

Abstract
Ischemic stroke is a major cause of death. Besides the direct damage resulting from oxygen and glucose deprivation, sterile inflammation plays a pivotal role in increasing cellular death. Damaged-associated molecular patterns (DAMPs) are passively released from dying cells and activate the innate immune system. Thus, they take part in the direct and rapid activation of the inflammatory response after stroke onset. In this review the role of the most important DAMPs, high mobility group box 1, heat and cold shock proteins, purines, and peroxiredoxins, are addressed. Moreover, intracellular pathways activated by DAMPs in microglia are illuminated.
Keywords: DAMPs, stroke, microglia, HMGB1, purines, Hsp, peroxiredoxin
Introduction
Stroke is a major issue in terms of an increasing incidence and considering disability-adjusted life years (DALYs) lost. In 2030 up to 12 million deaths are expected.1 Moreover, it is the leading cause of long-term disability.2 Approximately 2 million neurons per minute die when exposed to ischemia.3 Acute thromboembolic or local thrombotic occlusion of a cerebral artery accounts for 80% of strokes. Systemic thrombolysis and mechanic recanalization are so far the only acute treatments in these cases, but less than 20% of patients receive it due to restrictive inclusion criteria. Besides the direct brain injury resulting from ischemia and reperfusion, there is growing evidence that a rapid evolving sterile inflammation worsens infarct size in the first 1–3 days post stroke, and thus clinical outcome.4 In immune-deficient mice, about 20–30% smaller infarct sizes were detected after experimental stroke induction.5 Cells invade the ischemic brain in a specific temporal and spatial pattern.6 In the first couple of days, neutrophils, macrophages and microglia predominantly infiltrate the ischemic tissue in large numbers, reaching their maximum around 3 days post ischemia. Even though smaller in numbers, other potentially highly immune competent lymphocyte subpopulations and dendritic cells accumulate in the ischemic tissue during the first days after the ischemic insult. Overall, the initial cellular inflammatory reaction follows a highly conserved pattern of sterile inflammatory reactions, which are also known from other organs. Whereas the detrimental effects of proinflammatory signals released by the innate immune system occur in the early course of stroke,7 the role of the adaptive immune system is still controversial.3 Interestingly, the immunologic response not only accounts for detrimental effects, but it also influences neuronal regeneration in the following weeks and months after brain infarction.8 In the setting of infections by pathogens, the immune system protects the human body against danger in general, as introduced by Polly Matzinger in 1994.9 These warning mechanisms are not only released by pathogens, they also encompass released or secreted molecules from injured cells, which are called damaged-associated molecular patterns (DAMPs).10 In general, DAMPs can be divided into PAMPs (pathogen-associated molecular patterns) and alarmins, which categorize endogenous molecules.11 Both protein [e.g. high mobility group box 1 (HMGB1), heat shock proteins (Hsps)] and nonprotein alarmins (e.g. adenosine triphosphate (ATP)) exist. Overall, DAMPs are promising candidates in stroke pathophysiology as several experimental data indicate their importance for the activation of the immune system early following ischemia.2 In stroke, the sterile inflammation starts with the release of DAMPs from dying neuronal and non-neuronal brain cells, which then activate local immune cells (e.g. microglia) and perivascular endothelia cells via pattern recognition receptors (PRRs) including toll-like receptors (TLRs). The local immune reaction is amplified, which results in the massive invasion of systemic immune cells.3 Besides the local immune response, activation of the immune system also has systemic consequences, since the phenomenon of a systemic immune suppression can be observed in stroke patients. This is associated with an increased susceptibility to bacterial infections such as pneumonia or lower urinary infections.12 As depicted, the inflammatory response is a major potential target of future therapeutic treatments in stroke. In this review, we focus on the acute involvement of DAMPs as one of the first reactions after ischemia.
High mobility group box 1
The HMGB1 protein is a widely expressed nonhistone cytokine-like factor, which mainly stabilizes the chromosomes in the nucleus.13 Under physiological conditions, it has a proangiogenic effect on endothelial cells and neurite outgrowth is stimulated by HMGB1 during embryogenesis.14 HMGB1 is both passively released from necrotic cells and actively secreted by the innate immune system in case of infections. Interestingly, it does not diffuse in the extracellular matrix when cells undergo apoptosis because here HMGB1 binds irreversibly to the modified chromatin.15,16 HMGB1 appears early in sterile inflammation and initiates the production of proinflammatory mediators.14,17 Multiple extracellular receptors are activated by HMGB1, including the receptor for advanced glycation end products (RAGE) or TLR2 and TLR4, which are ubiquitously expressed by resident central nervous system (CNS) cells.18 Additionally, HMGB1 possesses divergent functions in different redox states.19 In positions 23, 45 and 106, three cysteine residues are located. A disulfide bond between C23 and C45 as well as the thiol form of C106 is obligatory for its tumor necrosis factor (TNF)-stimulating effects via TLR4.20,21 Overall, the reduced form is associated with chemotactic functions whereas the disulfide form is thought to be proinflammatory through the induction of nucleotide-binding domain, leucine-rich repeat, pyrin domain containing protein 3 (NLRP3) and nuclear factor κBIα (NFκBIα).22
HMGB1 also plays a pivotal role in ischemic stroke. High levels of systemic HGMB1 were measured in serums of patients with cerebral ischemia.23 Elevated HMGB1 levels correlate with severe stroke sizes.24–26 In the CNS, HMGB1 is upregulated after stroke and maintains the inflammatory process.27 It is secreted by activated astrocytes and microglia, and stimulates the release of interleukin (IL)-1, TNFα, IL-6 and IL-8, as well as inducible nitric oxide synthase (iNOS) expression.28–31 An increased vascular and blood–brain barrier permeability is observed.31 Moreover, matrix metalloproteinase 9 (MMP-9) is upregulated by HMGB1 via TLR4 and its induced cytokines as TNFα or IL-1β mediating cellular death after ischemic stroke.31 A maturation process of the passively released protein in the blood circulation to a cytokine-stimulating isoform has been described.32 Furthermore, HGMB1-induced activation of RAGE results in systemic lymphocytopenia after brain ischemia.32 Locally, the amplification of the post-ischemic inflammation is also maintained by HMGB1.33,34 Leucocyte infiltration is stimulated as well as activation of microglia and amplification of metalloproteinases. The late treatment with amlexanox or an anti-HMGB1 antibody after ischemia appears to protect the brain from stroke-induced brain damage by the inhibition of the release of HMGB1.35 Several studies demonstrated protective effects by pharmacological inhibition of HMGB1 or its pathway underlying the detrimental function of HMGB1 in the early stages following stroke.3,36–38 Reduced inflammation and stroke size are seen after inhibition of RAGE or TLR4.34,39 Contrary to that, HMGB1 also stimulates angiogenesis, neurite outgrowth and neuronal survival in the late period after stroke, and is actively secreted by astrocytes with positive signaling on endothelial progenitor cells.31 In summary, HMGB1 has mainly detrimental effects in the early post-ischemic period, though HMGB1 activation also occurs to take part in stroke recovery mechanisms.
