

Abstract
Background
Hard ticks are hematophagous ectoparasites characterized by their long-term feeding. The saliva that they secrete during their blood meal is their crucial weapon against host-defense systems including hemostasis, inflammation and immunity. The anti-hemostatic, anti-inflammatory and immune-modulatory activities carried out by tick saliva molecules warrant their pharmacological investigation. The Hyalomma dromedarii Koch, 1844 tick is a common parasite of camels and probably the best adapted to deserts of all hard ticks. Like other hard ticks, the salivary glands of this tick may provide a rich source of many compounds whose biological activities interact directly with host system pathways. Female H. dromedarii ticks feed longer than males, thereby taking in more blood. To investigate the differences in feeding behavior as reflected in salivary compounds, we performed de novo assembly and annotation of H. dromedarii sialotranscriptome paying particular attention to variations in gender gene expression.
Results
The quality-filtered Illumina sequencing reads deriving from a cDNA library of salivary glands led to the assembly of 15,342 transcripts. We deduced that the secreted proteins included: metalloproteases, glycine-rich proteins, mucins, anticoagulants of the mandanin family and lipocalins, among others. Expression analysis revealed differences in the expression of transcripts between male and female H. dromedarii that might explain the blood-feeding strategies employed by both genders.
Conclusions
The annotated sialome of H. dromedarii helps understand the interaction of tick-host molecules during blood-feeding and can lead to the discovery of new pharmacologically active proteins of ticks of the genus Hyalomma.
Electronic supplementary material
The online version of this article (10.1186/s13071-018-2874-9) contains supplementary material, which is available to authorized users.
Keywords: Hyalomma dromedarii tick, Camels, Tunisia, Sialotranscriptome, Gene expression, Gene enrichment
Background
Ticks are hematophagous arthropods that injure their hosts. More dangerously, they are vectors of many pathogens including arboviruses, spotted fever Rickettsia, Anaplasma, Borrelia, Babesia, that cause human and veterinary diseases worldwide [1–5]. Even though chemical acaricides proved to be effective against tick infestations [6], the problem of tick resistance is becoming a problem and a primary cause of growing economic losses [6]. Research on alternatives to the use of acaricides is strongly focused on the development of anti-tick vaccines that are considered to be a more cost-effective, environmentally safe strategy [7].
One of the most promising strategies of the synthesis of anti-tick vaccines is based on tick salivary molecules that have immunosuppressive properties that are secreted during blood-feeding [8]. Indeed, ticks have developed an arsenal of salivary molecules including anti-hemostatic, anti-inflammatory and immunomodulatory compounds that are involved in avoiding host defense, enabling them to remain safe while taking their blood meal [9]. In addition, bioactive molecules secreted in tick saliva are involved in the transimission of the pathogens to the host: this phenomenon also called saliva-assisted transmission (SAT) [10]. As they are long-term blood-feeders, ticks are constantly threated by host defense pathways that might interrupt the blood meal and even kill the tick [9]. Although male and female ticks are both hematophagous, they have very different feeding behaviors. For example, females have a longer blood-feeding process than males, and their body weight differences are more than 50-fold after feeding [11, 12].
The first tick Ixodes scapularis genome became available only in 2016 [13], as tick genomes are typically large, highly repetitive, and difficult to assemble. Recent advances in tick sialotranscriptomic analysis, combined with NGS projects and functional studies, have provided the genomic datasets needed for further research and have shed light on a large number of the active molecules that could explain tick salivary gland physiology and identify vaccine candidates [14]. RNA sequencing (RNAseq) is an excellent technique for investigating several non-model organisms, such as ticks, cost-effectively [15, 16]. For last three decades, given the valuable information provided by tick sialome data analysis, many tick salivary gland transcriptomes were generated from adult males and females as well as on other tick development stages [17–23]. These transcriptomes pointed out the complexity of salivary protein families in the different tick species and identified new candidate genes involved in feeding. However, no such in depth transcriptome has yet been generated for Hyalomma dromedarii Koch, 1844, despite it being one of the most economically important ticks especially in the Saharan regions.
Hyalomma dromedarii is closely associated with camels, which are the main hosts of the adult ticks [24]; nymphs and larvae are more ubiquitous and can parasitize rodents, hedgehogs and birds [25]. The species represents nearly 90% of ticks infesting camels and is distributed wherever camels are present, in southern Russia, the Far, Middle, and Near East, North Africa and south of the great northern deserts as far south as Somaliland and northeastern Kenya [24]. Hyalomma dromedarii is the vector of the life-threatening Crimean-Congo hemorrhagic fever virus [26]. Its association with camels has an economic importance as the health and reproduction of camels are affected by heavy tick infestations [27]. The long-term blood meal of H. dromedarii adult ticks implies the involvement of a large, diverse number of salivary gland components. However, to our knowledge, no report has described these important molecules in H. dromedarii. In the present work, we have therefore aimed to: (i) de novo assemble the sialotranscriptome of H. dromedarii that enriches sequences information available in gene databases; (ii) provide a high-quality annotation and characterization of tick secretory proteins; and (iii) specify genes putatively associated with tick blood meal by exploring the differential expression between tick genders.
Methods
Ticks and salivary gland collection
Hyalomma dromedarii ticks were collected from camels in the Saharan bioclimatic zone of southern Tunisia (33°25'908"N, 009°00'952"E). The camels were thoroughly inspected; especially the inguinal region and the legs, preferential attachment sites of this species. Partially engorged ticks, at different nearly feeding stages, were removed manually from the camel body, placed in flasks containing a piece of filter paper and then brought to the laboratory. Each tick was identified using a taxonomic key [28]. Within the first hour of collection, ticks were washed and fixed in paraffin by their legs and then lateral cuts were made with a scalpel to remove the scutum. Salivary glands (SG) were teased away from other organs using ultra fine forceps. SG were washed in ice-cold PBS, pH 7.2 and stored at -80 °C until RNA isolation. Salivary glands were pooled by sex, resulting in one sample for female and one sample for male ticks.
mRNA isolation, quantification and integrity
Total RNA was extracted from 50 pairs of H. dromedarii SG, of each gender, using Trizol total RNA extraction reagent (Ambion, Life Technologies, CA, USA). SG were homogenized and the total RNA was extracted and re-suspended in RNAase-free water according to the manufacturer's protocol. RNA integrity was assessed using 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The mRNA was prepared with magnetic beads with an oligo (dT) according to Dynabeads® mRNA DIRECT kit (Ambion, Life Technologies, CA, USA). mRNA was quantified by Quant-iT™ RiboGreen® RNA reagent and Kit (Invitrogen, Life Technologies Corp., Carlsbad , USA). The integrity of mRNA was evaluated in a 2100 Bioanalyzer (Agilent Technologies, CA, USA).
cDNA library preparation, sequencing and pre-processing analysis
Extracted mRNA was further processed for cDNA libraries constructions following the stranded TruSeq RNA Sample Prep Kit protocol (Illumina, San Diego, CA, USA). Briefly, selected poly-A-RNA was fragmented and primed with random hexamers. Fragmented RNA was reverse transcribed in order to generate first strand cDNA. Indexing adapters were ligated to cDNA for hybridization onto the flow cell of Illumina HiSeq 1500 sequencing machine. The size distribution of the cDNA libraries was measured by 2100 Bioanalyzer with DNA1000 assay (Agilent Technologies, CA, USA). An ABI Step One Plus Real-Time PCR System were used in quantification of the sample library before sequencing. The cDNA libraries were sequenced on the Illumina HiSeq 1500 System, in Rapid run mode, generating 2 × 150 bp paired-end reads, according to the standard manufacturer protocol.
Pre-processing of RNA-Seq raw data and de novo transcriptome assembly
Using Illumina Casava software (v1.8.2), with Illumina quality control QC > Q30, two paired-end fastq files were generated corresponding to each H. dromedarii gender. RNA-Seq raw data reads were filtered by PhiX contaminant, using the software bowtie2 version 2.2.3 [29] and by quality, read size (> 40 bp), homopolymer (> 90%), low complexity sequences (> 90%) and poly-A/T/N tails and adapters, using the software fastq-mcf version 1.04.662 [30]. Paired-end read sequences with good quality from the two genders were assembled to obtain one sialotransciptome, using the Trinity assembler using the paired-end option, with parameters CuffFly and in silico normalization of reads with a maximum of 50× coverage [31]. The assembled transcripts were filtered according to their sequence length and those lowly expressed (that likely represent artifacts) by filtering with a Fragments Per Kilobase of exon per Million fragments mapped (FPKM < 1) [32]. No assumptions are made about genes with low abundance levels, but they are not easily distinguishable from background noise. The completeness of the transcriptome was also estimated by the presence of sequences belonging to the set of ultra-conserved eukaryotic proteins, tested using the CEGMA pipeline [33] and BUSCO approach [34] based on eukaryotic database.
