184. Rescue assays indicated that UCA1 promoted the progression of cardiac hypertrophy through competitively binding with miR-184 to enhance the expression of HOXA9.
Keywords: lncRNA UCA1, miR-184, HOXA9, Cardiac hypertrophy
Introduction
Cardiac hypertrophy is associated with an adaptive response of the heart to fight against diverse pressures in order to maintain cardiac function in an early phase [1]. In addition, unremitting cardiac hypertrophy is usually concurrent with maladaptive cardiac remodeling, which can result in increased heart failure and cardiac death [2, 3, 4]. Despite the fact that a series of factors such as specific peptide hormones, growth factors, and microRNAs (miRNAs) have been determined to be responsible for cardiac hypertrophy, the potential molecular mechanisms for the progression of cardiac hypertrophy are still not clear [5, 6]. Therefore, the study of the pathogenesis of cardiac hypertrophy will benefit the diagnosis and treatment of cardiac hypertrophy.
Long noncoding RNAs (lncRNAs) belong to nonprotein-coding RNAs with a length longer than 200 nt. lncRNAs are popular in molecular studies due to their critical functions in regulating diverse biological behaviors [7]. In addition, it has been disclosed that lncRNAs are also key regulators in various cardiovascular diseases (heart failure in particular); these lncRNAs include lncRNA CHRF [8], lncRNA H19 [1], lncRNA HOTAIR [9], and others. lncRNA UCA1 is a well-known molecule in multiple cancers. However, studies about the role of UCA1 in cardiovascular diseases are quite few, only discussing the role of UCA1 in acute myocardial infarction [10], the contributions of UCA1 to apoptotic cardiomyocytes via inhibiting p27 [11], and the association of UCA1 with the poor prognosis of heart failure [12]. No further information is available concerning the role of UCA1 in cardiac hypertrophy with regard to other molecular mechanisms.
In addition, miRNAs also belong to nonprotein-coding RNAs with a length less than 22 nt, and numerous miRNAs have been reported to be critical participants in the initiation and development of various diseases including cardiac hypertrophy; for example, there is loss of miR-22 in cardiac hypertrophy [13], miR-301a is a novel cardiac regulator [14], and miR-133a impairs cardiac hypertrophy [15]. miR-184 has been reported to be a suppressor in various diseases including cardiovascular diseases, such as its suppressive role in colorectal cancer [16], lung cancer [17], and osteosarcoma [18], and the downregulation of miR-184 in cyanotic congenital heart diseases [19]. However, detailed information about the function of miR-184 in cardiac hypertrophy is unclear.
Here, we discussed the molecular correlation between lncRNAs and miRNAs, which is critically important for the development and progression of cardiac hypertrophy. It was discovered that lncRNA UCA1 was highly expressed in transverse aortic constriction (TAC)-processed mouse heart and phenylephrine (PE)-treated cardiomyocytes. In addition, the knockdown of UCA1 could narrow the PE-handled augmented cell surface area and weaken the mRNA and protein levels of ANP and BNP. Afterwards, it was found that miR-184 was downregulated in PE-triggered cardiomyocytes, and the enlarged cell surface area and upregulated expression of ANP and BNP induced by PE were attenuated by the overexpression of miR-184. miR-184 was confirmed to be negatively related with UCA1 in hypertrophic cardiomyocytes. Furthermore, bioinformatics software and the luciferase reporter analysis demonstrated that miR-184 was a target of UCA1 in cardiac hypertrophy. It was also pointed out that UCA1 actually functioned as a competing endogenous RNA (ceRNA) in cardiac hypertrophy to improve the expression of mRNA HOXA9 through competitively binding with miR-184. The regulatory pattern consisting of lncRNA UCA1, miR-184, and mRNA HOXA9 mediated the development of cardiac hypertrophy, which was expected to make contributions to the treatment of cardiac hypertrophy.
Materials and Methods
Animal Specimens
We obtained the 8-week-old male C57BL6 mice (SPF, 20–25 g) from Vital River Laboratory Animal Company (Beijing, China). The processes involved in this assay were carried out according to the guidelines of the Care and Use of Laboratory Animals published by the Committee of The Second People's Hospital of Hefei. TAC was applied to induce cardiac hypertrophy in the mouse model. In summary, following anesthetization by using intraperitoneal ketamine (100 mg/kg) and xylazine (10 mg/kg), the transverse thoracic aorta was dissected. Later, the mice were placed on a ventilator for recovery with the core temperature at 37°C. The mice with a pressure gradient of less than 60 mm Hg were excluded a week after TAC on the basis of echocardiography in this experiment. Anatomic M-mode echocardiography from mice with matched echocardiographic images of the aortic arch was used to detect stenosis of the transverse aorta. The sham operation for the corresponding matched mice experienced the same processes, without the seam for the aorta.
