

Abstract
An antibiotic resistance (AR) Dashboard application is being developed regarding the occurrence of antibiotic resistance genes (ARG) and bacteria (ARB) in environmental and clinical settings. The application gathers and geospatially maps AR studies, reported occurrence and antibiograms, which can be downloaded for offline analysis. With the integration of multiple data sets, the database can be used on a regional or global scale to identify hot spots for ARGs and ARB; track and link spread and transmission, quantify environmental or human factors influencing presence and persistence of ARG harboring organisms; differentiate natural ARGs from those distributed via human or animal activity; cluster and compare ARGs connections in different environments and hosts; and identify genes that can be used as proxies to routinely monitor anthropogenic pollution. To initially populate and develop the AR Dashboard, a qPCR ARG array was tested with 30 surface waters, primary influent from three waste water treatment facilities, ten clinical isolates from a regional hospital and data from previously published studies including river, park soil and swine farm samples. Interested users are invited to download a beta version (available on iOS or Android), submit AR information using the application, and provide feedback on current and prospective functionalities.
Keywords: AR dashboard database application, antibiotic resistance gene qPCR array, environmental resistome, mobile genetic elements, mapping genes
An antibiotic resistance Dashboard application is being developed to collect and share data regarding the occurrence of antibiotic resistance genes and bacteria in environmental and clinical settings.
INTRODUCTION
The emergence of antibiotic resistance (AR) in pathogenic bacteria (ARB) is a global problem (WHO 2014). While AR is increasing at an alarming rate (e.g. methicillin-resistant Staphylococcus aureus increased from less than 5% in the 1980s to 60% of cases today, (Cardo et al. 2004)), development of new antimicrobials is occurring at a much slower rate (Wright 2015). Links between humans and the environment can promote emergence, spread and transmission of AR infections. Development and implementation of surveillance tools are among the major goals identified in the National Action Plan for Combating Antibiotic-Resistant Bacteria (The White House, March 2015).
In response, numerous national and international networks are currently focusing on aspects of stewardship and surveillance (Peirano et al. 2014) including: World Health Organization's Global Report on Surveillance (WHO 2014), CDC's National Antimicrobial Resistance Monitoring System (NARMS; FDA 2010), European Antimicrobial Resistance Surveillance Network (EARS-Net; ECDC 2012), Canadian Antimicrobial Resistance Surveillance System (CARSS; PHAC 2015) and Asian Network for Surveillance of Resistant Pathogens (ANSORP; www.ansorp.org). Most of these programs have databases to disseminate and coordinate AR related information primarily focused on health care settings. However, AR associated risk assessment must include tracking AR bacteria and genes (ARG) in the environment (Berendonk et al. 2015). A curated database that combines healthcare and environmental ARB and ARG occurrence could bridge this gap. In addition, the availability of a comprehensive database to help differentiate the natural resistome from anthropogenic distribution of AR in the environment would identify where changes are likely to be most effective for containment, whether through public policies or community actions.
We are developing an AR Dashboard Application for geospatial mapping of ARGs, mobile genetic elements (MGEs) and ARB occurrence in environmental and clinical samples. The term ‘Dashboard’ relates to mapping functionality. The Dashboard App is well-timed with the increasing use of technologies including next generation sequencing (Li et al. 2015), bioinformatics tools to mine metagenomes (McArthur et al. 2013; Gupta et al. 2014; Rowe et al. 2015; Wang et al. 2015), qPCR arrays to survey hundreds of ARGs in parallel (Looft et al. 2012; Wang et al. 2014), the increasing availability of point of use tools to routinely track genetic markers (Stedtfeld et al. 2012; Kostic et al. 2015) and curated sequence database (e.g. Antibiotic Resistance Gene Database, ARDB; Liu and Pop 2009; and Comprehensive Antibiotic Resistance Database, CARD; McArthur et al. 2013; Structured ARG Database, SARG; Yang et al. 2015). The AR Dashboard is being developed to provide two key tools. The first will be in the form of an antibiogram dashboard, and the second for geographically linking information on ARG and ARB occurrence. Antibiograms, a matrix of the percent of infectious agents resistant to specific antimicrobials regionally encountered, enhance awareness of ARB present at a given location, and help clinicians formulate appropriate treatment strategies and inform policy formulation, implementation and evaluation. Our expectation is that by making the AR Dashboard freely available globally, regional antibiograms will be more accessible to all physicians, scientists and policy makers to observe clinical resistance trends at national and international scales. Similarly, mapping AR information geographically will allow for identification of regional hotspots and actions for containment. Together, the AR Dashboard may also provide baseline information necessary to link the environmental spread of AR to routes of transmission.
The AR dashboard is still under development and should be considered at beta stage. This manuscript is to demonstrate current and potential functionalities of the database and also invite interested physicians and researchers to (i) download a beta version of the application (publically available by March 2016, search respective application stores for keywords ‘AR Dashboard’), (ii) submit information using the application and (iii) provide feedback regarding current and future functionalities. Information including antibiograms, studies and ARG/ARB occurrence can currently be added using submission pages available on the app (Fig. 1). Our goal is to have a publically available beta version by March 2016 and a more complete application and database by September 2016. The application is cross platform and can be downloaded for free using respective stores available on iOS, Android or Windows smartphones.
