

SUMMARY
Glioblastomas (GBM) grow in a rich neurochemical milieu, but the impact of neurochemicals on GBM growth is largely unexplored. We interrogated 680 neurochemical compounds in patient-derived GBM neural stem cells (GNS) to determine the effects on proliferation and survival. Compounds that modulate dopaminergic, serotonergic, and cholinergic signaling pathways selectively affected GNS growth. In particular, dopamine receptor D4 (DRD4) antagonists selectively inhibited GNS growth and promoted differentiation of normal neural stem cells. DRD4 antagonists inhibited the downstream effectors PDGFRβ, ERK1/2, and mTOR and disrupted the autophagy-lysosomal pathway, leading to accumulation of autophagic vacuoles followed by G0/G1 arrest and apoptosis. These results demonstrate a role for neurochemical pathways in governing GBM stem cell proliferation and suggest therapeutic approaches for GBM.
In Brief
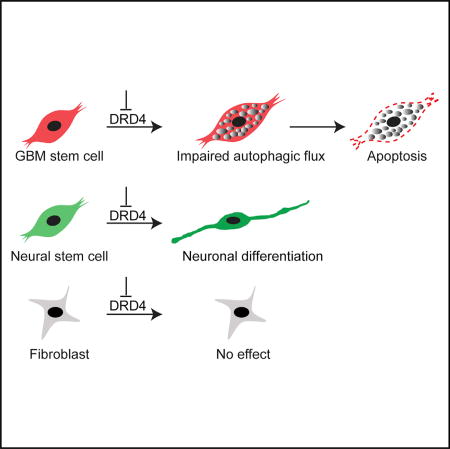
Dolma et al. show that compounds that modulate dopaminergic, serotonergic, and cholinergic signaling pathways selectively affected glioblastoma neural stem cells (GNS). In particular, dopamine receptor D4 antagonists disrupt the autophagy-lysosomal pathway of GNS, leading to growth arrest and apoptosis.
INTRODUCTION
Glioblastoma (GBM) is the most common malignant primary brain tumor in adults and has proved resistant to all therapeutic strategies attempted to date. The alkylating agent temozolomide (TMZ) is the only chemotherapeutic that yields any benefit, but its effects are transient and only in a subset of patients (Brennan et al., 2013; Hegi et al., 2005). Therefore, there is an urgent need for identification of improved therapeutic approaches for the treatment of GBM. A prerequisite to identifying more effective therapeutics is a better understanding of the diversity of mechanisms that govern GBM growth.
GBM growth is initiated and maintained by small subpopulations of tumorigenic cells termed GBM stem cells, which have a phenotype similar to normal neural stem cells (NS) (Galli et al., 2004; Singh et al., 2004). GBM stem cells contribute to tumor progression and resistance to therapy (Bao et al., 2006; Chen et al., 2012), such that long-term disease control is likely to require elimination of this driver cell population, in addition to the more differentiated tumor bulk. GBM stem cells are best prospectively identified from fresh tumors and interrogated directly in vivo, but tumorigenic cells that show similar properties to directly isolated cells (herein called GBM-derived neural stem cells, GNS) can be grown in a defined media allowing tractability for in vitro screening (Pollard et al., 2009). A deeper understanding of the regulatory mechanisms that govern the proliferation and survival of GNS will be essential to developing rational therapies. In a previous unbiased screen of a small-molecule library on mouse NS, we found that neurochemical signaling pathways can affect the proliferation and survival of normal NS populations (Diamandis et al., 2007). This observation raised the intriguing possibility that known neuromodulators might also affect tumorigenic GNS.
Neurotransmitters are endogenous chemical messengers that mediate the synaptic function of differentiated neural cells in the mature CNS. Recent studies suggest an important role of neurochemicals, for example γ-aminobutyric acid (GABA) and glutamate, in regulating NS fate in both early development (Andang et al., 2008; Schlett, 2006) and adult neurogenesis (Berg et al., 2013; Hoglinger et al., 2004; Song et al., 2012). These effects may reflect influences of local or more distant neuronal activity on the NS niche. Consistent with this idea, dopamine afferents project to neurogenic zones and depletion of dopamine decreases the proliferation of progenitor cells in the adult subventricular zone (SVZ) (Hoglinger et al., 2004). Dopamine is also detected during early neuronal development in the lateral ganglionic eminence (LGE), where it is known to modulate LGE progenitor cell proliferation (Ohtani et al., 2003).
Neurochemicals and their receptors have been implicated in the growth and progression of many non-CNS cancers (Dizeyi et al., 2004; Schuller, 2008). The mechanisms whereby neurochemicals affect cancer growth are not well understood, but given that GBM arises in the rich neurochemical milieu of the mature CNS it is plausible that neurochemical pathways may promote GBM growth and tumor progression. Consistent with this proposition, optogenetic manipulation of cortical neuronal activity in a mouse GBM xenograft model can influence GBM growth (Venkatesh et al., 2015). In addition, antidepressants may affect survival of lower-grade models of GBM (Shchors et al., 2015). We hypothesized that a systematic survey of known neuroactive compounds against GNS could reveal regulatory mechanisms and targets outside of traditional chemotherapies for GBM.
RESULTS
Identification of GNS-Selective Compounds
To identify compounds that selectively inhibit the growth of GNS, we established proliferation assays for three different human cell types: GNS, fetal NS, and the BJ fibroblast cell line. GNS are patient-derived tumor cells that display many characteristics of normal NS including expression of the stem cell markers Nestin and SOX2, and the ability to self-renew and partially differentiate (Lee et al., 2006; Pollard et al., 2009). Human NS serve as a well-matched control for their neoplastic GNS counterparts, while fibroblasts were used to eliminate compounds with non-specific cytotoxic effects. We defined NS-selective compounds as those that target both NS and GNS but not fibroblasts, and GNS-selective compounds as those with more activity toward GNS compared with NS.
We screened a library of 680 neuroactive compounds against three human GNS lines, two NS lines, and the BJ line at a concentration of 5 µM for 5 days (Figure 1A). We defined the primary hits as compounds that caused greater than 20% growth inhibition compared with the DMSO control. The total hit rate in all cell populations ranged from 2.6% to 6.5% (Figure 1A). Of all the neurochemical classes, compounds known to modulate dopaminergic (27%), cholinergic (17%), adrenergic (18%), and serotonergic (9%) pathways were enriched in the total hits, suggesting that these pathways may play a specific role in regulating NS growth (Figure 1B). These pathways were also the main enriched hits when normalized to number of hits per total number of compounds in each class (Figure 1C).
Figure 1. Identification of GNS-Selective Compounds.

(A) An outline of the primary and secondary screens. Primary screen data are shown as growth inhibition of each compound (5 µM) across the six cell lines screened. Secondary screens were done in dose series to determine the fold selectivity (IC50 of BJ/IC50 of any NS or GNS with the lowest IC50).
(B) Percentage of different neurochemical classes enriched in the total hits.
(C) Percent activity (hits) of each neurochemical class. Number of hits/total number of compounds present in the library of each class.
(D) The ten NS-selective compounds and their IC50 (µM) across different cell lines. The ten NS-selective compounds are grouped under their neurochemical classes.
See also Tables S1 and S2.