Cardiomyocyte Culture
The neonatal mice were prepared for cardiomyocyte isolation according to a preceding description [20]. The extracted cardiomyocytes were split into small pieces. Soon afterwards, the tissues were shifted to a digestion solution including 0.1% collagenase type IV, 0.1% trypsin, 15 μg/mL DNase I, and 1% chicken serum in HEPES-buffered saline at 37°C. The trypsin was later neutralized through a supplement of 10% calf serum. Following centrifugation, the resolved cells were resuspended in Dulbecco's modified Eagle's medium/F12 with supplements, followed by plating on the collagen-coated silicone sheet in preparation for cardiomyocyte extraction. Serum-free media were used instead of the cultivation media at 24 h after seeding and the cultivation continued for another 24 h before the assay. All the cells used in the assay were treated with PE in advance. Lipofectamine 2000 was used to transfect cardiomyocytes based on the manufacturer's suggestions.
Plasmid Construction and Transfection
Following construction for pcDNA3.0-UCA1 and pcDNA3.0-HOXA9 vectors on the basis of amplification with specific primers, cDNAs for UCA1 and HOXA9 experienced cloning into the mammalian expression vector pcDNA3.0 (Invitrogen, Carlsbad, CA, USA). The specific primers used in this process were purchased from Sangon Biotech (Shanghai, China). The EndoFree Mini Plasmid kit (Tiangen, China) was used to isolate the plasmid vectors. RiboBio Co. (Shanghai, China) provided us with the mimics and negative control (NC) mimics for miR-148. The pcDNA3.0-UCA1 and pcDNA3.0-HOXA9 vector plasmids and miR-148 mimics were transfected into cardiomyocytes using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's guidelines. After 48 h of transfection, we reaped the cardiomyocytes in preparation for the PCR and Western blot assays. All assays were carried out at 3 independent times.
qRT-PCR Analysis
All RNAs were isolated by means of the Trizol reagent (Invitrogen). DNase I (Invitrogen) was used to deliver 1 μg of RNA and the mRNA sample plate was used to synthesize the cDNA in vitro based on the SuperScript® III First-Strand Synthesis Kit (Invitrogen). The primers in the qPCR process were listed as the following: ANP (former): 5′-CTCCGATAGATCTGCCCTCTTGAA-3′ and (reverse): 5′-GGTACCGGAAGC TGTTGCAGCCTA-3′; BNP (former): 5′-GCTCTTGAAGGACCAAGGCCTCAC-3′ and (reverse): 5′-GATCCGATCCGGTCTATCTTGTGC-3′; lncRNA UCA1 (former): 5′-TTTGCCAGCCTCAGCTTAAT-3′ and (reverse): 5′-TTGTCCCCATTTTCCATCAT-3′; miR-184 (former): 5′-GCATGCCTAAATGTTGACAGCC-3′ and (reverse): 5′-GTGCAGGGTCCGAGGT-3′; HOXA9 (former): 5′-GTGGTTCTCCTCCAGTTGATAG-3′ and (reverse): 5′-AGTTGGCTGCTGGG TTATT-3′; GAPDH (former): 5′-CCACCCATGGCAAATTCCATGGCA-3′ and (reverse): 5′-TCTAGACGGCAGGTCAGGTCCACC-3′. The 2–ΔCt method was employed to measure the fold change of all genes, in which ΔCt = Cttarget – CtGAPDH for ANP, BNP, lncRNA UCA1, miR-184, and HOXA9.
Luciferase Reporter Analysis and Transfection in HeLa Cells
The luciferase reporter gene was cloned by using the specific plasmid for the luciferase reporter analysis. The luciferase reporter plasmid containing the lncRNA UCA1 sequence was constructed by Ribo Company (Guangzhou, China). Then, we took advantage of the Renilla luciferase plasmid to transfect the plasmids into the cells. Generally speaking, HeLa cells were co-transfected on the 12-well plates by means of 50 or 100 nM NC or mimics and 0.4 μg of the luciferase reporter vector. 48 h later, the cell lysates were ready. We then used a Monolight 3010 luminometer (Pharmingen, San Diego, CA, USA) to detect the luciferase activity. The transfections were performed in triplicate.
