

Abstract
Multiple myeloma is a fatal plasma cell neoplasm accounting for over 10,000 deaths in the United States each year. Despite new therapies, multiple myeloma remains incurable, and patients ultimately develop drug resistance and succumb to the disease. The response to selective CDK4/6 inhibitors has been modest in multiple myeloma, potentially because of incomplete targeting of other critical myeloma oncogenic kinases. As a substantial number of multiple myeloma cell lines and primary samples were found to express AMPK-related protein kinase 5(ARK5), a member of the AMPK family associated with tumor growth and invasion, we examined whether dual inhibition of CDK4 and ARK5 kinases using ON123300 results in a better therapeutic outcome. Treatment of multiple myeloma cell lines and primary samples with ON123300 in vitro resulted in rapid induction of cell-cycle arrest followed by apoptosis. ON123300-mediated ARK5 inhibition or ARK5-specific siRNAs resulted in the inhibition of the mTOR/S6K pathway and upregulation of the AMPK kinase cascade. AMPK upregulation resulted in increased SIRT1 levels and destabilization of steady-state MYC protein. Furthermore, ON123300 was very effective in inhibiting tumor growth in mouse xenograft assays. In addition, multiple myeloma cells sensitive to ON123300 were found to have a unique genomic signature that can guide the clinical development of ON123300. Our study provides preclinical evidence that ON123300 is unique in simultaneously inhibiting key oncogenic pathways in multiple myeloma and supports further development of ARK5 inhibition as a therapeutic approach in multiple myeloma.
Introduction
Multiple myeloma is a malignancy characterized by uncontrolled proliferation of clonal plasma cells with an incidence of about 20,000 per year in the United States (1, 2). The major clinical presentations of the disease include hypercalcemia, renal failure, anemia, and lytic bone destruction (3). Despite recent advances in new therapies for multiple myeloma, this disease remains incurable with a median overall survival of 7 to 8 years (4). The pursuit for drugs that inhibit cyclin-dependent kinases (CDK) has been an intense area of research (5). Despite cell-cycle dysregulation being prominent in myeloma pathogenesis, efficacy of CDK inhibitors as single agents has been modest (6, 7). Therefore, there is an urgent need to identify new myeloma targets for drug development.
To address these challenges, we recently described the development of ON123300 (8, 9), a second-generation, orally bioavailable CDK inhibitor that potently inhibits CDK4 as well as AMPK-related protein kinase 5 (ARK5). ARK5 (also known as NUAK1) is a member of the AMP-activated protein kinase (AMPK) catalytic subunit family and functions as a key regulator of cellular energy homeostasis (10). ARK5 expression is associated with increased tumor cell invasiveness in multiple myeloma, transcriptionally regulated by multiple myeloma oncogenes such as MMSET, c-MAF, and MAFB (11), and is also directly activated by Akt, thereby regulating Akt-dependent cell survival and migration activity (12, 13).
In this study, we sought to determine whether selective inhibition of ARK5 and CDK4 could be an effective way to target cellular proliferation in multiple myeloma. Our findings demonstrate specific and potent antimyeloma activity of ON123300, a dual ARK5/CDK4 inhibitor, and that ARK5 inhibition is lethal in multiple myeloma cells in vitro and in vivo while sparing normal healthy B cells. Our study also suggests a novel function for ARK5 in bridging the mTOR/Rb/MYC pathways. In summary, our study has revealed a new regulatory mechanism for controlling multiple myeloma with immediate translational relevance using small-molecule inhibitors of ARK5.
Materials and Methods
Cell lines, culture conditions, and drug treatment
Multiple myeloma cell lines MM.1R, KMS11, ARP1, RPMI-8226, MM1.S, EJM, JJN3, and NCI-H929 were cultured in RPMI1640 medium (CellGro) supplemented with 10% FBS (Gemini Bio Products), N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 100 U/mL penicillin G, and 100 μg/mL streptomycin (CellGro). ON123300 and PD-0332991 compounds were supplied by Dr. M.V. Reddy (Icahn School of Medicine, Mount Sinai, New York, NY). SRT1720 (cat# S1129) was purchased from Selleck Chemicals. All drugs were stored between 4°C and −20°C. Cells were treated in series of eight 100 μL wells for 48 hours for viability assessment and in 3 mL wells in triplicate, for 24 to 48 hours, to determine protein amounts. All cells were propagated in standard cell culture conditions (5% CO2, 37°C) in cell culture–treated T75/T150 flasks (Falcon). Once cells had reached 80% confluency, cells were replated in T75 flasks. After 10 to 12 passages, cells were discarded. All cell lines were authenticated and tested negative for mycoplasma.
Cell viability assay
Cell viability was determined by a fluorometric resazurin reduction method (CellTiter-Blue; Promega) according to the manufacturer’s instructions. The number of viable cells in each treated well was calculated 48 hours after treatment. Cells (100 μL; 105 cells per well) were plated in 96-well plates (8 replicates per condition), with 20 μL of CellTiter-Blue Reagent added to each well. After 1 hour of incubation with the dye, fluorescence (560Ex/590Em) was measured with the FLUOstar Microplate Reader (BMG LABTECH). The number of viable cells in each treated well was calculated based on the linear least-squares regression of the standard curve. Cell viability in drug-treated cells was normalized to their respective untreated controls. Cell counts were confirmed on the Countess Automated Cell Counter (Life Technologies).
Primary multiple myeloma cells and BMSCs
All studies involving human samples were performed under Mount Sinai Hospital (New York, NY) Institutional Review Board committee–approved protocols, through which informed consent was obtained, and deidentified samples were utilized. Mononuclear cells were isolated from aspirates as detailed in the Miltenyi MidiMACS bone marrow mononuclear cell protocol. Mononuclear cells isolated from multiple myeloma patient bone marrow aspirates were positively sorted using anti-CD138 magnetic microbeads and the MidiMACS cell sorting system following manufacturer’s protocol (Miltenyi Biotec) to >95% purity as determined by anti-CD138-PE staining and FACS analysis and cultured in 37°C/5% CO2 incubators in RPMI1640 medium. Human HS-5 bone marrow stromal cells (BMSC; ATCC CRL-11882) were obtained from ATCC. These cells were cultured in RPMI1640 medium containing 20% FBS. A total of 30,000 HS5 BMSCs per well were plated in 6-well dishes and allowed to form an adherent monolayer for more than 48 hours. A total of 90,000 multiple myeloma cells (MM1.S and NCI-H929 cells) were plated over the monolayer. The BMSC mono-layer remained undisturbed.
Apoptosis and cell-cycle analysis
MM1.S and NCI-H929 cells were treated with ON123300/PD-0332991 for 24 hours. Approximately 106 cells of each cell line were harvested (along with untreated controls) after 24 hours of treatment in 12-well plates. Cells were washed with PBS and resuspended in appropriate volume of Annexin binding buffer and then stained with Annexin V Pacific Blue/PI (BD Biosciences; Clontech), followed by quantification of apoptotic cells using FACSFortessa (BD Biosciences). Cell cycle was analyzed by using Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit, and cell-cycle distribution was assessed with FACSFortessa.
