

Abstract
Combinatorial post-translational modifications (PTMs), which can serve as dynamic “molecular barcodes”, have been proposed to regulate distinct protein functions. However, studies of combinatorial PTMs on single protein molecules have been hindered by a lack of suitable analytical methods. Here, we describe erasable single-molecule blotting (eSiMBlot) for combinatorial PTM profiling. This assay is performed in a highly multiplexed manner and leverages the benefits of covalent protein immobilization, cyclic probing with different antibodies, and single molecule fluorescence imaging. Especially, facile and efficient covalent immobilization on a surface using Cu-free click chemistry permits multiple rounds (>10) of antibody erasing/reprobing without loss of antigenicity. Moreover, cumulative detection of coregistered multiple data sets for immobilized single-epitope molecules, such as HA peptide, can be used to increase the antibody detection rate. Finally, eSiMBlot enables direct visualization and quantitative profiling of combinatorial PTM codes at the single-molecule level, as we demonstrate by revealing the novel phospho-codes of ligand-induced epidermal growth factor receptor. Thus, eSiMBlot provides an unprecedentedly simple, rapid, and versatile platform for analyzing the vast number of combinatorial PTMs in biological pathways.
Short abstract
An erasable single molecule blot (eSiMBlot) assay provides an unprecedentedly simple and versatile platform for analyzing the combinatorial post-translational modifications in biological pathways.
Introduction
Determining how proteins are regulated to generate diverse protein functions is an important topic of research in the postgenomic era. Protein function is coordinated by various multilayered and interconnected mechanisms, including transcription of new mRNA, alternative RNA splicing, and translation of the mature mRNA into protein.1 Among these diverse regulatory mechanisms, post-translational modifications (PTMs) provide enormous potential for indexing and exponential expansion of the protein repertoire,2 and also have the advantages of being highly dynamic and largely reversible.1 Accumulating evidence suggests that PTMs fine-tune protein functions to provide rapid responses to stimuli without requiring genomic, transcriptomic, or translational regulation.3 Multiple sites of individual proteins can be subjected to a wide range of covalent modifications to orchestrate an integrated response to environmental signals. Thus, combinatorial PTMs (“PTM codes”) such as the histone code can exert distinct biological effects and exponentially expand the diversity of possible proteoforms.4,5 Currently, conventional methods such as western blotting6 and mass spectrometry7,8 are widely used as “gold standards” for PTM studies. However, information regarding combinatorial PTM codes can be concealed by conventional ensemble-averaging measurements, especially when different sites on the same protein are simultaneously modified.5 Consequently, PTM codes contain a wealth of functional information that we are currently unable to access. There are several inherent limitations with the previously reported single-molecule PTM profiling techniques, which detract from their utility.9−12 Among these, the most critical is a low multiplexing capability which is limited to only di-post-translationally modified proteins.
Here, we developed an erasable single-molecule blot (eSiMBlot) assay using a Cu-free click reaction, which allows a single protein to be assayed, and reassayed, multiple times using several different antibodies to reveal PTM codes. This new assay consists of three parts. The first is the stable and robust immobilization of the protein onto a surface using a Cu-free click reaction; the second part is cyclic probing13,14 of the surface various antibodies; the third part is imaging of the bound antibodies with a single-molecule level fluorescence imaging. Since the proteins are stably anchored on the surface by the Cu-free click reaction, the surface can be subjected to multiple cycles of imaging and erasing, using site-specific anti-PTM antibodies in conjunction with single-molecule fluorescence microscopy. The eSiMBlot provides a simple, rapid, and direct method for unravelling the PTM codes of a single protein.
Results
Scheme of eSiMBlot
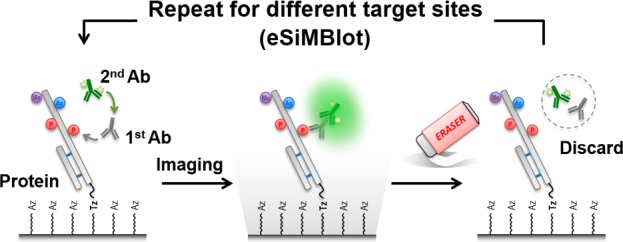
As schematically illustrated in Figure 1a, the eSiMBlot technology consists of three main steps. First, as in the SiMBlot assay, the protein of interest is securely immobilized on the imaging surface (hereafter, termed the single-molecule surface) and probed with a primary antibody specific for a modified site of interest (i.e., a site-specific modification antibody), followed by a fluorescently labeled secondary antibody (Figure 1a, left). In the second step, total internal reflection fluorescence microscopy is used to acquire fluorescence images in separate channels, yielding localization information for the fiducial marker and site-specifically modified proteins probed by immunofluorescence (IF) with a site-specific modification antibody (Figure 1a, middle). In the third step, without any disturbance of the immobilized antigen proteins, IF antibodies are specifically cleared from the single-molecule surface using the erasing buffer, and the immobilized proteins are reinitialized for the next round of IF with a different site-specific modification antibody targeting the same protein (Figure 1a, right). For N sequential cycles, the presence of each site-specific modification can be identified (Figure 1b), yielding N subsets of image data for the same localization-based single-molecule specimen. These data represent the molecule’s combinatorial PTM profile, consisting of N site-specific modifications (Figure 1c). For example, when the probing/imaging procedure is carried out for 10 cycles, 10 modifications can be studied within individual protein molecules. In the case of phosphorylation, this corresponds to a theoretical distribution of 210 = 1024 binary phosphorylation codes.
Figure 1.

Schematic model of erasable single-molecule blotting (eSiMBlot). (a) Post-translational modifications of the immobilized proteins are visualized using total internal reflection fluorescence (TIRF) microscopy, a site-specific anti-PTM antibody, and a fluorophore-labeled secondary antibody. After image acquisition, erasing is performed by removing the probing antibodies and restoring antigenicity for another probing cycle. (b) N images are acquired by sequential repetition of probing for different modifications over N cycles. (c) Following superimposition of N images, combinatorial PTM codes of immobilized proteins are deciphered via localization-based profiling of fluorescence signals.
