

Abstract
Background
Developmental delay (DD) and intellectual disability (ID) are frequently associated with a broad spectrum of additional phenotypes. Chromosomal microarray analysis (CMA) has been recommended as a first-tier test for DD/ID in general, whereas the diagnostic yield differs significantly among DD/ID patients with different comorbid conditions.
Methods
To investigate the genotype-phenotype correlation, we examined the characteristics of identified pathogenic copy number variations (pCNVs) and compared the diagnostic yields among patient subgroups with different co-occurring conditions.
Results
This study is a retrospective review of CMA results generated from a mixed cohort of 710 Chinese patients with DD/ID. A total of 247 pCNVs were identified in 201 patients (28%). A large portion of these pCNVs were copy number losses, and the size of copy number losses was generally smaller than gains. The diagnostic yields were significantly higher in subgroups with co-occurring congenital heart defects (55%), facial dysmorphism (39%), microcephaly (34%) or hypotonia (35%), whereas co-occurring conditions of skeletal malformation (26%), brain malformation (24%) or epilepsy (24%) did not alter the yield. In addition, the diagnostic yield nominally correlated with ID severity.
Conclusion
Varied yields exist in DD/ID patients with different phenotypic presentation. The presence of comorbid conditions can be among factors to consider when planning CMA.
Electronic supplementary material
The online version of this article (10.1186/s12920-018-0368-4) contains supplementary material, which is available to authorized users.
Keywords: Chromosomal microarray, Developmental delay, Intellectual disability, Pathogenic copy number variations, Comorbid conditions
Background
Developmental delay (DD) and intellectual disability (ID) are estimated to affect ~ 1% of the children across the world [1]. Genetic factors play a major part in DD/ID (up to ~ 47.5%) [2]. Identifying the genetic cause is crucial for accurate etiological diagnosis and refined clinical management. Chromosomal microarray analysis (CMA) has been recommended as a first-tier genetic test for unexplained DD/ID and congenital malformations [3, 4]. The reported diagnostic yields of clinical CMA vary between 12 and 20%, depending on the population and methods used [3, 5]. A wide spectrum of phenotypes can be present in the DD/ID cohorts, including different degrees of ID severity [1] and co-occurrence of other conditions, like epilepsy, autism or dysmorphic features [6]. The diagnostic yields in subgroups of patients with different clinical manifestation are not clear yet. Further assessment of the diagnostic yields in DD/ID patients with different ID severity and co-occurring condition is desired, for it could offer clinicians the phenotypic clues of pathogenic copy number variations (CNVs).
In this study, we reviewed CMA results generated from a Chinese cohort of 710 patients with DD/ID as the main manifestation. We characterized the property and physical distribution of pathogenic CNVs (pCNVs), and compared the yield of CMA among patients with different ID severity and comorbid conditions. Together we delineated the genotypes, diagnostic yields and phenotypes in a DD/ID cohort with heterogeneous manifestations.
Methods
Patients
This is a retrospective study conducted in Endocrine and Genetic Department, Xinhua Hospital and Shanghai Children’s Medical Center, China. CMA results from 710 patients (432 males and 278 females, age range from 1 month to 29 years old, average 4.2 years old, visited the clinic during the period of March 2011 to February 2016) were reviewed. Two criteria were met for inclusion in this study: 1) individuals who presented DD/ID as the main manifestation, with or without additional features such as congenital heart defects (CHD), autism, dysmorphism et al.; 2) individuals with whole-genome microarray analysis done. The exclusion criteria were individuals who had central nervous system infection, brain injury or intracranial tumor. This study was reviewed and approved by the ethical committee of Xinhua hospital and Shanghai Children’s Medical Center, China, and informed consent was obtained from the patients or parents (for patients under 18 years old).
The phenotypic information of patients were assessed routinely in the clinic, including family history, pre/peri-natal history, physical examination, standardized measure of intelligence/development, instrumental evaluations (brain MRI, EEG, ultrasound etc.). These clinical records were collected, categorized and accessed electronically in this study. Patient were classified based on ID severity: mild (IQ level 55–70), moderate (IQ level 40–55), severe (IQ level 25–40) and profound ID (IQ level below 25), when the standardized measure of intelligence was valid and available. For comparison of the diagnostic yields in Table 1.II/III, only patients with clinical record indicating negative for the specific condition is counted as “without select condition”. For example, in the group of “abnormal blood biochemistry”, only patients who went through tandem mass spectrometry of blood samples with a normal biochemical profile returned were counted as “without select condition”. For the groups of kidney/urinary tract, gastrointestinal or respiratory tract anomalies, clinicians did not perform detailed examination of these systems in most patients, thus we did not conduct statistical analysis in these groups. For “pre−/peri-natal problems”, conditions included intrauterine growth retardation, pre- or post-term birth and low APGAR score etc. “Family history” was considered positive when first- or second-degree relative with DD/ID was reported. For “karyotypical abnormalities”, patients with abnormal results of karyotype were counted as “with select condition”. These patients were included as they comprised a portion of DD/ID patients referred to CMA, with the main purpose to confirm the chromosomal abnormality and further delineate the spanning of gain or loss.