Transient transfection
For transfections, two Stealth siRNAs against human ARK5 (#1:5′-GAAGTTATGCTTTATTCAC-3′;#2:5′-CATCCTCTGATTCTAGGTG-3′) and a scrambled siRNA were synthesized by Life Technologies. Individual ON-TARGETplus SIRT1 siRNAs (#J-003540-09 and J-003540-10) were purchased from GE Dharmacon. MM1.S and NCI-H929 cells were transiently transfected with ARK5 siRNA and scrambled nontargeting siRNA using the cell line 4D Nucleofector Solution SG program EO100 (Amaxa Biosystems). Cells were harvested 24 hours after transfection, followed by analysis using immunoblotting, apoptosis, and cell viability assay.
Western blot analysis
Cells were lysed in modified RIPA buffer containing 50 mmol/L Tris-HCl (pH, 8), 300 mmol/L NaCl, 10% NP-40, 1% sodium deoxycholate, and 0.1% SDS and a protease inhibitor cocktail tablet (Roche Applied Science). Protein extracts, approximately 30 μg of each sample, were resolved by SDS-PAGE followed by immunoblotting with ARK5 antibody (Cell Signaling Technology #4458), total MYC (Cell Signaling Technology #9402), acetyl MYC lysine 323 (Millipore #ABE26), SIRT1 (Cell Signaling Technology # 2310), AMPK (Cell Signaling Technology #2532), total Rb (Santa Cruz Biotechnology #sc-74562), phosphoRb (Cell Signaling Technology #9308), total S6K (Cell Signaling Technology #9202), phosphoS6K (Cell Signaling Technology #9205), and actin antibody (C-11, horseradish peroxidase, goat polyclonal antibody; Santa Cruz Biotechnology) and detected by enhanced chemiluminescence (Santa Cruz Biotechnology). After treatment, cells were harvested and washed with ice-cold PBS and subsequently lysed with RIPA buffer with fresh protease and phosphatase inhibitors. Blot patterns were analyzed using ImageJ software (http://rsbweb.nih.gov/ij/), providing a quantitative measure of protein expression.
RNA sequencing and data analysis
Total RNA was isolated using PureLink RNA Mini Kit (Life Technologies). We extracted poly A–selected RNA from CD138+-selected primary multiple myeloma cells and NCI-H929 cell line. In total, 100 ng of total RNA input was used to construct RNASeq libraries using TruSeq RNA Sample Preparation Kit v2 (Illumina) following the manufacturer’s instructions. Sequencing was done on a HiSeq 2500 System using 100 bases and paired-end read sequencing for primary cells and two samples of ARK5 siRNA experiment with NCI-H929 cells. The data discussed in this article have been deposited in the sequence read archive of NCBI with study accession number SRP051819. The reads with quality Phred scores more than 30 were aligned to reference sequence database (UCSC hg19), as well as RefSeq exons, splicing junctions, and contamination databases, including ribosome and mitochondria sequences using the Burrows–Wheeler Aligner. To compare the expression levels, the read counts of transcripts in each sample were normalized by leveling the total read count in each sample to the maximum of the read counts in all samples. The differentially expressed transcripts were identified using LIMMA package in Bioconductor (14). Subsequent analysis of gene expression data was done in the freely available statistical computing language R (www.r-project.org) with packages available from the Bioconductor project (www.bioconductor.org). Further pathway analysis was carried out using MetaCore of GeneGo, Inc. MetaCore analyzes experimental high-throughput data in the context of pathways and networks that are ideal for data mining. This pathway analysis tool was used to obtain curated molecular interactions related to the differentially regulated genes. Gene set enrichment analysis (GSEA, version 2.0.14) was performed with the C6 oncogenic signature gene sets from the MSigDB version 4.0 (http://www.broadinstitute.org/gsea/msigdb; ref. 15) with pre-ranked genes (selected based on log-fold change and adjusted P < 0.05) that were differentially expressed between NCI-H929 ARK5 siRNA-depleted and control cells.
Statistical analysis
Comparisons of means between control and experimental groups were by Student t test. A P value of less than 0.05 was considered to indicate statistical significance, and all tests were two-tailed. All in vitro experiments were performed in triplicate and repeated at least three times. Results were expressed as mean + SE of the untransformed data. All the analyses were performed with packages in R unless otherwise specified.
In vivo tumor models
All animal studies were carried out in accordance with the guidelines of the Institute for Animal Studies at Mount Sinai (New York, NY). Two million of MM1.S and NCI-H929 cells were mixed with 50% Matrigel (BD Biosciences) and injected subcutaneously into the right flank of 4- to 5-week-old NSG female mice (Taconic). When the tumors approached 0.5 to 0.7 cm in diameter at approximately 10 to 14 days after injection of cancer cells, the mice were divided into 2 groups: (i) ON123300 group, which received a dose of 100 mg/kg i.p. every alternate day and (ii) control untreated group, which received saline every alternate day. Tumor volume and weight were assessed every 2 days. Tumor volumes were calculated using the following equation: (4/3) [(L+W)/4]3. The data were expressed as average tumor volume (mm3) per group as a function of time. Animals were sacrificed when tumor diameter exceeded 1 cm or after loss of greater than 10% body weight in accordance with the institutional guidelines.
Results
ON123300 is a multitargeted kinase inhibitor with potent in vitro activity against multiple myeloma cells
Our own efforts in anticancer drug discovery led to the development of ON123300 (Fig. 1A; Supplementary Table S1), a multitargeted kinase inhibitor with potent inhibitory activity against ARK5 and CDK4 (9). Our earlier studies using ON123300 in breast, glioma, and mantle cell lymphoma models had shown potent inhibition of these kinases in vitro and in vivo (9, 16, 17). Prior studies have also described that ARK5 is expressed in an MAF-dependent manner in newly diagnosed multiple myeloma patients (11). ARK5 expression in primary myelomas expressing c-MAF and MAFB suggested that ARK5 may be a transcriptional target of the large MAF family and that ARK5 might be involved in multiple myeloma pathogenesis. In addition, as a recent study (10) showed that ARK5 overexpression is required to prevent proapoptotic effects of MYC overexpression in tumor cells, we examined whether ARK5 could serve as a potential therapeutic target in multiple myeloma. To address this hypothesis, we evaluated the antiproliferative effect of the ARK5/CDK4 inhibitor ON123300 against a panel of multiple myeloma cell lines and primary multiple myeloma cells. Initially, we evaluated the levels of ARK5 in multiple myeloma by Western blot analysis. We examined 8 multiple myeloma cell lines and 3 primary samples from relapsed multiple myeloma patients for the expression of ARK5 protein levels. ARK5 was found to be overexpressed in all primary samples as well as 7 of the multiple myeloma cell lines with the exception of RPMI 8226 (Fig. 1B). ON123300 decreased cell viability by approximately 60% to 80% at a concentration of 50 nmol/L in 4 of 8 multiple myeloma cell lines (Fig. 1C). Similarly 4 of 6 primary multiple myeloma cells were sensitive to treatment with ON123300 at the same concentration (Fig. 1D). Normal peripheral blood cells were spared from the effects of this compound, confirming a potent and specific anti–multiple myeloma effect of ON123300.
Figure 1.