However, the successful implementation of multicycle probing technology depends heavily on overcoming several key challenges: (i) IF probing must be completely stripped after each imaging cycle to prevent signal carry-over to the next reprobing cycle; (ii) target antigenicity and specimen integrity of the immobilized protein must be retained throughout multiple probing/imaging/erasing cycles; and (iii) the detection rate of the specific antigen by the antibody, which is generally heterogeneous, must be brought up to a reliable level to avoid false negatives. These challenges potentially severely restrict the application of eSiMBlot to visualizing the profile of combinatorial PTM codes and must therefore be carefully considered.
Rapid and Stable Covalent Immobilization Using Cu-free Click Reaction
The key requirements for eSiMBlot are the complete erasing of IF labeling and the stable immobilization of proteins of interest in the single-molecule surface, which must be maintained during the repeated probing and erasing cycles. The antigen–antibody binding is well-known as a high affinity protein–protein interaction. Although Shema et al. reported single-nucleosome modification codes by the repeated probing and washing cycles,12 the passive washing is generally not enough to completely remove the IF antibodies bounded on the antigens (see Supporting Information, Figure S1). To strip the IF antibodies from the single-molecule surface, we applied a low-pH/detergent-based erasing buffer after SiMBlot assay of a model protein such as in vitro autophosphorylated epidermal growth factor receptor (EGFR, a transmembrane receptor protein for the epidermal growth factors (EGFs) family of extracellular ligands)15 with anti-pTyr primary antibody and an Alexa Fluor 555-labeled secondary antibody. As shown in Figure S2 (see Supporting Information), brief exposure to this buffer achieved quick and efficient erasing on the biotin-NeutrAvidin (Bt-NA) single-molecule surface. This efficiency was possible for two reasons. First, IF probing does not involve formation of large precipitates. Second, the immobilized proteins are generally denatured to their unfolded linear forms by exposure to low pH and ionic detergent, allowing more efficient immunolabeling; therefore, protein–protein complexes, including antigen–antibody complexes, can be easily disrupted by exposure to the erasing buffer. Thus, the erasing condition is successfully optimized to completely strip IF labeling.
Next, we tested whether the immobilized proteins could be repeatedly detected by IF relabeling with the same antibodies. The results revealed that signals from repeated IF gradually decreased during multiple probing/erasing cycles (see Supporting Information Figure S3). As shown in Figures S4 and S5 (see Supporting Information), this decrease arose from the loss of immobilized proteins from the Bt-NA single-molecule surface, rather than the surface-coating NA. Therefore, to stably retain immobilized proteins through multiple rounds of erasing and relabeling, it was necessary to develop a new single-molecule surface.
In place of Bt-NA pairing, which has been widely used for biomolecule immobilization on surfaces, we employed a form of copper-free click chemistry, the strain-promoted azide–alkyne cycloaddition (SPAAC) reaction,16 to covalently immobilize a protein of interest in the single-molecule surface17 (Figure 2a). Because Bt-NA pairing is a ligand–protein interaction, it is easily disrupted by exposure to the erasing buffer, resulting in loss of protein function due to denaturation (see Supporting Information Figures S3 and S5). However, the triazole linkage, a stable covalent bond generated by the SPAAC reaction, could provide highly selective and permanent immobilization of a protein of interest in the single-molecule surface. To determine whether the SPAAC reaction is suitable for the eSiMBlot assay system (i.e., for both immobilization of a specific protein on the single-molecule surface and resistance to multiple erasing procedures), we developed a click single-molecule surface (hereafter, termed the click surface) coated with azide-terminal polyethylene glycol to provide a bio-orthogonal reaction with dibenzocyclooctyl (DBCO)-tagged proteins and prevent nonspecific adsorption of antibodies, thereby minimizing false positives18 (Figure 2a). Next, we prepared DBCO-conjugated fluorescent IgG proteins and applied them to the click surface. The results revealed that the SPAAC reaction specifically immobilized DBCO-conjugated proteins on the click surface (see Supporting Information Figure S6 and Figure 2b). In addition, we examined the stability of covalent immobilization on the click surface over multiple cycles of erasing. Fluorescence signals (Nf = 1311 ± 135) from the immobilized proteins on the click surface were stably retained even after 10 exposures to the erasing buffer (see Supporting Information Figure S7). Furthermore, to determine whether the immobilized proteins could be repeatedly detected by probing antibody, we prepared and immobilized the DBCO-conjugated rabbit IgG protein on the click surface and performed the multicycle imaging procedure with fluorescently labeled antirabbit IgG secondary antibody (see Supporting Information Figure S8 and Figure 2c). In contrast to the gradual detachment of immobilized proteins on the Bt-NA single-molecule surface (see Supporting Information Figure S3), the immobilized proteins on the click surface (Nf = 330 ± 28) could be repeatedly and consistently probed with fluorescently labeled secondary antibody (Figure 2d), suggesting that covalent immobilization on the click surface by SPAAC reaction was immune to multiple erasing procedures. Therefore, unlike Bt-NA pairing, SPAAC reaction on the click surface enables stable immobilization of target proteins, and the click surface is well suited for implementation of eSiMBlot.
Figure 2.

Stable and robust protein immobilization on the imaging surface by copper-free click chemistry. (a, b) An azide glass surface is prepared by conjugation of azide-PEG-COOH to an amine-functionalized glass surface using conventional EDC coupling. Both DBCO- and dye-conjugated proteins are immobilized on the click surface by a strain-promoted azide–alkyne cycloaddition reaction. Scale bar, 5 μm. (c) DBCO-conjugated rabbit IgG proteins are immobilized on the click surface. After repetition of erasing (E) and reprobing (P) with Alexa Fluor 488-labeled antirabbit IgG antibody, representative TIRF images of Alexa Fluor 488 signals that remained were detected by immobilized rabbit IgG proteins. Scale bar, 10 μm. (d) Average numbers of fluorescent molecules per imaging area (Nf). Error bars denote standard deviation (n > 8). *P < 0.005, Student’s t-test.