Table 1.
Diagnostic yields in patients categorized by ID severity and co-occurring conditions
| I. Based on ID severity | ||||||||
| total | P# | Yield | P-value | |||||
| Mild | 140 | 27 | 19% | 0.08 | ||||
| Moderate | 105 | 23 | 22% | |||||
| Severe | 85 | 28 | 33% | |||||
| Profound | 15 | 2 | 13% | |||||
| Not categorized | 365 | 121 | 33% | / | ||||
| II. Based on co-occurring conditions present in > 50 patients, with statistical analysis performed | ||||||||
| With select condition | Without select condition | Odds ratio | P-value | |||||
| total | P# | Yield | total | P# | Yield | |||
| SSa | 201 | 65 | 32% | 136 | 30 | 22% | 1.69 | 0.048 |
| CHDb | 98 | 54 | 55% | 110 | 20 | 18% | 5.52 | 2.66E-08 |
| Gonadal dysplasia | 50 | 18 | 36% | 103 | 20 | 19% | 2.33 | 0.03 |
| Skeletal malformation | 62 | 16 | 26% | 24 | 3 | 13% | 2.44 | 0.251 |
| Facial dysmorphism | 201 | 79 | 39% | 194 | 39 | 20% | 2.57 | 4.27E-05 |
| Microcephaly | 128 | 43 | 34% | 140 | 25 | 18% | 2.33 | 0.0033 |
| Brain malformation | 147 | 36 | 24% | 148 | 29 | 20% | 1.33 | 0.328 |
| Epilepsy | 62 | 15 | 24% | 115 | 23 | 20% | 1.28 | 0.567 |
| Hypotonia | 54 | 19 | 35% | 113 | 18 | 16% | 2.87 | 0.00613 |
| Pre/peri-natal problems | 160 | 50 | 31% | 234 | 50 | 21% | 1.67 | 0.034 |
| SS + dysmorphism | 79 | 30 | 38% | 71 | 9 | 13% | 4.22 | 6.70E-04 |
| SS + microcephaly | 66 | 25 | 38% | 74 | 14 | 19% | 2.61 | 0.015 |
| SS + brain malformation | 56 | 17 | 30% | 45 | 9 | 20% | 1.74 | 0.261 |
| CHD + dysmorphism | 50 | 31 | 62% | 61 | 8 | 13% | 10.81 | 9.10E-08 |
| III. Based on co-occurring conditions present in < 50 patients | ||||||||
| total | P# | Yield | total | P# | Yield | |||
| Family history | 38 | 7 | 18% | Macrocephaly | 14 | 2 | 14% | |
| Autism | 20 | 3 | 15% | Hypertonia | 26 | 6 | 23% | |
| Muscle weakness | 10 | 4 | 40% | Cleft lip/palate | 8 | 7 | 88% | |
| Obesity | 16 | 10 | 63% | Low weight | 47 | 23 | 49% | |
| Ocular/auditory anomalies | 50 | 8 | 16% | Kidney/urinary tract anomalies | 12 | 5 | 42% | |
| Gastrointestinal anomalies | 10 | 3 | 30% | Respiratory tract anomalies | 6 | 4 | 67% | |
| Abnormal blood biochemistry | 33 | 9 | 27% | Karyotypical abnormalities | 26 | 24 | 92% | |
aSS-short stature; bCHD-congenital heart defects
P# number of patients with pCNVs identified, Odds ratio yielding pCNVs in patients with select condition versus without select condition, based on fisher’s exact; p-value < 0.01, two-tailed were displayed in bold
Chromosomal microarray analysis
500 μl peripheral venous blood was withdrawn to ethylenediamine tetra-acetic acid (EDTA) tubes. Genomic DNA was extracted with GentraPuregene Kit (Qiagen, Germany) or Lab-Aid 820 kit (ZSandx, China). Detection of genomic CNVs was performed with AffymetrixCytoScan HD or 750 K arrays (average probe spacing 1.1 kb and 4.1 kb, respectively) following the manufacturer’s instructions. Array results were visualized and analyzed by Chromosome Analysis Suite software (Affymetrix, USA). The parental origin of CNVs was examined by CMA or quantitative real-time polymerase chain reaction.