Figure 1.
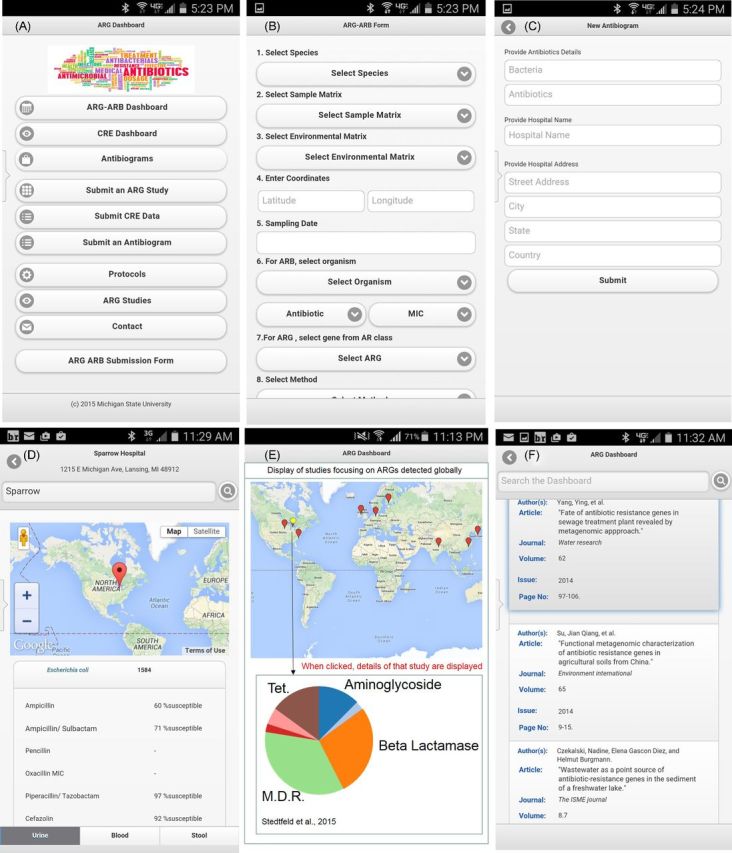
Screenshots of AR Dashboard functions including (A) home screen, (B) submitting ARG and ARB occurrence, (C) submitting antibiograms, (D) mapping results of antibiograms and providing data about antibiotic susceptibility, (E) mapping results of submitted AR occurrence and (F) curated AR studies.
To demonstrate utility, populate and develop the AR Dashboard Database, a qPCR array targeting ARGs and MGEs was tested with multiple samples including (i) 30 environmental surface waters throughout Michigan, (ii) primary influent from three waste water treatment facilities in mid-Michigan and (iii) ten clinical isolates from a regional hospital (Sparrow Hospital, Lansing, MI). Results of these samples were analyzed to demonstrate utility of the downloadable database for identifying ARG and AR hotspots, studying factors that influence levels of ARG in the environment, categorizing natural versus anthropogenic distributed ARGs and track ARB infections. Additional data from swine farm, park soil and river samples (Zhu et al. 2013; Wang et al. 2014; Ouyang et al. 2015) were assembled to demonstrate connections and clusters between sample types.
MATERIALS AND METHODS
AR dashboard application
The AR Dashboard App was developed using a cross platform application program interface (API) technology developed by Appery.io (www.appery.io). The platform allows the use of existing APIs with the ability to incorporate new programs using JavaScript. Additional features can be incorporated using Java, css and HTML5. Integration with Google Maps and potentially other powerful online databases such as Google Charts (for including, sorting and visualizing different levels of environmental factors with presence of AR) or CARD (for linking with sequence information) are an integral part of the Appery.io. Perhaps the greatest functionality of the Appery.io, is that developed applications are readily usable on Android, iOS and Windows operating systems. Thus it can be deployed on most smartphones and personal computers.
Collection and processing of environmental samples
Surface water samples (50 mL) were collected from 28 lakes and two different samples sites from a single river in the state of Michigan. Primary influent samples were also collected from three waste water treatment facilities (East Lansing, Lansing, Jackson, MI, USA). Samples were immediately frozen at −20°C until solid and shipped to the laboratory overnight in insulated containers. Received samples were stored at −20°C prior to processing. Sample processing included filtration, DNA extraction and whole-genome amplification. To concentrate biomass, 3 mL of each sample was passed through a 0.45 μm filter (Millipore, Darmstadt, Germany). DNA was extracted directly from filters using the MoBio PowerWater DNA extraction kit (Mo Bio Laboratories, Carlsbad, USA) following suggested protocols and a final elution volume of 50 μL. DNA extraction yields were measured using a Qubit fluorimeter and the Qubit dsDNA high sensitivity kit.