We chose 29 compounds that showed a selective effect on GNS and NS compared with fibroblasts and then retested each in a dose-response series (0.39–50 µM) in the same cell lines as in the primary screen (Table S1). From this secondary screen, we selected ten compounds that showed more than 8-fold selectivity toward GNS and NS compared with fibroblasts. These ten NS-selective compounds were PNU-96415E, L-741,742, ifenprodil tartrate, LY-165,163, MDL-72222, tropanyl 3,5-dimethylbenzoate, N,N-diethyl-2-(4-(phenylmethyl)phenoxy) ethanamine, (±)-tropanyl-2-(4-chlorophenoxy)butanoate, MG-624, and ivermectin (Figure 1D and Table S2). The ten NS-selective compounds were enriched for dopaminergic, serotonergic, and cholinergic classes (Figure 1D), suggesting these pathways as potential targets for GBM. To further validate selectivity, we tested each compound in three further non-NS control cell lines: Daoy (human medulloblastoma), U-2 OS (human osteosarcoma), and C8-D1A (mouse astrocytes). The ten compounds were 8- to 128-fold more active against NS or GNS compared with BJ or other non-NS controls (Table S2). Notably, PNU 96415E, L-741,742, and ifenprodil tartrate showed 8-fold selectivity against GNS compared with NS (Table S2) and were termed GNS-selective compounds. Two of these compounds, PNU 96145E and L-741,742, represent DRD4 antagonists and were chosen for further investigation.
DRD4 Antagonists Selectively Inhibit GNS Growth and Reduce Clonogenic Potential of Primary GBM Tumor Cells
We next retested PNU 96145E and L-741,742 along with other commercially available DRD4 antagonists (L-745,870 and PD 168568) for effects on a larger panel of six GNS and four NS lines. All of the DRD4 antagonists showed selectivity toward GNS with differing potency (IC50), in the order L-741,742 (1.5–6.2 µM) > L-745,870 (3.1–6.2 µM) > PNU 96415E (6.25 µM) > PD 168568 (25–50 µM). L-741,742 and PNU 96415E are both specific DRD4 antagonists and showed the greatest selectivity toward GNS (Figures 2A and 2B). PNU 96415E displayed robust selectivity toward GNS compared with NS and non-NS control cells, the latter of which were not sensitive even at the highest concentration tested (50 µM) (Figure 2A). L-741,742 was the most potent GNS inhibitor but showed variable effects on different GNS lines (Figure 2B). L-745,870 exhibited selectivity toward GNS but was less potent, while PD 168568 showed a modest selective effect at higher concentrations (Figures S1A and S1B).
Figure 2. DRD4 Antagonists Are GNS Selective and Show Synergistic Effect with TMZ.

(A) Percent cell viability of four non-NS control cell lines, three NS, and six GNS lines upon treatment with PNU 96415E in dilution series from 0.39 µM to 50 µM. Controls, n = 3, mean ± SEM; NS lines, n = 5–15, mean ± SEM; GNS lines, n = 3–12, mean ± SEM.
(B) Percent cell viability of BJ, three NS, and five GNS lines upon treatment with L-741,742 in dilution series from 0.39 µM to 50 µM. BJ, n = 3, mean ± SEM; NS lines, n = 3–11, mean ± SEM; GNS lines, n = 3–7, mean ± SEM.
(C) Linear regression plot of in vitro LDA for freshly dissociated patient tumor (GBM 686) treated with L-741,742 (10 µM), PNU 96415E (25 µM), and DMSO. Representative phase-contrast image of neurospheres at day 14 in a well seeded with 2,000 cells. Scale bars, 100 µm.
(D) Immunofluorescence staining for VGlut1, βIII-tubulin, and DAPI in NS (hf5205 and hf6539) differentiated in DMSO and L-741,742 for 3 weeks. Scale bars, 100 µm.
(E) Combination index plot for TMZ with L-741,742 or PNU 96415E in GNS. Combination index (CI) plotted against fractions affected (Fa) analyzed using COMPUSYN.
See also Figure S1.
To confirm that the effect of DRD4 antagonism was not merely specific to established GNS lines, we tested L-741,742 and PNU 96415E in freshly isolated GBM patient tumor cells using a primary in vitro limiting dilution assay (LDA), which measures stem cell potential from fresh tumors. We observed a massive reduction in the frequency of colony-forming cells after treatment with L-741,742 (40- to 83-fold reduction) and PNU 96415E (19- to 29-fold reduction) (Figures 2C, S1C, and S1D). These data strongly suggest that both L-741,742 and PNU 96415E inhibit the clonogenic potential of fresh primary tumor cells, and may therefore effectively target the stem cell population in each patient tumor.
DRD4 Antagonist Promotes Neuronal Differentiation in Normal NS
DRD4 antagonism in normal NS showed a mild effect on proliferation (Figures 2A and 2B) without any sign of apoptosis. However, we noted a striking change in cell morphology in treated cells, in particular striking elongated processes arranged in parallel fashion (Figure S1E). These observations led us to test whether DRD4 antagonism promotes neuronal differentiation in a non-neoplastic context. To this end, we treated two normal human NS lines (hf5205 and hf6539) with L-741,742 followed by sequential withdrawal of epidermal growth factor and fibroblast growth factor. The DRD4 antagonist dramatically enhanced neuronal differentiation of NS, characterized by the expression of VGlut1, a marker for mature glutamatergic neurons (Figure 2D).
DRD4 Antagonists Are Synergistic with TMZ
We next evaluated the effect of DRD4 antagonists in conjunction with the commonly used chemotherapeutic agent TMZ to assess the potential synergism of this drug pair combination. L-741,742 and PNU 9641E exhibited striking synergism with TMZ against GNS in vitro (Figure S1F). We quantified the degree of synergism using the combination index (CI) method (Chou, 2010), for which a CI value of 1 indicates an additive effect, a value of <1 indicates synergism, and a value >1 indicates antagonism. The lowest CI value for L-741,742 in combination with TMZ in G481 and G362 was 0.28 and 0.29, respectively, and for PNU 96415E in combination with TMZ was 0.32 and 0.56, respectively (Figure 2E). These in vitro data suggested that both DRD4 antagonists may enhance the therapeutic efficacy of TMZ in patients.
Primary GBM Tumor and GNS Express Functional DRD4 Receptor, with Higher Expression Linked to a Worse Prognosis
To determine whether the DRD4 antagonists exerted their effects directly through the DRD4 receptor, we first confirmed that DRD4 was expressed in both GNS and NS by western blot (Figure 3A). We further confirmed that patient GBM tissue samples also expressed DRD4 at varying levels by western blot (Figure 3B) and immunohistochemistry (Figures 3C and S2A). To assess DRD4 function in GNS, we measured a known downstream readout of receptor activity. DRD4 is a dopamine D2-like receptor that inhibits adenylate cyclase and decreases cyclic AMP (cAMP) levels (Rondou et al., 2010). We treated GNS with forskolin to activate adenylate cyclase and then assessed whether activation of the DRD4 receptor by the DRD4- specific agonist A412997 could block the forskolin-induced cAMP response. Forskolin treatment in GNS increased cAMP concentration by 2.1-fold and pretreatment with DRD4 agonist A412997 blocked this response by 31.3%, confirming that DRD4 elicits a canonical response in GNS (Figure 3D). Primary tumor and tumor-derived GNS thus express DRD4 and can respond to DRD4-dependent signals.