Variant filtering
Size threshold for CNV analysis was set at > 100 kb for gains, > 50 kb for losses and > 10 Mb for loss of heterozygosity. Next, analysis was restricted to rare CNVs - those with < 80% overlap of any common CNVs (< 1% frequency) in the DGV (Database of Genomic Variants) or a database of 2691 phenotypically normal controls (offered by Affymetrix). Interpretation and report of CNVs followed the ACMG guideline [7]. The CNVs deemed benign were not reported.
Statistical analysis
All statistical analyses were performed on Vassarstats (http://www.vassarstats.net/). The correlation between select condition and pCNV finding was analyzed by Fisher’s exact test on a 2 × 3 or 2 × 2 contingency table (row: the number of patients with or without certain condition; column: the number of patients with or without pCNVs identified). Analysis was only done in conditions with patients number above 50. Odds ratio was calculated, and statistical differences were defined as p < 0.01, two-tailed test.
Results
The clinical overview and overall diagnostic yield
In this retrospective study, a total of 710 patients with DD/ID as the main manifestation were included. Among this cohort, standardized measure of intelligence was available in 345 patients. Based on the degree of ID severity, the patients can be categorized to “mild” (140), “moderate” (105), “severe” (85) and “profound” (15) (Table 1I). Only 161 patients present DD/ID as the main manifestation without other phenotype reported, and the rest of the cohort were with one or more comorbid conditions, as listed in Table 1 part II/III. The most common conditions co-occurring with DD/ID were short stature (201 patients), facial dysmorphism (201 patients), pre/peri-natal problems (160 patients), brain malformation (147), microcephaly (128) and congenital heart defects (98).
The overall diagnostic rate was 28% - 201 patients were found to harbor pathogenic CNVs. One hundred nine patients (16%) were found with variants of uncertain clinical significance (VOUS), and 400 patients (56%) received a negative result (no CNV or only benign CNVs found). A total of 406 CNVs were reported to the patients (Fig. 1b), including 247 pathogenic CNVs (pCNVs, 61%), 18 VOUS-likely pathogenic (VOUS-LP, 4%), 136 VOUS (36%) and 5 VOUS-likely benign (VOUS-LB, 1%).
Fig. 1.
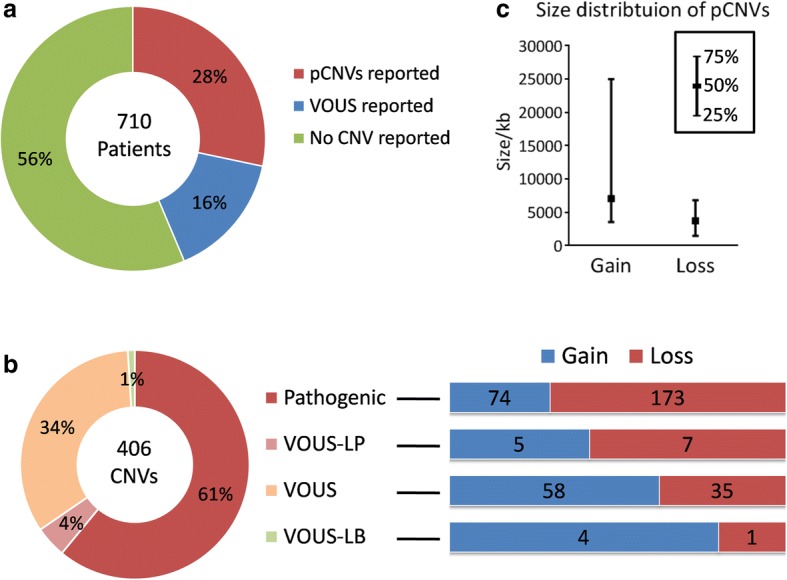
Characterization of CNVs identified in 710 patients with DD/ID. a Diagnostic yield of CMA. pCNVs: pathogenic CNVs; VOUS: variants of uncertain significance. b Interpretation of 406 rare CNVs identified, and the number of “Gain” and “Loss” in each category. VOUS-LP: variants of uncertain significance, likely pathogenic; VOUS-LB: variants of uncertain significance, likely benign. c Size distribution of 247 pCNVs
Characterization of pCNVs
We characterized the property, size and physical distribution of pCNVs. Among 247 pCNVs identified, 173 were losses and 74 were gains (Fig. 1b). A bias towards loss was observed in pCNVs - the proportion of loss increased accordingly with the interpretation of CNV towards pathogenicity, from “VOUS-LB”, “VOUS”, “VOUS-LP” to pathogenic (Fig. 1b). Regarding the size of CNVs, losses were generally smaller than gains (Fig. 1c), with a median size of 3724 kb compared to 7047 kb of gains. Regarding the physical distribution, pCNVs distributed over all chromosomes, and most terminals were covered by gains or losses found in these 201 patients (Fig. 2). Enrichment of pCNVs was found in chr7, chr15 and chr22, mainly due to a few common syndromes identified in our cohorts – William Beuren syndrome (29 patients), Prader-Willi/Angelmen syndrome (21 patients), 22q11.2 deletion (5 patients) and 22q13.3 deletion (7 patients), respectively. The chromosomes with less frequent CNVs (frequency below 5) were chr6, chr9, chr14, chr16, chr19, chr20 and chrY. A full list of pCNVs analyzed was included in Additional file 1: Table S1.