Community DNA extracted from surface water samples was enriched further using whole-genome amplification (GE Healthcare Life Sciences’ Illustra TempliPhi Little Chalfont, UK). Following the suggested protocol, 1 ng of extracted DNA was added to 5 μL of sample buffer followed by denaturation at 95°C for 3 min and cooling to room temperature. A premix containing 5 μL of reaction buffer and 0.2 μL enzyme mix was added to the DNA buffer mixture. Reaction mix was incubated at 30°C overnight. After incubation, the enzyme was heat-inactivated at 65°C for 10 min. The amplification product was fragmented via sonication and purified using QIAGEN's QIAquick PCR Purification kit. Amplified DNA concentrations were measured using the Qubit fluorimeter and the Qubit dsDNA broad range kit (Life Technologies, Carlsbad, USA). Preliminary experiments were performed to evaluate bias of whole-genome amplification prior to qPCR. A positive correlation of R2 = 0.74 was observed between threshold cycle of samples with and without whole-genome amplification (Fig. S1, Supporting Information).
Collection and processing of clinical isolates
Bacterial isolates (Table S1, Supporting Information) were collected at Sparrow Hospital's Microbiology, Immunology & Molecular Diagnostics Laboratory (Lansing, MI) using standard culture techniques in the Sparrow Clinical Microbiology Laboratory. Identification was performed using Siemens Microscan, BD Phoenix, and/or biochemical tests. Prior to revival, isolates were stored in 15% glycerol stocks at −80°C. Isolates were revived by growing on TSB media overnight at 37°C (no agitation). Genomic DNA was extracted from 1.5 mL of the revived stocks using the Qiagen Blood and Tissue kit (Qiagen, Hilden, Germany), following the protocol for lysing Gram-positive bacteria. DNA concentrations were measured using the Qubit fluorimeter and the Qubit dsDNA broad range kit (Life Technologies, Carlsbad, USA), and samples were run without previous whole-genome amplification.
qPCR array
The qPCR array contained 296 primer sets targeting AR mechanisms in all major classes of antibiotics. All primers sets were described in previous studies (Zhu et al. 2013; Wang et al. 2014). The intI1 gene primer proposed by Gillings and coauthors (Gillings et al. 2015) was also included (run separately on the Chromo4 BioRad (Hercules, USA) using reagents and cycling parameters recommended with the Power SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, USA)). Quantitative PCR reactions with environmental samples were performed using a Wafergen SmartChip Real-time PCR system (Wafergen Biosystems, Inc, Fremont, USA) as reported previously (Wang et al. 2014). Sample and primers were dispensed into the SmartChip using a Multisample Nanodispenser. PCR cycling conditions and initial data processing was performed as previously described (Wang et al. 2014), except that a threshold cycle of 28 was used as the detection limit, based on recommendations from Wafergen. Experiments with clinical isolates were performed on the OpenArray platform (Applied Biosystems, Carlsbad, USA) with reaction conditions and analysis as previously described (Stedtfeld et al. 2008; Looft et al. 2012). The OpenArray had the same primers as the 296 primer array, but contained 40 additional primer sets covering ARGs (these additional primers were not included in analysis). All qPCR reactions were run in triplicate reaction wells.
Data analysis
Estimated genomic copies and relative abundance were calculated as previously described (Looft et al. 2012; Wang et al. 2014). Relative abundance was calculated to normalize for inhibition and variations of bacterial DNA in each sample. The previously described (Looft et al. 2012) equation assumes an efficiency of one and estimates gene abundance. qPCR efficacy has been tested for our method of primer design and the small volume qPCR (Stedtfeld et al. 2008). To verify correct amplification of target genes, amplicons from ∼30 primer sets have been sequenced. Multivariate analysis of ARG profiles between samples was performed in R using the Vegan package (Oksanen et al. 2014). Log2 transformed values of the gene abundances relative to the 16S rRNA gene were used for Redundancy Analysis (RDA), which is a multivariate analysis tool for ecological studies (Oksanen et al. 2014). A heat-map was generated using TMEV (www.tm4.org/mev.html ). Venn diagrams were prepared using R Studio v.3.1.3 (RStudio, Boston, MA, 2012). Figure showing population density with level of ARGs in surface waters was made using Tableau Student Edition (Tableau Software, Seattle, WA). qPCR results from swine farms, park soils and river samples (Zhu et al. 2013; Wang et al. 2014; Ouyang et al. 2015) were included to compare sample type and connections in ARG occurrence. To demonstrate quantitative co-occurrence among various methods of measurement (e.g. qPCR and metagenomics), metagenomics data from two of the five primary influent waste water treatment plant (WWTP) samples (Li et al. 2015) were used for network analysis. Gene co-occurrence and RDA ordination were performed using the summed relative abundance for all primers targeting a given gene. Co-occurrence network analysis, based on Spearman correlation (Williams, Howe and Hofmockel 2014), was rendered using Cytoscape v. 3.3.0 (Institute for Systems Biology, Seattle, USA). Networks were organized by the prefuse force directed openCL layout based on correlation. Gene co-occurrence calls included detection in a majority of samples among a sample type (excluding lake samples in which the cutoff was 25%), false-discovery correction q-value < 0.05, ρ > 0.75 and P-value < 0.005. Node size was discretely mapped with number of connections between primers, and color of edges was continuously mapped with correlation (gray: ρ ∼ 0.75; black: ρ ∼ 1.0).