Figure 3. Primary GBM and GNS Express Functional DRD4 Receptor.

(A and B) Western blot analysis for anti-DRD4 and anti-β actin across different NS and GNS lines (A) and primary GBM patient tumor samples (B).
(C) Immunohistochemistry staining for DRD4 in patient tumor sample (GBM 742). Scale bars, 100 µm.
(D) Fold change of cAMP levels in G362 cells treated with forskolin (30 µM) alone and pretreatment with DRD4-specific agonist A412997 (30 µM) followed by forskolin treatment. n = 3, mean ± SEM.
(E) Western blot analysis for anti-DRD4 and anti-β actin in G362 cells 72 hr after transiently transfected with sh-DRD4 and sh-eGFP.
(F) Cell viability assay (Alamar blue) for G362 cells transiently transfected with sh-DRD4 and sh-eGFP measured over 5 days. n = 3, mean ± SEM. *p < 0.005, **p < 0.0005, unpaired one-tailed t test.
(G) Percent growth inhibition of G362 cells transiently transfected with sh-DRD4 and sh-eGFP treated with L-741,742 for 3 days. n = 3, mean ± SD.
See also Figure S2.
To probe the possible clinical relevance of DRD4 expression, we analyzed The Cancer Genome Atlas (TCGA) data on GBM gene expression and found that patients having tumors with high DRD4 expression have worse survival than those with low DRD4 expression (Figures S2B–S2D). A similar pattern was seen with TH (tyrosine hydroxylase) expression (Figure S2E), the rate-limiting enzyme for dopamine synthesis. Furthermore, the DRD4 gene was not methylated in GBM samples (Figure S2F), consistent with active expression in the primary tumor.
Loss of DRD4 Function Suppresses GNS Growth
To validate DRD4 as a therapeutic target in GBM and determine whether loss of its function phenocopies the effect of PNU 96415E and L-741,742, we performed short hairpin RNA (shRNA)-mediated knockdown experiments and measured the effect on cell proliferation. We tested five lentiviral sh-DRD4 constructs, out of which only one (sh-DRD4-4) caused consistent knockdown at 72 hr after transfection (Figure S2G). We confirmed reduced DRD4 expression after transduction of the sh-DRD4 construct in two separate GNS lines (Figures 3E and S2H). This knockdown was accompanied by a significant reduction in proliferation compared with control sh-eGFP transfected cells (Figures 3F and S2I). Importantly, the residual growth of DRD4 knockdown cells was less sensitive to DRD4 antagonists (L-741,742) than control sh-eGFP transfected cells, consistent with an on-target effect of the DRD4 antagonists (Figure 3G). These results confirm the inferred role for DRD4 function in GNS growth.
Effect of DRD4 Antagonism on Global Gene-Expression Patterns
To determine the potential mechanism of action of DRD4 antagonists on GNS proliferation and survival, we determined global gene-expression profiles with and without DRD4 inhibition. Two GNS lines (G362 and G411) were treated with PNU 96415E (25 µM) for 24 and 48 hr and analyzed for the effect of PNU 96415E on gene expression. Gene set enrichment analysis was used to identify pathways enriched in differentially regulated genes upon PNU 96415E treatment. Genes that were downregulated at 48 hr were enriched in 172 gene sets that are highly connected, as categorized into 25 main biological functions including DNA replication, chromatin remodeling, DNA repair, cell cycle, and RNA splicing (Figure 4A). Overlap of the top downregulated genes (fold change < −1.5) in both G362 and G411 cell lines revealed genes involved in DNA replication and cell-cycle phase transitions (Figure S3A). For genes upregulated upon PNU 96415E treatment, we observed enrichment in 45 gene sets (false discovery rate [FDR] ≤ 0.002) that comprised 14 main pathways including lipid/cholesterol biosynthesis, autophagic vacuoles, and lysosomes (Figure 4A). Overlap of the top upregulated genes (fold change >1.5) uncovered pathways involved in cholesterol biosynthesis after 24 hr and autophagic vacuole formation after 48 hr (Figure S3A). This expression analysis suggested that the genes involved in DNA replication and cell-cycle progression were inhibited by DRD4 antagonism, while genes involved in lipid metabolism and autophagy were activated.
Figure 4. Effect of DRD4 Antagonists on Global Gene Expression.

(A) Gene set enrichment map of pathways containing genes downregulated (blue) or upregulated (red) upon PNU 96415E treatment. Colored circles (nodes) represent gene sets (pathways) that were significantly enriched in the comparison treated versus control samples (FDR ≤ 0.002).
(B) Western blot analysis for anti-LC3B and anti-β-actin in G411 and G362 cells treated with PNU 96415E (25 µM) and L-741,742 (10 µM) at indicated time points.
(C) Immunofluorescence staining for LC3B+ puncta in G362 and G411 cells treated with PNU 96415E (25 µM) and L-741,742 (10 µM) at 48 hr. Scale bars, 17 µm. Quantification of LC3B+ cells in each group (cells counted >200 cells). n = 3, mean ± SEM, unpaired one-tailed t test.
(D) Transmission electron microscopy images showing large autophagic vacuoles in G362 and G411 cells treated with L-741,742 (10 µM) and PNU 96415E (25 µM) compared with control DMSO at 48 hr. Arrows indicate enlarged autophagic vacuoles. Scale bars, 100 nm.
(E) Filipin staining for free cholesterol in G362 and G411 cells treated with L-741,742 (10 µM) and PNU 96415E (25 µM) compared with control DMSO at 48 hr. Scale bars, 11 µm.
See also Figure S3.
DRD4 Antagonism Causes Massive Accumulation of Autophagic Vacuoles and Cholesterol
Prompted by the pronounced upregulation of autophagy genes in response to DRD4 inhibition, we assessed autophagy status in GNS. Conversion of LC3-I (microtubule-associated protein 1 light chain 3) to LC3-II serves as a hallmark for autophagosome formation. L-741,742 (10 µM) and PNU 96415E (25 µM) treatment of GNS (G411 and G362) caused an increase in levels of LC3B-II consistent with accumulation of autophagosomes (Figure 4B). We also observed increased LC3B+ puncta in GNS upon treatment, with more than 50% cells showing large LC3B+ puncta after 48 hr, indicating the presence of autophagosomes (Figures 4C and S3B). Accumulation of autophagosomes was further corroborated by transmission electron microscopy. L-741,742 and PNU 96415E treatment in both G411 and G362 caused the formation of large autophagic vacuoles containing various cellular fragments (Figures 4D and S3C).
As genes involved in cholesterol biosynthesis were also upregulated upon DRD4 inhibition, and since cholesterol accumulation is associated with autophagy impairment in Niemann-Pick type C disease (Vance, 2006), we next analyzed the cholesterol level in GNS with filipin, an antibiotic polyene that fluoresces upon interaction with cholesterol. Upon treatment with L-741,742 (10 µM) and PNU 96415E (25 µM), GNS showed accumulation of cholesterol in large puncta, compared with the diffuse pattern observed in control cells (Figure 4E). Together, these experiments revealed a massive accumulation of autophagic vacuoles and cholesterol in GNS after DRD4 receptor antagonism.