Heat shock proteins and cold shock domain proteins
Hsps belong to the group of molecular chaperones, which help guide the correct folding of proteins during their synthesis. They are produced when cells are exposed to elevated temperatures or any kind of injury as part of a feedback system, which detects misfolded proteins.40 The two major chaperones are Hsp60 and Hsp70, which are located in the cytosol. Hsp70 acts before the protein leaves the ribosome whereas Hsp60 interferes after the protein has been fully synthesized.40 The Hsp70 is the best studied Hsp in ischemic stroke. Depending on its location, opposite effects of Hsp70 have been described. Intracellular Hsp70 can decrease the signaling of proinflammatory factors such as the transcription factor NFκB, MMPs and reactive oxygen species (ROS). Located in the extracellular matrix, Hsp70 binds to TLRs on macrophages, dendritic cells and microglia, and thereby activates the proinflammatory NFκB and its associated pathways. It can also elicit the immune response of CD4+ und CD8+ T cells.41,42 Following brain infarction, Hsp70 can be detected in neurons, microglia, astrocytes and endothelial cells. TNFα and IL-1β were downregulated after Hsp70 overexpression in experimental stroke.41 Increased intracellular Hsp70 protein expression appears to be cerebro protective in stroke studies of viral overexpression in neurons and glia, whereas Hsp70 knockouts worsen outcome.42 Additionally, exogenously delivered Hsp70 has a neuroprotective function. This is demonstrated by intravenous injection of Hsp70 in rat, reducing the cerebral infarction zone.43–45 Dynamin is a downregulated protein by Hsp70, which increases after experimental stroke. It transfers the death receptor Fas to the cell surface, inducing apoptosis after its ligands have bonded. A selective dynamin inhibitor also showed neuroprotective effects and may serve as a new therapeutic target.46 Taken together, it seems that Hsp70 is mainly protective in stroke though detrimental effects such as the activation of the immune system.
It is the Y-box binding protein (YB-1), respectively DNA-binding protein B (DbpB), which is a prototype of the conserved cold shock domain proteins. It binds single-stranded RNA or DNA via two ribonucleoprotein domains (RNP-1 and RNP-2) to prevent annealing upon the induction of cold shock, and thus affects multiple cellular functions.47 YB-1 takes part in a set of bacterial and sterile inflammatory processes.47,48 It is upregulated in proliferating microglia in an experimental ischemia model.49 The cold-inducible RNA-binding protein (CIRP) also belongs to the group of stress-responsive proteins with RNPs. When it is released from microglia after experimental ischemic brain injury, it fosters neuroinflammation via CIRP-NFκB-mediated TNFα expression.50 Moreover, in CIRP-deficient mice, the brain infarct volume, TNFα levels and activation of microglia are decreased.51 Therapeutic hypothermia is discussed to be neuroprotective after stroke.52 In experimental stroke research, hypothermia upregulates CIRP and Hsp70.1 and decreases TNFα and IL-6.53 To sum up, both CIRP and YB-1 appear to have damaging functions in stroke.
Peroxiredoxins
Peroxiredoxin is a redox-active cytosolic DAMP, which is neuroprotective within cells by eliminating ROS, but loses its antioxidant capacity once released.54 The oxidized peroxiredoxin 2 (PRDX2) is a set free from macrophages under inflammatory conditions and stimulates the production of TNFα.55 Peroxiredoxin family proteins (Prxs) like Prx-1, Prx-2, Prx-5 and Prx-6 are released from necrotic brain cells 12 h after stroke onset and induce the production of proinflammatory cytokines like IL-23 from macrophages through TLR2 and TLR4 binding.54 IL-23 induces IL-17-producing γδT cells and is responsible for delayed brain damage as neutrophils are recruited.56,57 Concordantly, the application of anti-IL-17 antibodies reduces infarct sizes and improves neurological outcome.58,59 Prx antibodies can reduce brain damage up to 12 h after ischemia. More than HMGB1, Prxs activate infiltrating macrophages.54 Only exogenous Prx-1, Prx-2 and Prx-4 seem to act as proinflammatory in the ischemic brain through TLR4/NFκB signaling whereas Prx-3, Prx-5 and Prx-6 have no relevant effect on the production of NO, TNFα, IL-6, iNOS and TLR4 expression in macrophages.60 In humans, low plasma levels of Prx-5 but high levels of Prx-1 during the first hours after stroke onset are found.61,62 Treatment with a gastrodin derivative, a constituent from the medicinal plant Gastrodiae rhizoma, reduced the post-ischemic expression of Prx-1, Prx-2 and Prx-4 after middle cerebral artery occlusion (MCAO) and the overall inflammatory response.63 However, there is also evidence that Prx-2 can have neuroprotective effects in stroke as it inhibits both the poly (ADP-ribose) polymerase 1 (PARP1) and the p53-Bax-mediated death pathway.64 Overall, peroxiredoxins mainly have damaging effects after stroke onset.
Purines
Extracellular nucleotides such as ATP serve as a multifunctional intercellular signaling system. Not only are they actively secreted through plasma membrane proteins such as pannexin and connexin but also passively released after stress-induced cell injury.65 Ectonucleotidases such as CD39, CD73 and nucleoside diphosphate kinase degrade extracellular nucleotides in a stepwise process, eventually leading to adenosine. Extracellular nucleotides bind to the family of P2 receptors, which comprise both ligand-gated P2X cation channels and G-protein coupled P2Y receptors.65 In the first 20 min after in vivo ischemia, the ATP extracellular concentration rises. Simultaneously, the degradation of ATP by ectonucleotidases is initiated so extracellular adenosine increases. This excitotoxicity returns to its basal level after about 4 h.66 ATP concentration rises due to various mechanisms, for example, a vesicular release from neurons, exocytosis from astrocytes and microglia, or through membrane channels from glial cells and pyramidal neurons.66 ATP itself aggravates cerebral damage after ischemic stroke as high ATP concentrations induce cell death.67 All P1 and P2 receptor subtypes are expressed in neurons and glial cells, but also in lymphocytes and neutrophils.66 Binding of the receptor P2X7 stimulates the inflammasome and enhances caspase-1 activity and the release of proinflammatory cytokines from innate immune cells. The receptor activation happens on K+ efflux through the cell membrane.68 The highest concentrations of the purinergic receptor P2X7 can be measured on activated microglia, which leads to IL-1β release. The P2Y2 receptor is upregulated due to a rising IL1-β concentration after the P2X7 receptor is stimulated on microglia and astrocytes.68 CD39-deficient mice, leading to less ATP metabolization, show larger infarct areas and more neutrophils and macrophages enter the CNS.69 Hyman and colleagues have shown that CD39 on endothelial cells and leukocytes regulates their influx in the cerebral infarct zone.70 The usage of a P2X7 antagonist or P2X7R blockade by Brilliant Blue G leads to a decrease in cerebral damage after ischemia in vivo.71–73 In contrast, Masuch and colleagues demonstrated a neuroprotective effect of microglia in organotypic hippocampal slice cultures, which is mediated by the P2X7 activation and the induction auf TNFα.74 P2X7 activation also leads to alterations of the T-cell surface and can even cause T-cell death in the case of prolonged stimulation.75 In contrast to ATP, adenosine, the end product of CD39, is considered to be neuroprotective.66 Gaudin and colleagues created nanoparticles conjugated with adenosine, so that its metabolism was prolonged. As a result, decreased parenchymal damage after experimental cerebral ischemia was observed.76 Concerning CD39, pretreatment with the small molecule inhibitor 3-(2,4-dichloro-5-methoxyphenyl)-2-sulfanyl-4(3H)-quinazolinone (Mdivi-1) raises adenosine in a MCAO mouse model, consequently protecting against cerebral ischemia. CD39 level was also increased as well as the cAMP response element-binding protein (CREB), which regulates CD39 expression.77 In summary, purines are involved in the cellular reactions after ischemic stroke and it appears that ATP has a detrimental function whereas adenosine shows positive neuroprotective effects.