Ethics statement
Experiments with clinical isolates collected from human samples was conducted following the Human Research Protection Program and performed in accordance with institutional regulations after pertinent review and approval by the Institutional Review Board at Michigan State University (East Lansing, MI). Michigan State University and Sparrow Hospital Investigational Review Board human subjects exemption with MSU IRB# 12–706; i041390 determined to be non-human subject research.
RESULTS AND DISCUSSION
Wireframe and functionality of AR Dashboard
The AR Dashboard is being developed for participation and collaboration in tracking the occurrence, spread and emergence of ARB and ARGs (Fig. 1). Currently, the App provides a central repository, mapping visualization and the ability to submit new information such as studies, antibiograms and occurrence of AR events. It was developed with the emphasis that information can be gathered and made available to researchers and care givers globally.
In detail, users of the AR Dashboard can submit information on AR type, the sample matrix in which the AR was observed, information regarding biological and technical replication, method for detection, latitudinal and longitudinal coordinates of sampling area and the date of sample collection. For submission of multiple results in parallel, users can download a submission form to be filled in a tabular format. Once completed, information is uploaded via the application, and automatically added to the database. Mapping capabilities will allow for an assortment of viewing capabilities based on type of ARG or ARB, location, sampling matrices, sampling date and method used for measurement. The database will be freely available (downloadable within the application), in which users can perform additional offline analysis.
The Dashboard app also provides a mechanism to collect and disseminate clinical antibiograms. Antibiograms are developed and maintained by many well-equipped hospitals, community health organizations and laboratories; as a tool to inform and aid physicians with decisions on antibiotic therapies. Currently, antibiograms are only available locally, however, the AR Dashboard will provide a page for users to access antibiograms globally and submit new antibiograms. Users can simply search for antibiograms based on hospital name, location or by finding with a map. The program will allow for users to sort and graphically visualize antibiogram information based on location of antibiogram, antibiotics and bacteria type. Currently, the application only has two antibiograms from local hospitals in Lansing, MI, but we are inviting researchers and clinicians to use the application to upload additional antibiograms. Antibiograms can be submitted via tabular or graphic formats.
Currently, the application also includes a CRE Dashboard specific to carbapenem resistance Enterobactericea, a global threat that is being tracked by many countries due to current impact on human health. Due to the clinical relevance of this group of organisms, it can be handled independently of all other ARGs and ARB. A current version of the ARG array, not yet described in the literature also focuses on this group of organisms. In the future, this capability can be extended to other resistant pathogens of global concern. The Dashboard App also has a repository of key protocols for screening and tracking ARGs.
Prospective challenges of the AR Dashboard will include scaling-up with continual updates, participation and inconsistencies between measurement methods. Scaling challenges may require the use of additional database utilities. This was the reason for selection of the Appery.io, which can integrate other programs such as Goggle Maps and Charts. Oversight costs are also minimized using the Appery.io, which is a visual programming language that does not require developers to understand coding languages. As such, the Application can be maintained by relatively non-trained personnel.
For samples that require anonymity (e.g. detection of ARGs from a commercial farm or agricultural facilities), results do not need to be mapped with exact GPS coordinates. Submission of these occurrences can solely mention sample matrix and proximal location at a regional city or country level. In some cases, nomenclature of ARGs may not be consistent between reporting events (e.g. ARGs associated with aminoglycoside resistance); however, increased use of structured ARG databases should alleviate these issues (Yang et al. 2015).
Methods for detecting ARGs (e.g. metagenomics, qPCR, hybridization or susceptibility arrays) with varying levels of sensitivity, throughput and target coverage will influence quantitative comparisons between datasets. These variabilities may also cause discrepancies in reported occurrence, however, the database asks users to provide details on the tools used for testing, and different types of quantitative information. Relative abundance and concentrations of detected ARGs can be provided for all techniques to map results together. For example, abundance ratios can be calculated as previously described (Li et al. 2015) for metagnomics results. In detail, length of ARG reads and reference sequences and the number of sequence reads can be used to calculate abundance ratio, which provides results similar to qPCR relative abundance. Only occurrence or presence of ARG/ARB can also be submitted if a given technique is not quantitative. The application is being designed so that users can choose to examine or request occurrence results from varies methods individually or together.