To test whether autophagosome formation is specific to GNS, we assessed autophagosome formation by analyzing LC3-II levels of both fibroblasts and NS after treatment. While DRD4 antagonism at 48 hr did cause a slight increase in LC3-II level in both cases, this occurred to a much lesser extent compared with GNS (Figure S3D). We also note that fibroblast cells have a much higher basal level of autophagy under normal growth conditions compared with GNS (Figure S3E), which may explain the differential vulnerability and dependence on autophagy for survival.
Autophagosome Accumulation Is Due to an Inhibition in Autophagic Flux and Disruption in Lysosomal Function
An increase in LC3-II levels and autophagosome number can result from either the induction of autophagy or the inhibition of autophagic flux at a later stage in the pathway. Flux can be measured by assessing LC3-II turnover in the presence or absence of inhibitors of lysosomal degradation such as chloroquine. In chloroquine-treated cells an autophagy inducer will increase LC3-II levels, whereas an autophagy blocker will not change LC3-II levels. In the presence of chloroquine, L-741,742 and PNU 96415E treatment did not increase LC3-II levels compared with control, despite the fact that both drugs increased LC3-II levels when administered alone (Figures 5A and S4A). These data suggest that the effect of DRD4 antagonism on LC3-II levels is a result of impaired flux.
Figure 5. DRD4 Antagonism Impairs Autophagy-Lysosomal Degradation Pathway in GNS.

(A) Western blot analysis for anti-LC3B, anti-p62, and anti-β-actin in G411 cells treated with L-741,742 (10 µM) or PNU 96415E (25 µM) in the presence and absence of chloroquine (30 µM) at 48 hr. Western quantification for LC3B-II was done using β-actin as control. n = 3, mean ± SEM.
(B) Western blot analysis for corresponding anti-LC3B, anti-p62, anti-LAMP1, anti-mono- and polyubiquitin (Ub) protein conjugates, and anti-β-actin in G411 cells treated with L-741,742 (10 µM) and PNU 96415E (25 µM) at indicated time points.
(C) Confocal analysis of G411 cells expressing tandem mRFP-GFP-LC3 reporter treated with rapamycin (500 nM), chloroquine (30 µM), L-741,742 (10 µM), and PNU 96415E (25 µM) at 48 hr, and quantification of ratio of red puncta indicating autolysosome (AL) versus yellow puncta indicating autophagosome (AP). n = 3, mean ± SEM. **p = 0.0006 and p = 0.0003 in L-741,742 and PNU 96415E, respectively. Scale bars, 10 µm.
(D) Confocal analysis of fibroblast (BJ) and NS (hf5205) expressing tandem mRFP-GFP-LC3 reporter treated with L-741,742 (10 µM) and PNU 96415E (25 µM) at 48 hr. Scale bars, 7 µm.
(E) Western blot analysis for corresponding anti-DRD4, anti-LC3B, anti-p62, anti-mono- and polyubiquitin (Ub) protein conjugate, and anti-β-actin in transient transfected sh-DRD4 and sh-eGFP GNS after 72 hr.
(F) Confocal analysis for red puncta indicating dequenched BSA in GNS (G411) NS (hf5205) and fibroblast (BJ) treated with L-741,742 (10 µM) and PNU 96415E (25 µM) at 48 hr. Quantification of red puncta per cell in each treatment. n = 3, mean ± SEM, unpaired one-tailed t test. Scale bars, 13 µm.
See also Figure S4.
We then assessed autophagy turnover, first by assaying for the autophagy-specific substrate p62. As predicted for a block in autophagic flux, p62 accumulated along with the increase in LC3B-II in L-741,742- or PNU 96415E-treated GNS (Figures 5B and S4B). Consistent with an impairment in autophagy, we also observed an increase in undegraded ubiquitinated protein conjugates in treated cells (Figures 5B and S4B) and further noted an increased level of LAMP 1 (lysosomal-associated membrane protein 1) (Figures 5B and S4B) and LysoID reactivity (Figure S4C), both of which indicate an increase in lysosome accumulation due to impaired autophagic flux.
We further validated flux inhibition by performing an image-based colocalization analysis of a tandem mRFP-GFP-LC3 construct (Kimura et al., 2007). In this experiment, GFP fluorescence is quenched by the acidic pH within a lysosome, allowing differentiation between autophagosomes (GFP-positive and RFP-positive: yellow) and autolysosomes (GFP-negative and RFP-positive: red). Treatment of transfected GNS with DRD4 antagonists showed an accumulation of yellow puncta, indicating autophagosome accumulation (Figure 5C). As a negative control we used rapamycin, an autophagy inducer, which showed a higher ratio of red puncta, and as a positive control we used chloroquine, which led to a higher ratio of yellow puncta (Figure 5C). We also noted that the ratio of yellow versus red puncta at 48 hr (Figure 5C) increased from that seen at 24 hr after treatment (Figure S4D), suggesting an inhibition in autophagic flux over time during DRD4 antagonism. To validate the specificity to GNS, we further monitored flux inhibition in fibroblasts and NS with the same assay. Interestingly, we did not see a dramatic increase in yellow puncta upon DRD4 antagonist treatment in either case, suggesting no massive accumulation of autophagosomes in these cell types (Figure 5D).
To confirm that the impairment of the autophagy-lysosomal degradation pathway induced by L-741,742 and PNU 96415E was mediated through DRD4, we assessed autophagic flux after shRNA knockdown of DRD4 in GNS. We observed increased levels of LC3-II in DRD4 knockdown cells compared with sheGFP transduced controls (Figures 5E and S4E). This increase in LC3-II was accompanied by accumulation of p62 and ubiquitinated protein conjugates, all consistent with a block in autophagic flux (Figures 5E and S4E). We further validated this effect with another short hairpin from different source (Origene) and noted similar results (Figure S4F). Taken together, these data strongly suggest that the cytotoxicity observed after DRD4 antagonism in GNS is due to a block at a later stage of autophagy that results in massive accumulation of autophagic vacuoles.
To further understand the inhibition in autophagic flux, we assessed lysosomal function using DQ BSA, a derivative of BSA that is labeled with a self-quenched red fluorescent dye. Upon proteolysis by lysosomal proteases, the fluorescence is dequenched and can be detected by microscopy. Compared with control NS and fibroblasts, GNS treated with DRD4 antagonists displayed a reduction in red puncta per cell, suggesting very low dequenching of the fluorescent BSA due to compromised lysosomal function (Figure 5F). These data demonstrated that autophagic flux impairment in GNS treated with DRD4 antagonists was due to an inhibition in lysosomal function.
DRD4 Antagonism Causes a Disruption in PDGFRβ-ERK1/2 and mTOR Signaling
To determine the possible mechanism whereby DRD4 receptor antagonism in GNS causes the striking cellular effects observed, we studied the phosphorylation status of 43 kinases and substrates implicated in various signaling pathways in GNS versus NS using a dot blot assay. Cells were treated with L-741,742 (10 µM) and PNU 96145E (25 µM) for a period of 24 hr, and protein lysates were harvested and assessed with a phosphoprotein antibody array (Figures 6A and S5A). We identified 18 phosphoproteins that exhibited a decrease in phosphorylation upon treatment in GNS (Figures 6B and S5B). ERK1/2 was one of the top hits in the array, with a 40% reduction compared with the untreated control. DRD4 is known to activate ERK1/2 by transactivation of platelet-derived growth factor receptor β (PDGFRβ) (Gill et al., 2010; Oak et al., 2001). The selectivity of DRD4 antagonism to GNS was reflected in the much more modest changes in the phospho-profile of NS after treatment (Figure S5A).