Repetitive Reprobing on a Single Antigen Peptide
Complete removal of IF labeling and antigen regeneration of the immobilized proteins is essential for the success of the eSiMBlot assay system. To carefully validate reprobing ability, we used HA peptide N-terminally conjugated to a DBCO functional group as the single antigen molecule19 (Figure 3a), and DBCO- and Alexa Fluor 488-labeled goat IgG protein as the fiducial marker on the click surface (see Supporting Information Figures S6 and S9). After immobilization of the HA peptide and the fiducial marker, we performed eSiMBlot analysis with anti-HA primary antibody and Alexa Fluor 555-labeled secondary antibody (Figure 3b). In each cycle, the number of Alexa Fluor 555 signals remained constant (Nf = 422 ± 32, Figure 3c). Furthermore, to confirm whether unremoved primary or secondary antibody might persist between probing/erasing rounds, we performed IF labeling with only fluorescently labeled secondary antibody in the final round, and then counted the number of fluorescence molecules. As shown in Figure 3c, a small number of fluorescence signals was detected even after seven rounds of IF labeling of HA peptides. These observations suggested that the eSiMBlot assay eliminated the potential risk of continued occupancy by persistent antibodies or steric hindrance from multicolor IF labeling.
Figure 3.

Repetitive immunolabeling and imaging on a single antigen peptide molecule. (a) DBCO-labeled HA peptides were prepared by conjugation of NHS-PEG4-DBCO to N-terminal amine of HA peptides using a conventional NHS reaction. (b) DBCO-labeled HA peptides and fiducial marker proteins were immobilized on the click surface. Immobilized HA peptides were visualized using anti-HA tag antibody and Alexa Fluor 555-labeled secondary antibodies. Scale bar, 5 μm. (c) Average numbers of fluorescent molecules per imaging area (Nf). (d) Average colocalization ratio of fluorescence signals in images of two different cycles, which were corrected using fiducial markers. (e) Graph shows theoretical or experimental detection coverages using anti-HA antibody and Alexa Fluor 555-labeled secondary antibody, depending on the number of cycles. Error bars denote standard deviation (n > 8).
Next, we superimposed the multicycle fluorescence image data sets for immobilized HA peptides detected by IF labeling, using the data sets for the fiducial marker as a guide. We applied colocalization analysis to two images of multicycle data sets on the same imaging plane. The pairwise colocalization ratio of each image (Rd= P(x|y), where x, y cycles are different) was 27%, suggesting a detection rate of anti-HA antibody (Figure 3d). A low antigen detection rate by an antibody such as anti-HA could severely decrease confidence about the results of the eSiMBlot assay. To address this issue, we designed an experimental strategy of cumulative detection from multiple image data sets, with repetitive reversible labeling of the same antigens. This approach could achieve a higher antigen detection rate, as estimated by theoretical simulation for the cumulative detection rate (Tn= 1 – (1 – Rd)n, where n is the cumulative cycle number; Figure 3e). Surprisingly, we found that the experimental detection with anti-HA antibody following six rounds of probing/erasing reached 75%, with a correlation coefficient (r = 0.999) between experimental and theoretical coverages very close to 1, promising that cumulative detection from multicycle images could compensate for low or heterogeneous antigen detection rates among antibodies. Collectively, these results verified complete removal of antigen-specific IF labeling by the erasing buffer and showed that cumulative detection of a single antigen sequence could increase the antibody detection rate, decreasing the likelihood of false negatives.
Decoding of Multisite Phospho-EGFR
After addressing the key challenges of the eSiMBlot assay, we tested the power of this approach to directly visualize multisite PTM codes in individual protein molecules. For these proof-of-principle experiments, we used in vitro autophosphorylated recombinant EGFR (rEGFR) tagged with enhanced green fluorescence protein (eGFP), SNAP-tag,20 and FLAG antigen sequences,21 isolated by affinity purification with anti-FLAG beads (Figure 4a). EGFR is a distinct subfamily of receptor tyrosine kinases. When extracellular ligands such as EGFs bind to EGFR, autophosphorylations occur on multiple tyrosine residues in the cytoplasmic domain of EGFR to induce signal pathways in cells.15 We anticipated that most tyrosine residues of the purified rEGFR proteins would be fully phosphorylated in in vitro conditions, as confirmed by immunoblotting (see Supporting Information Figure S10). To immobilize rEGFR, a protein of interest, on the click surface, site-specific labeling of the fusion proteins with a DBCO functional group was necessary. Hence, we synthesized a benzylguanine (BG) derivative, BG-PEG13-DBCO, as a substrate of the SNAP-tag, leading to irreversible covalent labeling of the SNAP-tag with DBCO (see Supporting Information Figure S11, Methods).20 After immobilization of rEGFR on the click surface (see Supporting Information Figure S12), we performed the eSiMBlot assay with six different site-specific phospho-antibodies (pTyr845, pTyr992, pTyr998, pTyr1068, pTyr1082, and pTyr1173 of EGFR, Figure 4b) to individually examine each autophosphorylation site of rEGFR. In an analysis of more than 2500 autophosphorylated rEGFR molecules (Nf = 527 ± 22), we quantified each potential combinatorial phosphorylation (total: 26 – 1 = 63 binary codes) in individual rEGFR proteins (Figure 4c). As expected, the distribution of observed molecules over the possible binary phospho-codes (Figure 4d) revealed that the most abundant combinatorial code was fully autophosphorylated rEGFR (18%), whereas the average number of each of the other binary codes was significantly lower (1%). Moreover, the phospho-stoichiometry distribution demonstrated that the in vitro autophosphorylations dramatically reduced the level of the monophosphorylated forms from 95.1% to 22.5%, but increased the level of the multiphosphorylated forms from 4.9% to 77.5% (Figure 4e). Together, these results demonstrate that the eSiMBlot assay enabled direct and quantitative analysis of multisite PTM codes of individual protein molecules, information that had previously been hidden in ensemble results.