Other DAMPs in stroke
Apart from the discussed DAMPs, several more candidates have to be considered, including lipids and other proteins (S100A9; Mrp 8, Mrp9, CIRP, ASC specks).78 Controversial opinions exist on the exact functions of these molecules in the field of stroke (for a review see Shichita and colleagues78). In primary microglial culture, exogenous S100B appears to induce the expression of IL-1β, TNFα and iNOS, and in vivo injections of S100B activate microglia. These effects can be suppressed by the addition of poly (ADP-ribose) polymerase 1 (PARP-1).79 Moreover, levels of hyaluronan, a large glycosaminoglycan in the extracellular matrix, are increased in patients with acute stroke. Concerning patients with intracerebral hemorrhagia, serum hyaluronan levels can be used for a prediction of the 3-month functional outcome80 Cerebral hyaluronan expression is also induced by cortical ischemia, as shown in an experimental stroke mouse model.81 Other endogenous TLR ligands, such as fibronectin, defensin or heparin sulfate proteoglycan, also activate TLR2 and TLR4, and are potentially involved in the initiation of sterile inflammatory responses.82 Their role in stroke is still to be revealed. As a conclusion, various intracellular and also extracellular components might be involved as DAMPs in the inflammatory reactions after stroke, but until now their functions remain unclear.
DAMPs and microglia
Microglia are the resident immune cells of the cerebral nervous system and they are mainly responsible for the local immune surveillance.83,84 Overall, microglia is a heterogeneous population with a broad variety of functions and highly plastic.85 Recently, a microglia-focused multicolor reporter mouse model monitored microglia dynamics in different states of health, disease and recovery. A self-renewing network with regional differences was described as being capable of reacting to different environmental challenges. Selected clonal expansion was observed in response to acute neurodegeneration as well as selected apoptosis in means of restoring homeostasis.86 Neuronal cell injury alters the local microglia from a ramified phenotype to a macrophage-like one.87
Regarding brain ischemia, hypoxia affects local microglia, which changes either into a proinflammatory or anti-inflammatory phenotype characterized by different secreted mediators.88 There is no uniform response to injury as the relationship of microglia is complex.89 More precisely, a spatiotemporal relationship between microglia morphology and localization of ischemic injury is established.84 The activation of microglia is one of the first main responses to brain ischemia but continues for several weeks.6,88 In in vitro stroke models it was shown that microglia can worsen neuronal damage.90 On the contrary, Szalay and colleagues elucidated thee protective effects. In an experimental study the authors treated their animals with the colony-stimulating factor 1 receptor CSF1R antagonist PLX3397 for 21 days, which resulted in a 98% decrease of microglia. This selective elimination of microglia led to a 60% augmented neuronal death after MCAO in mice.91 Consequently, microglia are not yet completely understood as players in the concert of cellular reactions after ischemic stroke.
DAMPs interact with microglia and specific pathways are activated accounting for the polarization of microglia and their response to cell injury after stroke. One of the involved DAMPs is HMGB1. An autocrine loop includes the binding of glucocorticoids to microglia, which results in the release of the disulfide form of HMGB1 (dsHMGB1) and subsequent binding of dsHMGB1 to TLR2/4 as part of the priming process92 [Figure 1(a)]. Inescapable tail shocks (ISs) in rat lead to elevated levels of HMGB1, an upregulation of the transcription factor NFκB and its NLRP3 inflammasome in microglia.93 A second immune challenge results in a potentiated proinflammatory response of microglia with elevated levels of IL-1β.94 Here, it is again the disulfide form of HMGB1, which induces the priming process.22 Moreover, high concentrations of dsHMGB1 can be measured in the serum of patients with stroke.92 When it comes to future therapeutic targeting of HMGB1, inflachromene inhibits microglia activation, and thus the release of HMGB1 with a reduced proinflammatory response.95 As a result, HMGB1 is a main activator of microglia after ischemic stroke and its inhibition could possibly be a neuroprotective therapeutic option.
Figure 1.

DAMP associated pathways in microglia.
(a) Glucocorticoids (GCs) induce the production of the disulfide form of high mobility group box 1 (dsHMGB1), which secondarily binds to its pattern recognition receptor (PRR). Nuclear factor κB (NFκB) is activated, translocates to the nucleus and nucleotide-binding domain, leucine-rich repeat, pyrin domain containing protein 3 (NLRP3) components are transcribed as well as pro-interleukin-1β (pro-IL-1β). A second immune challenge results in higher levels of oligomerized NLRP3. The core structures of the NLRP3 inflammasome are the NOD-like receptor NALP3, the adaptor of the apoptosis associated speck-like protein (ASC) with a caspase recruitment domain (CARD) and the protein caspase 1. When an activating signal is sensed, the oligomerization of the inflammasome starts, which leads to the proteolytic cleavage of pro-IL-1β into the bioactive mature IL-1β through caspase 1 and thus, a potentiated inflammatory response.96 The described pathways are best demonstrated by toll-like receptor (TLR)-agonist lipopolysaccharide (LPS). (b) Heat shock protein (Hsp) binds to its TLR4 receptors and Hsp70 associates with Na+/H+ exchanger 1 (NHE1). Ca2+ streams into the cell activating both Ca2+-cal-modulin-dependent protein kinase II (CaMKII) and transforming growth factor β-activated kinase 1 (TAK1). IκB kinase β (IKKβ) initiates the degradation of Inhibitors-of-kappaBand liberates NFκB, resulting in a higher gene transcription of inducible nitric oxide synthase (iNOS) and other proinflammatory cytokines. Hsp also upregulates hypoxia inducible factor 1 (HIF-1), interferon regulator factor 1 (IFR1) and single transducer and activator of transcription 3 (STAT3) through TLR2 and -4. The described pathways are best demonstrated by TLR agonist LPS.