Figure 6. Phosphokinase Array Reveals Suppression of ERK1/2 and mTOR Pathway Upon DRD4 Antagonism.

(A) Dot blot containing 43 phosphoproteins in duplicates after exposure to lysate of G362 cells treated with L-741,742 (10 µM) and PNU 96415E (25 µM) and DMSO for 24 hr. Red arrow indicates one of a paired spot for ERK1/2.
(B) Signal intensity of each spot corresponding to each phosphoprotein (average of two spots) in (A) that changed upon treatment compared with DMSO. Signal intensity was quantified using ImageJ.
(C) Western blot analysis for anti-phospho-ERK1/2, anti-total ERK1/2, and anti-β-actin in G362 cells treated with L-741,742 (10 µM) and PNU 96415E (25 µM) at indicated time intervals.
(D) Western blot analysis for anti-phospho-ERK1/2 and anti-total ERK1/2, anti-phospho-S6 and anti-total S6, and anti-β-actin in GNS (G362 and G411), fibroblast (BJ), and NS (hf5205) treated with L-741,742 (10 µM) and PNU 96415E (25 µM) for 48 hr.
(E) Western blot analysis for anti-phospho-PDGFRβ, anti-total PDGFRβ, and anti-β-actin in G411 cells treated with L-741,742 (10 µM), PNU 96415E (25 µM), and L-745,870 (15 µM) for 48 hr.
(F and G) Western blot analysis for anti-phospho-ERK1/2 and anti-total ERK1/2 in G362 (F) and G481 (G) transiently transfected with sh-DRD4 and control sheGFP after 72 hr (same protein lysates from Figures 3E and S2H).
See also Figure S5.
We validated the effect of DRD4 antagonism on ERK1/2 phosphorylation by western blot at various time intervals and observed a decrease in ERK1/2 phosphorylation over time in GNS but not in NS and fibroblasts (Figures 6C, 6D, and S5C–S5E), along with a concordant decrease in PDGFRβ phosphorylation (Figure 6E). We also confirmed that transient DRD4 knockdown decreased ERK1/2 phosphorylation in GNS compared with control sh-eGFP transfected cells (Figures 6F and 6G). These biochemical data further suggest that the DRD4 antagonists act on target and that DRD4 regulates GNS growth in part through the central ERK1/2 pathway.
We further validated phospho-array data on downregulation of target of rapamycin (TOR) (Figures 6B and S5B), which is known to regulate autophagy at early stages as well as during later steps of lysosome biogenesis (Jewell et al., 2013). We observed a dramatic decrease in phospho-S6, a downstream effector of mammalian TOR (mTOR), in GNS after 48 hr of treatment, suggesting downregulation of the mTOR pathway (Figure 6D). We also observed no change in phospho-ERK1/2 and phospho-S6 abundance in treated fibroblasts and NS (Figures 6D and S5C), again consistent with the GNS specificity of DRD4 antagonists.
DRD4 Antagonists Trigger a G0/G1 Phase Arrest and Apoptosis
As DRD4 antagonists decreased the expression of cell-cycle genes (Figure S3A), we sought to determine the effect on the cell cycle. Flow cytometric analysis of DNA content in G411 and G362 cells treated with either L-741,742 or PNU 96415E revealed a G0/G1 arrest in a time-dependent manner (Figures 7A and S6A). After 48 hr of treatment with DRD4 antagonists, we also observed an induction of caspase 3/7 activity (Figure 7B), suggesting an increase in apoptosis subsequent to the earlier inhibition of autophagic flux. This result was corroborated by the presence of cleaved poly(ADP) ribose polymerase (PARP) at 48 hr (Figure 7C). Finally, GNS treated with an autophagy inhibitor (chloroquine) or ERK1/2 pathway inhibitor (PD 0325901) also underwent aG0/G1 arrest (Figure S6B), suggesting that both events downstream of DRD4 inhibition were sufficient to trigger cell-cycle arrest. Collectively, these data suggested that DRD4 inhibition ultimately results in cell-cycle arrest followed by apoptosis.
Figure 7. DRD4 Antagonists Induce G0/G1 Cell-Cycle Arrest and Apoptosis.

(A) Cell-cycle analysis of G411 and G362 cells after treatment with L-741,742 (10 µM) and PNU 96415E (25 µM) at 48 hr.
(B) Fluorescence readout for caspase 3/7 activity in G411cells treated with L-741,742 (10 µM) and PNU 96415E (25 µM) for 48 hr, and doxorubicin (1 µM) for 24 hr as positive control. n = 3, mean ± SD. Significance analyzed by unpaired one-tailed t test.
(C) Western blot analysis for apoptosis marker anti-cleaved PARP in G362 treated with L-741,742 (10 µM) and PNU 96415E (25 µM) at indicated time points, and doxorubicin (1 µM) at 24 hr as control.
See also Figure S6.
DRD4 Antagonists Inhibit GBM Xenograft Growth In Vivo
To probe the in vivo efficacy of L-741,742 and PNU 96415E on GBM growth, we first tested their effects in a subcutaneous tumor model. GNS were injected into the flanks of immunocompromised NOD scid gamma (NSG) mice and treated with PNU 96415E (20 mg/kg), L-741,742 (20 mg/kg), or vehicle until tumors reached the mandated institutional volumetric cutoff of 17 mm3 in any one mouse. Measurement of tumor volume over this time course revealed much slower growth in the treated groups compared with the vehicle control group (Figure 8A). The average tumor weight at the endpoint was reduced by 44.3% with PNU-96415E treatment and 40.9% with L-741,742 treatment (Figure 8B). Control and treated tumors were then dissociated and subjected to primary in vitro limiting dilution assays to determine whether L-741,742 and PNU-96415E affected the clonogenic capacity of the in vivo treated tumor cells. We observed a substantial reduction in the frequency of colony-forming cells in both treatment groups compared with controls (Figure 8C), demonstrating a reduction in the functional stem cell potential of the treated xenograft tumors. We further validated the effect of PNU 96415E in vivo using an independent GNS line (G411) and observed a similar reduction in tumor growth rate and endpoint size (Figures S7A–S7C). In addition, we observed increased LC3, p62, and ubiquitinated protein substrate in the treated tumors (Figure 8D), confirming induction of the same cellular response and mechanisms in an in vivo tumor context.
Figure 8. DRD4 Antagonists Inhibit GBM Xenograft Growth In Vivo.

(A) Growth curve of subcutaneous implanted tumor (G362) over a period of time. Control, n = 15, mean ± SEM; PNU 96415E, n = 16, mean ± SEM; L-741,742, n = 16, mean ± SEM. **p < 0.005, *p < 0.05, unpaired one-tailed t test.
(B) Dot plot showing tumor mass from the three treatment groups in (A) at endpoint. Control, n = 15, mean ± SEM; PNU 96415E, n = 16, mean ± SEM; L-741,742, n = 16, mean ± SEM. Significance analyzed by unpaired one-tailed t test.