Western Blot
The incubated cells and cardiac tissues were lysed on ice with mammalian PRO-PREP solution (Thermo Fisher Scientific, Waltham, MA, USA) as well as protease inhibitors (Sigma-Aldrich, St. Louis, MO, USA). Following centrifugation, SDS-PAGE was used to split the cell lysate protein (40 μg) followed by transfection onto PVDF membranes (Bio-Rad). Membranes were sealed by using 5% milk powder in TBST followed by immunoblotting with primary antibodies such as ANP (mouse monoclonal, dilution 1: 1,000; Santa Cruz, CA, USA) and BNP (mouse monoclonal, dilution 1: 1,000; Santa Cruz, CA, USA). GAPDH was applied as a loading control. The protein bands were determined by using HRP binate secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and improved chemiluminescence (Pierce, Rockford, IL, USA). Digital images of luminescence were detected by AlphaImager (FluorChem 5500TM, Alpha Innotech, San Leandro, CA, USA).
Immunofluorescence Staining
The cultivated cells were washed twice with PBS, followed by fixation with 4% paraformaldehyde for 20 min and further washing 3 times with PBS. Subsequently, 10% normal goat serum together with 1% BSA (Sigma-Aldrich) was used to seal the fixed cells for 45 min; anti-α-actinin (mouse monoclonal, dilution 1: 1,000; Sigma-Aldrich) was applied to culture the above cells at 4°C overnight. Having been washed with PBS 3 times, the cells were again cultivated by means of the secondary antibody for 1 h at 37°C. Next, the mounting medium (Dako) including DAPI was employed to coverslip the slides so as to counterstain the nuclei.
Statistical Analysis
Data are exhibited as the mean ± standard deviation (SD) from at least 3 independent assays in vitro. The statistical significance among groups in vivo was examined by the Student t test. One-way ANOVA and Tukey multiple comparison tests were used to analyze data from experiments in vitro (SPSS 20.0). A p value < 0.05 was identified as being statistically significant.
Results
The Expression of lncRNA UCA1 Was Elevated in in vivo Hypertrophic Cardiomyocytes
In order to clarify the function of lncRNA UCA1 in cardiac hypertrophy, we tested the expression level of lncRNA UCA1 in the hypertrophic cardiomyocytes of mice that had undergone TAC surgery in comparison with that in the healthy heart tissues of mice that had experienced the sham operation. As illustrated in Figure 1a, the expression level of UCA1 was obviously higher in the TAC group than that in the sham group. The upregulation of lncRNA UCA1 was accompanied by cardiac hypertrophy responses such as improved expression of the fetal genes ANP and BNP in cardiomyocytes after treatment with TAC, compared to the sham group (Fig. 1b, c). Such phenomena predicted that lncRNA UCA1 might stimulate cardiac hypertrophy.
Fig. 1.
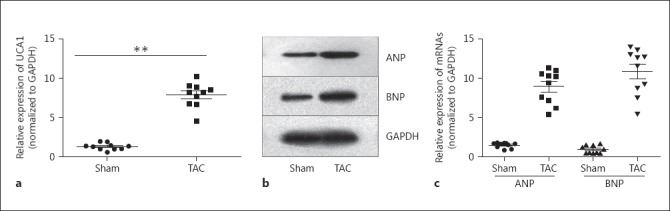
The expression of lncRNA UCA1 was elevated in in vivo hypertrophic cardiomyocytes. a The expression level of lncRNA UCA1 in the hypertrophic cardiomyocytes of mice was tested by qRT-PCR. b, c Western blot and qRT-PCR were applied to examine the protein and mRNA expressions of ANP and BNP in cardiomyocytes. Error bars represent the mean ± SD of at least 3 independent experiments. ** p < 0.01 versus control group. TAC, transverse aortic constriction.