Figure 4.

eSiMBlot enables decoding of multiple modifications at single-molecule resolution. (a) Illustration of experimental procedures. After starvation for 24 h, 293T cells expressing SNAP-EGFR-eGFP-flag (rEGFR) were extracted using lysis buffer containing 10 μM AG1478. For in vitro SNAP-tag labeling and autophosphorylation, affinity-purified rEGFR was labeled with 2 μM benzylguanine-derived reagent (BG-PEG13-DBCO), followed by incubation with 100 ng mL–1 EGF and 100 μM ATP for 1 h. Autophosphorylation sites of rEGFR immobilized on the click surface were quantified by the eSiMBlot assay with the indicated antibodies (pY845, pY992, pY998, pY1068, pY1086, and pY1173). (b) Representative eSiMBlot images of Alexa Fluor 555 signals generated by probing site-specific phosphorylation with the indicated primary and fluorescently labeled secondary antibodies in each cycle. Scale bar, 5 μm. (c) Average numbers of fluorescent molecules per imaging area (Nf). (d) Graph shows average numbers of the 63 (26 – 1) combinatorial PTM codes per imaging area (Nf). A computer-generated binary code is placed below the corresponding bar. (e) The phospho-stoichiometry distribution of each categorized combinatorial phospho-code. Error bars denote standard deviation (n > 8).
Ligand-Induced EGFR Phospho-Codes
Next, we examined the ability of the eSiMBlot assay to measure changes in combinatorial PTM codes in response to environmental stimuli. As noted above, we confirmed that the accumulation of tyrosine phosphorylation due to in vitro EGFR autophosphorylations, which are immune to dephosphorylation by phosphatases, would generate a fully phosphorylated form of EGFR (Figure 4). Moreover, Kleiman et al.22 reported that, in response to ligand, EGFR phosphorylations in cells have very short half-lives due to the potent activities of endogenous phosphatases. To determine whether monophosphorylated EGFRs on cells are derived from phosphatases that prevent the accumulation of EGFR phosphorylation, we used the eSiMBlot assay to thoroughly analyze the combinatorial phospho-codes of EGFR in response to treatment of living cells with EGF and/or pervanadate.23 In these experiments, COS7 cells ectopically expressing rEGFR were serum-starved for 12 h. After treating the cells with EGF (100 ng mL–1) and/or pervanadate (1 mM) for 10 min, we isolated rEGFR from crude cell extracts by affinity purification and labeled the SNAP-tag with BG derivatives (BG-PEG13-DBCO), as shown in Figure 5a. Next, the eluates were immobilized onto the single-molecule surface by the SPAAC reaction and probed with anti-pTyr primary antibody and Alexa Fluor 555-labeled secondary antibody (Figure 5b). In addition, we performed the eSiMBlot analysis with multiple site-specific phospho-antibody sets (pThr669, pTyr845, pTyr992, pTyr998, pTyr1068, pTyr1082, and pTyr1173 of EGFR, Figure 5c). Levels of overall tyrosine phosphorylation and each site-specific phosphorylation significantly increased in response to treatment with EGF and/or pervanadate.
Figure 5.

Profiling of ligand-induced EGFR phospho-codes by eSiMBlot assay. (a) Illustration of experimental procedures. After starvation for 24 h, COS7 cells ectopically expressing SNAP-EGFR-eGFP-Flag (rEGFR) were incubated with or without 100 ng mL–1 EGF and/or 1 mM pervanadate for 10 min. Proteins were extracted using lysis buffer containing 10 μM AG1478. rEGFR was isolated from cell lysates by affinity purification and in vitro labeled with 2 μM BG-PEG13-DBCO. The eluates were applied to the click surface. (b) Tyrosine phosphorylations of immobilized rEGFR proteins were probed by a primary antibody against tyrosine phosphorylation and an Alexa Fluor 555-labeled secondary antibody. Graphs show the ratio of pY on rEGFR and fold changes. (c) Immobilized rEGFR proteins were analyzed by the eSiMBlot assay with the indicated primary antibodies (pT669, pY845, pY992, pY998, pY1068, pY1086, and pY1173) and an Alexa555-labeled secondary antibody. Graph shows fold changes of ratios of each site-specific phosphorylation on rEGFR. (d) Phospho-stoichiometry distribution of each categorized combinatorial phospho-code. (e) Fold changes of multiphosphorylation ratios of each site-specific phosphorylation on rEGFR. Error bars denote standard deviation (n > 4). *P < 0.05, Student’s t-test.
Furthermore, we successfully coregistered all seven image data sets with site-specific IF labeling, using the data sets for the fiducial marker as a guide. From the analysis of over 10 000 rEGFR molecules, we could quantify the molecular ratio of 27 – 1 = 127 binary codes, corresponding to the possible combinatorial phosphorylations, in individual phosphorylated rEGFR proteins (see Supporting Information Figure S13). Ligand-induced multiple phosphorylations of receptor tyrosine kinases are widely believed to occur, based on the results of ensemble biochemical assays;24−26 however, our recently published results call into question the prevalent model that EGF induces multiple phosphorylations of EGFR on the cell membrane.11 Remarkably, we observed that the combinatorial PTM distribution of rEGFR molecules was strongly biased toward monophosphorylation codes (NT: 94.4%, EGF: 94.5%, Figure 5d). These results strongly support the idea that EGF-induced EGFR on the cell membrane is rarely multiphosphorylated.