(C) Linear regression plot of in vitro LDA for in vivo treated tumors. Average of each group was taken for the plot, neurospheres scored for 18 wells (six wells from each tumor).
(D) Immunohistochemistry staining and quantification for anti-p62 and anti-mono- and polyubiquitin (Ub) conjugate and LC3 in in vivo treated tumor. Scale bars, 11 µm (p62 and Ub conjugate) and 50 µm (LC3). n = 3 sections, mean ± SEM.
(E) Kaplan-Meier survival curve of immunodeficient mice injected intracranially with G362 cells and treated with L-741,742 (25 mg/kg). n = 6 per group. Significance was performed using log-rank (Mantel-Cox) test.
(F) Kaplan-Meier survival curve of immunodeficient mice injected with G362 cells treated with TMZ alone, or TMZ and L-741,742. Significance was performed using log-rank (Mantel-Cox) test. n = 18 per group and censorship/endpoint at day 182.
See also Figure S7.
We then tested the effect of L-741,742 in an intracranial xenograft model, with treatment commencing 1 week after engraftment once tumor size had become substantial (Figure S7D). The DRD4 antagonist alone had a modest but significant benefit in survival over control (Figure 8E). However, when combined with TMZ, data from two in vivo experiments demonstrated that L-741,742 significantly improved survival compared with TMZ alone (Figure 8F). Together, these data from two different human patient-derived xenograft models provide a strong basis for further exploration of DRD4 antagonism and/or inhibition of autophagic flux as an additional avenue for GBM therapy.
DISCUSSION
We investigated whether neurochemical signaling activity influences the growth and survival of human GNS, based on our previous studies in murine NS (Diamandis et al., 2007) and the work of others in neuronal development and adult neurogenesis (Berg et al., 2013; Martins and Pearson, 2008; Ohtani et al., 2003; Song et al., 2012). This study represents a systematic and unbiased interrogation of all neurochemical classes on human GBM cell survival. Of the 13 neurochemical classes, we found that modulation of dopaminergic, serotonergic, and cholinergic pathways predominantly affected GNS. Each of these neurochemical classes has been previously implicated in regulating NS during brain development and adult neurogenesis (Diaz et al., 1997; Hoglinger et al., 2004; Mohapel et al., 2005; Tong et al., 2014).
We found that specific DRD4 antagonists selectively inhibit GNS growth, mediated by on-target inhibition of the DRD4 receptor, which is expressed in both GNS and primary GBM patient samples, and concomitant suppression of the downstream effectors PDGFRβ/ERK1/2. PDGFRβ is expressed preferentially by tumorigenic GBM stem cells, and blocking PDGFRβ in this context inhibits self-renewal and tumor growth (Kim et al., 2012), further supporting our findings. At a cellular level, we find that DRD4 antagonism impairs the autophagy-lysosomal degradation pathway and that this effect is accompanied by a G0/G1 cell-cycle arrest and subsequent apoptosis, as mediated by inhibition of the ERK1/2 pathway and autophagy flux.
Dopamine is a catecholamine synthesized by neurons predominantly in the midbrain region with diffuse cortical projections. Notably, many GBMs arise in the projection field of dopaminergic neurons. Dopamine signals are transmitted through five G-protein-coupled receptors (D1–5). Dopamine signaling is dysregulated in diverse neurological and psychiatric diseases including Parkinson’s disease, schizophrenia, and drug addiction. In addition to serving a key neurotransmitter function, dopamine has been implicated in the regulation of endogenous neurogenesis during brain development (Borta and Hoglinger, 2007; Diaz et al., 1997; Ohtani et al., 2003) and adult neurogenesis in the SVZ by activating D2-like receptors on transit-amplifying progenitor cells (Coronas et al., 2004; Hoglinger et al., 2004; Lao et al., 2013). In postmortem brains of Parkinson’s disease patients, a disease characterized by depletion of dopamine, a reduction of proliferating cells in the SVZ has been observed (Hoglinger et al., 2004). These reports suggest an important role for dopamine signaling in the regulation of normal neurogenesis. Epidemiological evidence suggests that patients with various neuropsychiatric disorders, including Parkinson’s disease, have a reduced incidence of primary CNS tumors, consistent with the hypothesis that dopamine signaling may influence the emergence of these tumors (Diamandis et al., 2009).
Little is known about whether dopamine signaling affects GBM stem cell behavior. In addition to DRD4 antagonists, our primary screen also uncovered the DRD2-specific antagonists thioridazine and trifluoperazine as hits. Consistently, a recent genome-wide shRNA screen in a GBM cell line (U87MG) identified a role for DRD2 in GBM growth (Li et al., 2014). However, DRD4 antagonists showed better selectivity for GNS in our screen compared with DRD2 antagonists (data not shown). Although epigenetic suppression of DRD4 has been reported in pediatric CNS tumors (Unland et al., 2013), DRD4 is not methylated in TCGA GBM samples, and mutations in the DRD5 and DRD3 genes occur in a small fraction of GBM samples (Brennan et al., 2013). Finally, we note that dopamine receptor antagonists may have activity against other cancer stem cell types, including leukemia (Sachlos et al., 2012) and lung cancer (Yeh et al., 2012). In conjunction with our findings, these studies suggest that dopamine receptor antagonists will be useful probes for the further study of GBM growth and survival, and potentially other cancer types.
The autophagy-lysosomal degradation pathway system appears to be critical for the progression and/or maintenance of many cancer types. For example, autophagy is important for breast cancer stem cell maintenance (Choi et al., 2014; Gong et al., 2013). In a number of cancer studies, including for GBM, autophagy inhibition with chloroquine appears to augment the efficacy of anticancer therapies (Sotelo et al., 2006). Recent work suggests that the antidepressant imipramine can also modify autophagy mechanisms in GBM cells (Shchors et al., 2015).
Impairment of the autophagy-lysosomal pathway is also intimately associated with neurodegenerative diseases in which neurochemical signaling is dysregulated, including Parkinson’s disease, Alzheimer’s disease, and Niemann-Pick type C1 disease (Nixon, 2013). Autophagy has been implicated in the maintenance of adult NS (Wang et al., 2013), and suppression of autophagy in NS during development in mice causes neurodegenerative diseases (Hara et al., 2006). Our study provides additional insights into the potential role of DRD4 signaling in neurological disorders such as attention deficit/hyperactivity disorder, bipolar disorder, and schizophrenia, which are reported to be associated with DRD4 polymorphism (LaHoste et al., 1996; Rondou et al., 2010).
Our data suggest that autophagy plays an essential role in GNS growth and survival, and that dopamine signaling through the DRD4 receptor is required to maintain the autophagy-lysosomal degradation system. GNS are selectively vulnerable to disruption of this pathway, whereas NS undergo differentiation, and fibroblasts and other non-NS control cells are unaffected. We hypothesize that potential differences in the basal activity and dependence of GNS on the autophagy-lysosomal pathway compared with normal NS and fibroblasts may account for the susceptibility of GNS to DRD4 antagonists. Consistently, it has recently been reported that GBM cells are sensitive to the vacuolization agent vacquinols (Kitambi et al., 2014), and the breast cancer stem cell-selective compound salinomycin is also reported to confer selectivity through inhibiting autophagic flux (Yue et al., 2013). The identification of DRD4 antagonists as GBM-selective agents suggests that modulation of dopamine DRD4 signaling may hold therapeutic potential in GBM patients, although further clinical exploration is needed. More broadly, exploration of the role of additional neurochemical signaling pathways, including other dopaminergics, serotonergics, and cholinergics, in GBM may lead to additional CNS-accessible therapeutic approaches to treat this intractable disease.