Hsps also interact with microglia in stroke. In the case of danger, Hsp70 associates with the Na+/H+ exchanger 1 (NHE1), Ca2+ streams into the cell, which activates both the Ca2+-cal-modulin-dependent protein kinase II (CaMKII) and transforming growth factor β-activated kinase 1 (TAK1). IκB kinase β (IKKβ) initiates the degradation of IκB-α, and thus liberates NFκB resulting in a higher gene transcription of, for example, iNOS97 [Figure 1(b)]. Whereas extracellularly located, Hsp activates microglia through TLRs, upregulating NFκB, hypoxia inducible factor 1 (HIF-1), interferon regulator factor 1 (IFR1) and single transducer and activator of transcription 3 (STAT3). This results in the release of proinflammatory cytokines such as IL-1β, IL-6, IL-12 and TNFα.41,42,98 Another player in the fields of microglia activation after stroke is Hsp72. It is the major cytosolic form of the 70 kDa family of Hsps and its overexpression in microglia is described to be cytoprotective as it interferes with the IKK-IκB-NFκB pathway.99,100 Doeppner and colleagues fused Hsp70 to the transactivator of transcription (TAT) domain of the human immunodeficiency virus to pass the blood–brain barrier. Intravenous infusion of TAT-Hsp70 reduced post-ischemic brain injury among others by diminished microglial activation.44 Notably, Hsp90 inhibits 70 kDa inducible Hsp70 synthesis. This is why treatment with an Hsp90 inhibitor such as geldanamycin prevents lipopolysaccharide (LPS)-induced BV2-microglial cell death through subsequent Hsp70 induction.101
Regarding peroxiredoxins, Prx-1 is an antioxidant mainly expressed in microglia that diminishes microglial activation in proinflammatory processes.102 LPS is a randomly used inflammogen (PAMP) activating microglia in several mammalian model systems. TLR agonists such as LPS upregulate Prx-1 via a ROS/p38 mitogen-activated protein kinase (MAPK)-dependent pathway and suppresses NFκB signaling, and thus nitric oxide (NO) production103 (Figure 2). The Nox (membranous nicotinamide adenine dinucleotide phosphate (NADPH) oxidase)/ROS (reactive oxygen species)/JNK (c-jun N-terminal kinase) axis is also activated by LPS, and hence, NO as an inflammatory mediator rises. Prx-5 is upregulated by LPS and lowers intracellular ROS levels in microglia. Thereby, a balanced inflammatory response is generated.104 Furthermore, Prx-5 also reduces NFκB and MAPK activation in microglia after LPS application.105 When it comes to sterile inflammation after stroke, Prx-3 was significantly elevated from 3 days post ischemia in an ischemia/reperfusion stroke model (I/R). As a result, phagocytic processes in the subacute phase after stroke can be assumed.106 Prx-6 in microglia protects neurons against oxygen glucose deprivation and reoxygenation (OGD/R), showing a higher neuron viability.107 Exogenous intraventricular Prx-3 treatment decreases microglia activation after experimental cerebral ischemia induction in gerbils.106 Furthermore, spinal cord injury in rat increases Prx-1 expression in astrocytes and microglia, which also enhances microglia proliferation after inflammation.108 Prx-1 was also induced in surrounding microglia after hemorrhagic stroke in rat.109 Interestingly, it appears that microglia is not the target of Prxs but infiltrating immune cells releasing IL-17 and IL-23.110 As a promising therapeutic approach, the natural compound obovatol suppresses Prx-2-mediated microglial activation in a NFκB-dependent manner.111
Figure 2.
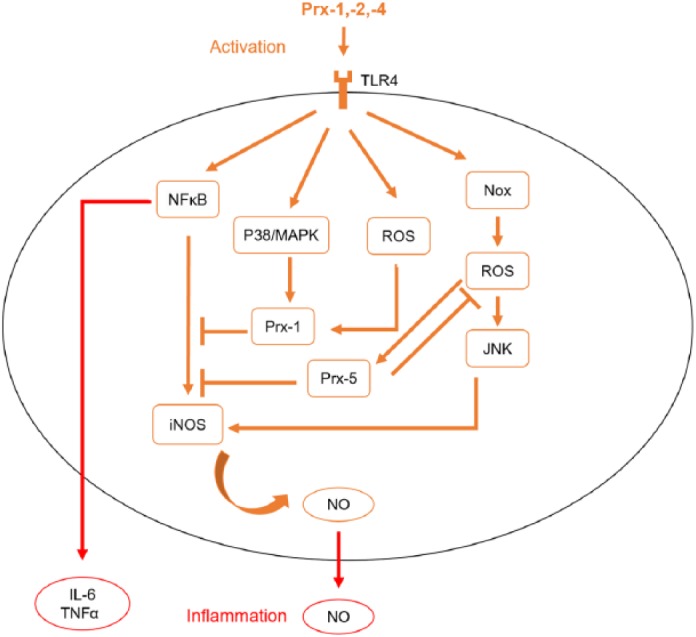
Peroxiredoxin-associated pathways in microglia. Damaged-associated molecular patterns (DAMPs) such as peroxiredoxin family proteins Prx-1, -2 and -4 activate the the Nox (membranous NADPH oxidase) /ROS (reactive oxygen species)/ JNK (c-jun N-terminal kinase) axis and the concentrations of Nuclear-Factor κB (NFκB), mitogen-activated protein kinase (P38/MAPK) and reactive oxygen species (ROS) increase. Both Prx-1 and Prx-5 are upregulated and diminish the proinflammatory response by lowering ROS and NFκB signaling. Described pathways are best demonstrated by toll-like receptor (TLR) agonist lipopolysaccharide (LPS).
ATP is a major modulator of microglial function with at least four different expressed ATP receptors (P2X4, P2X7, P2Y6, P2Y12) (Figure 3). P2X7 activation through ATP in cultured mouse primary microglia results in an increased release of IL-6, CCL2 and TNFα.112 Moreover, priming of cultured microglia with TLR isoform ligands such as LPS, a TLR4 ligand, lead to a release of IL-1β upon ATP application. The receptors TLR2, -3 and -4 are all found on microglia. The ATP-dependent release of IL-1β after pretreatment with TLR agonists can be blocked by a P2X7 antagonist A7410003 in primary cultures of microglia.113 Regarding the oxygen/glucose deprivation (OGD) model, an accepted cell culture stroke model, the ATP receptors P2X4 and P2X7 are upregulated on microglia, which accounts for cellular damage after OGD. Purinergic antagonists decreased cell injury under these conditions.114 The P2Y6 receptor on microglia is activated by uridine diphosphate (UDP) from damaged neurons, causing microglial phagocytosis, and thus limiting the inflammatory response after cell injury in in vitro and in vivo rat models.115 Phagocytosis is also enhanced by P2Y2/4 and P2Y12 receptors.116 In contrast to that, the P2Y12 receptor on microglia has a proinflammatory and chemotactic effect in brain ischemia models as inhibition improves neuron viability, which is interesting. As the common use of the antiplateleta agent clopidogrel targets P2Y12, an additional therapeutic side effect can be assumed.117 Moreover, P2Y-receptor-mediated Ca2+ signaling and migration of microglia, which are mainly dependent on P2Y1 and P2Y6 receptors, are altered by extracellular acidosis.118 On the other hand, 2’,3’-cAMP released from damaged cells is metabolized by an extracellular pathway on microglia to 2’- and 3’-AMP, which is then metabolized to adenosine. Adenosine has a neuroprotective role if bound to its A1 receptor on microglia. Therefore, this 2’,3’-cAMP-adenosine pathway functions as a limiting factor to the microglial response after cell injury.119 In LPS-stimulated microglia, a reduced number of A1 receptors can be observed whereby ATP limits its own activation potential.120
Figure 3.