Fig. 2.

Physical distribution of pCNVs on the chromosomes (only pCNVs with a size smaller than 30 Mb were displayed). Total count of pCNVs on each chromosome was labeled, and the most common pCNVs were located on chr7, 15 and 22. The main syndromes involved in these common pCNVs were annotated
Diagnostic yields in select conditions
When patients were divided to subgroups based on ID severity, the group of severe ID obtained the highest diagnostic yield of 33%, compared with 13–22% for the mild, moderate and profound group (Table 1a). The yield nominally increased with ID severity, though not statistically significant (p = 0.084, fisher’s exact, two-tailed).
To assess the diagnostic yields in DD/ID patients with co-occurring conditions, the cohort were divided to subgroups based on the presence of following conditions or their combination: pre/peri-natal problems, family history, short stature, congenital heart defects and twenty other conditions (Table 1, also see Methods for details). The number of patients with pCNVs identified and the diagnostic rate of each group was listed in Table 1 part II/III. Odds ratio (OR) of yielding a pathogenic finding in the presence of select condition was calculated (Table 1.II) and plotted (Fig. 3) when the number of patients with select condition was above 50. Four conditions when co-occurring with DD/ID showed a statistically higher chance of yielding pCNVs - congenital heart defects (55%, OR:5.52), facial dysmorphism (39%, OR:2.57), microcephaly (34%, OR:2.33), hypotonia (35%, OR:2.87) - markedly higher than the 28% overall diagnostic rate. In the presence of two comorbid conditions - short stature and facial dysmorphism, or congeital heart defects and facial dysmorphism, the yield was also markedly elevated (38%, OR:4.22; 62%, OR:10.81, respectively). Karyotypical abnormalities were known in 26 patients prior to CMA. Among these patients, 24 (92%) were identified with pCNVs, including 11 cases with the genomic content of marker chromosomes revealed, 3 cases with gain or loss found in “balanced rearrangement” (based on karyotype), 9 cases with gain/loss confirmed and spanning clarified, and 1 case with pCNV identified in a region other than the structural rearrangement site detected by karyotypical analysis. The rest 2 with no pCNV identified were patients with karyotypic results showing balanced rearrangement.
Fig. 3.
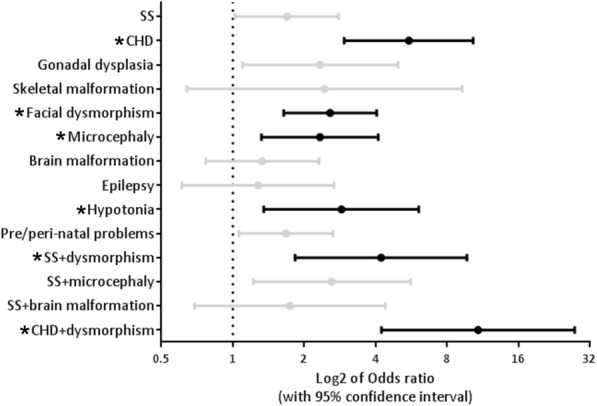
Odds ratio of yielding pCNVs in DD/ID patients with different co-occurring conditions. Log2 of odds ratio were displayed. Odds ratio with a p-value< 0.01, two tailed were displayed in black (also a “*” mark before the text), while others were shown in gray. SS:short stature; CHD:congenital heart defects
Discussion
More losses than gains in pCNVs
In this study, an increasing proportion of losses were observed with the pathogenic interpretation of CNVs. This is consistent with the notion that many gains present in the human genome are benign. Based on the CNV study on 59,898 exomes by Exome Aggregation Consortium, most phenotypically normal individuals possess higher number of duplications than deletions [8]. The proportion of losses in the pCNVs can also be influenced by the interpretation, as the evidence guiding copy number losses towards pathogenic interpretation is more readily available than gains (in 1247 genes curated by ClinGen expert team, 250 genes have been rated as “sufficient evidence for haploinsufficiency”, while only three genes were rated as “sufficient evidence for triplosensitivity”, https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/index.shtml).