The Knockdown of lncRNA UCA1 Suppressed Hypertrophic Responses in Cardiomyocytes Treated with PE
In order to obtain a clear understanding of the biological functions of UCA1 in the development and progression of cardiac hypertrophy, we detected the expression of UCA1 in a cell model of hypertrophic cardiomyocytes produced from the PE-treated neonatal cardiomyocytes. PE has been acknowledged to be able to trigger cardiac hypertrophy in vitro [21, 22]. The qRT-PCR assay demonstrated that the expression of UCA1 was dramatically enhanced in cardiac hypertrophy that had been treated with PE (Fig. 2a). Furthermore, data from the immunofluorescence staining and qRT-PCR disclosed that the cell surface area was outstandingly enlarged after PE stimulation in comparison with that in the control group (Fig. 2b, c). In the meantime, both the mRNA and protein levels of ANP and BNP were also markedly augmented in PE-triggered hypertrophic cardiomyocytes (Fig. 2d, e). Combined with the high expression of UCA1 in the mouse heart, the enhanced UCA1 in PE-induced cardiomyocytes indicated that dysregulated UCA1 might be a critical factor responsible for cardiac hypertrophy. Thus, the expression of UCA1 was silenced with the specific small interfering RNA (si-RNA). It was discovered that the level of UCA1 was obviously decreased after the transfection of si-UCA1 in PE-treated hypertrophic cardiomyocytes (Fig. 2a). With the knockdown of UCA1, the augmented cell surface area and the enhanced mRNA and protein levels of ANP and BNP caused by the PE stimulation were dramatically impaired compared to those in the si-RNA control group (Fig. 2b, c, d, e).
Fig. 2.
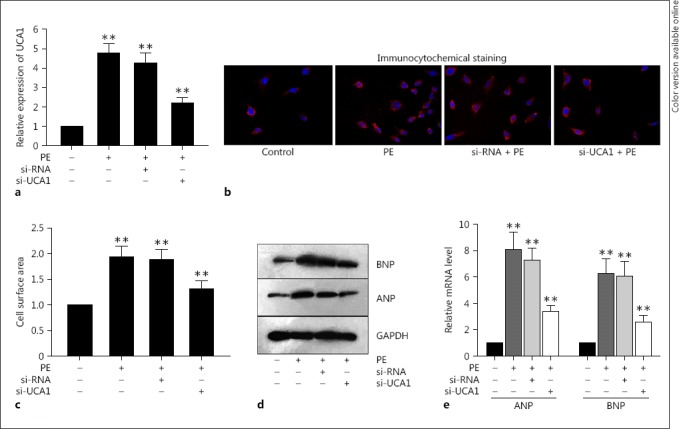
The knockdown of lncRNA UCA1 suppressed hypertrophic responses in cardiomyocytes treated with PE. a Highly expressed UCA1 in PE-treated hypertrophic cardiomyocytes was silenced by specific si-RNA according to qRT-PCR. b, c Immunofluorescence staining and qRT-PCR were performed to show that PE-induced enlarged cell surface area was decreased by the knockdown of UCA1. d, e Western blot and qRT-PCR were designed to exhibit that PE-triggered increased protein and mRNA expressions of ANP and BNP were lessened by si-UCA1. Error bars represent the mean ± SD of at least 3 independent experiments. ** p < 0.01 versus control group. PE, phenylephrine.
MiR-184 Functioned as the Target of UCA1 to Inhibit Cardiac Hypertrophy
To fully understand the development mechanism of cardiac hypertrophy, we aimed to figure out whether other molecules were involved in this process. Thus, miR-184 was chosen to be the object of this study, and we assessed the expression of miR-184 in the hypertrophic cardiomyocytes that had been treated with PE. It was found out that miR-184 was remarkably downregulated in PE-handled cardiomyocytes. For a better comprehension of the role of miR-184 in cardiac hypertrophy, the expression of miR-184 was upregulated with miR-184 mimics (Fig. 3a). Interestingly, the enlarged cell surface area and the elevated mRNA and protein expressions of ANP and BNP induced by PE were damaged by the upregulated miR-184 in hypertrophic cardiomyocytes (Fig. 3b, c, d). In consideration of the expression of UCA1 in cardiac hypertrophy under the same conditions, we conjectured that miR-184 was negatively correlated with UCA1 in cardiac hypertrophy, which was then verified by the Spearman correlation analysis (Fig. 3e). In order to clarify the relationship between the abovementioned two molecules in cardiac hypertrophy, bioinformatics software (http://carolina.imis.athena-innovation.gr) was applied, which searched out the binding sites between miR-184 and UCA1 sequences (Fig. 3f). Subsequently, in order to further confirm the binding of miR-184 to UCA1, the luciferase reporter analysis was designed. UCA1, having been transfected with miR-184 mimics, was cloned into cardiomyocytes. As a result, in comparison with the mutant UCA1 and the NC groups, the luciferase activity of wild-type UCA1 was attenuated by the overexpression of miR-184, ensuring the direct binding of UCA1 to miR-184 in cardiac hypertrophy (Fig. 3g).
Fig. 3.