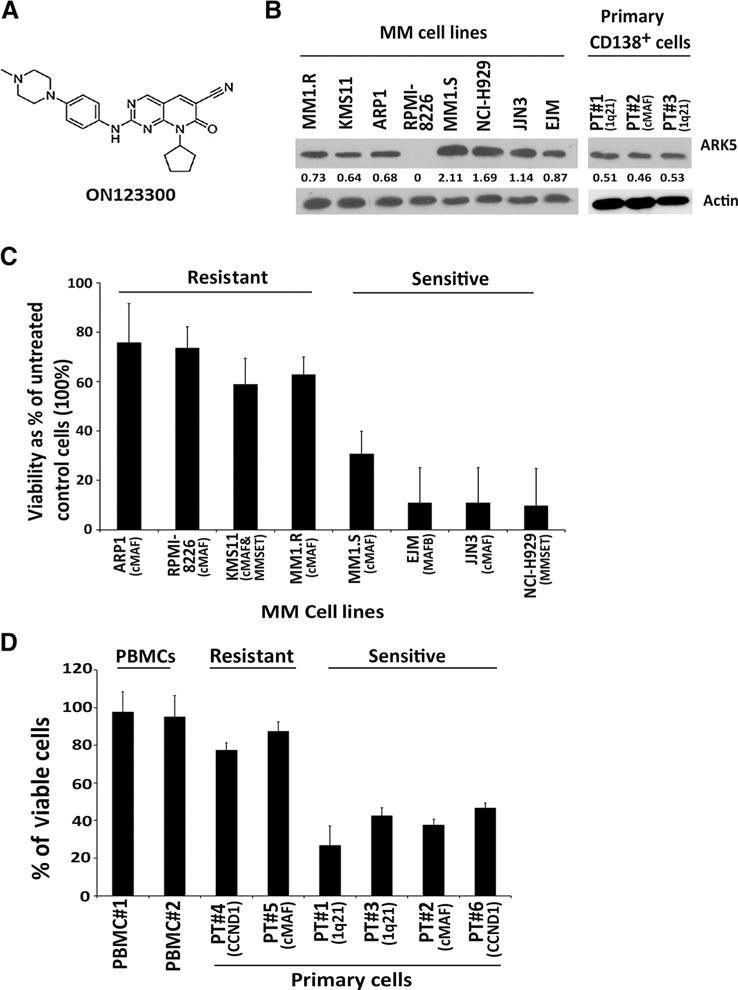
Anti–multiple myeloma activity of ON123300, a multitargeted kinase inhibitor. A, structure of ON123300-8-Cyclopentyl-2-[4-(4-methylpiperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile. B, ARK5 protein expression was confirmed by Western blot in multiple myeloma (MM) cell lines and primary cells. ARK5 level was significantly higher in the cell lines MM1.S, NCI-H929, JJN3, EJM, and in primary samples with 1q21 deletion and cMAF translocation. The numerical values indicate densitometric analysis of band intensity using ImageJ. C, ON123300 decreased cell viability by approximately 30% to 70% (IC50, 50–200 nmol/L) in 8/8 multiple myeloma cell lines examined. On the basis of IC50 of 50 nmol/L, we could separate the cell lines into two groups: four cell lines (MM.1R, KMS11, RPMI-8226, and ARP1) had IC50 of 50–150 nmol/L, and four cell lines (MM1.S, EJM, JJN3, and NCI-H929) were very sensitive (IC50 < 50 nmol/L). D, purified CD138+ patient multiple myeloma cells from six patients were treated with ON123300 at 50 nmol/L concentration for 48 hours, followed by assessment of viability using CellTiter-Blue assay. PBMCs from healthy donors treated with ON123300 for 48 hours showed no change in the cell viability, whereas there was a significant decrease in cell viability in primary multiple myeloma cells and in both cell lines.
ON123300 induces apoptosis, cell-cycle arrest, and negatively regulates mTOR/MYC pathways in multiple myeloma cells
To further understand the mechanism of action underlying ON123300-induced multiple myeloma cell death, MM1.S and NCI-H929 cells were treated with ON123300 (5 nmol/L, 10 nmol/L, and 50 nmol/L) for 24 hours and then stained with Annexin V/PI and subjected to flowcytometric analysis. ON123300 induced a significant increase in both early and late apoptotic cell populations (Fig. 2A and Supplementary Fig. S1). In contrast, the highly selective CDK4/6 inhibitor PD-0332991 (7, 18) that is currently in clinical trials showed no clear evidence of activation of apoptosis in multiple myeloma cells (Supplementary Fig. S2). In PD-0332991 treated MM1.S cells, the majority of cells were arrested in the G1 phase of the cell cycle, and in NCI-H929 cells, no significant changes were observed in response to PD-0332991 treatment (Supplementary Fig. S3). ON123300 induced a significant G1–S arrest in both cell lines treated as determined by 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay (Fig. 2B and Supplementary Fig. S4). Together, these data indicate that ON123300 inhibits an antiapoptotic program in multiple myeloma cells and that inactivation of CDK4 and ARK5 by ON123300 might lead to a rapid induction of apoptosis as compared with PD-0332991 in both MM1.S and NCI-H929 cells.
Figure 2.
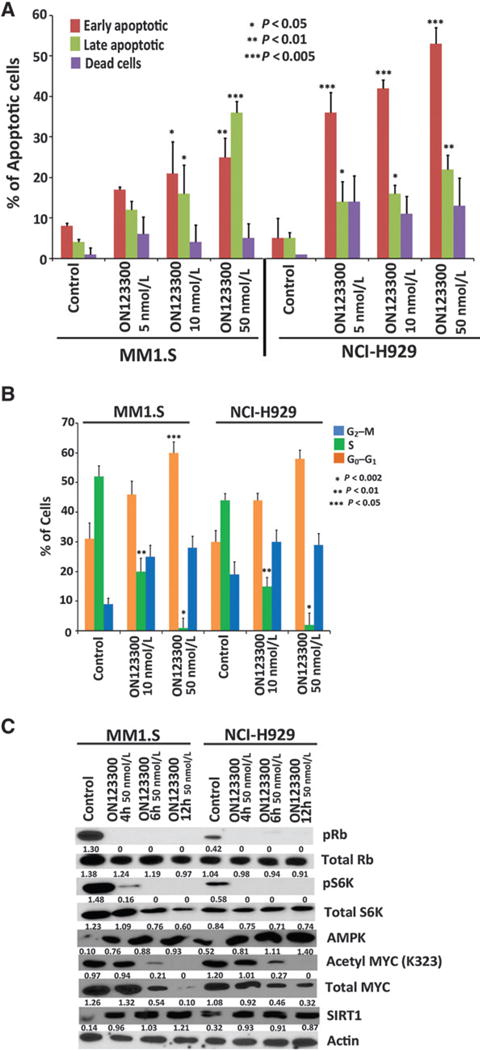
Dual ARK5 and CDK4 inhibitor ON123300 potently induces apoptosis and cell-cycle arrest and negatively regulates mTOR/MYC pathways in multiple myeloma cells. A, MM1.S and NCI-H929 cells were treated with ON123300 at three different concentrations (5 nmol/L, 10 nmol/L, and 50 nmol/L) for 24 hours, followed by analysis for apoptosis with Annexin V/PI double staining. ON123300 triggered a significant increase in both early (Annexin V+/PI−) and late (Annexin V+/PI−) apoptotic cell populations in both cell lines (mean ± SD; n = 3). B, MM1.S and NCI-H929 cells were treated with ON123300 (10 nmol/L and 50 nmol/L) for 24 hours, followed by analysis for cell proliferation by EdU incorporation assay with APC/PI double staining. Quantification of cell-incorporated (EdU) and total DNA content in control- and ON123300-treated cells showed a significant G0–G1 cell-cycle arrest in treated cells. C, MM1.S and NCI-H929 cells were treated with ON123300 (50 nmol/L) for 4, 6, and 12 hours, followed by Western blot analysis. As seen, treatment with ON123300 increased the expression of AMPK and SIRT1, resulting in decreased acetylation of MYC and total MYC. Alternatively, ON123300 also inhibits Rb/mTOR pathway via reduction in phosphoRb and phosphoS6K in both cell lines.