Functional annotation of H. dromedarii transcriptome
The transcriptome was annotated using two approaches, BLASTx similarity tool [35] (e-value < 1 × 10-5) to compare to the gene ontology (GO) database [36], the nucleotide sequences to the NCBI non-redundant protein database (NR), UniProt Knowledgebase translated EMBL-Bank predicted peptides from the I. ricinus genome, Animal Toxin Database (ATDB) [37] and search for Signal Peptide in the predicted proteins [38]. The second approach, dCAS (desktop annotation system), an automatic annotation server [39], was used to assign transcript to the Pfam [40], (SMART) [41], eukaryotic ortholog groups (KOG) [42] databases using rpsblast tool [35]. Results were mapped into an excel spreadsheet which was integrated to the first approach and is presented in Additional file 1: Table S1 as described in the dCAS software tool [39]. TransDecoder utility [43] was used to predict the Open Reading Frames (ORFs) from the assembled transcripts, and putative signal peptides were predicted using the software SignalP 4.0 [38]. The predicted amino acid sequences were BLASTp aligned against specific protein databases. A priority order of UniPro- tKB/TrEMBL, Pfam database and NR-NCBI was used for annotation and selection of best candidate for each transcript. Thus, transcripts were filtered according to the following steps: (i) blast against Uniprot-Acari with e-value < 0.05; (ii) blast against Pfam with e-value < 0.05; (iii) deletion of “unknown” and “uncharacterized” Pfam annotations; and (iv) the sum of female and male TPM must be greater than 1.
Predicted proteins were kept in the dataset if a significant BLASTp (e-value < 1 × 10-5) or domain-based match (e-value < 1 × 10-3) was obtained.
The annotation of KEGG pathways and KEGG orthology (KO) were assigned to assembled transcripts using the online KEGG Automatic Annotation Server (KAAS version and date) [44]. The Bi-directional Best Hit (BBH) method was used to obtain KEGG Orthology (KO) assignment with BHRs Score ≥ 60, and I. scapularis genome (version) was used as reference.
Phylogenetic analysis
The nucleotide sequences of each individual sequence were translated into amino acid sequences using transdecoder, only the Kunitz domain region (with the 6-cysteines motif) were aligned using ClustalW [45] with default parameters. Thereafter we manually edited the amino acid sequences using Seaview [46] and performed the phylogenetic analysis with this same tool. Next we selected the best evolutionary model with ProtTest 2.4 [47]. Prottest selected the protein evolution model that best fit in the monolaris sequence alignment using default parameters: WAG with site heterogeneity model gamma þ invariant sites. Bayesian analyses were carried out using Markov chain Monte Carlo (MCMC) implemented in BEAST 1.7.5 software [48]. We ran four independent MCMC searches using distinct randomly generated starting trees. Each run consisted of 50,000,000 generations, and the trees were sampled every 1000 generations. Convergence was inspected in Tracer v1.5 [48], and uncertainties were addressed as 95% HPD intervals. All runs reached a stationary level after 10% 'burn in' with a large effective sample size. Trees obtained after the ‘burn in’ step were used to generate a maximum clade credibility tree with TreeAnnotator v1.7.5 [48], using a majority rule. The obtained tree was visualized and edited using FigTree v1.4.0 (available at http://tree.bio.ed.ac.uk/software/Figtree).
Transcripts expression analysis
For the expression profile analysis, the assembled transcripts were filtered by putative ribosomal genes using as reference the Metazoa rRNA database [49] and BLASTn alignment with parameters of e-value < 1 × 10-20, query coverage > 60% and query identity > 70%. In order to estimate transcript abundance we aligned each set of reads back to the H. dromedarii assembled transcriptome and maximum likelihood abundance estimates were obtained using the RSEM method [50]. Final abundance estimates were calculated as Fragments Per Kilobase of exon per Million fragments mapped (FPKM) and Transcripts Per Million (TPM) values. We subsequently identified differentially expressed genes between the male and female tick samples with the EdgeR Bioconductor software package [51], a preferred methodology for studies lacking biological replicates, and extract those transcripts that are at least four fold differentially expressed with false discovery-corrected statistical significance of at most 0.001.
For the identification of gender-specific transcripts the sample reads of male and female were mapped individually to the transcriptome assembly and the transcripts were classified in each group (male, female, both) based on the transcripts abundance with FPKM > 1. All plots have been developed using R and ggplot2 [52]. Statistics analysis for enzyme families and classes were done using chi-square statistics [53].
Transcriptome enrichment analysis
The identification and categorization of metabolic pathways in H. dromedarii based on KEGG database and integration with the lists of differentially expressed genes in each gender was possible using “the Enrichment analysis for the metabolic pathways” based on GeneMerge software v1.4 [54]. The GeneMerge uses a hypergeometric distribution and apply a Bonferroni correction for a more appropriated and significant identification of enriched pathways. The analyses were divided into two sets, a group of 557 differentially expressed genes in Tick Female and another group of 353 differentially expressed genes in Tick Male, and a filter was applied for the identification of the most enriched pathways with corrected P-value ≤ 1 × 10-3 and FDR ≤ 1%.
Results and discussion
Overview of the sialotranscriptome of Hyalomma dromedarii
Next-generation sequencing using HiSeq 1500 Illumina technology was conducted to determine mRNA sequences of the salivary glands of male and female H. dromedarii collected in south Tunisia. A total of 330,285,649 paired-end reads were generated for H. dromedarii salivary glands having an average size of 808.74 nucleotides. Adequate adapter trimming and quality filtering discarded about 5,076,399 (2.5%) and 3,255,818 (2.4%) reads for male and female, respectively. Details of filtered RNA-Seq raw data results are shown in Table 1. Only sequencing reads longer than 80 nucleotides were used to assemble primary transcripts that were classified according to their putative functions and plotted in hyperlinked excel spreadsheets (available in Additional file 1: Table S1). The combined assembly of the sequences from male and female led to the extraction of 15,342 transcripts after PFAM filter. Such a high number of transcripts was expected, and it confirms the complexity of ixodid sialotranscriptomes already examined in other studies [17–23, 55]. Indeed, tick saliva is known for the complexity of its molecules, and in many cases, their redundancy [14].
Table 1.
Filter of Hyalomma dromedarii RNA-Seq raw data results
| Female | Male | |||
|---|---|---|---|---|
| No. of reads | % of reads | No. of reads | % of reads | |
| Total reads (raw data) | 133,633,885 | – | 196,651,764 | – |
| Total good reads (approved) | 13,037,8067 | 97.56 | 191,575,365 | 97.42 |
| Total filtered reads (removed) | 3,255,818 | 2.44 | 5,076,399 | 2.58 |
| Too short after clip | 2,723,639 | 2.04 | 4,198,316 | 2.13 |
| Filtered on quality | 488,212 | 0,37 | 796,059 | 0.4 |
| Filtered on homopolymer | 39,730 | 0.03 | 75,314 | 0.04 |
| Filtered on low complexity | 4,237 | 0 | 6,710 | 0 |
| Trimmed reads (not totally removed) | 22,868,516 | 17.11 | 32,950,850 | 16.76 |
For the past three decades, a reiterated transcript classification was followed for nearly all described tick sialotranscriptomes according to the function of proteins regarding tick-host interactions [18, 19, 21–23, 55, 56]. Following the same approach, the transcripts of the H. dromedarii were classified into 4 main categories: housekeeping, secreted, transposable elements, and transcripts with unknown function classes (Fig. 1a). A total of 1749 transcripts (11.4% of the whole transcriptome) were associated with the secreted class, while 8063 transcripts (52.56% of the whole transcriptome) belonged to the housekeeping class. Transposable elements represented almost 7.12% of all transcripts, while the rest of the reads were mapped to transcripts of unknown function (28.93%). All transcripts and their matches to several databases are available in Additional file 1: Table S1.
Fig. 1.

Functional classification of Hyalomma dromedarii tick transcripts. a Sialotranscriptome of H. dromedarii was divided into 4 categories: Housekeeping, Uncharacterized, Transposable elements and Secreted. b Housekeeping class of transcripts found in the sialotranscriptome of Hyalomma dromedarii (only families with high transcript count were represented)
It is worth mentioning that the sialotranscrpitome was obtained from ticks collected on camels. These ticks might be infected by several pathogens that can affect their transcriptome. While our data need further validation using quantitative (q) RT-PCR, they do provide important information on H. dromedarii sialotranscriptome.
Housekeeping class
A total of 8063 transcripts (52.56% of the total numbers of transcripts) were attributed to the housekeeping class, expressed in the SG from H. dromedarii. The group of housekeeping genes was divided among 156 more detailed functional subgroups of which 8033 matched the KOGG database (Table 2; Fig. 1b; Additional file 2: Table S2). The largest transcript count was attributed to the signal transduction mechanisms family, post-translational modification, protein turnover, the chaperones family, transcription and RNA processing and modification families with transcript counts of 858 (10.7% of housekeeping transcripts), 708 (8.8%) and 596 (7.4%), respectively. The presence of these major subgroups was expected as the major role of salivary glands is the secretion of the saliva. In addition, the following proteins involved in host immunity and inflammation, such as enzymes related to detoxification and oxidative metabolism, were identified: sulfotransferases, selenoproteins, superoxide dismutase and peroxidases (Additional file 2: Table S2). Similar results were reported for Amblyomma maculatum [57] and Rhipicephalus pulchellus [21]. The high amount of housekeeping/intracellular detoxification enzymes may be due to their unconventional secretion to the extracellular medium, where they play more extracellular functions that are not usually associated with their intracellular functions [58]. Furthermore, they may play different roles in the tick-host interface [59]. This distribution resembles that of previously described sialotranscriptomes [17–23].