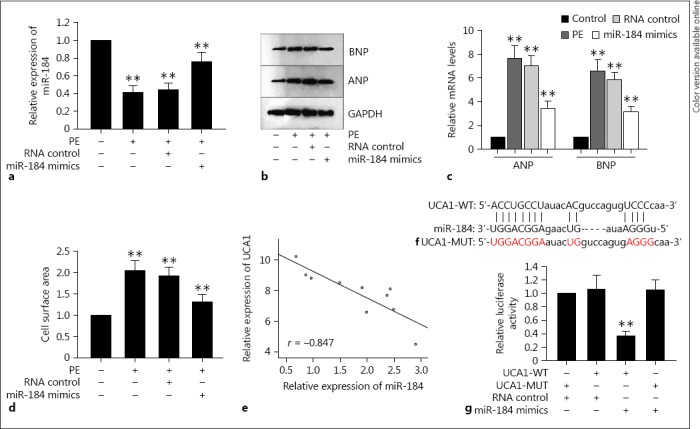
miR-184 functioned as the target of UCA1 to inhibit cardiac hypertrophy. a The miR-184 mimics improved the expression of miR-184 in PE-treated cardiomyocytes according to qRT-PCR. b–d The enlarged cell surface area and the elevated mRNA and protein expressions of ANP and BNP induced by PE were damaged by upregulated miR-184 in hypertrophic cardiomyocytes according to immunofluorescence staining, Western blot, and qRT-PCR. e The Spearman correlation analysis demonstrated the negative relationship between UCA1 and miR-184. f The binding sites between UCA1 and miR-184. g The luciferase reporter analysis was used to prove the binding of UCA1 to miR-184. Error bars represent the mean ± SD of at least 3 independent experiments. ** p < 0.01 versus control group. PE, phenylephrine.
LncRNA UCA1 Competitively Sponged with miR-184 to Enhance HOXA9 in Cardiac Hypertrophy
The above findings together with the data in Figures 1 and 2 elucidated that miR-184 was capable of suppressing the UCA1-mediated stimulus in cardiac hypertrophy. In order to determine how UCA1 interacted with miR-184 in cardiac hypertrophy, we made use of the bioinformatics software miRanda (http://www.microrna.org) to seek out the underlying mRNAs and we finally selected mRNA HOXA9 to be studied in this paper because of its association with hypertension, which was correlated with cardiac hypertrophy [23]. Originally, after treatment with PE in cardiac hypertrophy, the cell surface area was augmented and both the mRNA and protein levels of ANP and BNP were also elevated significantly. However, the downregulated HOXA9 considerably decreased the abovementioned improved hypertrophic responses (Fig. 4a, b, c). In combination with the changes of hypertrophic responses induced by miR-184, we hypothesized that miR-184 was also inversely related to HOXA9 in hypertrophic cardiomyocytes, which was confirmed by the Spearman correlation analysis (Fig. 4d). These findings implied that mRNA HOXA9 might participate in the development of cardiac hypertrophy mediated by lncRNA UCA1.
Fig. 4.
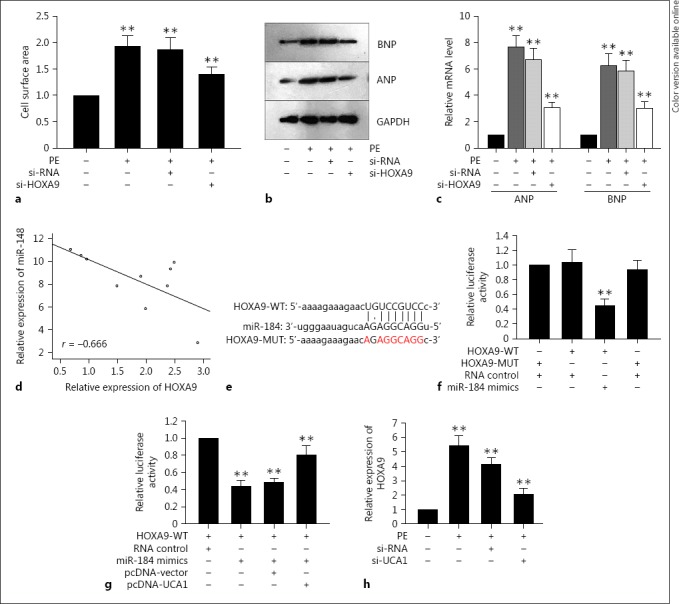
LncRNA UCA1 enhanced HOXA9 in cardiac hypertrophy by competitively binding with miR-184. a–c The silence of HOXA9 outstandingly decreased the improved hypertrophic responses including the cell surface area, the mRNA and protein levels of ANP and BNP. d Spearman's correlation analysis confirmed the negative correlation between HOXA9 and miR-184. e The binding sites between HOXA9 and miR-184. f The luciferase reporter analysis was used to determine the binding of HOXA9 to miR-184. g The luciferase reporter analysis was applied to show that overexpressed UCA1 rescued the decreased luciferase activity of HOXA9 caused by miR-184 mimics. h qRT-PCR showed that the high expression of HOXA9 in hypertrophic cardiomyocytes caused by PE was decreased by the down-regulation of UCA1. Error bars represented the mean ± SD of at least 3 independent experiments. ** p < 0.01 versus control group. PE, phenylephrine.