EXPERIMENTAL PROCEDURES
Primary Tissue Samples and Cell Culture
All human tissue samples were procured following informed consent, and all experimental procedures were approved by the Research Ethics Board of The Hospital for Sick Children and Toronto Western Hospital. Primary tissue samples were dissociated in artificial cerebrospinal fluid followed by treatment with enzyme cocktail at 37°C. GNS and NS lines were grown as adherent monolayer culture in serum-free medium as described previously (Pollard et al., 2009).
Compound Library
The neurochemical library was purchased from BIOMOL International (now Enzo Life Sciences). All compounds for retesting were purchased from Tocris Bioscience.
Tandem mRFP-GFP-LC3 Reporter Assay
Cells were transfected with mRFP-GFP-LC3 construct using Amaxa Nucleofector kit (VPG-1004). After 24 hr, cells were treated with compounds as indicated. Live cells were imaged using Quorum spinning disc confocal microscopy. Autophagy flux was assessed by counting cells mRFP+GFP+LC3 (yellow puncta), which represents autophagosomes, and mRFP+GFP−LC3 (red puncta), which represents autolysosomes.
DQ-Red BSA Assay
Cells were pulsed with DQ-Red BSA (Molecular Probes, D-12051) for 1 hr and chased for 4 hr before imaging live cells using Quorum spinning disc confocal microscopy.
Patient-Derived Xenografts
All mouse procedures were approved by the Hospital for Sick Children’s Animal Care Committee. For subcutaneous xenograft, 2 × 105 GNS were injected into flanks of NSG female mice. L-741,742 and PNU 96415E were dissolved in 40% 2 hydroxy β-cyclodextrin (Sigma). Mice were treated 3 days after tumor implantation. Both L-741,742 and PNU 96415E were treated at 20 mg/kg intraperitoneally for 5 days on, 2 days off until the endpoint. Tumor growth was monitored with microcalipers until tumor volume reached 17 mm3 in any one tumor from any group, and all mice were euthanized at the same endpoint. For intracranial xenograft experiments, 5 × 103 GNS (G362) were injected in the forebrain of NSG mice. Mice were treated 1 week after tumor implantation with L-741,742 (25 mg/kg) 5 days on, 2 days off for 2 weeks. TMZ was administered during the first week through gavage, whereby TMZ was administered alone (25 or 12.5 mg/kg) or together with L-741,742 (25 mg/kg). Kaplan-Meier curves show time (in days) elapsed to death due to the assigned event (development of a brain tumor with dome head and neurological symptoms) or to censorship. In both experiments, there was no significant difference between the treatment and control groups in the frequency of censored animals versus deaths (p > 0.05, data not shown).
Statistical Analysis
All grouped data are presented as mean ± SEM unless otherwise stated. Differences between groups were assessed using a Student’s t test.
Further experimental procedures are detailed in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Dopaminergic, serotonergic, and cholinergic pathways affect GNS survival
GBM tumors and patient-derived GNS express functional DRD4 receptor
DRD4 antagonism selectively targets GNS growth in vitro and in vivo
GNS are vulnerable to DRD4-mediated disruption of autophagy-lysosomal pathway
Significance.
Neurochemicals that mediate synaptic communication between mature neurons have recently been shown to govern aspects of normal neurogenesis. Given the close similarities between GNS and NS, and their existence in the rich brain neurochemical milieu, neurochemical signaling may profoundly affect GBM stem cell growth. We tested this hypothesis and found that modulation of the dopaminergic, serotonergic, and cholinergic pathways affects GNS growth, particularly dopamine signaling via the DRD4 receptor. GNS express functional DRD4 and its inhibition causes impairment in the endolysosomal system, a block in autophagic flux, and eventual cell death. The vulnerability of GNS to disruption in the autophagy-lysosomal system by neurochemical signaling modulation opens up additional avenues of therapeutic investigation for this incurable tumor.
Acknowledgments
P.B.D. holds a Garron Family Research Chair in Childhood Cancer Research and is supported by grants from the Canadian Institutes of Health Research (CIHR), the Ontario Institute for Cancer Research (OICR), the Canadian Cancer Society Research Institute, and Genome Canada. P.B.D. also receives support from the Hospital for Sick Children Foundation, Jessica’s Footprint, Hopeful Minds, and B.R.A.I.N.Child. A portion of this work was supported by a research grant from Primera Biosciences. M.T. was supported by a Canada Research Chair in Systems and Synthetic Biology, and by grants from the CIHR (MOP 119572), the European Research Council (233457), and Genome Quebec. J.H.B. holds the Pitblado Chair in Cell Biology at the Hospital for Sick Children. The authors thank the donors, the RCWIH BioBank, the Lunenfeld-Tanenbaum Research Institute, and the Mount Sinai Hospital/UHN Department of Obstetrics and Gynecology for human tissue.
Footnotes
ACCESSION NUMBERS
The GenBank accession number for the PNU 96415E-treated GNS microarray data described in this manuscript is GEO: GSE62714.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2016.05.002.
AUTHOR CONTRIBUTIONS
S.D., P.B.D., and M.T. conceived the project, designed experiments, analyzed data, and wrote the manuscript. H.J.S. critically evaluated data and helped write the manuscript. S.D., L.L., X.L., M.K., H.W., R.H., M.S., X.Z., and K.R. performed experiments. N.P., V.V., C.X., and G.B. performed bioinformatics analysis. L.H. performed statistical analysis and revised the manuscript. M.B. provided samples. J.H.B. helped devise and analyze experiments and revised the manuscript. T.A. and I.D.C. provided intellectual contributions to experimental design and data analysis.