As ON123300 is a CDK4 inhibitor, we evaluated its effect on the Rb and mTOR pathways in ON123300-treated multiple myeloma. MM1.S and NCI-H929 cells were treated at 3 different time points (4, 6, and 12 hours treated with 50 nmol/L concentration of the compound) and showed decreased levels of phosphoRb as well as phosphoS6K as compared with untreated control cells (Fig. 2C). Although PD-0332991 showed a similar level of phosphoRb inhibition when MM1.S and NCI-H929 cell lines were treated for similar time points at two different concentrations of 250 nmol/L and 1 μmol/L (Supplementary Fig. S5), there was no significant change in the phosphoS6K levels when treated with PD-0332991. These results suggest that ON123300 regulates the expression of phosphoRb and phosphoS6K via the Rb/mTOR signaling pathways. Similarly, we examined the expression of proteins downstream of mTOR such as MYC for any significant changes in response to ON123300. Western blot analyses of MYC in MM1.S and NCI-H929 cell lines showed a significant decrease of MYC levels in both the cell lines (Fig. 2C).
Our Western blot analysis of cell lysates from ON123300 treated multiple myeloma cells showed a strong upregulation of AMPK (Fig. 2C), another member of the AMP protein kinase cascade that plays a key role in energy homoeostasis (19). As AMPK has been shown to regulate the activity of SIRT1, a NAD-dependent class III histone deacetylase (20), we next investigated whether the increase in AMPK levels upon ON123300 treatment resulted in an induction of SIRT1 protein levels. Our results (Fig. 2C) show that the treatment of multiple myeloma cell lines with ON123300 results in increased levels of SIRT1 protein. As SIRT1 has been shown to decrease the MYC protein levels (21), we next examined whether increased SIRT1 levels seen in ON123300-treated cells resulted in the deacetylation of MYC and decreased MYC stability. The results shown in Fig. 2C suggest that ARK5 inhibition (through increased levels of AMPK) indeed results in the activation of SIRT1, leading to a reduction in MYC protein levels. These results strongly imply that AMPK is an important positive regulator of SIRT1 in multiple myeloma cells and support the relevance of AMPK/SIRT1 as a conserved signaling axis that is activated upon ON123300 treatment.
ARK5 depletion using siRNAs results in inhibition of Rb/mTOR/MYC pathways and induction of apoptosis and cell-cycle arrest
As ON123300 is a multikinase inhibitor, we wanted to verify that the effects described above are due to inhibition of ARK5 kinase activity and whether knockdown of ARK5 by siRNAs phenocopies the effects seen with ON123300. Depletion of ARK5 expression in the MM1.S and NCI-H929 multiple myeloma cell lines using two different siRNAs resulted in a significant inhibition of cell viability, whereas treatment of cells with scrambled siRNA had a minimal effect (Supplementary Fig. S6). ARK5 siRNA–transfected cells also exhibited reductions in the levels of phosphoRb and phosphoS6K compared with control cells transfected with scrambled siRNA (Fig. 3A). These data suggest that ARK5 depletion significantly downregulates expression of phosphoRb and phosphoS6K via Rb and mTOR signaling pathways.
Figure 3.
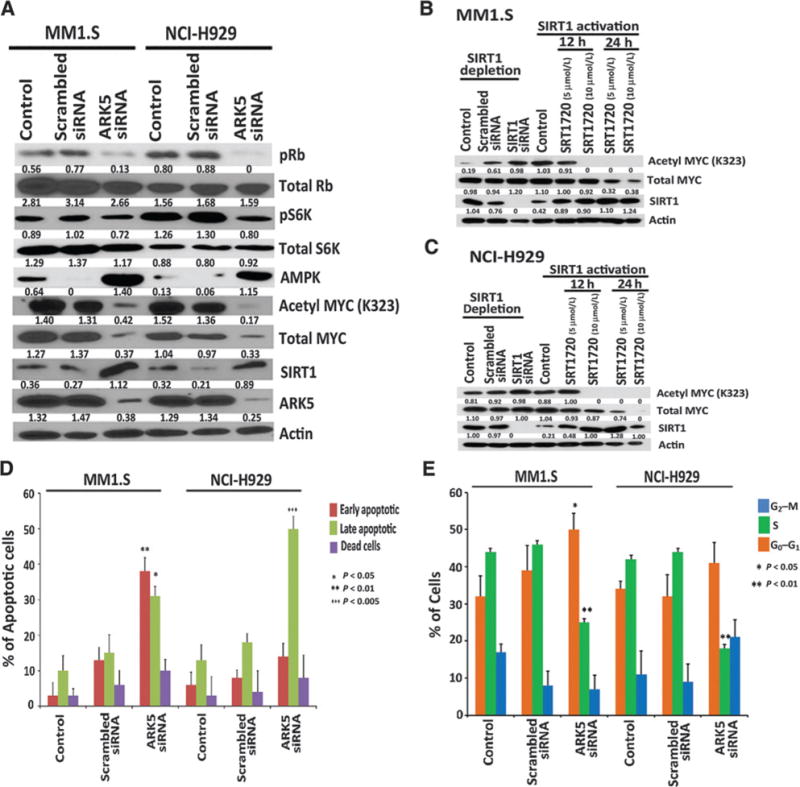
ARK5 depletion inhibits multiple myeloma cell growth via Rb/mTOR/MYC pathways and induces apoptosis and cell-cycle arrest. A, ARK5 repression significantly attenuated phosphoS6K, phosphoRb, MYC, and acetyl MYC activities in multiple myeloma. Conversely, depletion of ARK5 increased AMPK and SIRT1. Western blot analysis showing the levels of ARK5, SIRT1, MYC, acetyl MYC, total S6K, Rb, phosphoS6K, and phosphoRb proteins after 24-hour transfection in MM1.S and NCI-H929 siRNA–transfected cells. There was no difference in total Rb and total S6K between the groups. B and C, MM1.S and NCI-H929 cells were subjected to SIRT1 depletion by siRNA and SIRT1 activation with SRT1720. Western blotting confirmed that depletion of SIRT1 increased the acetylation activities of MYC, whereas the activation with SRT1720 resulted in decreased acetylation of MYC in a dose- and time-dependent manner. D, transfection in MM1.S and NCI-H929 cells induced apoptosis in ARK5-depleted cells as compared with nontransfected cells after 24-hour transfection confirmed with Annexin V/PI double staining. E, transfection in MM1.S and NCI-H929 cells also induced cell-cycle arrest in ARK5-depleted cells confirmed by EdU cell proliferation assay. Forty percent of MM1.S cells transfected with scrambled control siRNA showed EdU incorporation. EdU incorporation in cells transfected with ARK5 siRNA was approximately half that of the negative control. In NCI-H929 cells, 50% of cells transfected with negative control siRNA showed EdU incorporation, and EdU incorporation in cells transfected with ARK5 siRNA was less than half that.