Afterwards, we searched out the binding sites between miR-184 and HOXA9 on the basis of the miRanda software (http://www.microrna.org/microrna/getGeneForm.do) (Fig. 4e). Subsequently, the luciferase reporter analysis was designed with the construction of the 3′-UTR segment of HOXA9, containing the binding sites of both the wild-type HOXA9 and the mutant HOXA9. It was discovered that the overexpression of miR-184 repressed the luciferase activity of wild-type HOXA9 rather than that of mutant HOXA9, compared to the NC group (Fig. 4f), indicating that HOXA9 was a target of miR-184 in cardiac hypertrophy.
Immediately after the above assay, the 3′-UTR of HOXA9 was co-transfected with miR-184 mimics and pcDNA-UCA1 for the next luciferase reporter analysis. It was shown that the attenuated luciferase activity of HOXA9 induced by miR-184 mimics was slightly recovered by the upregulation of UCA1, whereas no obvious variation occurred in the control group (Fig. 4g). Next, to obtain further affirmation of the effect of UCA1 on HOXA9 in cardiac hypertrophy that had been treated with PE, we weakened the expression of UCA1 to observe the expression response of HOXA9. Consistent with the luciferase activity, the high expression of HOXA9 in hypertrophic cardiomyocytes caused by PE was decreased by the downregulation of UCA1 (Fig. 4h). In general, all of the above phenomena elucidated that UCA1 functioned as a ceRNA to enhance the expression of HOXA9 through binding with miR-184 in cardiac hypertrophy.
Discussion
Cardiac hypertrophy is related to a wide range of pathological and physiological elements, and its growth is closely correlated with the expression of the embryonic genes including ANP and BNP and improved protein synthesis [9, 24]. In addition, it has been reported that lncRNAs and miRNAs are key participants in the nosogenesis of cardiovascular diseases containing cardiac hypertrophy [25]. A series of researches discussing the molecular mechanisms of these lncRNAs and miRNAs have made clear the underlying effects of these molecules on clinical application. And despite lncRNA UCA1 has been studied to be a poor prognosis marker in Chronic Heart Failure [12], and miR-184 was an effective suppressive factor in cyanotic congenital heart diseases [19], the potential mechanism aiming at the biological roles of UCA1 and miR-184 in cardiac hypertrophy is still blurry. In present article, it was demonstrated that UCA1 was highly expressed in the mice heart with hypertrophic cardiomyocytes after the operation of TAC, compared to the sham operation group. And the knockdown of UCA1 dwindled the enlarged cell surface area and cut down the elevated expressions of ANP and BNP even after the treatment of PE in cardiomyocytes, implying the growth-stimulation function of UCA1 in cardiac hypertrophy.
Apart from lncRNAs, miRNAs have been also comprehensively studied to discuss the nosogenesis of cardiac hypertrophy due to their diverse biological functions. A large number of miRNAs have been detected to be dysregulated in hypertrophic cardiomyocytes. Among these miRNAs, some are highly expressed in hypertrophic cardiomyocytes, such as miR-21, miR-208, and miR-499, while others are underexpressed in cardiac hypertrophy, such as miR-1, miR-133, and miR-30 [26, 27]. However, information about miR-184 in cardiac hypertrophy is not available. Here, it was verified that miR-184 was downregulated in PE-induced hypertrophic cardiomyocytes, and the overexpression of miR-184 decreased the enlarged cell surface area and the enhanced mRNA and protein levels of the embryonic genes (ANP/BNP), which had been treated with PE in advance. Moreover, miR-184 was demonstrated to be a direct target of UCA1 in cardiac hypertrophy, and was negatively correlated with UCA1 in the progression of hypertrophic cardiomyocytes.