Table 2.
Number of transcripts, KEGG orthologs (KOs) and enzymes summarized by class
| Transcripts | KOs | Enzymes | |
|---|---|---|---|
| Oxidoreductases | 263 | 137 | 101 |
| Transferases | 628 | 365 | 209 |
| Hydrolases | 357 | 197 | 123 |
| Lyases | 79 | 50 | 39 |
| Isomerases | 52 | 32 | 24 |
| Ligases | 128 | 65 | 45 |
| No Enzymes | 1625 | 960 | 0 |
Transposable elements
In our study, 1092 transcripts (7.12% of the sialotranscriptome) were annotated as transposable elements (TEs) (Fig. 1a) belonging to different types such as retrovirus-like element (class I) and a group of coding solely for a transposase protein with inverted terminal repeats (class II). Our results revealed the existence of both types of TE retrotransposon (gypsy, bell, outcast, Jockey, L1) and DNA transposon (PiggyBac, Mariner) (Additional file 3: Table S3). TEs are DNA sequences that can be integrated elsewhere in a genome and with few exceptions, have been identified in all eukaryotic genomes sequenced to date [60]. TEs have the potential to provide regulatory and/or protein coding sequences at a new integration site [61] and were described in nearly all published tick sialotranscriptomes.
Secreted proteins
In order to classify annotated transcripts to a secreted class, we referred to a previous catalog of tick proteins [62] and to the recently published tick sialomes [18, 19, 21, 22]. A total of 1749 (11.4%) transcripts were classified further into 11 families (Fig. 2) including enzymes, lipocalin, protease inhibitors, glycine-rich, metastriate specific, immunity-related, mucin, ixodegrin, ixostatin and antigen 5 and other secreted proteins. We also calculated the relative abundance of each secreted protein family in the H. dromedarii sialotranscriptome. All results are discussed in the following sections.
Fig. 2.
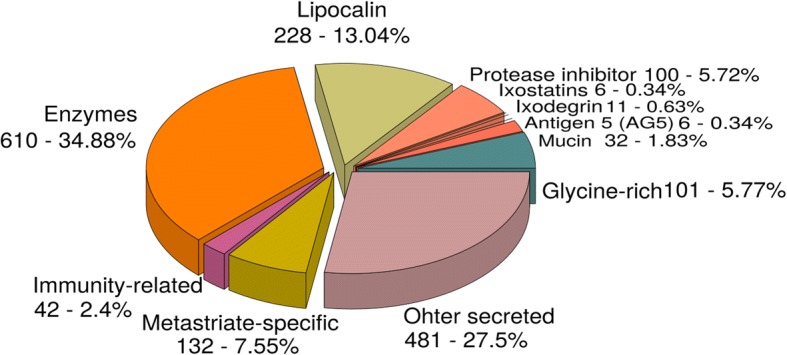
Secreted class of transcripts of the sialotranscriptome of Hyalomma dromedarii
Enzymes
Our results show that enzymes are the most abundant group in the annotated dataset of H. dromedarii: they total 610 (34.9%) transcripts (Fig. 2), including serine protease, metalloprotease (ADAM and M13/neprilysindomain), lipase, endonuclease, 5'-nucleotidase/apyrase, ectonucleotide pyrophosphatase/phosphodiesterase and other proteases. The relative abundance of enzymes was higher for H. dromedarii male SG transcriptome (Fig. 3). During blood-feeding, the mass of tick salivary glands increases due to the proteolytic activation required for countering host mechanism defenses. Indeed, proteases are powerful putative weapons of hematophagous ectoparasites [63]. Although enzymes play a very important role in the success of a tick achieving its blood meal by maintaining the feeding pool, and contribute to keeping the gut contents in liquid form [64], we can make no assumptions about whether the difference in relative abundance reflects differences in the physiological behavior of males and females. Therefore we studied gene expression for each enzyme family separately by calculating Log fold change (LFC) (log2 (TPM female/TPM male)). Relative Abundance for each secreted protein family. To compare relative abundance between females and males a chi-square test was performed given χ2 = 131810, df = 13, and P < 0.001, therefore we can infer that secreted protein profile between genders are statistically different. Details on the most important families are described below.
Fig. 3.

Relative abundance for each secreted protein family
Metalloproteases
Metalloproteases are proteases requiring a metal ion, usually Zn2+, for catalysis [65]. They often have extra domains that can interact with matrix proteins. Their importance and pluripotency makes it unsurprising to find 145 of transcripts (23.8% of enzymes) related to the metalloprotease family compared to other enzymes found in the sialotranscriptome of H. dromedarii. Transcripts of this family were expressed in both male and female ticks. While only 64 transcripts were overexpressed in H. dromedarii male SG, of which 41 had significant LFC, 81 transcripts were highly expressed in female ticks, of which 54 had a significant LFC. This protein family, which is commonly found in tick saliva, salivary glands, ovaries and the mid-gut, plays an important role in countering host inflammation, immunomodulation, fibrinolysis, blood protein digestion, nociception, vitellogenesis, remodeling the extracellular matrix and pathogen transmission [21, 66]. Metalloproteases were characterized in several tick species: I. scapularis, A. americanum, Haemaphysalis longicornis, Rh. microplus and Ornithodoros savignyi [56, 67, 68]. It has also been shown that metalloproteases are involved not only in avoiding host defense mechanisms, but also for spermiogenesis and fertilization [68, 69]. Of metalloproteases, our results showed a total of 9 transcripts from H. dromedarii sialotranscriptome, which was revealed to be members of the ADAM family, of which 6 were expressed exclusively in H. dromedarii male ticks. ADAMs, originally known as MDC proteins (metalloproteinase/disintegrin/cysteine-rich), belong to the Metzincins superfamily of metalloproteases and display a series of biological functions including inhibition of cell adhesion, migration and angiogenesis [70]. It was suggested that members of this family included a sperm surface enzyme important for normal fertility [71]. A total of 38 transcripts of M13/neprilysin were identified in H. dromedarii sialotranscriptome; 20 transcripts were overexpressed in female ticks and 18 in male ticks, of which only 14 had significant LFC. Recent studies have highlighted the involvement of this family in engorgement, tick egg hatching success, and changes in their 16S-rRNA-based microbial loads [72]. These could function by destroying inflammatory peptidic mediators such as cytokines, anaphylatoxins or bradykinin from the hosts [21].
Endonucleases
Our analysis of the H. dromedarii sialotranscriptome revealed 135 transcripts that were assigned to endonucleases, which expressed significantly more in male (76 vs 2) than female salivary glands (Fig. 4). Endonucleases in H. dromedarii might play an important role in destroying neutrophils extracellular traps (NET) and therefore in enhancing parasite infectivity as reported in a previous study [73]. Expression of such enzymes by salivary glands enhances the host-parasite interaction and could be lysosomal or have another housekeeping function [62, 74]. Secreted endonucleases were identified, for the first time, in the mosquito Culex quinquefasciatus salivary glands and proven to play a role in blood-feeding by diffusing pharmacologic components through the host dermis that lowers the viscosity of the lacerated skin matrix [75].
Fig. 4.

Differentially expressed enzyme families in H. dromedarii male and female ticks
Serine proteases
Twenty-one transcripts of serine proteases were found in H. dromedarii sialotranscriptome after our analysis (Fig. 4). Eleven transcripts were overexpressed in male H. dromedarii SG, of which 8 have statistically significant LFC while 10 were expressed exclusively in female SG, of which 8 have statistically significant LFC. Serine protease overexpression in males has been previously reported and these differences have been associated with tick reproductive biology. In fact, males have specific seminal fluid serine proteases that play in important role in spermatozoid survival [76]. Serine proteases interfere in many biological processes including cytogenesis, apoptosis, angiogenesis, neuronal plasticity, zymogen processing, matrix remodeling, immune response, inflammation, blood coagulation, and fibrinolysis [77–79]. Several serine proteases that enable ticks to establish blood pools have been described, such as longistatin in Haemaphysalis longicornis [80]. Serine carboxypeptidase from the tick midgut can hydrolyze bovin hemoglobin supporting serine proteases role in tick feeding success [81]. Serine proteases were also identified in male ticks such as the Rhipicephalus pulchellus and in other arthropods such as Drosophila and bumblebees [22, 69, 70, 82, 83]. They may also play a role in the reproductive biology of ticks [84].
5'-nucleotidase/apyrase
Twelve transcripts were annotated in H. dromedarii as 5'-nucleotidase/apyrases with 3 transcripts overexpressed specifically in each gender (Fig. 4). 5'-nucleotidase/apyrase are very common in the saliva of hematophagous arthropods where they hydrolyze ATP or ADP to AMP. These enzymes can be involved in decreasing local host hemostasis when ADP is released by damaged cells and inhibits host platelet aggregation and inflammation [85]. Previous reports showed that 5'-nucleotidase/apyrase proteins expression decrease significantly after the blood meal, underscoring the important role of these proteins in the tick feeding process [86]. Similarly, in the soft tick Ornithodoros savignyi, apyrases were demonstrated to disaggregate platelets, confirming this protein family’s role in avoiding host platelet aggregation [87].
