

Abstract
This report details a palladium-catalyzed process to access highly functionalized optically active allylic aryl ethers. A number of electron-deficient alkenyl triflates underwent enantio- and site-selective coupling with acyclic aryl enol ethers in the presence of a chiral palladium catalyst. This transform provides chiral allylic ether products in high yields and excellent enantiomeric ratios furnishing a unique disconnection to incorporate heteroatoms at a stereocenter. Finally, the applicability of the products to target synthesis was demonstrated through the formation of a chiral allylic alcohol and the generation of a flavone-inspired product.
Graphical Abstract
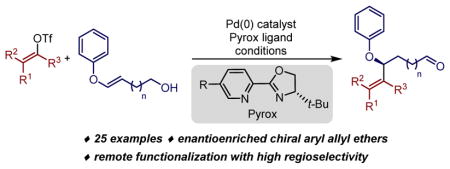
Access to enantiomerically enriched aryl allyl ethers is of high synthetic utility as they are represented in numerous ether containing natural products and pharmaceuticals.1 Substantial progress has been reported for their synthesis using transition-metal catalysis.2 Most methods strategically rely on the reaction of symmetric π-allyl intermediates of Pd,3 Rh,4 and Ir5 using phenoxides or excess phenol as the trapping exogenous nucleophile. Alternatively, branched allylic ethers can be accessed in high enantioselectivity using a distinct Pd(II)-catalyzed SN2′ reaction of allylic trichloroacetamidates.6 In spite of these advances, the alkenyl components installed using these methods are relatively simple inspiring us to consider new reactions to obtain such products. Herein we describe a palladium-catalyzed redox-relay enantioselective Heck reaction of a new class of substrates, namely acyclic aryl enol ethers. As illustrated below, this reaction provides facile access to a range of diversely functionalized aryl allyl ethers, especially in terms of the alkenyl component, in high enantioselectivity.
In previous reports, the redox-relay Heck strategy has led to the successful enantioselective coupling of acyclic disubstituted alkenols with a range of aryl, alkenyl, and alkynyl groups (Figure 1a).7 The site-selectivity observed in the migratory insertion step is sensitive to the polarization of the alkene carbons in the transition state, which is proposed to be stabilized by the CO dipole of the alcohol moiety.8 Although the measured site-selectivity for allylic alcohols is generally high, it decreases substantially with the increasing chain-length between the alkene and the alcohol.9 To presumably further bias the migratory insertion improving site selectivity of more remote functionalization reactions, we envisioned the coupling of alkenyl electrophiles with acyclic enol ether substrates (Figure 1b). This is proposed to be due to increased nucleophilicity of carbon distal to the oxygen in the enol ether substrate (2, Figure 1c).10 Furthermore, high site-selectivity was anticipated irrespective of the number of carbons between the alkene and alcohol in the enol ether substrate. Successful implementation incorporates a heteroatom at the stereocenter formed in the migratory insertion process to access enantioenriched aryl allyl ether products (3).
Figure 1.
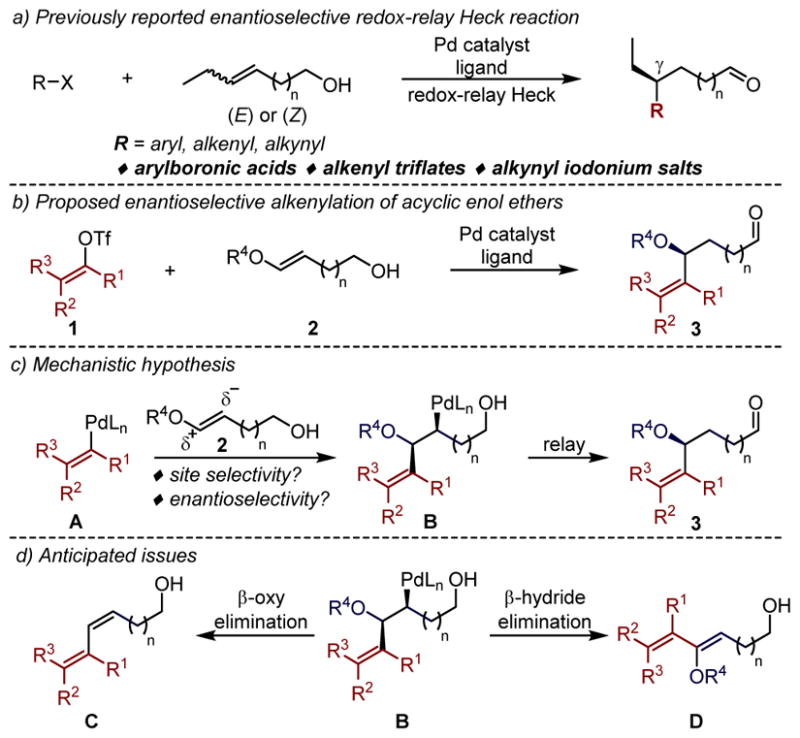
(a) Previously reported enantioselective redox-relay Heck reaction. (b) Proposed enantioselective alkenylation of acyclic enol ether. (c) Mechanistic hypothesis. (d) Anticipated issues.