Furthermore, the ratios of multiphosphorylated EGFR forms, which were unchanged by EGF treatment alone, significantly increased from 5.6% to 8.3% or 13.9% of total phosphorylated EGFR molecules upon treatment with pervanadate in the absence or presence of EGF, respectively (Figure 5d). Moreover, all multiphosphorylation ratios of each site-specific phosphorylation of EGFR increased upon treatments with pervanadate, with or without EGF (see Supporting Information Figure S14 and Figure 5e). In particular, cotreatment with EGF and pervanadate significantly increased the multiphosphorylation ratio of pY1173 of EGFR, which recruits SHP1 phosphatase for EGFR dephosphorylation,27 2-fold relative to pervanadate treatment alone. However, most ratios were slightly decreased by EGF treatment alone (Figure 5e), even though the level of each site-specific phosphorylation was elevated (Figure 5c). These outcomes may be due to inhibition of endogenous phosphatase activities, which enables the accumulation of phosphorylation on individual EGFR molecules by preventing dephosphorylation, as in the in vitro environment (Figure 4). Accordingly, in contrast to conventional methods, the detailed analysis of combinatorial phospho-codes of EGFR by eSiMBlot assay suggested that the balance between kinase and phosphatase activities is intimately involved in the control of ligand-dependent combinatorial phosphorylation patterns of EGFR in living cells (see Supporting Information Figure S15). Although we examined only ligand-induced EGFR phospho-codes in our investigation of physiological combinatorial PTMs (Figure 5), we followed routine and well-established biochemical procedures for the sample preparation, e.g., affinity purification and in vitro SNAP-tag labeling. Therefore, we are confident that the eSiMBlot assay is well suited to the study of combinatorial PTM codes in general, allowing unprecedented straightforward visualization of combinatorial site-specific PTMs within individual signaling molecules.
Discussion
We have established a platform technology for proteomic approaches, based on multiplexed single-molecule imaging techniques, for analysis of the combinatorial PTM codes of diverse cellular proteins. The platform relies on the combination of Cu-free click chemistry, cyclic probing with different antibodies and single molecule fluorescence imaging. In some sense, the eSiMBlot technique can be viewed as an extension of serial stripping and reprobing in western blots, but the technical evolution from ensemble to single molecules confers several incomparable advantages, and also enjoys the merits of previous single-molecule isolation techniques such as a rapid, sensitive experimental procedure and the ability to use small amounts of specimen.11,28 First, eSiMBlot can easily and rapidly provide exquisite quantitative data regarding the status of multiple PTMs at a single-molecule level, without any limitations on the number of colors or antibody host species. Second, the erasing process completely eliminates concerns about steric hindrance (also known as epitope occlusion) due to multicolor IF labeling on a single polypeptide, as well as overcoming issues related to the heterogeneous sensitivity of primary antibodies by allowing superimposition of images from repeated rounds of probing. Third, if proper site-specific modification antibodies for a protein of interest are available, the method can provide an unprecedented level of information about combinatorial PTM codes that had previously been concealed in ensemble results. This detailed information on different PTM code states in subpopulations sheds new light on the events and the regulation of intracellular signaling11,12 and may help molecular diagnostics by identifying clinical biomarkers for precision medicine.29,30 Moreover, in combination with microfluidics platforms for single-cell study,31 eSiMBlot could be extended to measurement of cell-to-cell variations in combinatorial PTM codes. Alternatively, in conjunction with automated imaging systems,32 the technique could liberate researchers from the need to perform labor-intensive manual experiments.
PTM codes can modulate protein activity, directional and dynamic protein–protein interactions, or allosteric effects as if they were digital logic gates,33 and these phenomena can in turn affect PTM codes.34 In particular, PTM codes are closely linked with interactome profiles;24,35 however, it remains difficult to determine how PTM codes are related with multiple protein–protein interactions. In addition, the influence of PTM codes on protein functions, including enzyme activity and conformational changes, has barely been elucidated. Because eSiMBlot can double as a preparatory tool for analyzing individual complex proteins at the single-molecule level,28 it could help elucidate these relationships by sequential analysis of protein complexes and PTM codes on the same molecule, which is currently impossible using conventional methods.
Methods
No unexpected or unusually high safety hazards were encountered.
Antibodies and Reagents
Antibodies against EGFR and phospho-EGFR (pThr669, pTyr998, pTyr1068, and pTyr1173) were purchased from Cell Signaling Technology (Cat. #2239, #3056, #2641, #2236, and #4407). Monoclonal anti-pTyr845 EGFR antibody and centrifugal filter unit were purchased from Millipore (Cat. #04-283, #UFC503024). Polyclonal anti-pTyr992 EGFR antibody was purchased from Abcam (Cat. #ab5638). Polyclonal anti-pTyr1086 EGFR antibody, fluorescently labeled secondary antibodies and Lipofectamine were purchased from Invitrogen Life Technology (Cat. #44790G, #A21110, #A11001, #A11034, #A21424, #A21429, and #18324). Recombinant human EGF was purchased from R&D Systems (Cat. #236-EG). Sulfo-NHS-DBCO and NHS-PEG13-DBCO were purchased from Click Chemistry Tools (Cat. #A124 and #1015). Anti-FLAG M2 affinity gel, 3X FLAG peptide, HA peptide, and Phosphatase Inhibitor Cocktails 2&3 were purchased from Sigma-Aldrich (Cat. #A2220, #F4799, #I2149, #5726, and #P0044).
Cell Culture and Transfection
COS7 cells were maintained in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum at 37 °C in a humidified CO2-controlled (5%) incubator. For transfection and transient expression of recombinant proteins, cells were transfected with plasmids encoding rEGFR using Lipofectamine and then cultured for an additional 48 h to achieve ectopic expression of rEGFR.