The phenotypes associated with pCNV finding
Though CMA has been recommended as the first-tier genetic test for DD/ID, it remains costly and majority of patients could not obtain a diagnosis after the test [9]. Development of next-generation sequencing offers another option for genetic diagnostics of DD/ID. Based on a recent study, whole exome sequencing identified 29.3% conclusive diagnoses in a cohort of 150 patients with complex pediatric neurological conditions [10]. In the foreseeable future, the option between CMA and whole exome sequencing for DD/ID is likely to be put into discussion. Delineating the phenotypic clues of pathogenic CNVs can offer hints for the best cost-effectiveness, though a definite answer should come from the direct comparison of diagnostic yields between next-generation sequencing and CMA.
In our study, diagnostic yield of CMA appeared to correlate positively with ID severity (mild:19%, moderate:22% and severe:33%, Table 1.I), though the correlation was not statistically significant (p = 0.08, Fisher’s exact). The number of profound ID cases in our cohort was small to generate a reliable conclusion. A study based on 349 individuals in Italy reported a higher detection rate of causative CNVs in mixed ID (21.5%, IQ < 70) than borderline ID (8.8%, IQ:70–85), but no further categorization of severity in the mixed ID cohort was assessed [11]. Larger datasets are needed to warrant the correlation between ID severity and CMA yield, ideally in a non-syndromic ID cohort.
The comorbidity of congenital heart defects (CHD) in DD/ID is the strongest single phenotype associated with pCNV finding in our study, with an odds ratio of 5.52. When additional comorbid condition is present, the effect size can be even larger - 62% yield was found in DD/ID patients with co-occurring CHD and facial dysmorphism (OR:10.81). This is consistent with a prior study by Shoukier et al., based on CMA results from 342 children with unexplained DD/ID in Europe - they found CHD was more frequently seen in children with pCNVs compared to those with normal array CGH results [12]. There are also reports about higher yields in syndromic CHD with additional phenotypic indications [13, 14]. Geng et al. reported 22.7% detection rate of pathogenic CNVs in CHD patients with co-occurring DD/ID or ASD, compared to 4.3% in isolated cases [15]. Together with our finding, elevated CMA yield was found in comorbidity of CHD and DD/ID, and resorting to CMA is appropriate in such conditions.
Short stature is another comorbid condition frequently seen in our cohort (201/710). The overall yield of DD/ID with short stature was 32%, and increased to 38% when additional feature of dysmorphism or microcephaly was present (Table 1.II). Though the statistical power was less than adequate (p = 0.048, Fisher’s exact) in our study, short stature has been reported to be more frequently seen in DD/ID children with pCNVs [12], thus it is a possible indication of pCNV finding. The yield of CMA reported in short stature was between 4% [16] -10% [17], and the difference could be attributed to the varied proportion of syndromic patients.
In cases of DD/ID comorbid with other neurological abnormalities, we found hypotonia (35%, OR:2.87, p = 0.006) and microcephaly (34%, OR:2.33, p = 0.003) was associated with higher CMA yield, but not epilepsy (24%, p = 0.567), brain malformations (24%, p = 0.328) or autism (15%) (Table 1, all statistical comparisons were based on fisher’s exact). This is consistent with previous reports - microcephaly and hypotonia, but not epilepsy, were more frequently seen in DD/ID patients harboring pathogenic CNVs than those with normal array results [12, 18]. The documented yield of CMA in patients comorbid of DD/ID and autism varied (12.7% [19], 14% [11], 22% [20] and 26.1% [21]), and in our small subset of 20 patients, three were found to harbor pCNVs, resulting an intermediate yield of 15%.