Mapping ARG hotspots and factors promoting distribution and persistence
One utility of the downloadable database is to help identify hotspots and factors contributing to AR occurrence. For samples tested within this study, ARG from nine major antimicrobial classes were detected. Initial analysis of these samples included a RDA of ARG relative abundance (Fig. S2A, Supporting Information). The RDA, which is a multivariate analysis (Oksanen et al. 2014), placed lake and primary influent samples into four groups. The three primary influent samples clustered into group 1, and the surface water samples with varying quantities of ARGs clustered into groups 2–4 (Fig. S2B, Supporting Information). A river sample collected one mile downstream of a waste water treatment facility (GR Walker, Fig. 2A) contained the highest detected number of ARGs (n = 37) and clustered into group 2. The surface water sample with the lowest number of detected ARG (1) clustered into group 4. On average, 161 ARG targeted primers amplified in the primary influent samples (Fig. S2B, Supporting Information), which was 4–20 times higher than the average number of ARGs detected in the environmental waters (groups 2–4).
Figure 2.

Mapping ARG database to identify hotspots and examine occurrence, co-occurrence and persistence of ARGs. (A) Total ARG copies versus population density for all 30 surface water samples, size of dot relates to total number of detected ARG copies, red, blue and green dots indicate samples from group 2, 3 and 4, respectively. Green portions of the map indicate no population information was available. (B) Trophic state of environmental surface waters, based on sample grouping via RDA (Fig. S2, Supporting Information).
As expected, the river sample collected near the effluent line of the waste water treatment facility was a hotspot for ARGs, which has been previously observed (Xu et al. 2015). The level of ARGs detected in the group 4 samples may potentially provide a baseline for background abundance levels in lakes (Czekalski et al. 2015). The elevated level of ARG observed in the other surface water samples (groups 2–3) may be due to a number of factors such as proximity, type and density of agricultural facilities (Pruden, Arabi and Storteboom 2012; Hsu et al. 2014), hospitals (Rodriguez-Mozaz et al. 2015), pharmaceutical and municipal landfills (Wu et al. 2015). Mapping the database of ARG and AR with these facilities may help identify factors potentially contributing to ARG abundance. For example, the environmental water samples were mapped with population density (Fig. 2A). A number of samples with higher levels of ARG were from urban or more densely populated regions, which has previously been described (Ouyang et al. 2015). As mentioned above, the sample with the highest quantity of ARGs were collected from the river in Grand Rapids (MI), which was one mile downstream from the effluent line of the WWTP. However, one of the samples collected from an area with lower human population showed a high level of ARGs (Hubbard Lake, northeast Lower Peninsula) indicating additional influencing factors.
Surface water ARG clusters were examined further using trophic conditions (Fig. 2B) reported by the USGS (not available for four sampled water bodies). A higher percentage (55%) of surface water bodies clustered within group 2 (highest level of ARG) were eutrophic, and a lesser percentage of water bodies were eutrophic in group 3 (33%) and group 4 (27%).
AR Dashboard database to examine natural and anthropogenic distribution, clustering and connections among ARGs
Within the limited set of samples tested to develop the application, inferences can be made regarding detection of ARGs from the natural resistome, and genes that may serve to indicate human fecal contamination in surface water samples. Concerning Michigan surface water and primary influent samples, 190 ARG primers amplified in one or more of the tested samples. A total of 60 primers amplified in one or more of the surface water samples (Fig. S3, Supporting Information), and 178 primers amplified in one or more of the three primary influent samples (Fig. 3A). In detail, 27 primers amplified in both primary influent samples and one or more group 2 surface water samples. Two primer sets (targeting the fox5 and blaOXY genes) appeared in all three primary influent waste water samples, seven or more of the surface water samples, and had a high Pearson correlation (>0.90) to total ARG copies. In addition, neither of these genes were detected in the group 4 surface water samples (with lowest level of ARGs). The fox5 and blaOXY genes are both associated with beta-lactam resistance (Arakawa et al. 1989; Gonzalez et al. 1994) and are typically observed in gram negative enteric organisms (e.g. E. coli and Klebsiella). In addition, the primer targeting the integron-integrase (intI1) gene, had a Pearson correlation of 0.76 compared to total abundance of genomic copies within all tested samples (Fig. 3B). The correlation between total ARGs and the intI1 gene in all water samples indicate potential for quantifying overall ARG abundance. Studies have examined and suggested using the intI1 (Gillings et al. 2015) or other genes (e.g. tetM; Li et al. 2015) as a potential proxy for human pollution due to widespread occurrence, and co-occurrence with other ARGs in environmental, human and animal fecal samples. Confidence in selection of certain ARGs as proxies for human activity can be heightened using the populated AR Dashboard Database. Once selected, markers can be used to routinely monitor environmental samples via point-of-use tools (e.g. Stedtfeld et al. 2012; Kostic et al. 2015; Shu, Zhang and Xing 2015).
Figure 3.

Utility of comprehensive database for differentiating ARGs from natural resistome, fecal contamination and identifying proxies for anthropogenic activity. (A) Number of amplified ARG primers clustered by RDA, group 1 includes primary influent samples, and groups 2–4 are surface water samples with varying levels of ARGs. (B) Correlation between total detected ARG copies (x-axis) and intI1 gene copies (y-axis), gray circles are the three primary influent samples, and black circles are 30 surface water samples.