References
- Andang M, Hjerling-Leffler J, Moliner A, Lundgren TK, Castelo-Branco G, Nanou E, Pozas E, Bryja V, Halliez S, Nishimaru H, et al. Histone H2AX-dependent GABA(A) receptor regulation of stem cell proliferation. Nature. 2008;451:460–464. doi: 10.1038/nature06488. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radio-resistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Berg DA, Belnoue L, Song H, Simon A. Neurotransmitter-mediated control of neurogenesis in the adult vertebrate brain. Development. 2013;140:2548–2561. doi: 10.1242/dev.088005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borta A, Hoglinger GU. Dopamine and adult neurogenesis. J. Neurochem. 2007;100:587–595. doi: 10.1111/j.1471-4159.2006.04241.x. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DS, Blanco E, Kim YS, Rodriguez AA, Zhao H, Huang TH, Chen CL, Jin G, Landis MD, Burey LA, et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells. 2014;32:2309–2323. doi: 10.1002/stem.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- Coronas V, Bantubungi K, Fombonne J, Krantic S, Schiffmann SN, Roger M. Dopamine D3 receptor stimulation promotes the proliferation of cells derived from the post-natal subventricular zone. J. Neurochem. 2004;91:1292–1301. doi: 10.1111/j.1471-4159.2004.02823.x. [DOI] [PubMed] [Google Scholar]
- Diamandis P, Wildenhain J, Clarke ID, Sacher AG, Graham J, Bellows DS, Ling EK, Ward RJ, Jamieson LG, Tyers M, Dirks PB. Chemical genetics reveals a complex functional ground state of neural stem cells. Nat. Chem. Biol. 2007;3:268–273. doi: 10.1038/nchembio873. [DOI] [PubMed] [Google Scholar]
- Diamandis P, Sacher AG, Tyers M, Dirks PB. New drugs for brain tumors? Insights from chemical probing of neural stem cells. Med. Hypotheses. 2009;72:683–687. doi: 10.1016/j.mehy.2008.10.034. [DOI] [PubMed] [Google Scholar]
- Diaz J, Ridray S, Mignon V, Griffon N, Schwartz JC, Sokoloff P. Selective expression of dopamine D3 receptor mRNA in proliferative zones during embryonic development of the rat brain. J. Neurosci. 1997;17:4282–4292. doi: 10.1523/JNEUROSCI.17-11-04282.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizeyi N, Bjartell A, Nilsson E, Hansson J, Gadaleanu V, Cross N, Abrahamsson PA. Expression of serotonin receptors and role of serotonin in human prostate cancer tissue and cell lines. Prostate. 2004;59:328–336. doi: 10.1002/pros.10374. [DOI] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Gill RS, Hsiung MS, Sum CS, Lavine N, Clark SD, Van Tol HH. The dopamine D4 receptor activates intracellular platelet-derived growth factor receptor beta to stimulate ERK1/2. Cell Signal. 2010;22:285–290. doi: 10.1016/j.cellsig.2009.09.031. [DOI] [PubMed] [Google Scholar]
- Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261–2272. 2272e.1–11. doi: 10.1038/onc.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I, Hirsch EC. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 2004;7:726–735. doi: 10.1038/nn1265. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013;14:133–139. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, Serwanski D, Sloan AE, Weil RJ, Lee J, et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Development. 2012;26:1247–1262. doi: 10.1101/gad.193565.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescenttagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Kitambi SS, Toledo EM, Usoskin D, Wee S, Harisankar A, Svensson R, Sigmundsson K, Kalderen C, Niklasson M, Kundu S, et al. Vulnerability of glioblastoma cells to catastrophic vacuolization and death induced by a small molecule. Cell. 2014;157:313–328. doi: 10.1016/j.cell.2014.02.021. [DOI] [PubMed] [Google Scholar]
- LaHoste GJ, Swanson JM, Wigal SB, Glabe C, Wigal T, King N, Kennedy JL. Dopamine D4 receptor gene polymorphism is associated with attention deficit hyperactivity disorder. Mol. Psychiatry. 1996;1:121–124. [PubMed] [Google Scholar]
- Lao CL, Lu CS, Chen JC. Dopamine D3 receptor activation promotes neural stem/progenitor cell proliferation through AKT and ERK1/2 pathways and expands type-B and -C cells in adult subventricular zone. Glia. 2013;61:475–489. doi: 10.1002/glia.22449. [DOI] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Li J, Zhu S, Kozono D, Ng K, Futalan D, Shen Y, Akers JC, Steed T, Kushwaha D, Schlabach M, et al. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget. 2014;5:882–893. doi: 10.18632/oncotarget.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins RA, Pearson RA. Control of cell proliferation by neurotransmitters in the developing vertebrate retina. Brain Res. 2008;1192:37–60. doi: 10.1016/j.brainres.2007.04.076. [DOI] [PubMed] [Google Scholar]
- Mohapel P, Leanza G, Kokaia M, Lindvall O. Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiol. Aging. 2005;26:939–946. doi: 10.1016/j.neurobiolaging.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat. Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Oak JN, Lavine N, Van Tol HH. Dopamine D(4) and D(2L) receptor stimulation of the mitogen-activated protein kinase pathway is dependent on trans-activation of the platelet-derived growth factor receptor. Mol. Pharmacol. 2001;60:92–103. doi: 10.1124/mol.60.1.92. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Goto T, Waeber C, Bhide PG. Dopamine modulates cell cycle in the lateral ganglionic eminence. J. Neurosci. 2003;23:2840–2850. doi: 10.1523/JNEUROSCI.23-07-02840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Rondou P, Haegeman G, Van Craenenbroeck K. The dopamine D4 receptor: biochemical and signalling properties. Cell Mol. Life Sci. 2010;67:1971–1986. doi: 10.1007/s00018-010-0293-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012;149:1284–1297. doi: 10.1016/j.cell.2012.03.049. [DOI] [PubMed] [Google Scholar]
- Schlett K. Glutamate as a modulator of embryonic and adult neurogenesis. Curr. Top. Med. Chem. 2006;6:949–960. doi: 10.2174/156802606777323665. [DOI] [PubMed] [Google Scholar]
- Schuller HM. Neurotransmission and cancer: implications for prevention and therapy. Anti Cancer Drugs. 2008;19:655–671. doi: 10.1097/CAD.0b013e3283025b58. [DOI] [PubMed] [Google Scholar]
- Shchors K, Massaras A, Hanahan D. Dual targeting of the autophagic regulatory circuitry in gliomas with repurposed drugs elicits cell-lethal autophagy and therapeutic benefit. Cancer Cell. 2015;28:456–471. doi: 10.1016/j.ccell.2015.08.012. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, et al. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 2012;489:150–154. doi: 10.1038/nature11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J, Briceno E, Lopez-Gonzalez MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, doubleblind, placebo-controlled trial. Ann. Intern. Med. 2006;144:337–343. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- Tong CK, Chen J, Cebrian-Silla A, Mirzadeh Z, Obernier K, Guinto CD, Tecott LH, Garcia-Verdugo JM, Kriegstein A, Alvarez-Buylla A. Axonal control of the adult neural stem cell niche. Cell Stem Cell. 2014;14:500–511. doi: 10.1016/j.stem.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unland R, Kerl K, Schlosser S, Farwick N, Plagemann T, Lechtape B, Clifford SC, Kreth JH, Gerss J, Muhlisch J, et al. Epigenetic repression of the dopamine receptor D4 in pediatric tumors of the central nervous system. J. Neurooncol. 2013;116:237–249. doi: 10.1007/s11060-013-1313-1. [DOI] [PubMed] [Google Scholar]
- Vance JE. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 2006;580:5518–5524. doi: 10.1016/j.febslet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 2015;161:803–816. doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liang CC, Bian ZC, Zhu Y, Guan JL. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci. 2013;16:532–542. doi: 10.1038/nn.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CT, Wu AT, Chang PM, Chen KY, Yang CN, Yang SC, Ho CC, Chen CC, Kuo YL, Lee PY, et al. Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am. J. Respir. Crit. Care Med. 2012;186:1180–1188. doi: 10.1164/rccm.201207-1180OC. [DOI] [PubMed] [Google Scholar]
- Yue W, Hamai A, Tonelli G, Bauvy C, Nicolas V, Tharinger H, Codogno P, Mehrpour M. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy. 2013;9:714–729. doi: 10.4161/auto.23997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.