One of the largest genotype-phenotype analysis so far - Cooper et al. investigated the rare CNV burden on 15,767 individuals in a mixed ID cohorts, and found greater enrichment of CNVs in patients with craniofacial anomalies and heart defects compared to those with epilepsy and autism [22]. Though the burden of rare CNVs cannot directly translate to clinically relevant findings, the overall trend should be informative. Our findings generally agreed that facial dysmorphism and CHD were more indicative of pCNV findings than epilepsy and autism. We additionally found gonadal dysplasia, skeletal malformation and pre/peri-natal problems were not associated with increased yield. The literature on CMA yield in DD/ID comorbid with these conditions is not sufficient, and relevant findings are subjected to further investigation.
A few other conditions, not commonly seen in DD/ID, were reviewed in this study but without statistical comparison performed (Table 1.III). Cleft lip/palate was found in 8 DD/ID patients, and CMA revealed pCNVs in 7 out of 8 patients (88%). These pCNVs were located at different regions, including 7p21 deletion (Saethre–Chotzen syndrome), partial trisomy of chromosome 9, 7q11.23 deletion, 10q26 deletion and 8q22-q24 duplication etc.. Since the reported yield of CMA in cleft lip/palate was between 11 and 14.8% based on larger cohorts [23, 24], our finding of high yield could be incidental as the number of patients was small. Another condition -abnormal biochemical profile - was not associated with elevated diagnostic yield in our DD/ID patients (yield: 27%, versus 29% in patients with normal biochemical results). Recent studies report that inborn errors of metabolism contribute to 1–5% of ID etiology [25]. American Pediatric Association also recommended considering the metabolic screening for children presenting with DD/ID [5]. In our study, patients referred to CMA after metabolic screening were mostly those without clear indication of a monogenic metabolic disorder. Our results did not support the atypical biochemical profile as a phenotypic indication of pCNV finding.
Karyotypical abnormality referred to CMA
Karyotypical abnormalities were known in some DD/ID patients prior to CMA. They were still referred mainly for further delineating the intervals of genomic aberration. Notably, in the 5 patients with karyotypically balanced rearrangement, 2 were identified to harbor micro-deletion/duplications with clinically relevant CNVs based on CMA. This highlights the possibility of genomic content loss/gain in those karyotypically balanced structural variations.
Limitations of this study
There are a number of limitations in our study: 1. the sample size is modest, especially those patients with fully accessible clinical information, which limited the statistical power in the comparison of yields based on disease severity and comorbid conditions. 2. Certain conditions can be a matter of clinical judgment like facial dysmorphism, and certain defects may be overlooked in patients without comprehensive evaluation of multiple systems. 3. The diagnostic yield in our study was overall higher than reported, which could be accounted by the patient selection. Patients with karyotypical abnormality were not excluded. In addition, metabolic screening was routinely done in our clinic, which excludes those with identifiable inborn errors of metabolism due to monogenic variants. Nonetheless, the patients in our study, with broad range of phenotypes, can be representative of DD/ID cohorts.
Conclusions
In conclusion, this study assessed the yield of CMA based on phenotypic features in a highly heterogeneous DD/ID cohort. The results suggest a disparity of gains and losses in identified pCNVs, and varied yields exist in patients with different phenotypic presentation. Congenital heart defects, microcephaly, hypotonia and facial dysmorphism co-occurring with DD/ID associate with an increased probability of pCNV finding. The presence of these comorbid conditions can be among factors to consider when planning CMA on DD/ID patients.
Additional file
Table S1. The chromosomal location, type and size of pathogenic CNVs reported in this study. (XLSX 31 kb)
Acknowledgements
We acknowledge all the patients and families. We thank Dr.Yiping Shen (Boston children’s hospital) and Dr.Yu Sun (Xinhua hospital, Shanghai) for constructive comments and discussion. We are also grateful for the expert team of Society of Medical Genetics, Chinese Medical Doctor Association in the following institutions for enrollment and referral of patients - Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region; Beijing Children’s Hosptial; Shengjing Hospital; Children’s Hospital of Fudan University; Guangzhou Women and Children’s Medical Center; Shanghai Children’s Hospital; Maternal and Child Health Hospital of Hunan Province; Nanjing Children’s Hospital; Wenzhou Medical College.
Funding
Funding for this project was from the National Natural Science Foundation of China (No. 81500972, to YF; No. 81670812, to YGY), the Shanghai Municipal Education Commission (No.15CG14, to YF), the Shanghai Municipal Science and Technology Commission (No.15YF1409600, to YF), and Shanghai Jiao Tong University School of Medicine (No.15ZH3003, to YGY), Shanghai Health Bureau (20134005 to WJQ) and Precision Medical Research of National Key Research and Development Program (2016YFC0905100 to WJQ and XFG). The funding organizations played no role in the design of the study, collection, analysis and interpretation of data and in writing the manuscript.