The database could also be used to differentiate naturally occurring ARGs that would only be expected in unsullied environments, and should not be included in screening or tracking studies. In our sample set, 12 primers (e.g. ermC, tetK) amplified in one or more surface water samples, but were not detected in any of the primary influent samples. These ARG may occur more often in natural environments. In fact, absence of these ARGs may indicate human activity, in which bacteria harboring them are deselected. Nine primer sets (e.g. mexF and oprD genes) amplified in all four clusters also indicating natural occurrence or genes that were less recently distributed. Studies of pristine samples from Antarctic soils also detected the mexF gene in multiple samples (Wang et al., in preparation).
The inclusion of additional sample data sets and quantitative information will also help identify supplemental factors related to presence and persistence of AR including co-occurrence and co-selection of ARGs in different environments and hosts. To demonstrate, qPCR data from previously described studies were assembled including eight park soils (Wang et al. 2014), three river samples (Ouyang et al. 2015) and nine pig farm samples (Zhu et al. 2013). Ordination of relative abundance revealed significant (P = 0.001) clusters related to sample type (Fig. 4A). The cluster spread associated with a given sample type varied, with river samples showing the largest cluster, which may be expected as some of the samples would be considered pristine, while other samples, such as the site downstream from the WWTP effluent had a higher number of ARGs. Pig farm samples also had a wide cluster, which is expected as these samples consisted of manure, compost and soil from three different farms (Zhu et al. 2013).
Figure 4.

Clustering and co-occurrence of ARGs with multiple sample types including previously published data (Zhu et al. 2013; Wang et al. 2014; Ouyang et al. 2015). (A) RDA of ARG Log2 transformed relative abundance. (B) Co-occurrence network with primer names as nodes constructed using Log2 transformed relative abundance values. Nodes connected by an edge have a statistically significant Spearman correlation and are co-occurring (q value < 0.05, and ρ > 0.75). Node color indicates sample type in which a higher majority of connections occur (gray from lake, red from pig farm, green from river, dark blue from primary influent of waste water treatment facility, light blue from park soils). Node size indicates number of connections, and edge color maps level of correlation (darker indicate higher ρ).
Using assembled qPCR data and results observed via metagenomics from the primary influent of two waste water treatment facilities (Li et al. 2015), networks of connected ARGs that co-occur within tested sample types was also observed (Fig. 4B). Co-occurrence was based on a significant Spearman correlation (ρ > 0.75, P-value < 0.005) of ARGs that amplified in 50% or more for a given sample type (excluding a cutoff of 25% for lake samples). The co-occurrence network contained 79 nodes, 302 edges and three modules containing more than four ARGs. Larger nodes within the two largest modules (aadA1 and tetA in one module, and mphA and acrA in the second module) indicate that the occurrence and abundance of these genes may serve as indicators for ARG abundance (Li et al. 2015) within these sample types. Larger acrA and mphA nodes with the second module (with a majority of connections between lake samples) target sequence alleles from E. coli, which is routinely used as indicator of fecal pollution in environmental waters.
Mapping occurrence and abundance over space and time, the database can potentially be used to establish connections and risk considerations among environments, animals and humans. As reviewed previously (Allen et al. 2010; Huijbers et al. 2015), some environments will harbor ARGs (in some instances irrespective of anthropogenic influence due to spread via environment or wild animals), and more studies are necessary to quantity contributions of varying environments to exposure and transmission. It should be mentioned that some ARGs have functional roles that are independent of resistance, such as efflux and electron transport, and will not always be associated with human activity (Dietrich et al. 2008). With culture-based AR analysis accurately identifying sources of host fecal contamination (Park, Lee and Kim 2015), molecular screening of ARGs may also provide a means of source tracking (Li et al. 2015). Thus, the AR Dashboard Database could be used to aid in differentiation of genes for various environmental and clinical screening applications.
Linking regional occurrence of AR infection with ARG
A vital utility of the AR Dashboard Database includes potentially linking occurrence with spread and transmission of AR infections on a regional and global scale via the integration of hospital generated antibiograms, ARGs in the environment, and ARGs in clinical infections. For demonstration, the ARG array was tested with ten clinical isolates. The distribution of AR classes in ARGs detected from isolates and the different surface and waste water samples from Michigan were mapped (Fig. 5). Genes detected with the ARG array were also compared with hospital tested culture based susceptibility (Table S1, Supporting Information), in which the array successfully identified 40 of 52 AR events. The AR events that were not detected are thought to be due to incomplete coverage of ARGs and other resistance mechanisms (e.g. sequence mutations). Molecular based assays of course cannot determine whether that trait is functionally expressed, but larger data sets of ARGs should provide information on population selection which is a complementary route into understanding ARG and ARB ecology and aid improvements in surveillance and stewardship. With global participation, the AR Dashboard could perhaps bridge this gap and help identify links between emergence, spread and transmission of AR pathogen infections.