There are numerous research studies reporting the mechanism of lncRNAs in various diseases, including cardiac hypertrophy, through functioning as a ceRNA. For instance, lncRNA HOTAIR might function as a ceRNA in later-stage gastric cancer [28]; lncRNA H19 acted as a competing endogenous RNA in cardiac fibrosis [29]; HOTAIR played its function in cardiac hypertrophy as a ceRNA [9]. In terms of this paper, it was discovered that mRNA HOXA9 was upregulated in PE-treated cardiomyocytes and the silence of HOXA9 attenuated the expressions of ANP and BNP and reduced the cell surface area, all of which were treated with PE. Highly expressed HOXA9 was determined to be reversely associated with miR-184 in hypertrophic cardiomyocytes, and was a target of miR-184 as proved by the luciferase reporter analysis. However, the overexpression of UCA1 rescued the attenuated luciferase activity of HOXA9 mediated by miR-184 mimics, exhibiting that UCA1 modulated the expression of HOXA9 through competitively binding with miR-184 in cardiac hypertrophy.
In brief, this paper put forward powerful explanations to illustrate the stimulative role of UCA1 in cardiac hypertrophy via boosting the expression of HOXA9 through competitively binding with miR-184. It was expected that such a study could bring some novel insights for the diagnosis and therapy of cardiac hypertrophy.
Disclosure Statement
The authors have no conflicts of interest to disclose.
Acknowledgments
The authors express their appreciation to all members involved in this study.
References
- 1.Liu L, An X, Li Z, Song Y, Li L, Zuo S, Liu N, Yang G, Wang H, Cheng X, Zhang Y, Yang X, Wang J. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc Res. 2016;111:56–65. doi: 10.1093/cvr/cvw078. [DOI] [PubMed] [Google Scholar]
- 2.Creemers EE, Wilde AA, Pinto YM. Heart failure: advances through genomics. Nat Rev Genet. 2011;12:357–362. doi: 10.1038/nrg2983. [DOI] [PubMed] [Google Scholar]
- 3.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 4.Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194:355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116:1462–1476. doi: 10.1161/CIRCRESAHA.116.304937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015;385:812–824. doi: 10.1016/S0140-6736(14)61889-4. [DOI] [PubMed] [Google Scholar]
- 7.Da Sacco L, Baldassarre A, Masotti A. Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis. Int J Mol Sci. 2012;13:97–114. doi: 10.3390/ijms13010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, Liu CY, Sun T, Zhang XJ, Li PF. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 9.Lai Y, He S, Ma L, Lin H, Ren B, Ma J, Zhu X, Zhuang S. HOTAIR functions as a competing endogenous RNA to regulate PTEN expression by inhibiting miR-19 in cardiac hypertrophy. Mol Cell Biochem. 2017;432:179–187. doi: 10.1007/s11010-017-3008-y. [DOI] [PubMed] [Google Scholar]
- 10.Yan Y, Zhang B, Liu N, Qi C, Xiao Y, Tian X, Li T, Liu B. Circulating long noncoding RNA UCA1 as a novel biomarker of acute myocardial infarction. Biomed Res Int. 2016;2016:8079372. doi: 10.1155/2016/8079372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Zhou D, Li G, Ming X, Tu Y, Tian J, Lu H, Yu B. Long non-coding RNA-UCA1 contributes to cardiomyocyte apoptosis by suppression of p27 expression. Cell Physiol Biochem. 2015;35:1986–1998. doi: 10.1159/000374006. [DOI] [PubMed] [Google Scholar]