Nucleotide-associated pathways in microglia. A toll-like receptor (TLR) agonist such as lipopolysaccharide (LPS) leads to the release of proinflammatory mediators such as interleukin (IL)-6 in an ATP-dependent manner. Signaling through P2X4 receptors enhances migration of microglia. Uridine triphosphate (UTP) binds to P2Y2/4, Uridine diphosphate (UDP) to P2Y6 and adenosine diphosphate (ADP) to P2Y12, which results in a higher rate of phagocytosis. 2’,3’-cyclic adenosine monophosphate (cAMP) is released from damaged cells and is metabolized by an extracellular pathway to 2’- and 3’-adenosine monophosphate (AMP), which is then metabolized to adenosine. After adenosine has activated its A1 receptor, it possesses neuroprotective functions.
In summary, DAMPs play a major role in microglia activation and possess both neuroprotective and inflammatory functions.
DAMPs and other cell types
Besides microglia, other brain resident cells can also be activated through DAMPs.
Concerning astrocytes, the expression of TLRs like TLR2 and -4 can be induced by DAMPs and other soluble factors, including interferon γ.121 In an oxygen-glucose deprivation/reoxygenation (OGD/R) model, the pathologic swelling of astrocytes can be mediated through TLR4, myeloid differentiation primary response gene 88 (MyD88) and NFκB.122 Further, it is known that astrocytes exhibit a PI3K/Akt-mediated upregulation of HMGB1 and IL-6 in the ischemic brain, which is crucial for their proangiogenic function.123 Moreover, a feedback loop between microglia and astrocytes exists. Following activation of microglia in primary cocultures, microglia-derived ATP recruits astrocytes, which in turn are a potential source of neurotoxic glutamate.124
Regarding endothelial cells, it has recently been shown that HMGB1 activates monocytes and endothelia via RAGE after stroke, resulting in atheroprogression, and thus vascular inflammation.125 It is described that polarized peroxiredoxin 1 localizes to endothelial cells from ipsilateral brain microvessels 12 h after transient middle cerebral artery occlusion (tMCAO) induction in vivo, which might be involved in its neuroprotective effect on tight junction proteins.126 Additionally, it has been shown that in the early phase after ischemia, peroxiredoxin 1 is upregulated and owns neuroprotective functions whereas in the later stages the degradation of peroxiredoxin 1, mediated by the E3 ubiquitin ligase E6-associated protein (E6AP), results in neurovascular damage.127
When it comes to oligodendrocytes, an autocrine HMGB1-TLR2 axis is hypothesized. Following ischemia, released HMGB1 binds to TLR2, which results in the activation of protective signaling pathways.128 In this context, HMGB1 acts as an endogenous trophic factor for oligodendrocytes, supporting white matter integrity following ischemia.129
In summary, the role of DAMPs on other cell types is diverse and further attempts are necessary to elucidate the exact involvement in means of stroke.
Conclusions and perspectives
A set of DAMPs were outlined in this review. Besides the neuroprotective effects of cold domain proteins or adenosine, proinflammatory reactions were described as induced by ATP or HMGB1. Therefore, DAMPs possess a wide variety of functions with context-specific effects dependent on cell type or intra- versus extracellular location. As released by necrotic cells after brain ischemia, they take part in the early reactions after stroke onset. Both the innate and the innate immune system are substantial players in the cellular concert after ischemia. The list of DAMPs can be extended by numerous other protein DAMPs, such as S100, serum amyloid A, histones and nonprotein DAMPs such as heparin sulfate, DNA or RNA.10 Their function is still to be further distinguished. Moreover, we shed light on the involvement of microglia, which play a major role in the acute innate immune reaction after brain ischemia. Microglia are affected by DAMPs but also secrete them actively. What is more, initial therapeutic strategies targeting DAMPs were discussed highlighting new methods in stroke therapy.
Footnotes
Funding: This work was supported by a grant from the Hermann and Lilly Schilling foundation.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Eileen Gülke, Department of Neurology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Mathias Gelderblom, Department of Neurology, University Medical Center Hamburg-Eppendorf, Martinistraße 52, 20246 Hamburg, Germany.
Tim Magnus, Department of Neurology, University Medical Center Hamburg-Eppendorf, Martinistraße 52, 20246 Hamburg, Germany.