Plasmids
A plasmid encoding C-terminal Flag-tagged eGFP was constructed by insertion of the corresponding cDNA sequences of eGFP into the XbaI/AgeI sites of the pcDNA 3.1 myc/his vector (Invitrogen). The inserted DNA fragment encoding C-terminally FLAG-tagged eGFP was prepared by PCR using vector pEGFP-C1 (Clontech Laboratory Inc.) as the template and the following primers: forward (XbaI): 5′-GCTCTAGAGGAGGG ATGGTGAGCAAGGGCGAGGAG-3′; reverse (AgeI): 5′-C ACCGGTTCA CTTGTCGTCATCGTCTTTGTAGTC CTTGTACAGCTCGTC-3′. The WT construct was cloned by inserting the N-terminus of the cDNA sequence of full-length EGFR lacking a stop codon into the NotI/XbaI sites of pcDNA3.1 eGFP-FLAG. To insert two restriction sites (AscI and SacII) between the signal peptide (SP) and membrane protein sequence (MP) of EGFR, two DNA fragments encoding the N-terminal SP or C-terminal MP sequences with two restriction sites were prepared by PCR using pcDNA3.1 EGFR WT-eGFP-FLAG as the template and the following primer sets: N-terminal SP sequence, forward: 5′-CGCAAATGGGCGGTAGGCGTG-3′ and reverse (SacII/AscI): 5′-CCGCGGTTGGCGCGCC AGCCCGACTCGCCGGGCAGAG-3′; C-terminal MP sequence, forward (AscI/SacII): 5′-GGCGCGCCAACCGCGG CTGGAGGAAAAGAAAGTTTGC-3′ and reverse (XbaI): 5′-GC TCTAGA TGCTCCAATAAATTCACTGCT-3′. The DNA fragment encoding EGFR WT (AscI/SacII) was prepared by overlapping PCR using the two DNA fragments as the template and the following primers: forward: 5′-CGCAAATGGGCGGTAGGCGTG-3′; reverse: 5′-GC TCTAGA TGCTCCAATAAATTCACTGCT-3′. The construct for EGFR WT-eGFP-FLAG (AscI/SacII) was cloned by inserting the integrated DNA fragment into pcDNA3.1 eGFP-FLAG at the NotI/XbaI sites. In addition, a plasmid encoding N-terminal SNAP-tagged EGFR WT-eGFP-Flag was constructed by insertion of the cDNA sequences encoding the SNAP-tag into the AscI/SacII sites of the modified EGFR WT-eGFP-FLAG construct. The SNAP-tag DNA fragment was prepared by PCR using vector pSNAPf (NEB) as the template and the following primers: forward (AscI): 5′-GGCGCGCCACATCATCACCATCACCAT ATGGACAAAGACTGCGAAATG-3′; reverse (SacII): 5′-TCC CCGCGG CCCTCCACTCCCACT ACCCAGCCCAGGCTTGCCCAG-3′.
Synthesis of BG-PEG13-DBCO
O6-[4-(Aminomethyl)benzyl]guanine (Matrix Scientific, 2.7 mg, 0.01 mmol) was suspended in dry DMF (0.7 mL) under an argon atmosphere, and trimethylamine (10 μL) and DBCO-PEG13-NHS (Click Chemistry Tools, 10 mg, 0.01 mmol) in dry DMF (0.3 mL) were added. After being stirred for 24 h at room temperature, the crude product was purified by HPLC. The purified compound was dried under reduced pressure to yield BG-PEG13-DBCO (5.5 mg, 47%). MS(ESI): m/z calculated for [M + H+]: 1201.6; observed, 1201.6.
Synthesis of DBCO-Conjugated HA Peptide
Influenza hemagglutinin (HA) peptide (Sigma-Aldrich, 0.1 mg, 0.09 μmol) was suspended in dry DMF (0.2 mL) under an argon atmosphere, and trimethylamine (2 μL) and DBCO-PEG4-NHS (Click Chemistry Tools, 1 mg, 1.54 μmol) in dry DMF (0.1 mL) were added. After being stirred for 24 h at room temperature, the crude product was purified by HPLC. The purified compound was dried under reduced pressure to yield DBCO-PEG4-HA peptide (65 μg, 44%). MS(ESI): m/z calculated for [M + H+]: 1636.7; observed, 1636.3.
Preparation of Both DBCO- and Dye-Conjugated Proteins
Alexa Fluor 488-labeled antihamster goat IgG (Invitrogen, 0.2 mg) or bovine serum albumin (Invitrogen, #A13100, 0.2 mg) was suspended in 0.3 mL of PBS. DBCO-PEG4-NHS (Click Chemistry Tools, 0.1 mg) was suspended in PBS (0.2 mL). After 1 h of stirring at room temperature, the crude product was purified by diafiltration using a centrifugal filter (Millipore, 30 kDa MWCO). Samples were subjected to single-molecule imaging or immunoblotting.
Preparation of in Vitro Autophosphorylated EGFR
COS7 cells ectopically expressing rEGFR were serum-starved for 12 h. After labeling of cell surface proteins with Sulfo-NHS-DBCO and preparation cell lysate using lysis buffer containing AG1478 (10 μM), rEGFR was affinity-purified using anti-FLAG M2-conjugated agarose, followed by three washes with lysis buffer (AG1478-free) and two washes with kinase buffer (25 mM HEPES, pH 7.4, 20 mM MgCl2, 5 mM β-glycerophosphate, 0.5 mM dithiothreitol, and 0.1 mM sodium orthovanadate). Immunoprecipitates were eluted from beads with 0.1 mg mL–1 3× FLAG peptide at 4 °C for 30 min. Purified rEGFR was incubated for 1 h at 30 °C with shaking. EGFR was activated by addition of ATP (0.1 mM) and EGF (100 ng mL–1) prepared in kinase buffer. Samples were subjected to single-molecule imaging or immunoblotting.