Protease inhibitors
Protease inhibitors are abundantly expressed in tick sialotranscriptomes as members of a large gene family. In H. dromedarii, 100 transcripts (5.72% of secreted category) were associated with genes encoding for this protein family (Fig. 5). More details are shown below.
Fig. 5.

Differentially expressed protease inhibitor families in H. dromedarii male and female ticks
Kunitz domain-containing proteins
The Kunitz domain is 50–60 amino acid residues and its fold is highly conserved, resembling the first Kunitz-domain protein, the bovine pancreatic trypsin inhibitor (BPTI).
Apart from the serine protease inhibitor function, Kunitz-domain proteins can also inhibit ion channels [88]. Kunitz-domain transcripts are one of the most abundant protein families in tick SGs. They are sub-classified by the number of Kunitz domains in each sequence (i.e. Monolaris, Bilaris, Trilaris). Interestingly, our study shows that 48 transcripts were related to Kunitz domain-containing proteins, of which 17 were Monolaris that were overexpressed exclusively in females (Fig. 6b). These proteins were described as having an anti-thrombin and an anti-factor Xa activity [89, 90]. Several Monolaris proteins were described in ticks, such as tryptogalinin, which may facilitate tick blood-feeding given that it inhibits several serine proteases involved in inflammation and vertebrate immunity [91]. Eight contigs of 17 were chosen for phylogenetic analysis, based on significant amino acid sequence difference, complete domain sequence and high expression values. We distinguished three major groups that share similarities (Fig. 6a). The clade indicated in blue is the most representative and the proteins assigned are mostly multifunctional Kunitz-type inhibitors acting mainly as coagulation enzymes, suggesting that they play an important role in maintaining blood fluidity during feeding of these parasites [92, 93]. The clade indicated in pink contains four contigs of putative Monolaris from H. dromedarii that seems to be exclusive from the genus Hyalomma, and is represented by a potential BPTI-Kunitz (E2J6Q5) [19]. The clade indicated in green contains three contigs of Monolaris from H. dromedarii and a putative Monolaris from Rhipicephalus pulchellus. Apparently, it does not contain molecules that clearly function to inhibit hemostatic processes already described. For a better understanding of the phylogenetic relationships of this family, it would be necessary to carry out further investigations for more species of high throughput data.
Fig. 6.

a Bayesian phylogenetic analysis of putative monolaris based on the Kunitz domain. The sequences from Hyalomma dromedarii were obtained in this study and sequences from other ticks species and vertebrates are indicated and referred to their GenBank accession numbers. b LFC between genders, for Monolaris, only female significant transcripts were found
Our analysis shows that Bilaris, another Kunitz-family proteins, were overexpressed in H. dromedarii female ticks as compared to male ticks (8 vs 2 transcripts). Previous studies showed that three Bilaris proteins (Monobin, Ornithodorin and Savignin) have been characterized in the salivary glands from the soft ticks A. monolakensis, O. moubata and O. savignyi, respectively, and all were thrombin inhibitors [81, 86, 87, 94, 95].
Cystatins
In H. dromedarii sialotranscriptome, 14 transcripts were identified with 9 overexpressed in female and two in males (Fig. 5). This is the largest number of transcripts expressed in a tick species to date [22, 96]. The overexpression in males was attributed to the supposed role played by cystatins in reproduction as they are abundant in seminal fluid [21] but there is no evidence on the targets of the different types of cystatins or their involvement in blood-feeding or other processes. These cysteine protease inhibitors have been found previously in both hard and soft tick sialotranscriptomes and have been detected in several tick tissues [57, 97]. The family comprises large reversible and tight-binding inhibitors of papain-like enzymes and legumain [98]. There are four cystatin subgroups: type 1 (stefins), type 2, type 3 (kininogens) and type 4 cystatins (fetuins) [99]. Tick cystatins are either secreted as immunomodulators into the host with saliva or regulate hemoglobin digestion, which is driven by cathepsins [100]. Most tick cystatin transcripts are conserved across tick species and belong to the extracellular group, which suggests that their role is predominantly immunomodulatory [101].
Madanin
We annotated nine transcripts from H. dromedarii sialotranscriptome, seven of which were highly expressed in female versus two in male. Madanin, isolated for the first time, from the tick Hae. longicornis salivary glands, has an antithrombin activity [102]. This protein family was identified in the sialomes of other Hyalomma species such as H. excavatum and H. rufipes [19, 23]. In addition, variegin and chimadanin were isolated from A. variegatum and Hae. longicornis, respectively, as antithrombin peptides that act directly to inhibit blood clotting, making them very important for the blood-feeding process [103, 104].
Serine protease inhibitors
We identified 19 serpins in the H. dromedarii SG transcriptome of which five were significantly overexpressed in male H. dromedarii ticks and seven in females (Fig. 5). These different levels of expression could be partially explained by the ability of H. dromedarii males to feed on several hosts, require serpins expression that are different from the H. dromedarii female ticks [24]. Due to their abundance in the secretions of several organisms, serine protease inhibitors are the best-characterized family of protease inhibitors [105–107] and able to counterbalance host response to injury by inhibiting clotting and chymase [108, 109].
Lipocalin
Our analysis of H. dromedarii sialotranscriptome showed that 228 (13.04% of secreted class) transcripts were assigned to the lipocalin family, of which 139 were overexpressed in females and 89 in male H. dromedarii (Fig. 7b). Our results corroborate previously published data as lipocalins were found in almost all other tick sialomes for both genders [18, 19, 21, 74]. The widely spread lipocalin family is abundantly expressed in ticks and triatomine insects sialotranscriptomes and belong to a diverse gene family [57]. They are a family of small proteins (∼20 kDa) characterized by an eight-stranded antiparallel β-barrel fold with a repeated +1 topology, typically preceded by a short N-terminal 310-helix and followed by a C-terminal α-helix. They frequently have one or more binding pocket(s) for small molecule ligand(s) [110]. In ticks, lipocalins were assigned to control inflammatory processes and interference with host homeostatic functions [63]. They were also found in nymph and adult tick saliva and are upregulated in response to injury and to viral or bacterial infections [111, 112].
Fig. 7.

Calculated LFC = log2 (TPMfemale / TPM male) of H. dromedarri SG transcripts for different families. a Lipocalin. b Glycine-rich. Only transcripts with absolute value of LFC greater equal to 1 are plotted
Glycine-rich
The present study demonstrated that the sialotranscriptome from H. dromedarii presents 101 transcripts related to glycine-rich proteins (Fig. 7a). A total of 68 transcripts were overexpressed in H. dromedarii female, while only 61, with statistically significant LFC, were overexpressed in male (Fig. 7b). The relative abundance of glycine was higher in female H. dromedarii ticks (41.63%) than in males (11.39%). This high abundance may reflect the fact that female H. dromedarii remain on the host for longer periods, making them more sensitive to removal. They therefore may secrete a cement in response to environmental threats such as host scratching. Glycine-rich proteins containing more than 60% glycine have been found in different tissues from many eukaryotic species [113] and in ticks, glycine-rich with other proteins constitute the cement that enables them to attach to their host [114]. These proteins, which resemble spider silk proteins, have been used as anti-tick vaccines [107–109].
Mucin
Mucins are heavily glycosylated proteins with numerous functions including lubrication, cell signaling and host defense against pathogens [115]. Thirty transcripts of mucin were identified in H. dromedarii sialotranscriptome, which is higher than the number recorded in H. excavatum and lower than in Rhipicephalus pulchellus, which expresses 118 mucin-coding sequences [21, 96]. The involvement of mucins in the feeding process is not well elucidated making it difficult to explain the diversity of encoded mucins. They may function in tick feeding by coating the chitinous feeding mouthparts or the feeding lesion [62].
Metastriate-specific
Proteins belonging to this family are specific to metastriatie ticks; no similar proteins exist in other arthropods. Thirty-five families of proteins totaling 196 sequences were found exclusively in metastriate arthropod genera. We found 132 transcripts related to metastriate-specific proteins, and discuss their sub-families below.
Evasin
Evasins are chemokine-binding proteins (CKBP) that differ from other CKBPs and whose molecular masses range from 7–11 kDa. In this study, 38 transcripts related to evasins were expressed in H. dromedarii SG. Their presence probably reflects the role in the inhibition of the recruitment of immune cells and therefore in reducing the risks that the host rejects the tick. Evasins are expressed more in H. dromedarii SG than in H. excavatum and Rhipicephalus pulchellus but less than in Rh. appendiculatus (34, 22 and 72, respectively). They were isolated from Rh. sanguineus for the first time and were also found in other metastriate sialotranscriptomes [74, 116].