In addition, ARK5-depleted cells showed a strong upregulation of AMPK in multiple myeloma cells (Fig. 3A), accompanied by increased levels of SIRT1 protein and a reduction in MYC protein levels. In addition, treatment of multiple myeloma cells with two different siRNAs directed against SIRT1 resulted in an overall increase in MYC acetylation and stabilization of the protein (Fig. 3B and C). The effect of SIRT1 on MYC described above raised the possibility that SIRT1 might negatively regulate the proliferation of multiple myeloma cells. To test this theory, we treated MM1.S and NCI-H929 cells with a small molecule agonist of SIRT1, SRT1720. Accordingly, treatment of multiple myeloma cells with SRT1720 significantly decreased cell viability of multiple myeloma cells as observed previously in another study (Supplementary Fig. S7; ref. 22). In addition, treatment of cells with SRT1720 led to increased SIRT1 expression, decreased acetylation of MYC, and reduced MYC stability (Fig. 3B and C). Taken together, our results provide mechanistic evidence that SIRT1 is downstream of ARK5 and that elevated expression of ARK5 in multiple myeloma cell lines correlated with decreased SIRT1 protein levels, suggesting a potential role for ARK5 in the regulation of MYC levels by suppressing SIRT1 expression.
To confirm that the decrease in cell number and viability is associated with apoptosis, we subjected ARK5-depleted MM1.S and NCI-H929 ARK5 cells to Annexin V/PI staining followed by flow cytometric analysis. There was a significant increase in the fraction of Annexin V–positive cells in ARK5 siRNA–treated cells compared with controls, consistent with the induction of apoptosis (Fig. 3D and Supplementary Fig. S8). The Rb tumor suppressor gene plays a major role in regulating the G1–S transition (23). Because ARK5 depletion suppressed cell growth, we next examined cellular incorporation of EdU using flow cytometry. Both ARK5 siRNA–transfected cell lines showed reduced levels of EdU incorporation that were approximately 50% of that of the scrambled control (Fig. 3E). Flow cytometric analysis also revealed that ARK5-depleted multiple myeloma cells underwent G1 arrest as evidenced by an increased frequency of cells in the G1 phase and a concomitant decrease of S-phase cells (Fig. 3E and Supplementary Fig. S9). Taken together, these results indicated that ARK5 depletion induces cell-cycle arrest in the G1–S phase followed by induction of apoptosis.
ON123300 overcomes bone marrow stromal protection
The bone marrow microenvironment is essential in multiple myeloma disease progression and drug resistance (24). To determine whether ON123300 can overcome the protective effects of the microenvironment and induce cytotoxicity in multiple myeloma cells in vitro, myeloma cells were grown in the presence of BMSCs. Although the BMSCs themselves did not show sensitivity in response to a range of ON123300 concentrations, the myeloma cell lines remained highly sensitive to ON123300 in the presence or absence of BMSCs as confirmed by Annexin V/PI staining (Fig. 4A and Supplementary Fig. S10). As a control, MM1.S and NCI-H929 cells were treated with proteasome inhibitor bortezomib in varying concentrations in the presence or absence of BMSCs for 48 hours, and the level of cytotoxicity was determined using a cell viability assay. In contrast to the results obtained with ON123300, bortezomib-treated MM1.S and NCI-H929 cells displayed higher levels of viability in the presence of stromal cells, confirming that the percentage of bortezomib-induced cytotoxicity in stroma context was significantly lower than that in stroma-free conditions (Fig. 4B). This result is consistent with previous studies suggesting that BMSCs induce resistance against cytotoxic and apoptotic effects of bortezomib in multiple myeloma cells (25). Therefore, our results suggest that ON123300 not only directly targets multiple myeloma cells but also overcomes the cytoprotective effects of the multiple myeloma–host bone marrow microenvironment.
Figure 4.
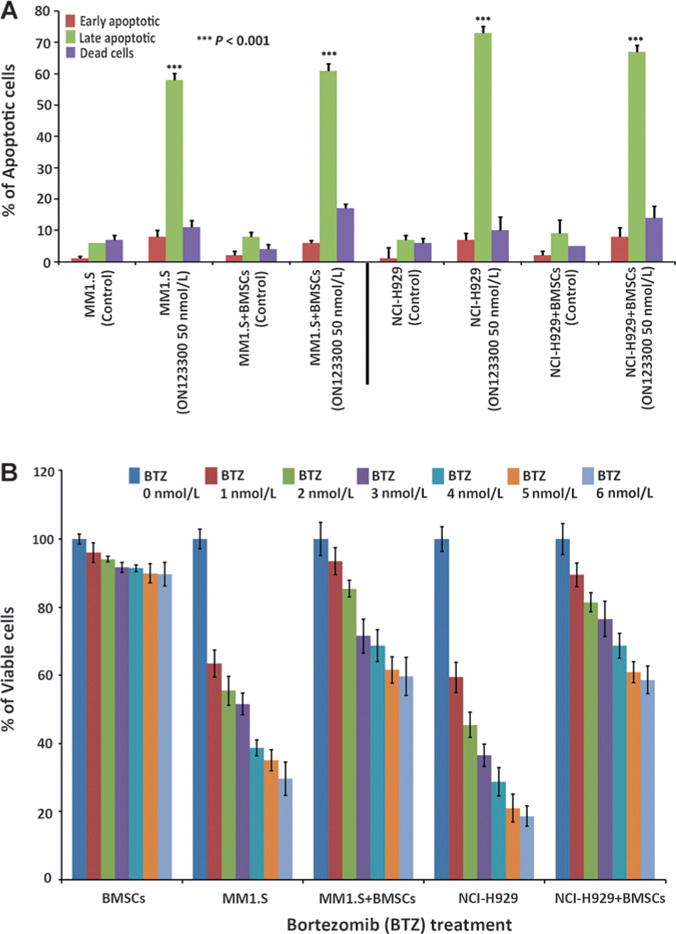
ON123300 overcomes bone marrow stromal protection. A, ON123300 overcomes the cytoprotective effects of the multiple myeloma–host bone marrow microenvironment as observed in MM1.S and NCI-H929 cells. MM1.S cells were cultured alone or with BMSCs for 48 hours in the presence or absence of ON123300 (50 nmol/L) and confirmed with Annexin V/PI double staining. As shown, there was significant inhibition of BMSC-induced proliferation of multiple myeloma cells in response to ON123300 treatment. B, MM1.S and NCI-H929 cells were treated with proteasome inhibitor bortezomib at different concentrations (1–6 nmol/L) in the presence or absence of BMSCs for 48 hours and then analyzed for cytotoxicity by CellTiter-Blue assay. As shown, there was no significant inhibition of BMSC-induced proliferation of multiple myeloma cells in response to bortezomib treatment.
Transcriptomic profiling reveals ARK5 depletion results in the suppression of cell-cycle regulating genes in multiple myeloma cell lines
Transcriptomic sequencing analysis of NCI-H929 cells transfected with ARK5 and scrambled siRNAs identified 894 differentially expressed genes (DEG) and their associated enriched pathways (Supplementary Table S2). Key cell-cycle regulators such as MYC, cyclins D1, E, and CDK4 were significantly suppressed as a function of ARK5 depletion (Fig. 5A). GSEA (15) with a rank ordered list of these 894 DEGs was performed to determine enrichment scores and significance values across the MSigDB (C6 oncogenic signatures) gene sets (15). Cell-cycle–regulating genes were significantly repressed following ARK5 depletion (Fig. 5B), further supporting our observations described above. As shown in the schematic (Fig. 5C), our results are consistent with a model whereby ARK5 is upstream of the AMPK-SIRT1 and Rb/mTOR/MYC pathways.
Figure 5.