At the outset, we were concerned that the migratory insertion event would be followed by β-alkoxy elimination instead of β-hydride elimination towards the alcohol (Figure 1d) as these processes could be competitive on the basis of previous efforts of our group.11 Additionally, the β-alkoxy aldehyde products 3 (n=0) may not survive the relatively acidic conditions and undergo an E1cB to form the α,β-unsaturated carbonyl product.12 Consistent with this, early experiments of enol ethers 2a and 2b, easily prepared through a two-step sequence from the propiolate ester,13 with the electron deficient vinylogous lactone triflate 1a in the presence of a Pd(0) catalyst and a chiral pyridine-oxazoline ligand L1 (Figure 2a), exclusively led to the undesired anticipated E1cB product 4 in 70% yield. Considering the relatively Brønsted/Lewis acidic conditions of this process, it was hypothesized that lowering the basicity of the oxygen on the enol ether may slow the presumed E1cB process.
Figure 2.
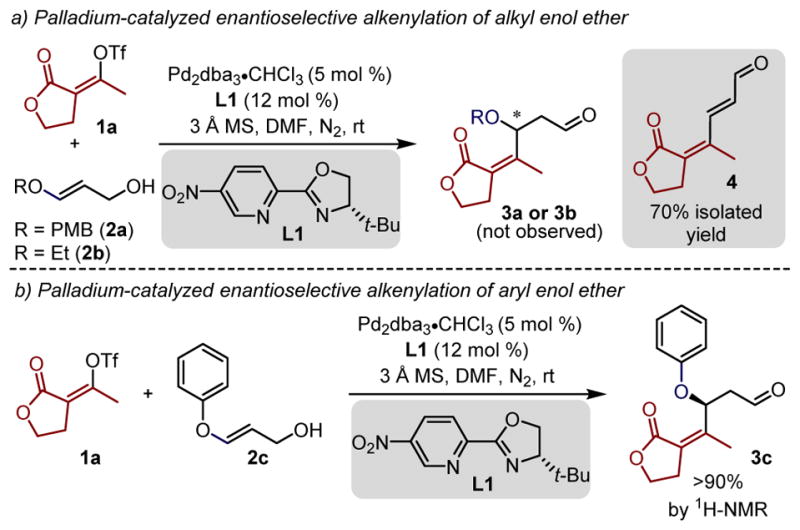
(a) Palladium-catalyzed enantioselective alkenylation of alkyl enol ether (b) Palladium-catalyzed enantioselective alkenylation of acyclic aryl enol ether.
To test this hypothesis, an enol ether substrate 2c containing an aryl substituent, instead of an alkyl group, was prepared and reacted with alkenyl triflate 1a under the standard redox-relay Heck conditions (Figure 2b). The evaluation of the crude reaction mixture by 1H NMR indicated the formation of the desired β-alkenylated aldehyde product 3c in high yield (> 90% NMR yield). However, an attempt to purify the β-phenoxy aldehyde product 3c via silica gel chromatography resulted in partial decomposition of the product and formation of product 4, previously observed. Thus, for characterization purposes and ease of handling, the obtained aldehydes were transformed to the corresponding alcohols upon reduction of the crude mixture with sodium borohydride. This allowed the resultant alcohol 5a to be isolated in 90% yield and an excellent enantiomeric ratio (er) of 99:1 (Table 1, 5a).
Table 1.
Scope of alkenyl triflates in the redox-relay Heck reaction of acyclic aryl enol ether.