Availability of data and materials
To ensure patient confidentiality, data containing potentially identifiable information was not shared. All data generated or analysed during this study, without identifiable information, is available from the corresponding author upon reasonable request.
Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- CHD
Congenital heart defects
- CMA
Chromosomal microarray analysis
- DD
Developmental delay
- DGV
Database of genomic variants
- ID
Intellectual disability
- LB
Likely benign
- LP
Likely pathogenic
- pCNVs
Pathogenic copy number variations
- VOUS
Variants of uncertain clinical significance
Authors’ contributions
YF and YW1 performed the analysis, prepared the figures and tables and wrote the manuscript. LW, YW2 and ZG performed the CMA experiments. WQ, JW, HZ, XJ1, JY, LH, XJ2, YS, FL, BX and LL recruited the patients and collected clinical information. XZ and XL reviewed and analyzed the clinical data. XG and YY recruited patients and supervised the study. All authors have read and approved the manuscript.
Ethics approval and consent to participate
This study was reviewed and approved by the ethical committee of Xinhua hospital and Shanghai Children’s Medical Center, China. Informed consent was obtained from the patients, and for those patients under 18 years old, consent was obtained from the parents on their behalf.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Yanjie Fan and Yanming Wu contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1186/s12920-018-0368-4) contains supplementary material, which is available to authorized users.
Contributor Information
Yanjie Fan, Email: fanyanjie@shsmu.edu.cn.
Yanming Wu, Email: amywu1982@163.com.
Lili Wang, Email: leafrose210@126.com.
Yu Wang, Email: wangyu.uu@163.com.
Zhuwen Gong, Email: jun_gong123@163.com.
Wenjuan Qiu, Email: qiuwenjuanxh@163.com.
Jingmin Wang, Email: wang66jm@163.com.
Huiwen Zhang, Email: huiwenzhang2010@163.com.
Xing Ji, Email: jxkshch@126.com.
Jun Ye, Email: yejun2314@163.com.
Lianshu Han, Email: xhhanls@163.com.
Xingming Jin, Email: zhujing7@public7.sta.net.cn.
Yongnian Shen, Email: syn14401@hotmail.com.
Fei Li, Email: lifei5861_cn@163.com.
Bing Xiao, Email: xiaobingdoctor@163.com.
Lili Liang, Email: lianglili0310@163.com.
Xia Zhang, Email: zhangxiapr@163.com.
Xiaomin Liu, Email: zhengdaliuxiaomin@163.com.
Xuefan Gu, Email: guxuefan@xinhuamed.com.cn.
Yongguo Yu, Email: yuyongguo@shsmu.edu.cn.
References
- 1.Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011;32(2):419–436. doi: 10.1016/j.ridd.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 2.Moeschler JB, Shevell M. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics. 2006;117(6):2304–2316. doi: 10.1542/peds.2006-1006. [DOI] [PubMed] [Google Scholar]
- 3.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.[Expert consensus on the clinical application of chromosomal microarray analysis in pediatric genetic diseases]. Zhonghua er ke za zhi = Chinese J Pediatr. 2016;54(6):410–3. [DOI] [PubMed]
- 5.Moeschler JB, Shevell M. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014;134(3):e903–e918. doi: 10.1542/peds.2014-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arvio M, Sillanpaa M. Prevalence, aetiology and comorbidity of severe and profound intellectual disability in Finland. J Intellect Disabil Res. 2003;47(Pt 2):108–112. doi: 10.1046/j.1365-2788.2003.00447.x. [DOI] [PubMed] [Google Scholar]
- 7.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13(7):680–685. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- 8.Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Daly MJ, MacArthur DG, Fromer M, Purcell SM. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet. 2016;48(10):1107–1111. doi: 10.1038/ng.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17(1):9–18. doi: 10.1038/nrg3999. [DOI] [PubMed] [Google Scholar]
- 10.Vissers LE, van Nimwegen KJ, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, Pfundt R, van der Wilt GJ, Krabbenborg L, Brunner HG, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017; [DOI] [PMC free article] [PubMed]