Ixodegrin
Ixodegrin is a cysteine-rich family of proteins that was identified firstly in I. pacificus and I. scapularisticks [56, 117]. This protein family has a predicted RGD or lysine, glycine, aspartic acid (KGD) domain indicative that interferes with fibrinogen binding to platelets [117]. Eighteen transcripts related to ixodegrin were assembled after reads sequencing and genes encoding for this family were expressed almost twice as much in male as in female ticks. As mentioned above, hemostasis starts within seconds of tissue injury and ticks face the launch of blood clotting cascades involving fibrinogen and leading to the platelet plug formation [118]. The expression of this family of proteins would therefore be expected for both genders of H. dromedarii; ixodegrin-like molecules are likely to be present in the saliva to inhibit the formation of platelet clot so as to facilitate the hematophagous feeding of fluid blood.
Da-p36
Our analysis showed that 10 transcripts were exclusively expressed in females of H. dromedarii compared to only one in males. DA-p36 was isolated for the first time from Dermacentor andersoni and it is a 36 kDa immunosuppressive protein that was widely found in metastriate ticks [119]. The presence of this protein family almost exclusively in H. dromadriii ticks can be related to the fact that female ticks are more exposed to host immune system because of their long-term feeding process.
Gene enrichment analysis
In the functional annotation of genes based on KAAS, we identified 121 Metabolic pathways with 2966 transcripts in the Transcriptome Assembly, corresponding to 1710 unique orthologs and 621 enzymes (Table 2). The identification and categorization of The GeneMerge uses a hypergeometric distribution and applies a Bonferroni correction for a more appropriate and significant identification of enriched pathways. The analysis was divided into two sets with a group of 557 differentially expressed genes in females and another group of 353 differentially expressed genes in males. These two sets served as a filter to identfy the most enriched pathways with P-value ≤ 1 × 10-3 and FDR ≤ 1%.
Gene enrichment, KEGG pathway analysis and enzyme classification
After the gene enrichment protocol, 7823 transcripts were identified exclusively in tick males and 4441 in tick females and 54,149 in both males and females. To evaluate the quality and coverage of H. dromedarii transcriptome assembly, we used the CEGMA pipeline to accurately annotate core genes [33], which showed that 248 (100%) of the Core Eukaryotic Genes (CEGs) were identified in the transcriptome and 241 (97.2%) of the CEGs were complete. Using the BUSCO core gene set which is based on orthologous genes from OrthoDBv9 [120], 937 (95.8%) proteins were identified from 978 core genes set, and only 41 conserved genes are missing (Table 3). In order to categorize and identify the biological pathways in H. dromedarii, the assembled contigs were used to obtain the Metabolic Pathways and Enzyme Commission (EC) when annotated against the KEGG database. A total of 2966 transcripts were assigned to 1710 unique KOs, 621 EC (Additional file 4: Table S4), and were summarized the number of specific and common transcripts in males and females in each enzyme classes (Additional file 5: Table S5). The ECs were subsequently grouped into 121 biochemical pathways. Only 4 pathways were not found based on 126 pathways from Ixodes scapularis genome (Glycosphingolipid biosynthesis-globo series, Glycosylphosphatidylinositol (GPI)-anchor biosynthesis, Mucin type O-Glycan biosynthesis, Regulation of autophagy). The enzyme sub-classes were distributed by the number of gender-specific and common transcripts, and the most representative sub-class “2.7 Transferring phosphorus-containing groups” has 224 classified transcripts and one of the most important enzyme sub-classes “3.4 Acting on peptide bonds (peptidases)” has 52 classified transcripts. After the assignment of KEGG pathways annotation to the assembled transcripts, the potential enzymes were further characterized using the predictions of Enzyme Commission (EC) numbers for each transcript (Additional file 5: Table S5). Enzyme classification revealed that transferases are the largest group of H. dromedarii enzymes (40.58%, 252 enzymes), followed by hydrolases (20.93%, 130 enzymes), oxidoreductases (18.84%, 117 enzymes), ligases (8.2%, 51 enzymes), lyases (7.2%, 45 enzymes) and isomerases (4%, 25 enzymes) (Fig. 8). The 1341 sequences having EC numbers were further characterized by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Interestingly, a large number of transcripts were found to be associated with Biosynthesis of antibiotics (197 transcripts), possibly indicating interesting genes for future drug target discovery studies.
Table 3.
Identification of ultra-conserved eukaryotic proteins from BUSCO
| Description | Total | % of total |
|---|---|---|
| Complete single-copy BUSCOs | 525 | 53.7 |
| Complete duplicated BUSCOs | 393 | 40.2 |
| Fragmented BUSCOs | 19 | 1.9 |
| Missing BUSCOs | 41 | 4.2 |
| Total BUSCO groups searched | 978 | – |
Fig. 8.

Enzyme Classification (EC) analysis of the transcriptome of H dromedarii. Number of EC number distribution of H. dromedarii compared with the number of transcripts, KEGG orthologs and enzymes
Conclusions
Transcript expression differed for male and female H. dromedarii ticks, which might be related to their feeding behaviors. The complexity and diversity of H. dromedarii transcriptome corroborated previous studies and may potentially reflect adaptation to the complexity of the host’s defense mechanisms. Our results contribute to the understanding of the tick-host molecules interaction during blood-feeding and the discovery of new pharmacologically active proteins of Hyalomma ticks. Our study has clearly enabled the creation of a database that will serve further proteomic and functional studies. The development of approaches to the identification of tick salivary proteins points the way to several directions in the areas of biomedical, veterinary and pharmacological work identifying vaccine targets that would disrupt the blood meal and/or the transmission of pathogens.
Additional files
Table S1. Annotated transcripts of H. dromedarii salivary glands. (XLSX 17611 kb)
Table S2. Housekeeping class transcripts and their annotations. (XLSX 775 kb)
Table S3.Transposable elements transcripts and their annotations. (XLS 2170 kb)
Table S4. Distribution of putative transcripts in KEGG pathways. (XLSX 18 kb)
Table S5. Enzyme Classification (EC) analysis of the transcriptome of H. dromdarii. Distribution of EC subClasses number in general EC terms compared to the number of specific transcripts in male, female and in common. (XLSX 10 kb)
Acknowledgements
We would like to thank Dr Deborah Glassman for her English corrections and constructive comments on the manuscript. The authors (Milton Yutaka Nishiyama Jr., Flavio Lichtenstein, Ursula Castro de Oliveira, Fernanda Faria, Inácio Loiola Meirelles Junqueira-de-Azevedo and Ana Marisa Chudzinski-Tavassi) received financial support from CAPES (Auxpe-Toxinologia 1207/2011) and FAPESP (2013/07467-1).
Funding
This work was conducted with financial support from the Collaborative internal project PCI-05/2012-2015 IPT.
Availability of data and materials
We deposited raw sequence reads in the NCBI under Bioproject accession number PRJNA358517, BioSample Accession: SAMN06174748. The Short read project were deposited under the SRA Accession: SRR5175867. The Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GFGI00000000. The version described in this paper is the first version, GFGI01000000.
Abbreviations
- ADAM
A disintegrin and metalloprotease
- ADP
Adenosine diphosphate
- AMP
Adenosine monophosphate
- ATDB
Animal Toxin Database
- ATP
Adenosine triphosphate
- BBH
Bi-directional best hit
- BPTI
Bovine pancreatic trypsin inhibitor
- cDNA
complementary DNA
- CEGs
Core eukaryotic genes
- CKBP
Chemokine-binding proteins
- dCAS
Desktop annotation system
- EC
Enzyme Commission
- FPKM
Fragments per kilobase of exon per million fragments mapped
- GO
Gene Ontology
- GPI
Glycosylphosphatidylinositol
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KGD
lysine, glycine, aspartic acid
- KO
KEGG Orthology
- KOG
Eukaryotic ortholog groups
- LFC
Log fold change
- MCMC
Markov chain Monte Carlo
- MDC
Metalloproteinase/disintegrin/cysteine-rich
- mRNA
Messenger RNA
- NCBI
National Center for Biotechnology Information
- NET
Neutrophils extracellular traps
- NGS
Next-generation sequencing
- NR
Non-redundant protein database
- ORF
Open reading frame
- PBS
Phosphate-buffered saline
- RGD
Arginylglycylaspartic acid
- RNAseq
RNA sequencing
- rRNA
Ribosomal RNA
- RT-PCR
Reverse transcription polymerase chain reaction
- SAT
Saliva-assisted transmission
- SG
Salivary glands
- TEs
Transposable elements
- TPM
Transcripts per million
Authors' contributions
CB contributed to the collection and identification of ticks, performing the experiments drafting the manuscript. MYNJR contributed in the data analysis and drafting the paper. CBH contributed in the data analysis. FL contributed in the data analysis. UCO contributed in the performing of the molecular experiment and phylogenetic analysis. FF contributed to the design of work, supervision of the work and correction of the manuscript. IMLJA contributed to the design and the supervision of the work. KG contributed to the supervision of the work. AB contributed to correction of the manuscript. YM contributed to the design and supervision of the work and correction of the manuscript. AMC contributed to the supervision of the work. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Commission on Ethics and Animal Welfare of the Institute Pasteur of Tunis - University of Tunis El Manar, Tunisia, with the given number IPT/LR03/Project PCI/05/2012. All technical procedures were in accordance with the National and the European legislation regarding animal welfare and have met the International Guiding Principles for Biomedical Research Involving Animals by the Council for the International Organizations of Medical Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13071-018-2874-9) contains supplementary material, which is available to authorized users.