References
- 1. Feigin VL, Forouzanfar MH, Krishnamurthi R, et al. Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet 2014; 383: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chamorro A, Dirnagl U, Urra X, et al. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol 2016; 15: 869–881. [DOI] [PubMed] [Google Scholar]
- 3. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nature Med 2011; 17: 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin R, Liu L, Zhang S, et al. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res 2013; 6: 834–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yilmaz G, Arumugam TV, Stokes KY, et al. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006; 113: 2105–2112. [DOI] [PubMed] [Google Scholar]
- 6. Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009; 40: 1849–1857. [DOI] [PubMed] [Google Scholar]
- 7. Planas AM, Gorina R, Chamorro A. Signalling pathways mediating inflammatory responses in brain ischaemia. Biochem Soc Trans 2006; 34: 1267–1270. [DOI] [PubMed] [Google Scholar]
- 8. Hu X, Leak RK, Shi Y, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol 2015; 11: 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12: 991–1045. [DOI] [PubMed] [Google Scholar]
- 10. Huang J, Xie Y, Sun X, et al. DAMPs, ageing, and cancer: the ‘DAMP Hypothesis.’ Ageing Res Rev 2015; 24: 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007; 81: 1–5. [DOI] [PubMed] [Google Scholar]
- 12. van de Beek D, Wijdicks EF, Vermeij FH, et al. Preventive antibiotics for infections in acute stroke: a systematic review and meta-analysis. Arch Neurol 2009; 66: 1076–1081. [DOI] [PubMed] [Google Scholar]
- 13. Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol 2015; 6: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 2011; 29: 139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005; 5: 331–342. [DOI] [PubMed] [Google Scholar]
- 16. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195. [DOI] [PubMed] [Google Scholar]
- 17. Qiu J, Nishimura M, Wang Y, et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab 2008; 28: 927–938. [DOI] [PubMed] [Google Scholar]
- 18. Venereau E, Schiraldi M, Uguccioni M, et al. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol 2013; 55: 76–82. [DOI] [PubMed] [Google Scholar]
- 19. Lorenzen I, Mullen L, Bekeschus S, et al. Redox regulation of inflammatory processes is enzymatically controlled. Oxid Med Cell Longev 2017; 2017: 8459402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang H, Lundback P, Ottosson L, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 2012; 18: 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A 2010; 107: 11942–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frank MG, Weber MD, Fonken LK, et al. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: a role for the NLRP3 inflammasome. Brain Behav Immun 2016; 55: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goldstein RS, Gallowitsch-Puerta M, Yang L, et al. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock 2006; 25: 571–574. [DOI] [PubMed] [Google Scholar]
- 24. Huang JM, Hu J, Chen N, et al. Relationship between plasma high-mobility group box-1 levels and clinical outcomes of ischemic stroke. J Crit Care 2013; 28: 792–797. [DOI] [PubMed] [Google Scholar]
- 25. Schulze J, Zierath D, Tanzi P, et al. Severe stroke induces long-lasting alterations of high-mobility group box 1. Stroke 2013; 44: 246–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sapojnikova N, Kartvelishvili T, Asatiani N, et al. Correlation between MMP-9 and extracellular cytokine HMGB1 in prediction of human ischemic stroke outcome. Biochim Biophys Acta 2014; 1842: 1379–1384. [DOI] [PubMed] [Google Scholar]
- 27. Tian X, Liu C, Shu Z, et al. Therapeutic targeting of HMGB1 in stroke. Curr Drug Deliv 2017; 14: 785–790. DOI: 10.2174/1567201813666160808111933 [DOI] [PubMed] [Google Scholar]
- 28. Semino C, Angelini G, Poggi A, et al. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood 2005; 106: 609–616. [DOI] [PubMed] [Google Scholar]
- 29. Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003; 22: 5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dumitriu IE, Bianchi ME, Bacci M, et al. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol 2007; 81: 84–91. [DOI] [PubMed] [Google Scholar]
- 31. Richard SA, Sackey M, Su Z, et al. Pivotal neuroinflammatory and therapeutic role of high mobility group box 1 in ischemic stroke. Biosci Rep 2017; 37: pii: BSR20171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liesz A, Dalpke A, Mracsko E, et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. T Neurosci 2015; 35: 583–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sundberg E, Fasth AE, Palmblad K, et al. High mobility group box chromosomal protein 1 acts as a proliferation signal for activated T lymphocytes. Immunobiology 2009; 214: 303–309. [DOI] [PubMed] [Google Scholar]
- 34. Kim JB, Sig Choi J, Yu YM, et al. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 2006; 26: 6413–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Halder SK, Ueda H. Amlexanox inhibits cerebral ischemia-induced delayed astrocytic high-mobility group box 1 release and subsequent brain damage. J Pharmacol Exp Ther. Epub ahead of print 12 January 2018. DOI: 10.1124/jpet.117.245340. [DOI] [PubMed] [Google Scholar]
- 36. Kikuchi K, Tancharoen S, Ito T, et al. Potential of the angiotensin receptor blockers (ARBs) telmisartan, irbesartan, and candesartan for inhibiting the HMGB1/RAGE axis in prevention and acute treatment of stroke. Int J Mol Sci 2013; 14: 18899–18924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gong G, Xiang L, Yuan L, et al. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS One 2014; 9: e89450 DOI: 10.1371/journal.pone.0089450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tang SC, Wang YC, Li YI, et al. Functional role of soluble receptor for advanced glycation end products in stroke. Arteriosclerosis, Thromb Vasc Biol 2013; 33: 585–594. [DOI] [PubMed] [Google Scholar]
- 39. Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A 2007; 104: 13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alberts B. Molecular biology of the cell. New York, NY: Garland Science Taylor & Francis, 2008. [Google Scholar]
- 41. Kim JY, Yenari MA. The immune modulating properties of the heat shock proteins after brain injury. Anat Cell Biol. 2013; 46: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sharp FR, Zhan X, Liu DZ. Heat shock proteins in the brain: role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential. Transl Stroke Res 2013; 4: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shevtsov MA, Nikolaev BP, Yakovleva LY, et al. Neurotherapeutic activity of the recombinant heat shock protein Hsp70 in a model of focal cerebral ischemia in rats. Drug Des Devel Ther 2014; 8: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doeppner TR, Kaltwasser B, Fengyan J, et al. TAT-Hsp70 induces neuroprotection against stroke via anti-inflammatory actions providing appropriate cellular microenvironment for transplantation of neural precursor cells. J Cereb Blood Flow Metab 2013; 33: 1778–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhan X, Ander BP, Liao IH, et al. Recombinant Fv-Hsp70 protein mediates neuroprotection after focal cerebral ischemia in rats. Stroke 2010; 41: 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim JY, Kim N, Zheng Z, et al. 70-kDa heat shock protein downregulates dynamin in experimental stroke: a new therapeutic target? Stroke 2016; 47: 2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lindquist JA, Brandt S, Bernhardt A, et al. The role of cold shock domain proteins in inflammatory diseases. J Mol Med (Berl) 2014; 92: 207–216. [DOI] [PubMed] [Google Scholar]