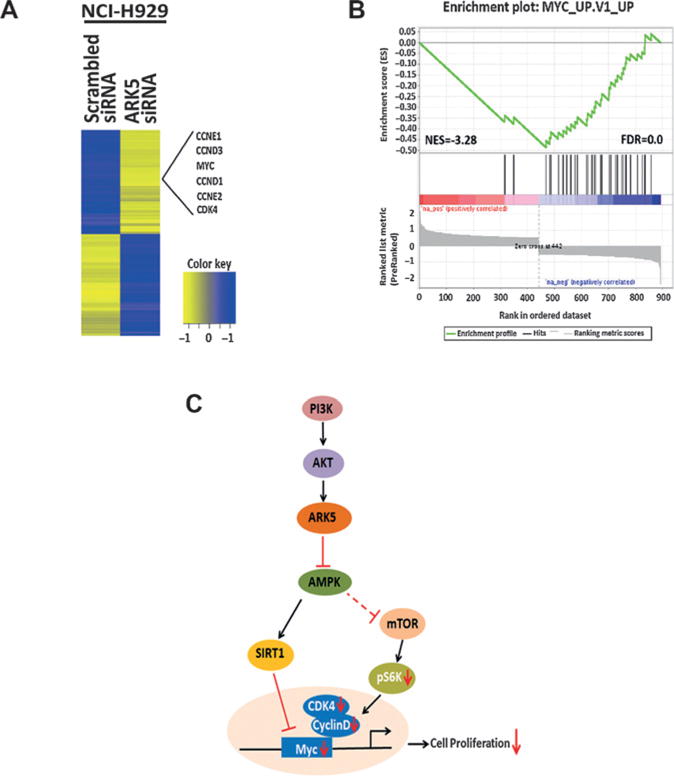
Transcriptomic profiling reveals ARK5 depletion results in the suppression of cell cycle regulating genes in multiple myeloma cell lines. A, RNA sequencing data from ARK5-depleted NCI-H929 cells identified 894 DEGs between ARK5 siRNA and scrambled siRNA. Cell-cycle pathway was enriched in ARK5-depleted cells with cell-cycle regulators like MYC, cyclin D and E, and CDK4 genes significantly repressed after ARK5 depletion as shown. B, enrichment score plot by GSEA showed a strong negative enrichment of MYC target genes in ARK5 depleted NCI-H929 cells. Preranked list of 894 DEGs was used for GSEA in the C6 oncogenic signatures. The distribution of MYC target genes (black lines) is presented as a function of the change in expression between ARK5 siRNA and scrambled siRNA, from highly upregulated (red) to highly downregulated (blue). Normalized enrichment score (NES) and FDR q values for MYC target gene set are shown. C, schematic representing the downstream signaling events of ARK5 is shown.
Transcriptomic profiling reveals cell-cycle–regulating genes are highly enriched in ON123300-sensitive multiple myeloma cell lines
Using RNA sequencing data from 8 multiple myeloma cell lines (see Fig. 1B), we identified gene signatures associated with ON123300 sensitivity. By using LIMMA (14), a total of 180 genes were differentially expressed in cell lines (Fig. 6A) with FDR < 0.05 and log2 ratio ≥ 2 between the ON123300-sensitive and -resistant cell lines. We next assessed whether certain biologic pathways and mechanisms featured more prominently according to the DEG. Biologic interpretation of these 180 DEGs was further annotated by GeneGo pathway analysis. Key cell-cycle regulators cyclin D1, cyclin E, CDK4, CDC25A, and RelA were significantly upregulated in the ON123300-sensitive cells (Fig. 6B). The most significantly top 10 enriched pathways for these upregulated genes are shown in Supplementary Table S3. Cell-cycle arrest inducing genes CDKN2A, CDKN1C and, p14ARF1 and apoptosis inducing genes APAF1, FADD, and caspase-9 were seen downregulated in the ON123300-sensitive cells (Fig. 6C). The most significantly top 10 enriched pathways for these downregulated genes are shown in Supplementary Table S4. Taken together, the gene signatures from multiple myeloma cell lines identified several cell cycle and apoptosis-related pathways as relevant to ON123300 sensitivity.
Figure 6.
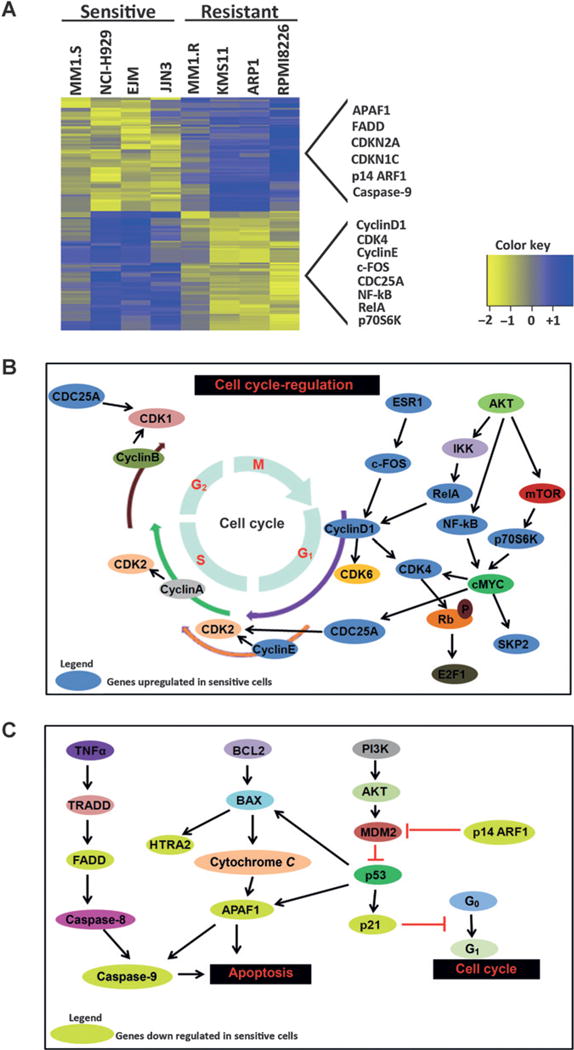
Transcriptomic profiling reveals cell cycle–regulated genes are highly enriched in ON123300-sensitive multiple myeloma cell lines. A, heatmap of 180 DEGs together with the indication about enrichment of MYC target genes in ON123300-sensitive multiple myeloma cell lines. B, molecular processes mediated by DEGs displaying altered expressions were sought using GeneGo program. Significant molecular biologic pathways were determined based on the analysis of significant biologic processes and expressions of the genes involved in these processes. As shown in the schematic, key cell-cycle regulators, with most of them being cell-cycle genes, have increased gene expression levels in ON123300 cells. Genes that are upregulated are denoted in blue ovals. C, conversely, apoptotic inducing genes are downregulated in ON123300 cells as shown in the schematic. Genes that are downregulated are denoted in lime ovals.
ON123300 inhibits multiple myeloma cell growth in vivo in multiple myeloma xenograft mouse models
We next examined the in vivo efficacy of ON123300 using mouse xenograft models. MM1.S and NCI-H929 cells were subcutaneously implanted into NSG female mice at 4 to 5 weeks of age. ON123300 (100 mg/kg) was administered on alternate days via intraperitoneal injection. Treatment of multiple myeloma tumor–bearing mice with ON123300 significantly decreased the growth of tumors as opposed to control-treated mice (Fig. 7A and B) and was well tolerated, with no significant weight loss (Supplementary Fig. S11). These data show potent in vivo activity of ON123300 against multiple myeloma cells.
Figure 7.