Figure 5.

Demonstration using database to regionally or globally study AR infection with environmental distribution of ARGs. Map shows distribution of AR classes in ARGs detected from different surface water throughout MI, the primary influent waste water samples, and from 10 isolates collected from patients at a local hospital. MDR: multiple drug resistance, MSLB: macrolide, lincosamide and streptogramin B.
Supplementary Material
Acknowledgments
We would like to thank Dr Tong Zhang and his group for providing ARG relative abundance results from their metagenomics studies (Li et al. 2015).
SUPPLEMENTARY DATA
FUNDING
The research was funded in part by the Pharmaceuticals in the Environment Initiative, the Center for the Health Impacts of Agriculture (CHIA), Michigan State University Clinical Translational Science Institute Seed Grant App ID 224, and the Environmental Protection Agency Great lakes Restoration Initiative (GL-00E01127-0), the Superfund Research Program (2 P42 ES004911-22A1) from the National Institute for Environmental Health Sciences.
Conflict of interest. None declared.
REFERENCES
- Allen HK, Donato J, Wang HH, et al. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol. 2010;8:251–9. doi: 10.1038/nrmicro2312. [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Ohta M, Kido N, et al. Chromosomal beta-lactamase of Klebsiella oxytoca, a new class A enzyme that hydrolyzes broad-spectrum beta-lactam antibiotics. Antimicrob Agents Ch. 1989;33:63–70. doi: 10.1128/aac.33.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendonk TU, Manaia CM, Merlin C, et al. Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol. 2015;13:310–7. doi: 10.1038/nrmicro3439. [DOI] [PubMed] [Google Scholar]
- Cardo D, Horan T, Andrus M, et al. National Nosocomial Infections Surveillance (NNIS) System Report, data summary from January 1992 through June 2004. Natl J Infect Control. 2004;32:470–85. doi: 10.1016/S0196655304005425. [DOI] [PubMed] [Google Scholar]
- Czekalski N, Sigdel R, Birtel J, et al. Does human activity impact the natural antibiotic resistance background? Abundance of antibiotic resistance genes in 21 Swiss lakes. Environ Int. 2015;81:45–55. doi: 10.1016/j.envint.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Dietrich LEP, Teal TK, Price-Whelan A, et al. Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science. 2008;321:1203–6. doi: 10.1126/science.1160619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ECDC. Antimicrobial resistance surveillance in Europe 2012. Annual report of the European Antimicrobial Resistance Surveillance Network (EARS-Net) Stockholm, Sweden: European Centre for Disease Prevention and Control; 2012. [Google Scholar]
- FDA. National Antimicrobial Resistance Monitoring System – enteric bacteria (NARMS): 2010 executive report. Rockville, Maryland: US Food and Drug Administration; 2010. [Google Scholar]
- Gillings M, Gaze W, Pruden A, et al. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 2015;9:1269–79. doi: 10.1038/ismej.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez LM, Perez-Diaz JC, Ayala J, et al. Gene sequence and biochemical characterization of FOX-1 from Klebsiella pneumoniae, a new AmpC-type plasmid-mediated beta-lactamase with two molecular variants. Antimicrob Agents Ch. 1994;38:2150–7. doi: 10.1128/aac.38.9.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SK, Padmanabhan BR, Diene SM, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Ch. 2014;58:212–20. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J-T, Chen C-Y, Young C-W, et al. Prevalence of sulfonamide-resistant bacteria, resistance genes and integron-associated horizontal gene transfer in natural water bodies and soils adjacent to a swine feedlot in northern Taiwan. J Hazard Mater. 2014;277:34–43. doi: 10.1016/j.jhazmat.2014.02.016. [DOI] [PubMed] [Google Scholar]
- Huijbers PM, Blaak H, de Jong MC, et al. Role of the environment in the transmission of antimicrobial resistance to humans: a review. Environ Sci Technol. 2015;49:11993–2004. doi: 10.1021/acs.est.5b02566. [DOI] [PubMed] [Google Scholar]
- Kostic T, Ellis M, Williams MR, et al. Thirty-minute screening of antibiotic resistance genes in bacterial isolates with minimal sample preparation in static self-dispensing 64 and 384 assay cards. Appl Microbiol Biot. 2015;99:7711–72. doi: 10.1007/s00253-015-6774-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Yang Y, Ma L, et al. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015;9:1–13. doi: 10.1038/ismej.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Pop M. ARDB- Antibiotic resistance genes database. Nucleic Acids Res. 2009;37:D443–7. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looft T, Johnson TA, Allen HK, et al. In-feed antibiotic effects on the swine intestinal microbiome. P Natl Acad Sci USA. 2012;109:1691–6. doi: 10.1073/pnas.1120238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AG, Waglechner N, Nizam F, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Ch. 2013;57:3348–57. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet F, Kindt R, et al. Vegan: Community Ecology Package. R package version 3.1.3. 2014. http://CRAN.R-project.org/package=vegan (12 February 2016, date last accessed) [Google Scholar]