- 48. Hanssen L, Alidousty C, Djudjaj S, et al. YB-1 is an early and central mediator of bacterial and sterile inflammation in vivo. J Immunol 2013; 191: 2604–2613. [DOI] [PubMed] [Google Scholar]
- 49. Keilhoff G, Titze M, Esser T, et al. Constitutive and functional expression of YB-1 in microglial cells. Neuroscience 2015; 301: 439–453. [DOI] [PubMed] [Google Scholar]
- 50. Zhu X, Buhrer C, Wellmann S. Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell Mol Life Sci 2016; 73: 3839–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou M, Yang WL, Ji Y, et al. Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim Biophys Acta 2014; 1840: 2253–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kurisu K, Yenari MA. Therapeutic hypothermia for ischemic stroke; pathophysiology and future promise. Neuropharmacology 2017: pii: S0028-3908(17)30392-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kaneko T, Kibayashi K. Mild hypothermia facilitates the expression of cold-inducible RNA-binding protein and heat shock protein 70.1 in mouse brain. Brain Res 2012; 1466: 128–136. [DOI] [PubMed] [Google Scholar]
- 54. Shichita T, Hasegawa E, Kimura A, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat med 2012; 18: 911–917. [DOI] [PubMed] [Google Scholar]
- 55. Salzano S, Checconi P, Hanschmann EM, et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A 2014; 111: 12157–12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Konoeda F, Shichita T, Yoshida H, et al. Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model. Biochem Biophys Res Commun 2010; 402: 500–506. [DOI] [PubMed] [Google Scholar]
- 57. Shichita T, Sugiyama Y, Ooboshi H, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med 2009; 15: 946–950. [DOI] [PubMed] [Google Scholar]
- 58. Thom V, Arumugam TV, Magnus T, et al. Therapeutic potential of intravenous immunoglobulin in acute brain injury. Front Immunol 2017; 8: 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gelderblom M, Weymar A, Bernreuther C, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012; 120: 3793–3802. [DOI] [PubMed] [Google Scholar]
- 60. Zhao LX, Du JR, Zhou HJ, et al. Differences in proinflammatory property of six subtypes of peroxiredoxins and anti-inflammatory effect of ligustilide in macrophages. PLoS One 2016; 11: e0164586 DOI: 10.1371/journal.pone.0164586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kunze A, Zierath D, Tanzi P, et al. Peroxiredoxin 5 (PRX5) is correlated inversely to systemic markers of inflammation in acute stroke. Stroke 2014; 45: 608–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Richard S, Lapierre V, Girerd N, et al. Diagnostic performance of peroxiredoxin 1 to determine time-of-onset of acute cerebral infarction. Sci Rep 2016; 6: 38300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mao XN, Zhou HJ, Yang XJ, et al. Neuroprotective effect of a novel gastrodin derivative against ischemic brain injury: involvement of peroxiredoxin and TLR4 signaling inhibition. Oncotarget 2017; 8: 90979–90995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leak RK, Zhang L, Luo Y, et al. Peroxiredoxin 2 battles poly(ADP-ribose) polymerase 1- and p53-dependent prodeath pathways after ischemic injury. Stroke 2013; 44: 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hechler B, Gachet C. Purinergic receptors in thrombosis and inflammation. Arterioscler Thromb Vasc Biol 2015; 35: 2307–2315. [DOI] [PubMed] [Google Scholar]
- 66. Pedata F, Dettori I, Coppi E, et al. Purinergic signalling in brain ischemia. Neuropharmacology 2016; 104: 105–130. [DOI] [PubMed] [Google Scholar]
- 67. Cisneros-Mejorado A, Perez-Samartin A, Gottlieb M, et al. ATP signaling in brain: release, excitotoxicity and potential therapeutic targets. Cell Mol Neurobiol 2015; 35: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Beamer E, Goloncser F, Horvath G, et al. Purinergic mechanisms in neuroinflammation: an update from molecules to behavior. Neuropharmacology 2016; 104: 94–104. [DOI] [PubMed] [Google Scholar]
- 69. Kanthi YM, Sutton NR, Pinsky DJ. CD39: Interface between vascular thrombosis and inflammation. Curr Atheroscler Rep 2014; 16: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hyman MC, Petrovic-Djergovic D, Visovatti SH, et al. Self-regulation of inflammatory cell trafficking in mice by the leukocyte surface apyrase CD39. J Clin Invest 2009; 119: 1136–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Arbeloa J, Perez-Samartin A, Gottlieb M, et al. P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol Dis 2012; 45: 954–961. [DOI] [PubMed] [Google Scholar]
- 72. Burnstock G. Purinergic signalling: therapeutic developments. Front Pharmacol 2017; 8: 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cisneros-Mejorado A, Gottlieb M, Cavaliere F, et al. Blockade of P2X7 receptors or pannexin-1 channels similarly attenuates postischemic damage. J Cereb Blood Flow Metab 2015; 35: 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Masuch A, Shieh CH, van Rooijen N, et al. Mechanism of microglia neuroprotection: involvement of P2X7, TNFalpha, and valproic acid. Glia 2016; 64: 76–89. [DOI] [PubMed] [Google Scholar]
- 75. Rissiek B, Haag F, Boyer O, et al. P2X7 on mouse T cells: one channel, many functions. Front Immunol 2015; 6: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gaudin A, Yemisci M, Eroglu H, et al. Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat Nanotechnol 2014; 9: 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cui M, Ding H, Chen F, et al. Mdivi-1 Protects against ischemic brain injury via elevating extracellular adenosine in a cAMP/CREB-CD39-dependent manner. Mol Neurobiol 2016; 53: 240–253. [DOI] [PubMed] [Google Scholar]
- 78. Shichita T, Ito M, Yoshimura A. Post-ischemic inflammation regulates neural damage and protection. Front Cell Neurosci 2014; 8: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xu J, Wang H, Won SJ, et al. Microglial activation induced by the alarmin S100B is regulated by poly(ADP-ribose) polymerase-1. Glia 2016; 64: 1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tang SC, Yeh SJ, Tsai LK, et al. Association between plasma levels of hyaluronic acid and functional outcome in acute stroke patients. J Neuroinflammation 2014; 11: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lindwall C, Olsson M, Osman AM, et al. Selective expression of hyaluronan and receptor for hyaluronan mediated motility (Rhamm) in the adult mouse subventricular zone and rostral migratory stream and in ischemic cortex. Brain Res 2013; 1503: 62–77. [DOI] [PubMed] [Google Scholar]
- 82. Arumugam TV, Okun E, Tang SC, et al. Toll-like receptors in ischemia-reperfusion injury. Shock 2009; 32: 4–16. [DOI] [PubMed] [Google Scholar]
- 83. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol 2014; 32: 367–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 2013; 10: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 2013; 39: 3–18. [DOI] [PubMed] [Google Scholar]
- 86. Tay TL, Mai D, Dautzenberg J, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 2017; 20: 793–803. [DOI] [PubMed] [Google Scholar]
- 87. Biber K, Owens T, Boddeke E. What is microglia neurotoxicity (Not)? Glia 2014; 62: 841–854. [DOI] [PubMed] [Google Scholar]