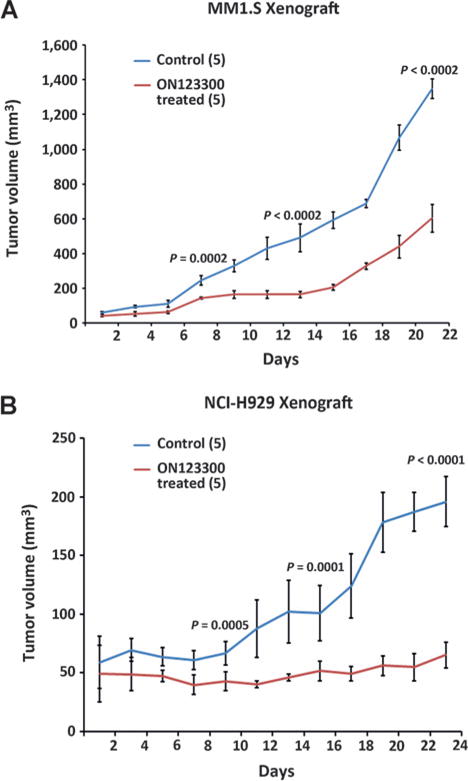
ON123300 inhibits multiple myeloma cell growth in vivo in multiple myeloma xenograft mouse models. A and B, ON123300 inhibits growth of MM1.S cells and NCI-H929 in NOD mice (Taconic). NCI-H929 cells and MM1.S (2 × 106 cells/mouse) were implanted subcutaneously in female mice. Mice were randomized to control and treatment groups and treated intraperitoneally with vehicle and ON123300 (100 mg/kg) every other day. Treatment with intraperitoneal injections of ON123300 slowed the growth of tumors compared with increase in tumors in control mice.
Discussion
In this communication, we have examined the effects of ON123300, a dual inhibitor of CDK4/6 and ARK5 kinases on the proliferation and survival of multiple myelomas. It is now well established that a large percentage of multiple myelomas over express cyclin D1 (CCND1), but this alone is insufficient to drive the cell cycle (26, 27). Because overexpression of CCND1 was found to result in high levels of CDK4/6 kinase activity, inhibition of CDK4/6 is an emerging therapeutic approach for multiple myelomas (28). Numerous CDK inhibitors with differing mechanistic profiles are currently being preclinically and clinically evaluated but have not as of yet resulted in a drug approval mainly because of lack of selectivity and high toxicity (29, 30). To explain the modest activity for the current class of CDK4/6 inhibitors, we hypothesized that this could be due to the narrow kinase inhibition profile of CDK4/6 inhibitors, such as PD-0332991, and incomplete targeting of additional myeloma-specific oncokinases may result in cytostatic rather than cytotoxic effects.
ARK5, a fifth member of the AMPK family has been identified as a transcriptional target of the large MAF family in multiple myeloma (11). In addition, a recent study (10) showed that ARK5 overexpression is required to prevent proapoptotic effects of MYC overexpression in tumor cells, suggesting that this kinase might play a critical role in multiple myeloma pathogenesis. The development of ON123300, a potent inhibitor of ARK5 allowed us to test the effects of ARK5 inhibition on the growth and survival potential of multiple myeloma cell lines and primary cells derived from patients who are refractory to bortezomib, both of which appear to express high levels of ARK5. Our results show that treatment of ARK5 overexpressing multiple myelomas with ON123300 results in cell-cycle arrest and apoptotic death of multiple myeloma cells. Interestingly, we observed that treatment of multiple myelomas with this compound results in the upregulation of AMPK and SIRT1 proteins, leading to increased acetylation and degradation of MYC protein. As ON123300 inhibits multiple kinases, we examined whether ARK5-specific siRNAs can phenocopy the effect of ON123300 seen on AMPK, SIRT1, and MYC levels. Our results clearly confirm that knockdown of ARK5 with siRNAs leads to similar modulation of AMPK, SIRT1, and MYC protein levels. These results demonstrate that ARK5 is critical for multiple myeloma survival and reveal new insights into the regulation of MYC via AMPK–SIRT1 signaling axis. Our findings are consistent with a model where AMPK-dependent phosphorylation provides a direct link between cellular energy metabolism and regulation of gene expression (31). When the energy levels are low and the intracellular AMP/ATP ratio elevated, AMPK becomes activated, resulting in switching off energy-consuming pathways to restore cellular ATP levels. This drop in cellular ATP and concomitant increase in AMP levels trigger phosphorylation-dependent activation of AMPK. These results suggest that MYC acetylation and stability is regulated by ARK5 upstream of SIRT1-MYC.
In addition, we provide evidence that the proliferation of multiple myeloma cells is positively regulated by ARK5, which coordinates the expression of mTOR, CCND1/Rb, in addition to MYC. A direct link between MYC and the cell-cycle machinery exists since the CCND1 and CCNE genes have been shown to be transcriptional targets of MYC (32, 33). Deregulated MYC expression is also linked to increased cyclin A and cyclin E expression and activation of CDK4 via its direct target gene CDC25 (34). CDK4 has also been identified in Drosophila as a MYC target gene (35), and the proliferative effect of MYC in multiple myeloma cells also appears to be dependent of CDK4 activation as evidenced by the down regulation of MYC and CDK4 upon ARK5 depletion (see Fig. 5A). Therefore, our data support a synthetic lethal interaction between CDK4 and MYC in multiple myeloma. One potential mechanism for this synthetic lethal interaction could be that loss of ARK5 subsequently activates apoptotic signaling events, resulting in cell death. Subsequently, apoptotic pathways appear to be involved in CDK4 and ARK5 inhibitor ON123300-induced cell death, providing mechanistic insight into MYC-CDK4 synthetic lethality in multiple myeloma cells.
Moreover, treatment of multiple myeloma cells that express higher levels of ARK5 with ON123300 also resulted in downregulation of phosphoS6K, which does not occur in cells that have been treated with PD-0332991 in multiple myeloma (Supplementary Fig. S5). Given that this is the first report showing that dual inhibition of ARK5 and CDK4 by ON123300 has significant effects on mTOR and MYC activities, these studies suggest that the proliferation of multiple myeloma cells is activated by ARK5, which coordinates the expression of CCND1 and Rb, potentially through mTOR. Inhibitors targeting the mTOR pathway are in clinical development as potential chemotherapeutic agents (36), and there is an increasing body of evidence implicating the mTOR signaling pathway (36), and more specifically phosphoS6K (37), in regulating multiple myeloma cell proliferation. Because our results suggest that ARK5, via its ability to regulate mTOR and MYC signaling, is a novel therapeutic target in multiple myeloma, our data provide the foundation for further development of ARK5 inhibitors and second-generation CDK inhibitors, such as ON123300, that target both ARK5 and CDK4 for the treatment of multiple myeloma.
Supplementary Material
Acknowledgments
The authors thank Jonathan Keats for providing RNA sequencing data on multiple myeloma cell lines and Leif P. Bergsagel for critical advice and discussion. Assistance of the shared resources from Mount Sinai Genomics core, flow cytometry core, and the scientific computing core is gratefully acknowledged.