Contributor Information
Chaima Bensaoud, Email: bensaoud.chaima@gmail.com.
Milton Yutaka Nishiyama, Jr, Email: yutakajr@gmail.com.
Cherif Ben Hamda, Email: cherifbenhamda@gmail.com.
Flavio Lichtenstein, Email: flalix@gmail.com.
Ursula Castro de Oliveira, Email: ursula.castrolive@gmail.com.
Fernanda Faria, Email: fernanda.faria@butantan.gov.br.
Inácio Loiola Meirelles Junqueira-de-Azevedo, Email: inacio.azevedo@butantan.gov.br.
Kais Ghedira, Email: ghedirakais@gmail.com.
Ali Bouattour, Email: ali.bouattour@pasteur.tn.
Youmna M’Ghirbi, Email: youmna.mgh@gmail.com.
Ana Marisa Chudzinski-Tavassi, Email: ana.chudzinski@butantan.gov.br.
References
- 1.Kocan KM, Blouin EF, Barbet AF. Anaplasmosis control: Past, present, and future. Ann N Y Acad Sci. 2000;916:501–509. doi: 10.1111/j.1749-6632.2000.tb05329.x. [DOI] [PubMed] [Google Scholar]
- 2.Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(Suppl.):S3–14. [DOI] [PubMed]
- 3.Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, et al. Update on tick-borne rickettsioses around the world: a geographic approach. Clin Microbiol Rev. 2013;26:657–702. doi: 10.1128/CMR.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brackney DE, Armstrong PM. Transmission and evolution of tick-borne viruses. Curr Opin Virol. 2016;21:67–74. doi: 10.1016/j.coviro.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Caimano MJ, Drecktrah D, Kung F, Samuels DS. Interaction of the Lyme disease spirochete with its tick vector. Cell Microbiol. 2016;18:919–927. doi: 10.1111/cmi.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbas RZ, Zaman MA, Colwell DD, Gilleard J, Iqbal Z. Acaricide resistance in cattle ticks and approaches to its management: the state of play. Vet Parasitol. 2014;203:6–20. doi: 10.1016/j.vetpar.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Domingos A, Antunes S, Borges L, Rosario VE, Domingos A, Antunes S, et al. Approaches towards tick and tick-borne diseases control. Rev Soc Bras Med Trop. 2013;46:265–269. doi: 10.1590/0037-8682-0014-2012. [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, Bruno JF, Luft BJ. Identification of novel tick salivary gland proteins for vaccine development. Biochem Biophys Res Commun. 2005;326:901–904. doi: 10.1016/j.bbrc.2004.11.127. [DOI] [PubMed] [Google Scholar]
- 9.Kazimírová M, Stibrániová I. Tick salivary compounds: their role in modulation of host defences and pathogen transmission. Front Cell Infect Microbiol. 2013;3:43. doi: 10.3389/fcimb.2013.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nuttall PA, Labuda M. Saliva-assisted transmission of tick-borne pathogens. In: Bowman AS, Nuttall PA, editors. Ticks. Cambridge: Cambridge University Press; 2008. pp. 205–219. [Google Scholar]
- 11.Zheng H, Yu Z, Chen Z, Zhou L, Zheng B, Ma H, et al. Development and biological characteristics of Haemaphysalis longicornis (Acari: Ixodidae) under field conditions. Exp Appl Acarol. 2011;53:377–388. doi: 10.1007/s10493-010-9415-3. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Li Y, Liu Z, Yang J, Yin H. The life cycle of Hyalomma rufipes (Acari: Ixodidae) under laboratory conditions. Exp Appl Acarol. 2012;56:85–92. doi: 10.1007/s10493-011-9490-0. [DOI] [PubMed] [Google Scholar]
- 13.Wespiser AR, Sánchez-Gracia A, Nuss AB, Walenz BP, Bissinger BW, Birren B, et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat Commun. 2016;7:10507. doi: 10.1038/ncomms10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chmelař J, Kotál J, Karim S, Kopacek P, Francischetti IMB, Pedra JHF, et al. Sialomes and Mialomes: a systems-biology view of tick tissues and tick-host interactions. Trends Parasitol. 2016;32:242–254. doi: 10.1016/j.pt.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins LJ, Biggs PJ, Voelckel C, Joly S. An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Inform. 2008;21:3–14. [PubMed] [Google Scholar]
- 16.Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ribeiro JMC, Alarcon-Chaidez F, Francischetti IMB, Mans BJ, Mather TN, Valenzuela JG, et al. An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochem Mol Biol. 2006;36:111–129. doi: 10.1016/j.ibmb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Anatriello E, Ribeiro JM, de Miranda-Santos IK, Brandão LG, Anderson JM, Valenzuela JG, et al. An insight into the sialotranscriptome of the brown dog tick, Rhipicephalus sanguineus. BMC Genomics. 2010;11:450. doi: 10.1186/1471-2164-11-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francischetti IMB, Anderson JM, Manoukis N, Pham VM, Ribeiro JMC. An insight into the sialotranscriptome and proteome of the coarse bontlegged tick, Hyalomma marginatum rufipes. J Proteomics. 2011;74:2892–2908. doi: 10.1016/j.jprot.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia GR, Gardinassi LG, Ribeiro JM, Anatriello E, Ferreira BR, Moreira HNS, et al. The sialotranscriptome of Amblyomma triste, Amblyomma parvum and Amblyomma cajennense ticks, uncovered by 454-based RNA-seq. Parasit Vectors. 2014;7:430. doi: 10.1186/1756-3305-7-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan AWL, Francischetti IMB, Slovak M, Manjunatha KR, Ribeiro JMC. Sexual differences in the sialomes of the zebra tick, Rhipicephalus pulchellus. J Proteomics. 2015;117:120–144. doi: 10.1016/j.jprot.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Castro MH, de Klerk D, Pienaar R, Latif AA, Rees DJG, Mans BJ. De novo assembly and annotation of the salivary gland transcriptome of Rhipicephalus appendiculatus male and female ticks during blood feeding. Ticks Tick Borne Dis. 2016;7:536–548. doi: 10.1016/j.ttbdis.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Ribeiro JMC, Slovák M, Francischetti IMB. An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis. 2016;8:201–207. doi: 10.1016/j.ttbdis.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoogstraal H. African Ixodoidea. l. Ticks of the Sudan. United States. Naval Medical Research Unit: Washington; 1956. [Google Scholar]
- 25.Apanaskevich DA, Schuster AL, Horak IG. The genus Hyalomma: VII. Redescription of all parasitic stages of H.(Euhyalomma) dromedarii and H. (E.) schulzei (Acari: Ixodidae) J Med Entomol. 2008;45:817–831. doi: 10.1093/jmedent/45.5.817. [DOI] [PubMed] [Google Scholar]
- 26.Chisholm K, Dueger E, Fahmy NT, Samaha HAT, Zayed A, Abdel-Dayem M, et al. Crimean-Congo hemorrhagic fever virus in ticks from imported livestock, Egypt. Emerg Infect Dis. 2012;18:181–182. doi: 10.3201/eid1801.111071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.ElGhali A, Hassan SM. Life cycle of the camel tick Hyalomma dromedarii (Acari: Ixodidae) under field conditions in Northern Sudan. Vet Parasitol. 2010;174:305–312. doi: 10.1016/j.vetpar.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 28.Bouattour A. Cle dichotomique et identification des tiques (Acari: Ixodidae) parasites du betail au Maghreb. Arch Inst Pasteur Tunis. 2002;79:43–50. [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aronesty E. Comparison of sequencing utility programs. Open Bioinforma J. 2013;7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 31.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 33.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 34.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 35.Altschul S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He Q-Y, He Q-Z, Deng X-C, Yao L, Meng E, Liu Z-H, et al. ATDB: a uni-database platform for animal toxins. Nucleic Acids Res. 2008;36:D293–D297. doi: 10.1093/nar/gkm832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 39.Guo Y, Ribeiro JMC, Anderson JM, Bour S. dCAS: a desktop application for cDNA sequence annotation. Bioinformatics. 2009;25:1195–1196. doi: 10.1093/bioinformatics/btp129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bateman A. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schultz J. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc. 2013;8:1494–512. [DOI] [PMC free article] [PubMed]
- 44.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 46.Gouy M, Guindon S, Gascuel O. SeaView Version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 47.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 48.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guillou L, Bachar D, Audic S, Bass D, Berney C, Bittner L, et al. The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013;41:D597–D604. doi: 10.1093/nar/gks1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.R: The R Project for Statistical Computing. https://www.r-project.org/. Accessed 28 Nov 2017.
- 53.Wickham H. ggplot2 - Elegant Graphics for Data Analysis. 2009. https://www.springer.com/us/book/9780387981413. Accessed 28 Nov 2017.