- 11.Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, Filippi T, Carey JC. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. 2013;17(6):589–599. doi: 10.1016/j.ejpn.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Shoukier M, Klein N, Auber B, Wickert J, Schroder J, Zoll B, Burfeind P, Bartels I, Alsat EA, Lingen M, et al. Array CGH in patients with developmental delay or intellectual disability: are there phenotypic clues to pathogenic copy number variants? Clin Genet. 2013;83(1):53–65. doi: 10.1111/j.1399-0004.2012.01850.x. [DOI] [PubMed] [Google Scholar]
- 13.Richards AA, Santos LJ, Nichols HA, Crider BP, Elder FF, Hauser NS, Zinn AR, Garg V. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatr Res. 2008;64(4):358–363. doi: 10.1203/PDR.0b013e31818095d0. [DOI] [PubMed] [Google Scholar]
- 14.Syrmou A, Tzetis M, Fryssira H, Kosma K, Oikonomakis V, Giannikou K, Makrythanasis P, Kitsiou-Tzeli S, Kanavakis E. Array comparative genomic hybridization as a clinical diagnostic tool in syndromic and nonsyndromic congenital heart disease. Pediatr Res. 2013;73(6):772–776. doi: 10.1038/pr.2013.41. [DOI] [PubMed] [Google Scholar]
- 15.Geng J, Picker J, Zheng Z, Zhang X, Wang J, Hisama F, Brown DW, Mullen MP, Harris D, Stoler J, et al. Chromosome microarray testing for patients with congenital heart defects reveals novel disease causing loci and high diagnostic yield. BMC Genomics. 2014;15:1127. doi: 10.1186/1471-2164-15-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Duyvenvoorde HA, Lui JC, Kant SG, Oostdijk W, Gijsbers AC, Hoffer MJ, Karperien M, Walenkamp MJ, Noordam C, Voorhoeve PG, et al. Copy number variants in patients with short stature. Eur J Hum Genet. 2014;22(5):602–609. doi: 10.1038/ejhg.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahnleiter D, Uebe S, Ekici AB, Hoyer J, Wiesener A, Wieczorek D, Kunstmann E, Reis A, Doerr HG, Rauch A, et al. Rare copy number variants are a common cause of short stature. PLoS Genet. 2013;9(3):e1003365. doi: 10.1371/journal.pgen.1003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caballero Perez V, Lopez Pison FJ, Miramar Gallart MD, Gonzalez Alvarez A, Garcia Jimenez MC, Garcia Iniguez JP, Orden Rueda C, Gil Hernandez I, Fuertes Rodrigo C, Fernando Martinez R, et al. Phenotype in patients with intellectual disability and pathological results in array CGH. Neurologia. 2017;32(9):568–78. doi: 10.1016/j.nrl.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Nicholl J, Waters W, Mulley JC, Suwalski S, Brown S, Hull Y, Barnett C, Haan E, Thompson EM, Liebelt J, et al. Cognitive deficit and autism spectrum disorders: prospective diagnosis by array CGH. Pathology. 2014;46(1):41–45. doi: 10.1097/PAT.0000000000000043. [DOI] [PubMed] [Google Scholar]
- 20.Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ, Frazier JA, Silverstein I, Picker J, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125(4):e727–e735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oikonomakis V, Kosma K, Mitrakos A, Sofocleous C, Pervanidou P, Syrmou A, Pampanos A, Psoni S, Fryssira H, Kanavakis E, et al. Recurrent copy number variations as risk factors for autism spectrum disorders: analysis of the clinical implications. Clin Genet. 2016;89(6):708–718. doi: 10.1111/cge.12740. [DOI] [PubMed] [Google Scholar]
- 22.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emy Dorfman L, Leite JC, Giugliani R, Riegel M. Microarray-based comparative genomic hybridization analysis in neonates with congenital anomalies: detection of chromosomal imbalances. J Pediatr. 2015;91(1):59–67. doi: 10.1016/j.jped.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 24.Cao Y, Li Z, Rosenfeld JA, Pursley AN, Patel A, Huang J, Wang H, Chen M, Sun X, Leung TY, et al. Contribution of genomic copy-number variations in prenatal oral clefts: a multicenter cohort study. Genet Med. 2016;18(10):1052–1055. doi: 10.1038/gim.2015.216. [DOI] [PubMed] [Google Scholar]
- 25.Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: genetic and metabolic testing on children with global developmental delay: report of the quality standards Subcommittee of the American Academy of neurology and the practice Committee of the Child Neurology Society. Neurology. 2011;77(17):1629–1635. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The chromosomal location, type and size of pathogenic CNVs reported in this study. (XLSX 31 kb)
Data Availability Statement
To ensure patient confidentiality, data containing potentially identifiable information was not shared. All data generated or analysed during this study, without identifiable information, is available from the corresponding author upon reasonable request.