- 12.Yu X, Zou T, Zou L, Jin J, Xiao F, Yang J. Plasma long noncoding RNA urothelial carcinoma associated 1 predicts poor prognosis in chronic heart failure patients. Med Sci Monit. 2017;23:2226–2231. doi: 10.12659/MSM.904113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diniz GP, Huang ZP, Liu J, Chen J, Ding J, Fonseca RI, Barreto-Chaves ML, Donato J, Jr, Hu X, Wang DZ. Loss of microRNA-22 prevents high-fat diet induced dyslipidemia and increases energy expenditure without affecting cardiac hypertrophy. Clin Sci (Lond) 2017;131:2885–2900. doi: 10.1042/CS20171368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rangrez AY, Hoppe P, Kuhn C, Zille E, Frank J, Frey N, Frank D. MicroRNA miR-301a is a novel cardiac regulator of Cofilin-2. PLoS One. 2017;12 doi: 10.1371/journal.pone.0183901. e0183901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SY, Lee CY, Ham O, Moon JY, Lee J, Seo HH, Shin S, Kim SW, Lee S, Lim S, Hwang KC. microRNA-133a attenuates cardiomyocyte hypertrophy by targeting PKCδa and Gq. Mol Cell Biochem. 2018;439:105–115. doi: 10.1007/s11010-017-3140-8. [DOI] [PubMed] [Google Scholar]
- 16.Wu G, Liu J, Wu Z, Wu X, Yao X. MicroRNA-184 inhibits cell proliferation and metastasis in human colorectal cancer by directly targeting IGF-1R. Oncol Lett. 2017;14:3215–3222. doi: 10.3892/ol.2017.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tung MC, Lin PL, Cheng YW, Wu DW, Yeh SD, Chen CY, Lee H. Reduction of microRNA-184 by E6 oncoprotein confers cisplatin resistance in lung cancer via increasing Bcl-2. Oncotarget. 2016;7:32362–32374. doi: 10.18632/oncotarget.8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin BC, Huang D, Yu CQ, Mou Y, Liu YH, Zhang DW, Shi FJ. MicroRNA-184 modulates doxorubicin resistance in osteosarcoma cells by targeting BCL2L1. Med Sci Monit. 2016;22:1761–1765. doi: 10.12659/MSM.896451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang J, Li X, Li H, Su Z, Wang J, Zhang H. Down-regulation of microRNA-184 contributes to the development of cyanotic congenital heart diseases. Int J Clin Exp Pathol. 2015;8:14221–14227. [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Lin ZQ, Long B, Li JH, Zhou J, Li PF. Cardiac hypertrophy is positively regulated by MicroRNA miR-23a. J Biol Chem. 2012;287:589–599. doi: 10.1074/jbc.M111.266940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue TL, Gu JL, Wang C, Reith AD, Lee JC, Mirabile RC, Kreutz R, Wang Y, Maleeff B, Parsons AA, Ohlstein EH. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]
- 22.Hilfiker-Kleiner D, Hilfiker A, Castellazzi M, Wollert KC, Trautwein C, Schunkert H, Drexler H. JunD attenuates phenylephrine-mediated cardiomyocyte hypertrophy by negatively regulating AP-1 transcriptional activity. Cardiovasc Res. 2006;71:108–117. doi: 10.1016/j.cardiores.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 23.Pirro M, Schillaci G, Menecali C, Bagaglia F, Paltriccia R, Vaudo G, Mannarino MR, Mannarino E. Reduced number of circulating endothelial progenitors and HOXA9 expression in CD34+ cells of hypertensive patients. J Hypertens. 2007;25:2093–2099. doi: 10.1097/HJH.0b013e32828e506d. [DOI] [PubMed] [Google Scholar]
- 24.Jiang F, Zhou X, Huang J. Long Non-Coding RNA-ROR mediates the reprogramming in cardiac hypertrophy. PLoS One. 2016;11 doi: 10.1371/journal.pone.0152767. e0152767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res. 2015;116:751–762. doi: 10.1161/CIRCRESAHA.116.303549. [DOI] [PubMed] [Google Scholar]
- 26.Wang N, Zhou Z, Liao X, Zhang T. Role of microRNAs in cardiac hypertrophy and heart failure. IUBMB Life. 2009;61:566–571. doi: 10.1002/iub.204. [DOI] [PubMed] [Google Scholar]
- 27.Da Costa Martins PA, De Windt LJ. MicroRNAs in control of cardiac hypertrophy. Cardiovasc Res. 2012;93:563–572. doi: 10.1093/cvr/cvs013. [DOI] [PubMed] [Google Scholar]
- 28.Cheng C, Qin Y, Zhi Q, Wang J, Qin C. Knockdown of long non-coding RNA HOTAIR inhibits cisplatin resistance of gastric cancer cells through inhibiting the PI3K/Akt and Wnt/β-catenin signaling pathways by up-regulating miR-34a. Int J Biol Macromol. 2018;107((pt B)):2620–2629. doi: 10.1016/j.ijbiomac.2017.10.154. [DOI] [PubMed] [Google Scholar]
- 29.Huang ZW, Tian LH, Yang B, Guo RM. Long noncoding RNA H19 acts as a competing endogenous RNA to mediate CTGF expression by sponging miR-455 in cardiac fibrosis. DNA Cell Biol. 2017;36:759–766. doi: 10.1089/dna.2017.3799. [DOI] [PubMed] [Google Scholar]