- 54.Castillo-Davis CI, Hartl DL. GeneMerge - post-genomic analysis, data mining, and hypothesis testing. Bioinformatics. 2003;19:891–2. [DOI] [PubMed]
- 55.Batista IF, Chudzinski-Tavassi AM, Faria F, Simons SM, Barros-Batestti DM, Labruna MB, et al. Expressed sequence tags (ESTs) from the salivary glands of the tick Amblyomma cajennense (Acari: Ixodidae) Toxicon. 2008;51:823–834. doi: 10.1016/j.toxicon.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 56.Valenzuela JG, Francischetti IMB, Pham VM, Garfield MK, Mather TN, Ribeiro JMC. Exploring the sialome of the tick Ixodes scapularis. J Exp Biol. 2002;205:2843–2864. doi: 10.1242/jeb.205.18.2843. [DOI] [PubMed] [Google Scholar]
- 57.Karim S, Singh P, Ribeiro JMC. A deep insight into the sialotranscriptome of the Gulf Coast tick, Amblyomma maculatum. PLoS One. 2011;6:e28525. doi: 10.1371/journal.pone.0028525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Radisky DC, Stallings-Mann M, Hirai Y, Bissell MJ. Single proteins might have dual but related functions in intracellular and extracellular microenvironments. Nat Rev Mol Cell Biol. 2009;10:228–234. doi: 10.1038/nrm2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tirloni L, Reck J, Terra RMS, Martins JR, Mulenga A, Sherman NE, et al. Proteomic analysis of cattle tick Rhipicephalus (Boophilus) microplus saliva: a comparison between partially and fully engorged females. PLoS One. 2014;9:e94831. doi: 10.1371/journal.pone.0094831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang CRL, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet. 2012;46:651–675. doi: 10.1146/annurev-genet-110711-155616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016;351:aac7247. doi: 10.1126/science.aac7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Francischetti IMB, Sa-Nunes A, Mans BJ, Santos IM, Ribeiro JMC. The role of saliva in tick feeding. Front Biosci. 2009;14:2051–2088. doi: 10.2741/3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beaufays J, Adam B, Menten-Dedoyart C, Fievez L, Grosjean A, Decrem Y, et al. Ir-LBP, an Ixodes ricinus tick salivary LTB4-binding lipocalin, interferes with host neutrophil function. PLoS One. 2008;3:e3987. doi: 10.1371/journal.pone.0003987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steen NA, Barker SC, Alewood PF. Proteins in the saliva of the Ixodida (ticks): pharmacological features and biological significance. Toxicon. 2006;47:1–20. [DOI] [PubMed]
- 65.Rawlings ND, Barrett AJ. Evolutionary families of metallopeptidases. Methods Enzymol. 1995;248:183–228. doi: 10.1016/0076-6879(95)48015-3. [DOI] [PubMed] [Google Scholar]
- 66.Ali A, Khan S, Ali I, Karim S, da Silva VI, Termignoni C. Probing the functional role of tick metalloproteases. Physiol Entomol. 2015;40:177–188. doi: 10.1111/phen.12104. [DOI] [Google Scholar]
- 67.Ribeiro JMC, Mather TN. Ixodes scapularis: salivary kininase activity is a metallo dipeptidyl carboxypeptidase. Exp Parasitol. 1998;89:213–221. doi: 10.1006/expr.1998.4296. [DOI] [PubMed] [Google Scholar]
- 68.Packila M, Guilfoile PG. Mating, male Ixodes scapularis express several genes including those with sequence similarity to immunoglobulin-binding proteins and metalloproteases. Exp Appl Acarol. 2002;27:151–160. doi: 10.1023/A:1021507404506. [DOI] [PubMed] [Google Scholar]
- 69.Ali A, Tirloni L, Isezaki M, Seixas A, Konnai S, Ohashi K, et al. Reprolysin metalloproteases from Ixodes persulcatus, Rhipicephalus sanguineus and Rhipicephalus microplus ticks. Exp Appl Acarol. 2014;63:559–578. doi: 10.1007/s10493-014-9796-9. [DOI] [PubMed] [Google Scholar]
- 70.Giebeler N, Zigrino P. A Disintegrin and Metalloprotease (ADAM): historical overview of their functions. Toxins. 2016;8:122. [DOI] [PMC free article] [PubMed]
- 71.Zhu G-Z, Gupta S, Myles DG, Primakoff P. Testase 1 (ADAM 24) a sperm surface metalloprotease is required for normal fertility in mice. Mol Reprod Dev. 2009;76:1106–1114. doi: 10.1002/mrd.21076. [DOI] [PubMed] [Google Scholar]
- 72.Bullard RL, Williams J, Karim S. Temporal gene expression analysis and rna silencing of single and multiple members of gene family in the lone star tick Amblyomma americanum. PLoS One. 2016;11:e0147966. doi: 10.1371/journal.pone.0147966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chagas AC, Oliveira F, Debrabant A, Valenzuela JG, Ribeiro JMC, Calvo E. Lundep, a sand fly salivary endonuclease increases Leishmania parasite survival in neutrophils and inhibits XIIa contact activation in human plasma. PLoS Pathog. 2014;10:e1003923. doi: 10.1371/journal.ppat.1003923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ribeiro JM, Anderson JM, Manoukis NC, Meng Z, Francischetti IM. A further insight into the sialome of the tropical bont tick, Amblyomma variegatum. BMC Genomics. 2011;12:136. doi: 10.1186/1471-2164-12-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Calvo E, Ribeiro JMC. A novel secreted endonuclease from Culex quinquefasciatus salivary glands. J Exp Biol. 2006;209:2651–2659. doi: 10.1242/jeb.02267. [DOI] [PubMed] [Google Scholar]
- 76.Veveris-Lowe TL, Kruger SJ, Walsh T, Gardiner RA, Clements JA. Seminal fluid characterization for male fertility and prostate cancer: kallikrein-related serine proteases and whole proteome approaches. Semin Thromb Hemost. 2007;33:87–99. doi: 10.1055/s-2006-958467. [DOI] [PubMed] [Google Scholar]
- 77.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci USA. 1999;96:14694–9. [DOI] [PMC free article] [PubMed]
- 78.Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307–321. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 79.Ekici ÖD, Paetzel M, Dalbey RE. Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17:2023–2037. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anisuzzaman, Islam MK, Alim MA, Miyoshi T, Hatta T, Yamaji K, et al. Longistatin is an unconventional serine protease and induces protective immunity against tick infestation. Mol Biochem Parasitol. 2012;182:45–53. [DOI] [PubMed]
- 81.Motobu M, Tsuji N, Miyoshi T, Huang X, Islam MK, Alim MA, et al. Molecular characterization of a blood-induced serine carboxypeptidase from the ixodid tick Haemaphysalis longicornis. FEBS J. 2007;274:3299–3312. doi: 10.1111/j.1742-4658.2007.05852.x. [DOI] [PubMed] [Google Scholar]
- 82.LaFlamme BA, Ravi Ram K, Wolfner MF. The Drosophila melanogaster seminal fluid protease “seminase” regulates proteolytic and post-mating reproductive processes. PLoS Genet. 2012;8:e1002435. doi: 10.1371/journal.pgen.1002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brabcová J, Kindl J, Valterová I, Pichová I, Zarevúcka M, Brabcová J, et al. Serine protease from midgut of Bombus terrestris males. Arch Insect Biochem Physiol. 2013;82:117–128. doi: 10.1002/arch.21075. [DOI] [PubMed] [Google Scholar]
- 84.Kiszewski AE, Matuschka FR, Spielman A. Mating strategies and spermiogenesis in ixodid ticks. Annu Rev Entomol. 2001;46:167–182. doi: 10.1146/annurev.ento.46.1.167. [DOI] [PubMed] [Google Scholar]
- 85.Ribeiro JM. Blood-feeding arthropods: live syringes or invertebrate pharmacologists? Infect Agents Dis. 1995;4:143–152. [PubMed] [Google Scholar]
- 86.Sor-suwan S, Jariyapan N, Roytrakul S, Paemanee A, Phumee A, Phattanawiboon B, et al. Identification of salivary gland proteins depleted after blood feeding in the malaria vector Anopheles campestris-like mosquitoes (Diptera: Culicidae) PLoS One. 2014;9:e90809. doi: 10.1371/journal.pone.0090809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mans BJ, Coetzee J, Louw AI, Gaspar AR, Neitz AW. Disaggregation of aggregated platelets by apyrase from the tick, Ornithodoros savignyi (Acari: Argasidae) Exp Appl Acarol. 2000;24:271–282. doi: 10.1023/A:1006440714276. [DOI] [PubMed] [Google Scholar]
- 88.Lucchesi KJ, Moczydlowski E. On the interaction of bovine pancreatic trypsin inhibitor with maxi Ca(2+)-activated K+ channels. A model system for analysis of peptide-induced subconductance states. J Gen Physiol. 1991;97:1295–1319. doi: 10.1085/jgp.97.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Waxman L, Smith D, Arcuri K, Vlasuk G. Tick anticoagulant peptide (TAP) is a novel inhibitor of blood coagulation factor Xa. Science. 1990;248:593–596. doi: 10.1126/science.2333510. [DOI] [PubMed] [Google Scholar]
- 90.Mans BJ, Louw AI, Neitz AWH. Amino acid sequence and structure modeling of savignin, a thrombin inhibitor from the tick, Ornithodoros savignyi. Insect Biochem Mol Biol. 2002;32:821–828. doi: 10.1016/S0965-1748(01)00169-2. [DOI] [PubMed] [Google Scholar]