- Ouyang W, Huang F, Zhao Y, et al. Increased levels of antibiotic resistance in urban stream of Jiulongjiang River, China. Appl Microbiol Biot. 2015;99:5697–707. doi: 10.1007/s00253-015-6416-5. [DOI] [PubMed] [Google Scholar]
- Park S, Lee S, Kim M. Modified multiple antibiotic resistance analysis for the nonpoint source tracking of fecal pollution. KSCE J Civ Eng. 2015;19:1–7. [Google Scholar]
- Peirano G, Lascols C, Hackel M, et al. Molecular epidemiology of Enterobacteriaceae that produce VIMs and IMPs from the SMART surveillance program. Diagn Micr Infec Dis. 2014;78:277–81. doi: 10.1016/j.diagmicrobio.2013.11.024. [DOI] [PubMed] [Google Scholar]
- PHAC. Canadian Antimicrobial Resistance Surveillance System report 2015. Ottawa, Ontario: Public Health Agency of Canada; 2015. [Google Scholar]
- Pruden A, Arabi M, Storteboom H. Correlation between upstream human activities and riverine antibiotic resistance genes. Environ Sci Technol. 2012;16:11541–9. doi: 10.1021/es302657r. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Mozaz S, Chamorro S, Marti E, et al. Occurrence of antibiotics and antibiotic resistance genes in hospital and urban wastewaters and their impact on the receiving river. Water Res. 2015;69:234–42. doi: 10.1016/j.watres.2014.11.021. [DOI] [PubMed] [Google Scholar]
- Rowe W, Baker KS, Verner-Jeffreys D, et al. Search engine for antimicrobial resistance: a cloud compatible pipeline and web interface for rapidly detecting antimicrobial resistance genes directly from sequence data. PLoS One. 2015;10:e0133492. doi: 10.1371/journal.pone.0133492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu B, Zhang C, Xing D. A handheld flow genetic analysis system (FGAS): towards rapid, sensitive, quantitative, multiplex molecular diagnosis at the point-of-care level. Lab Chip. 2015;15:2597–605. doi: 10.1039/c5lc00139k. [DOI] [PubMed] [Google Scholar]
- Stedtfeld RD, Baushke SW, Tourlousse DM, et al. Development and experimental validation of a predictive threshold cycle equation for quantification of virulence and marker genes by high-throughput nanoliter-volume PCR on the OpenArray platform. Appl Environ Microb. 2008;74:3831–8. doi: 10.1128/AEM.02743-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedtfeld RD, Tourlousse DM, Seyrig G, et al. Gene-Z: a device for point of care genetic testing using a smartphone. Lab Chip. 2012;12:1454–62. doi: 10.1039/c2lc21226a. [DOI] [PubMed] [Google Scholar]
- The White House. National Action action plan for combating antibiotic resistant bacteria. Washington DC: The White House; 2015. [Google Scholar]
- Wang F-H, Qiao M, Su J-Q, et al. High throughput profiling of antibiotic resistance genes in urban park soils with reclaimed water irrigation. Environ Sci Technol. 2014;48:9079–85. doi: 10.1021/es502615e. [DOI] [PubMed] [Google Scholar]
- Wang Q, Fish JA, Gilman M, et al. Xander: employing a novel method for efficient gene-targeted metagenomic assembly. Microbiome. 2015;3:1. doi: 10.1186/s40168-015-0093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Antimicrobial resistance: global report of surveillance. Geneva, Switzerland: World Health Organization; 2014. [Google Scholar]
- Williams RJ, Howe A, Hofmockel KS. Demonstrating microbial co-occurrence pattern analyses within and between ecosystems. Front Microbiol. 2014;5:358. doi: 10.3389/fmicb.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GD. Solving the antibiotic crisis. ACS Infect Dis. 2015;1:80–4. doi: 10.1021/id500052s. [DOI] [PubMed] [Google Scholar]
- Wu D, Huang Z, Yang K, et al. Relationships between antibiotics and antibiotic resistance gene levels in municipal solid waste leachates in Shanghai, China. Environ Sci Technol. 2015;49:4122–8. doi: 10.1021/es506081z. [DOI] [PubMed] [Google Scholar]
- Xu J, Xu Y, Wang H, et al. Occurrence of antibiotics and antibiotic resistance genes in a sewage treatment plant and its effluent-receiving river. Chemosphere. 2015;119:1379–85. doi: 10.1016/j.chemosphere.2014.02.040. [DOI] [PubMed] [Google Scholar]
- Yang Y, Jiang XT, Chai B, et al. ARGs-OAP: online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinformatics. 2015 doi: 10.1093/bioinformatics/btw136. (Submitted) [DOI] [PubMed] [Google Scholar]
- Zhu Y-G, Johnson TA, Su J-Q, et al. Diverse and abundant antibiotic resistance genes in Chinese swine farms. P Natl Acad Sci USA. 2013;110:3435–40. doi: 10.1073/pnas.1222743110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.