Preparation of EGFR Proteins from Cells
COS7 cells ectopically expressing rEGFR were serum-starved for 12 h. Cells were treated with a ligand such as EGF at 37 °C for 10 min and washed with ice-cold PBS. Cells were lysed in lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 5% glycerol, 1% NP-40, 10 μM AG1478, and phosphatase inhibitor cocktails) by sonication, and cell lysates were centrifuged at 13 500 rpm at 4 °C for 10 min. From supernatants, rEGFR was affinity-purified using anti-FLAG M2-conjugated agarose, followed by three washes with lysis buffer (AG1478-free) and one wash with PBS. Immunoprecipitates were labeled with BG derivatives (BG-PEG13-DBCO) for 30 min at 30 °C, and then washed four times with ice-cold PBS. The immunoprecipitates were eluted from beads with 0.1 mg mL–1 3× FLAG peptide for 30 min at room temperature. Eluates were subjected to single-molecule imaging or immunoblotting.
Flow Chambers and Single-Molecule Immobilization
Extensively cleaned cover glasses were prepared by washing with H2O and 1 M KOH for 2 h or longer, and then were treated with 3-(2-aminoethylamino)-propyltrimethoxysilane (Tokyo Chemical Industry Co., Cat. #A0774) and doped with azide-PEG (Laysan Bio, Inc., azide-PEG-COOH-5000) or a mixture of mPEG and biotin-PEG (Laysan Bio, Inc., mPEG-SVA-5000 and Biotin-PEG-SVA-5000) at a mass ratio of 25:1 (mPEG:biotin-PEG). In the case of the biotin surface, the flow chambers were coated with NeutrAvidin (Thermo Scientific, Cat. #31000).28
Before immobilization on the single-molecule surface, samples were serially diluted to obtain well-isolated spots on the single-molecule surface after incubation for 10 min. All dilutions were made immediately before experiments with PBS (0.1 mg mL–1 bovine serum albumin). Unbound antibodies and samples were removed from the channels by washing twice with buffer SB18 (40 mM HEPES, 105 mM NaCl, 5 mM KCl, 5 mM MgCl2, 0.05% [vol/vol] Tween-20, and 0.1 mg mL–1 bovine serum albumin, pH 8.0). The channels were blocked with SB18 (1.0 mg mL–1 bovine serum albumin was added). For IF detection, immobilized protein molecules were incubated with a primary antibody (13.3 nM) against the prey protein for 10 min, and with a fluorescent dye-labeled secondary antibody (2.66 nM) for 10 min immediately before imaging. After image acquisition, the previous antibodies were removed from the channels by washing twice with erasing buffer (IgG elution buffer added with 2% SDS and adjusted pH 1.85) and incubating for 5 min. For multiple rounds of relabeling, the flow channels were neutralized by washing three times with PBS, and then blocked with SB18 containing bovine serum albumin (1.0 mg mL–1).
Single-Molecule Imaging Analysis
An objective-type TIRF microscope (Nikon Ti-E, Hamamatsu EM-CCD) was used to acquire single-molecule data.28 eGFP and Alexa Fluor 488 were excited at 488 nm, and Alexa Fluor 555 was excited at 532 nm. Narrow band-pass filters (Chroma) were used to avoid crosstalk between channels. All experiments were performed at room temperature unless otherwise specified. Single-molecule analysis was performed as previously described.11 The mean spot count per image (imaging area, 3000 μm2) and standard deviation were calculated from images of five or more different regions.
Colocalization between different rounds of relabeling was assessed as described previously, with minor modifications.28 Briefly, two separate movies of the same region were taken using Alexa Fluor 488 as a fiducial marker and Alexa Fluor 555 to detect site-specific modification on a single polypeptide molecule. The fluorescent spots in both images were fitted with Gaussian profiles to determine the center positions of the molecules to half-pixel accuracy. Next, the positions of the fluorescent spots between different rounds of relabeling, which drifted on the X–Y stage, were corrected using the positions of the fiducial markers. The fluorescent spots between different rounds of relabeling, whose center was within a distance of three pixels (∼300 nm), were determined as colocalization spots. The ratio of combinatorial PTM codes was calculated as the number of colocalized molecules divided by the total number of fluorescent signals.
Statistical Analysis
All data are presented as means ± standard deviation, or as images representative of at least three sets of independent experiments. When necessary, data were statistically analyzed using Student’s t-test.
Acknowledgments
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. NRF-2015R1A2A1A13001834) and the Institute for Basic Science (IBS) [IBS-R007-D1].
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00114.
Figures for washing and erasing conditions for single-molecule surfaces (NeutrAvidin and Cu-free click surfaces), a synthetic scheme for BG-PEG13-DBCO, schemes and figures for analysis of PTM codes of rEGFR immobilized on a Cu-free click surface using eSiMBlot (PDF)
Author Contributions
K.L.K. conceived this project. K.L.K. designed and conducted all the experiments and prepared the manuscript. K.M.P. suggested the important idea of the imaging surface design and helped to characterize synthetic chemical compounds. K.M.P., J. M., K.K., and S.H.R. edited the manuscript. All authors discussed the results and approved the final version of the manuscript. S.H.R. and K.K. supervised the research.
The authors declare no competing financial interest.