Grant Support
This work was supported by The Chemotherapy Foundation and NIH grants 5R21CA169487 (S. Parekh), P01CA130821 (E.P. Reddy), and R01CA158209 (E.P. Reddy).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
H.J. Cho reports receiving commercial research grants from Agenus Inc. and Janssen. E.P. Reddy is a board director at, reports receiving commercial research grants from, has ownership interest (including patents) in, and is a consultant/advisory board member for Onconova Therapeutics Inc. S. Jagannath has received speakers bureau honoraria from and is a consultant/advisory board member for Bristol-Myers Squibb, Celgene, and Janssen. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: D. Perumal, S.K.A. Divakar, J. Brody, E.P. Reddy, S. Jagannath, S. Parekh
Development of methodology: D. Perumal, P.-Y. Kuo, S.K.A. Divakar, J. Brody, M.V.R. Reddy, S. Jagannath, S. Parekh
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): D. Perumal, P.-Y. Kuo, Z. Jiang, H.J. Cho, A. Chari, J. Brody, E.P. Reddy, S. Jagannath, S. Parekh
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): D. Perumal, V.V. Leshchenko, J. Brody, W. Zhang, E.P. Reddy, S. Parekh
Writing, review, and/or revision of the manuscript: D. Perumal, A. Chari, W. Zhang, E.P. Reddy, S. Parekh
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): D. Perumal, E.P. Reddy, S. Parekh
Study supervision: D. Perumal, E.P. Reddy, S. Parekh
Other (design and synthesis of ON123300 base and lactate forms used in this study): M.V.R. Reddy
References
- 1.Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122:3456–63. doi: 10.1172/JCI61188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Dolloff N, Talamo G. Targeted therapy of multiple myeloma. In: El-Deiry WS, editor. Impact of genetic targets on cancer therapy Advances in experimental medicine and biology. New York, NY: Springer; 2013. pp. 197–221. [DOI] [PubMed] [Google Scholar]
- 4.Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28:1122–8. doi: 10.1038/leu.2013.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malumbres M, Pevarello P, Barbacid M, Bischoff JR. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci. 2008;29:16–21. doi: 10.1016/j.tips.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 6.Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, et al. Prolonged early G1 arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle–coupled loss of IRF4. Blood. 2012;120:1095–106. doi: 10.1182/blood-2012-03-415984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niesvizky R, Badros AZ, Costa LJ, Ely SA, Singhal SB, Stadtmauer EA, et al. Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/ refractory multiple myeloma. Leuk Lymphoma. 2015;56:3320–8. doi: 10.3109/10428194.2015.1030641. [DOI] [PubMed] [Google Scholar]
- 8.Reddy EP, Divakar SA, Reddy MVR, Cosenza SC, Baker SJ, Akula B, et al. Abstract 4519: targeting of cyclin D/Rb/E2F and PI3K/AKT/MTOR pathways with ON 123300 as a therapeutic strategy for mantle cell lymphoma. Cancer Res. 2014;74:4519. [Google Scholar]
- 9.Reddy MVR, Akula B, Cosenza SC, Athuluridivakar S, Mallireddigari MR, Pallela VR, et al. Discovery of 8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carboni-trile (7x) as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and AMPK-related kinase 5 (ARK5) J Med Chem. 2014;57:578–99. doi: 10.1021/jm401073p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–12. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki A, Iida S, Kato-Uranishi M, Tajima E, Zhan F, Hanamura I, et al. ARK5 is transcriptionally regulated by the Large-MAF family and mediates IGF-1-induced cell invasion in multiple myeloma: ARK5 as a new molecular determinant of malignant multiple myeloma. Oncogene. 2005;24:6936–44. doi: 10.1038/sj.onc.1208844. [DOI] [PubMed] [Google Scholar]
- 12.Kusakai G, Suzuki A, Ogura T, Miyamoto S, Ochiai A, Kaminishi M, et al. ARK5 expression in colorectal cancer and its implications for tumor progression. Am J Pathol. 2004;164:987–95. doi: 10.1016/s0002-9440(10)63186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki A, Lu J, Kusakai G-I, Kishimoto A, Ogura T, Esumi H. ARK5 is a tumor invasion-associated factor downstream of Akt signaling. Mol Cell Biol. 2004;24:3526–35. doi: 10.1128/MCB.24.8.3526-3535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smyth Gordon K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Divakar SKA, Ramana Reddy MV, Cosenza SC, Baker SJ, Perumal D, Antonelli AC, et al. Dual inhibition of CDK4/Rb and PI3K/AKT/mTOR pathways by ON123300 induces synthetic lethality in mantle cell lymphomas. Leukemia. 2016;30:86–93. doi: 10.1038/leu.2015.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X, Lv H, Zhou Q, Elkholi R, Chipuk JE, Reddy MVR, et al. Preclinical pharmacological evaluation of a novel multiple kinase inhibitor, ON123300, in brain tumor models. Mol Cancer Ther. 2014;13:1105–16. doi: 10.1158/1535-7163.MCT-13-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66:7661–7. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 19.Carling D. The AMP-activated protein kinase cascade – a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Imai S-I, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 21.Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc–SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol. 2009;185:203–11. doi: 10.1083/jcb.200809167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chauhan D, Bandi M, Singh AV, Ray A, Raje N, Richardson P, et al. Preclinical evaluation of a novel SIRT1 modulator SRT1720 in multiple myeloma cells. Br J Haematol. 2011;155:588–98. doi: 10.1111/j.1365-2141.2011.08888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 24.Shain K, Dalton W. The bone marrow microenvironment: Novel targets to circumvent minimal residual disease and drug resistance in multiple myeloma. In: Munshi NC, Anderson KC, editors. Advances in biology and therapy of multiple myeloma. New York, NY: Springer; 2013. pp. 141–68. [Google Scholar]
- 25.Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E, et al. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014;124:555–66. doi: 10.1182/blood-2014-03-562439. [DOI] [PubMed] [Google Scholar]
- 26.Athanasiou E, Kaloutsi V, Kotoula V, Hytiroglou P, Kostopoulos I, Zervas C, et al. Cyclin D1 overexpression in multiple myeloma: a morphologic, immunohistochemical, and in situ hybridization study of 71 paraffin-embedded bone marrow biopsy specimens. Am J Clin Pathol. 2001;116:535–42. doi: 10.1309/BVT4-YP41-LCV2-5GT0. [DOI] [PubMed] [Google Scholar]
- 27.Specht K, Haralambieva E, Bink K, Kremer M, Mandl-Weber S, Koch I, et al. Different mechanisms of cyclin D1 overexpression in multiple myeloma revealed by fluorescence in situ hybridization and quantitative analysis of mRNA levels. Blood. 2004;104:1120–6. doi: 10.1182/blood-2003-11-3837. [DOI] [PubMed] [Google Scholar]
- 28.Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476–87. doi: 10.1093/jnci/djs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McInnes C. Progress in the evaluation of CDK inhibitors as anti-tumor agents. Drug Discov Today. 2008;13:875–81. doi: 10.1016/j.drudis.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–69. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 31.Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I, et al. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci. 2009;106:17781–6. doi: 10.1073/pnas.0909873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jansen-Dürr P, Meichle A, Steiner P, Pagano M, Finke K, Botz J, et al. Differential modulation of cyclin gene expression by MYC. Proc Natl Acad Sci U S A. 1993;90:3685–9. doi: 10.1073/pnas.90.8.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000;97:2229–34. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101–14. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Zhu J, Cao B, Mao X. The mTOR signaling pathway is an emerging therapeutic target in multiple myeloma. Curr Pharm Des. 2014;20:125–35. doi: 10.2174/13816128113199990638. [DOI] [PubMed] [Google Scholar]
- 37.Pene F, Claessens Y-E, Muller O, Viguie F, Mayeux P, Dreyfus F, et al. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 2002;21:6587–97. doi: 10.1038/sj.onc.1205923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.