- 88. Lee Y, Lee SR, Choi SS, et al. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Res Int 2014; 2014: 297241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science 2013; 339: 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Huang WC, Qiao Y, Xu L, et al. Direct protection of cultured neurons from ischemia-like injury by minocycline. Anat Cell Biol 2010; 43: 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Szalay G, Martinecz B, Lenart N, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016; 7: 11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Frank MG, Weber MD, Watkins LR, et al. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun 2015; 48: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Weber MD, Frank MG, Tracey KJ, et al. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. Journal Neurosci 2015; 35: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Weber MD, Frank MG, Sobesky JL, et al. Blocking toll-like receptor 2 and 4 signaling during a stressor prevents stress-induced priming of neuroinflammatory responses to a subsequent immune challenge. Brain Behav Immun 2013; 32: 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee S, Nam Y, Koo JY, et al. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat Chem Biol 2014; 10: 1055–1060. [DOI] [PubMed] [Google Scholar]
- 96. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009; 27: 229–265. [DOI] [PubMed] [Google Scholar]
- 97. Huang C, Lu X, Wang J, et al. Inhibition of endogenous heat shock protein 70 attenuates inducible nitric oxide synthase induction via disruption of heat shock protein 70/Na(+) /H(+) exchanger 1-Ca(2+) -calcium-calmodulin-dependent protein kinase II/transforming growth factor β-activated kinase 1-nuclear factor-kappaB signals in BV-2 microglia. J Neurosci Res 2015; 93: 1192–1202. [DOI] [PubMed] [Google Scholar]
- 98. Kim N, Kim JY, Yenari MA. Anti-inflammatory properties and pharmacological induction of Hsp70 after brain injury. Inflammopharmacology 2012; 20: 177–185. [DOI] [PubMed] [Google Scholar]
- 99. Sheppard PW, Sun X, Khammash M, et al. Overexpression of heat shock protein 72 attenuates NF-kappaB activation using a combination of regulatory mechanisms in microglia. PLoS Comput Biol 2014; 10: e1003471 DOI: 10.1371/journal.pcbi.1003471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zheng Z, Kim JY, Ma H, et al. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab 2008; 28: 53–63. [DOI] [PubMed] [Google Scholar]
- 101. Kacimi R, Yenari MA. Pharmacologic heat shock protein 70 induction confers cytoprotection against inflammation in gliovascular cells. Glia 2015; 63: 1200–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jin MH, Lee YH, Kim JM, et al. Characterization of neural cell types expressing peroxiredoxins in mouse brain. Neurosci Lett 2005; 381: 252–257. [DOI] [PubMed] [Google Scholar]
- 103. Kim SU, Park YH, Min JS, et al. Peroxiredoxin I is a ROS/p38 MAPK-dependent inducible antioxidant that regulates NF-kappaB-mediated iNOS induction and microglial activation. J Neuroimmunol 2013; 259: 26–36. [DOI] [PubMed] [Google Scholar]
- 104. Sun HN, Kim SU, Huang SM, et al. Microglial peroxiredoxin V acts as an inducible anti-inflammatory antioxidant through cooperation with redox signaling cascades. J Neurochem 2010; 114: 39–50. [DOI] [PubMed] [Google Scholar]
- 105. Park J, Choi H, Kim B, et al. Peroxiredoxin 5 (Prx5) decreases LPS-induced microglial activation through regulation of Ca(2+)/calcineurin-Drp1-dependent mitochondrial fission. Free Radic Biol Med 2016; 99: 392–404. [DOI] [PubMed] [Google Scholar]
- 106. Hwang IK, Yoo KY, Kim DW, et al. Changes in the expression of mitochondrial peroxiredoxin and thioredoxin in neurons and glia and their protective effects in experimental cerebral ischemic damage. Free Radic Biol Med 2010; 48: 1242–1251. [DOI] [PubMed] [Google Scholar]
- 107. Tan L, Zhao Y, Jiang B, et al. [Knockdown of PRDX6 in microglia reduces neuron viability after OGD/R injury]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2016; 32: 1014–1020. [PubMed] [Google Scholar]
- 108. Huang S, Liu X, Zhang J, et al. Expression of peroxiredoxin 1 after traumatic spinal cord injury in rats. Cell Mol Neurobiol 2015; 35: 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nakaso K, Kitayama M, Mizuta E, et al. Co-induction of heme oxygenase-1 and peroxiredoxin I in astrocytes and microglia around hemorrhagic region in the rat brain. Neurosci Lett 2000; 293: 49–52. [DOI] [PubMed] [Google Scholar]
- 110. Garcia-Bonilla L, Iadecola C. Peroxiredoxin sets the brain on fire after stroke. Nat Med 2012; 18: 858–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Suk K, Ock J. Chemical genetics of neuroinflammation: natural and synthetic compounds as microglial inhibitors. Inflammopharmacology 2012; 20: 151–158. [DOI] [PubMed] [Google Scholar]
- 112. Shieh CH, Heinrich A, Serchov T, et al. P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-alpha in cultured mouse microglia. Glia 2014; 62: 592–607. [DOI] [PubMed] [Google Scholar]
- 113. Facci L, Barbierato M, Marinelli C, et al. Toll-like receptors 2, -3 and -4 prime microglia but not astrocytes across central nervous system regions for ATP-dependent interleukin-1β release. Sci Rep 2014; 4: 6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cavaliere F, Dinkel K, Reymann K. Microglia response and P2 receptor participation in oxygen/glucose deprivation-induced cortical damage. Neuroscience 2005; 136: 615–623. [DOI] [PubMed] [Google Scholar]
- 115. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007; 446: 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Castellano B, Bosch-Queralt M, Almolda B, et al. Purine signaling and microglial wrapping. Adv Exp Med Biol 2016; 949: 147–165. [DOI] [PubMed] [Google Scholar]
- 117. Webster CM, Hokari M, McManus A, et al. Microglial P2Y12 deficiency/inhibition protects against brain ischemia. PLoS One 2013; 8: e70927 DOI: 10.1371/journal.pone.0070927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Langfelder A, Okonji E, Deca D, et al. Extracellular acidosis impairs P2Y receptor-mediated Ca(2+) signalling and migration of microglia. Cell Calcium 2015; 57: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Verrier JD, Exo JL, Jackson TC, et al. Expression of the 2’,3’-cAMP-adenosine pathway in astrocytes and microglia. J Neurochem 2011; 118: 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Luongo L, Guida F, Imperatore R, et al. The A1 adenosine receptor as a new player in microglia physiology. Glia 2014; 62: 122–132. [DOI] [PubMed] [Google Scholar]
- 121. Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 2014; 20: 160–172. [DOI] [PubMed] [Google Scholar]
- 122. Sun L, Li M, Ma X, et al. Inhibition of HMGB1 reduces rat spinal cord astrocytic swelling and AQP4 expression after oxygen-glucose deprivation and reoxygenation via TLR4 and NF-kappaB signaling in an IL-6-dependent manner. J Neuroinflammation 2017; 14: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chen JY, Yu Y, Yuan Y, et al. Enriched housing promotes post-stroke functional recovery through astrocytic HMGB1-IL-6-mediated angiogenesis. Cell Death Discov 2017; 3: 17054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pascual O, Ben Achour S, Rostaing P, et al. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A 2012; 109: E197–E205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Roth S, Singh V, Tiedt S, et al. Brain-released alarmins and stress response synergize in accelerating atherosclerosis progression after stroke. Sci Transl Med 2018; 10: pii: eaao1313. [DOI] [PubMed] [Google Scholar]
- 126. Ye WF, Tao RR, Jiang Q, et al. Peroxiredoxin 1 participates in ischemia-triggered endothelial polarization. CNS Neurosci Ther 2014; 20: 791–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tao RR, Wang H, Hong LJ, et al. Nitrosative stress induces peroxiredoxin 1 ubiquitination during ischemic insult via E6AP activation in endothelial cells both in vitro and in vivo. Antioxid Redox Signal 2014; 21: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Choi JY, Kim BG. Toll-like receptor 2: a novel therapeutic target for ischemic white matter injury and oligodendrocyte death. Exp Neurobiol 2017; 26: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Choi JY, Cui Y, Chowdhury ST, et al. High-mobility group box-1 as an autocrine trophic factor in white matter stroke. Proc Natl Acad Sci U S A 2017; 114: E4987–E4995. [DOI] [PMC free article] [PubMed] [Google Scholar]