Supplementary Material
References
- Deribe Y. L.; Pawson T.; Dikic I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17 (6), 666. 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- Sims R. J. 3rd; Reinberg D. Is there a code embedded in proteins that is based on post-translational modifications?. Nat. Rev. Mol. Cell Biol. 2008, 9 (10), 815. 10.1038/nrm2502. [DOI] [PubMed] [Google Scholar]
- Minguez P.; Letunic I.; Parca L.; Bork P. PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic Acids Res. 2012, 41 (D1), D306. 10.1093/nar/gks1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. The language of covalent histone modifications. Nature 2000, 403 (6765), 41. 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Thomson M.; Gunawardena J. Unlimited multistability in multisite phosphorylation systems. Nature 2009, 460 (7252), 274. 10.1038/nature08102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Ponten F. Antibody-based proteomics for human tissue profiling. Mol. Cell. Proteomics 2005, 4 (4), 384. 10.1074/mcp.R500009-MCP200. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Mann M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 2013, 12 (12), 3444. 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Wu S.; Stenoien D. L.; Pasa-Tolic L. High-throughput proteomics. Annu. Rev. Anal. Chem. 2014, 7, 427. 10.1146/annurev-anchem-071213-020216. [DOI] [PubMed] [Google Scholar]
- Ohshiro T.; Tsutsui M.; Yokota K.; Furuhashi M.; Taniguchi M.; Kawai T. Detection of post-translational modifications in single peptides using electron tunnelling currents. Nat. Nanotechnol. 2014, 9 (10), 835. 10.1038/nnano.2014.193. [DOI] [PubMed] [Google Scholar]
- Rosen C. B.; Rodriguez-Larrea D.; Bayley H. Single-molecule site-specific detection of protein phosphorylation with a nanopore. Nat. Biotechnol. 2014, 32 (2), 179. 10.1038/nbt.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. L.; Kim D.; Lee S.; Kim S. J.; Noh J. E.; Kim J. H.; Chae Y. C.; Lee J. B.; Ryu S. H. Pairwise detection of site-specific receptor phosphorylations using single-molecule blotting. Nat. Commun. 2016, 7, 11107. 10.1038/ncomms11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema E.; Jones D.; Shoresh N.; Donohue L.; Ram O.; Bernstein B. E. Single-molecule decoding of combinatorially modified nucleosomes. Science 2016, 352 (6286), 717. 10.1126/science.aad7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrazhevskiy P.; Gao X. Quantum dot imaging platform for single-cell molecular profiling. Nat. Commun. 2013, 4, 1619. 10.1038/ncomms2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray E.; Cho J. H.; Goodwin D.; Ku T.; Swaney J.; Kim S. Y.; Choi H.; Park Y. G.; Park J. Y.; Hubbert A.; et al. Simple, Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems. Cell 2015, 163 (6), 1500. 10.1016/j.cell.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N.; Endres N. F.; Engel K.; Deindl S.; Das R.; Lamers M. H.; Wemmer D. E.; Zhang X.; Kuriyan J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009, 137 (7), 1293. 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126 (46), 15046. 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Aleman E. A.; Pedini H. S.; Rueda D. Covalent-bond-based immobilization approaches for single-molecule fluorescence. ChemBioChem 2009, 10 (18), 2862. 10.1002/cbic.200900640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Hohng S.; Ha T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5 (6), 507. 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J.; Nikawa J.; Broek D.; MacDonald B.; Rodgers L.; Wilson I. A.; Lerner R. A.; Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol. 1988, 8 (5), 2159. 10.1128/MCB.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A.; Gendreizig S.; Gronemeyer T.; Pick H.; Vogel H.; Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21 (1), 86. 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Hunter M. R.; Grimsey N. L.; Glass M. Sulfation of the FLAG epitope is affected by co-expression of G protein-coupled receptors in a mammalian cell model. Sci. Rep. 2016, 6, 27316. 10.1038/srep27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman L. B.; Maiwald T.; Conzelmann H.; Lauffenburger D. A.; Sorger P. K. Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell 2011, 43 (5), 723. 10.1016/j.molcel.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G.; Liu S.; Kelly J.; Moffat J.; Payette P.; Kennedy B.; Tsaprailis G.; Gresser M. J.; Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997, 272 (2), 843. 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- Seet B. T.; Dikic I.; Zhou M. M.; Pawson T. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 2006, 7 (7), 473. 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141 (7), 1117. 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtwistle M. R.; Hatakeyama M.; Yumoto N.; Ogunnaike B. A.; Hoek J. B.; Kholodenko B. N. Ligand-dependent responses of the ErbB signaling network: experimental and modeling analyses. Mol. Syst. Biol. 2007, 3, 144. 10.1038/msb4100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyati M. K.; Morgan M. A.; Feng F. Y.; Lawrence T. S. Integration of EGFR inhibitors with radiochemotherapy. Nat. Rev. Cancer 2006, 6 (11), 876. 10.1038/nrc1953. [DOI] [PubMed] [Google Scholar]
- Jain A.; Liu R.; Ramani B.; Arauz E.; Ishitsuka Y.; Ragunathan K.; Park J.; Chen J.; Xiang Y. K.; Ha T. Probing cellular protein complexes using single-molecule pull-down. Nature 2011, 473 (7348), 484. 10.1038/nature10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Guy M. J.; Norman H. S.; Chen Y. C.; Xu Q.; Dong X.; Guner H.; Wang S.; Kohmoto T.; Young K. H.; et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J. Proteome Res. 2011, 10 (9), 4054. 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dazert E.; Colombi M.; Boldanova T.; Moes S.; Adametz D.; Quagliata L.; Roth V.; Terracciano L.; Heim M. H.; Jenoe P.; et al. Quantitative proteomics and phosphoproteomics on serial tumor biopsies from a sorafenib-treated HCC patient. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (5), 1381. 10.1073/pnas.1523434113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y.; Choi P. J.; Li G. W.; Chen H.; Babu M.; Hearn J.; Emili A.; Xie X. S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 2010, 329 (5991), 533. 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert W.; Bonnekoh B.; Pommer A. J.; Philipsen L.; Bockelmann R.; Malykh Y.; Gollnick H.; Friedenberger M.; Bode M.; Dress A. W. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006, 24 (10), 1270. 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- Creixell P.; Linding R. Cells, shared memory and breaking the PTM code. Mol. Syst. Biol. 2012, 8, 598. 10.1038/msb.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivomagi M.; Valk E.; Venta R.; Iofik A.; Lepiku M.; Balog E. R.; Rubin S. M.; Morgan D. O.; Loog M. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 2011, 480 (7375), 128. 10.1038/nature10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P.; Albanese V.; Kenner L. R.; Swaney D. L.; Burlingame A.; Villen J.; Lim W. A.; Fraser J. S.; Frydman J.; Krogan N. J. Systematic functional prioritization of protein posttranslational modifications. Cell 2012, 150 (2), 413. 10.1016/j.cell.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.