- 91.Valdés JJ, Schwarz A, Cabeza de Vaca I, Calvo E, JHF P, Guallar V, et al. Tryptogalinin is a tick Kunitz serine protease inhibitor with a unique intrinsic disorder. PLoS One. 2013;8:e62562. doi: 10.1371/journal.pone.0062562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Assumpção TC, Ma D, Mizurini DM, Kini RM, Ribeiro JMC, Kotsyfakis M, et al. In vitro mode of action and anti-thrombotic activity of boophilin, a multifunctional Kunitz protease inhibitor from the midgut of a tick vector of babesiosis, Rhipicephalus microplus. PLoS Negl Trop Dis. 2016;10:e0004298. [DOI] [PMC free article] [PubMed]
- 93.Liao M, Zhou J, Gong H, Boldbaatar D, Shirafuji R, Battur B, et al. Hemalin, a thrombin inhibitor isolated from a midgut cDNA library from the hard tick Haemaphysalis longicornis. J Insect Physiol. 2009;55:164–173. doi: 10.1016/j.jinsphys.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 94.Am J, Louw AI, Joubert F, Neitz AW. Cloning, nucleotide sequence and expression of the gene encoding factor Xa inhibitor from the salivary glands of the tick, Ornithodoros savignyi. Exp Appl Acarol. 1998;22:603–619. doi: 10.1023/A:1006198713791. [DOI] [PubMed] [Google Scholar]
- 95.Mans BJ, Andersen JF, Schwan TG, Ribeiro JM. Characterization of anti-hemostatic factors in the argasid, Argas monolakensis: implications for the evolution of blood-feeding in the soft tick family. Insect Biochem Mol Biol. 2008;38:22–41. doi: 10.1016/j.ibmb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.JMC R, Slovák M, IMB F. An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis. 2017;8:201–207. doi: 10.1016/j.ttbdis.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schwarz A, Valdés JJ, Kotsyfakis M. The role of cystatins in tick physiology and blood feeding. Ticks Tick Borne Dis. 2012;3:117–127. doi: 10.1016/j.ttbdis.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Abrahamson M, Alvarez-Fernandez M, Nathanson C-M. Cystatins. Biochem Soc Symp. 2003;70:179–199. doi: 10.1042/bss0700179. [DOI] [PubMed] [Google Scholar]
- 99.Rawlings ND, Barrett AJ. Evolution of proteins of the cystatin superfamily. J Mol Evol. 1990;30:60–71. doi: 10.1007/BF02102453. [DOI] [PubMed] [Google Scholar]
- 100.Horn M, Nussbaumerová M, Sanda M, Kovárová Z, Srba J, Franta Z, et al. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol. 2009;16:1053–1063. doi: 10.1016/j.chembiol.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ibelli AMG, Hermance MM, Kim TK, Gonzalez CL, Mulenga A. Bioinformatics and expression analyses of the Ixodes scapularis tick cystatin family. Exp Appl Acarol. 2013;60:41–53. doi: 10.1007/s10493-012-9613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Iwanaga S, Okada M, Isawa H, Morita A, Yuda M, Chinzei Y. Identification and characterization of novel salivary thrombin inhibitors from the ixodidae tick, Haemaphysalis longicornis. Eur J Biochem. 2003;270:1926–1934. doi: 10.1046/j.1432-1033.2003.03560.x. [DOI] [PubMed] [Google Scholar]
- 103.Mulenga A, Sugino M, Nakajima M, Sugimoto C, Onuma M. Tick-encoded serine proteinase inhibitors (serpins); potential target antigens for tick vaccine development. J Vet Med Sci. 2001;63:1063–1069. doi: 10.1292/jvms.63.1063. [DOI] [PubMed] [Google Scholar]
- 104.Koh CY, Kazimirova M, Trimnell A, Takac P, Labuda M, Nuttall PA, et al. Variegin, a novel fast and tight binding thrombin inhibitor from the tropical bont tick. J Biol Chem. 2007;282:29101–29113. doi: 10.1074/jbc.M705600200. [DOI] [PubMed] [Google Scholar]
- 105.Mourão CBF, Schwartz EF. Protease inhibitors from marine venomous animals and their counterparts in terrestrial venomous animals. Mar Drugs. 2013;11:2069–2112. doi: 10.3390/md11062069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chalaire KC, Kim TK, Garcia-Rodriguez H, Mulenga A. Amblyomma americanum (L.) (Acari: Ixodidae) tick salivary gland serine protease inhibitor (serpin) 6 is secreted into tick saliva during tick feeding. J Exp Biol. 2011;214:665–673. doi: 10.1242/jeb.052076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Espinoza-Gomez F, Newton-Sanchez O, Flores-Cazares G, De la Cruz-Ruiz M, Melnikov V, Austria-Tejeda J, et al. Tick paralysis caused by Amblyomma maculatum on the Mexican Pacific Coast. Vector Borne Zoonotic Dis. 2011;11:945–946. doi: 10.1089/vbz.2010.0154. [DOI] [PubMed] [Google Scholar]
- 108.Meekins DA, Kanost MR, Michel K. Serpins in arthropod biology. Semin Cell Dev Biol. 2017;62:105–119. doi: 10.1016/j.semcdb.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chmelar J, Oliveira CJ, Rezacova P, Francischetti IMB, Kovarova Z, Pejler G, et al. A tick salivary protein targets cathepsin G and chymase and inhibits host inflammation and platelet aggregation. Blood. 2011;117:736–744. doi: 10.1182/blood-2010-06-293241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Flower DR. The lipocalin protein family: structure and function. Biochem J. 1996;318:1–14. doi: 10.1042/bj3180001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim HJ, Je HJ, Cheon HM, Kong SY, Han J, Yun CY, et al. Accumulation of 23kDa lipocalin during brain development and injury in Hyphantria cunea. Insect Biochem Mol Biol. 2005;35:1133–1141. doi: 10.1016/j.ibmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 112.Konnai S, Nishikado H, Yamada S, Imamura S, Ito T, Onuma M, et al. Molecular identification and expression analysis of lipocalins from blood-feeding taiga tick, Ixodes persulcatus Schulze. Exp Parasitol. 2011;127:467–474. doi: 10.1016/j.exppara.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 113.Mousavi A, Hotta Y. Glycine-rich proteins: a class of novel proteins. Appl Biochem Biotechnol. 2005;120:169–174. doi: 10.1385/ABAB:120:3:169. [DOI] [PubMed] [Google Scholar]
- 114.Maruyama SR, Anatriello E, Anderson JM, Ribeiro JM, Brandão LG, Valenzuela JG, et al. The expression of genes coding for distinct types of glycine-rich proteins varies according to the biology of three metastriate ticks, Rhipicephalus (Boophilus) microplus, Rhipicephalus sanguineus and Amblyomma cajennense. BMC Genomics. 2010;11:363. doi: 10.1186/1471-2164-11-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Belley A, Keller K, Göttke M, Chadee K, Göettke M. Intestinal mucins in colonization and host defense against pathogens. Am J Trop Med Hyg. 1999;60:10–15. doi: 10.4269/ajtmh.1999.60.10. [DOI] [PubMed] [Google Scholar]
- 116.Vancová I, Hajnická V, Slovák M, Kocáková P, Paesen GC, Nuttall PA. Evasin-3-like anti-chemokine activity in salivary gland extracts of ixodid ticks during blood-feeding: a new target for tick control. Parasite Immunol. 2010;32:460–463. doi: 10.1111/j.1365-3024.2010.01203.x. [DOI] [PubMed] [Google Scholar]
- 117.Francischetti IMB, Pham VM, Mans BJ, Andersen JF, Mather TN, Lane RS, et al. The transcriptome of the salivary glands of the female western black-legged tick Ixodes pacificus (Acari: Ixodidae) Insect Biochem Mol Biol. 2005;35:1142–1161. doi: 10.1016/j.ibmb.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22:1381–1389. doi: 10.1161/01.ATV.0000031340.68494.34. [DOI] [PubMed] [Google Scholar]
- 119.Bergman DK, Palmer MJ, Caimano MJ, Radolf JD, Wikel SK. Isolation and molecular cloning of a secreted immunosuppressant protein from Dermacentor andersoni salivary gland. J Parasitol. 2000;86:516–525. doi: 10.1645/0022-3395(2000)086[0516:IAMCOA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 120.Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41:D358–D365. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Annotated transcripts of H. dromedarii salivary glands. (XLSX 17611 kb)
Table S2. Housekeeping class transcripts and their annotations. (XLSX 775 kb)
Table S3.Transposable elements transcripts and their annotations. (XLS 2170 kb)
Table S4. Distribution of putative transcripts in KEGG pathways. (XLSX 18 kb)
Table S5. Enzyme Classification (EC) analysis of the transcriptome of H. dromdarii. Distribution of EC subClasses number in general EC terms compared to the number of specific transcripts in male, female and in common. (XLSX 10 kb)
Data Availability Statement
We deposited raw sequence reads in the NCBI under Bioproject accession number PRJNA358517, BioSample Accession: SAMN06174748. The Short read project were deposited under the SRA Accession: SRR5175867. The Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GFGI00000000. The version described in this paper is the first version, GFGI01000000.