|
Yields are reported as an average of two parallel experiment. Enantioselectivily determined by SFC equipped with a chiral column. Reaction performed on 0.5 mmol scale. Absolute configuration determined to be (S) for a synthetic derivative of 5z (see Supporting Information) arising from the (E)-alkene. All other products were assigned by analogy.
NaBH(HFIP)3 in HFIP was used for the reduction of aldehyde to alcohol.
MnO2 oxidaticn of the diol yields the ketone 5e (overall yield reported).
With the optimal conditions determined, we began the assessment of a variety of electron poor alkenyl triflates (Table 1). The cyclic five and seven-membered alkenyl triflates performed well in the redox-relay Heck reaction to generate 5b and 5c in high yields and high enantiomeric ratio of 98:2 and 94:6, respectively (see Supporting Information for the determination of absolute configuration of compounds in Table 1, Table 2 and Table 3). The β-alkenylated product 5d containing a protected amine was also formed in good yield and high enantioselectivity, although sodium tris(1,1,1,3,3,3-hexafluoroisopropoxy)borohydride14 was used instead of sodium borohydride during the reduction step. The relay Heck reaction also tolerated a hindered tetrasubstituted cyclohexenone triflate. Unfortunately, in this case, a selective reduction of the aldehyde in the presence of the enone proved to be challenging. Thus, over reduction afforded a diastereomeric mixture of diols (see Supporting Information), which was treated with MnO2 to ultimately afford the chiral allylic ether 5e in good yield and 93:7 er.15 Next, a variety of acyclic alkenyl triflates, conveniently prepared from β–ketoesters in a stereodefined manner, were subjected to the redox-relay Heck reaction.16 Both (E) and (Z)-alkenyl triflates were added to enol ether 2c, forming 5f and 5g in high yield and enantiomeric ratio of 93:7 and 92:8, respectively. Additionally, acyclic alkenyl triflates having a benzyl substitution, a remotely positioned phthalimide, and an alkyl silane were successfully integrated to yield 5h–5j in modest to good yields and generally good enantioselectivity. Importantly, the olefins incorporated in 5f–5j did not undergo isomerization, thereby providing a means to generate stereodefined tetrasubstituted alkenes adjacent to a stereocenter. Lastly, a dihydrobenzofuran derived alkenyl triflate was also incorporated in 5k, albeit in low yield but high enantioselectivity. It is noteworthy that di- and tri-substituted alkenyl triflates performed poorly in this reaction with low conversion of 2c (see Supporting Information).17 Although, the precise cause for this difference in reactivity is unknown, it is assumed that the tetrasubstituted alkenyl triflates may slow β-hydride elimination to generate the diene.7b
Table 2.
Scope of allylic aryl enol ethers.

|
Yields are reported as an average of two parallel experiments. Enantioselectivity determined by SFC equipped with a chiral column. Reaction performed on 0.25 mmol scale. Absolute configuration determined to be (S) for a synthetic derivative of 5z (see Supporting Information) arising from the (E)-alkene. All other products were assigned by analogy.
THF and L2 ligand used instead of DMF and L1 ligand.
Table 3.
Evaluation of aryl enol ether with varying chain lengths.
|
Yields are reported as an average of two parallel experiments. Enantioselectivity determined by SFC equipped with a chiral column. Reaction performed on 0.25 mmol scale. Absolute configuration determined to be (S) for a synthetic derivative of 5z (see Supporting Information) arising from the (E)-alkene. All other products were assigned by analogy.
Next, we evaluated a range of electronically diverse aryl enol ethers. Electron-rich substituents on the phenol provided the desired allylic ether products 5l–5o in good yields and high enantioselectivity (Table 2). The hindered ortho-substitution in 5m showed no deleterious effects on either the yield or the enantioselectivity of the reaction. Interestingly, the use of an electron-poor group (for instance a p-Br substitution) on the phenyl ring of the acyclic enol ether did not undergo reaction under these conditions. Consequently, to tackle this issue, new conditions were identified empirically using THF as solvent, and ligand L2 (-CF3 substituted) instead of L1. Thereby, using the newly optimized reaction conditions, a number of enol ethers having an electron poor substituent on the phenyl ring, underwent the Heck reaction, and resulted in the formation of alcohols 5r–5t, upon reduction of the aldehydes, in consistently high yield and enantioselectivity. The reason for such disparity in the reactivity of electronically different arenes is not currently understood.
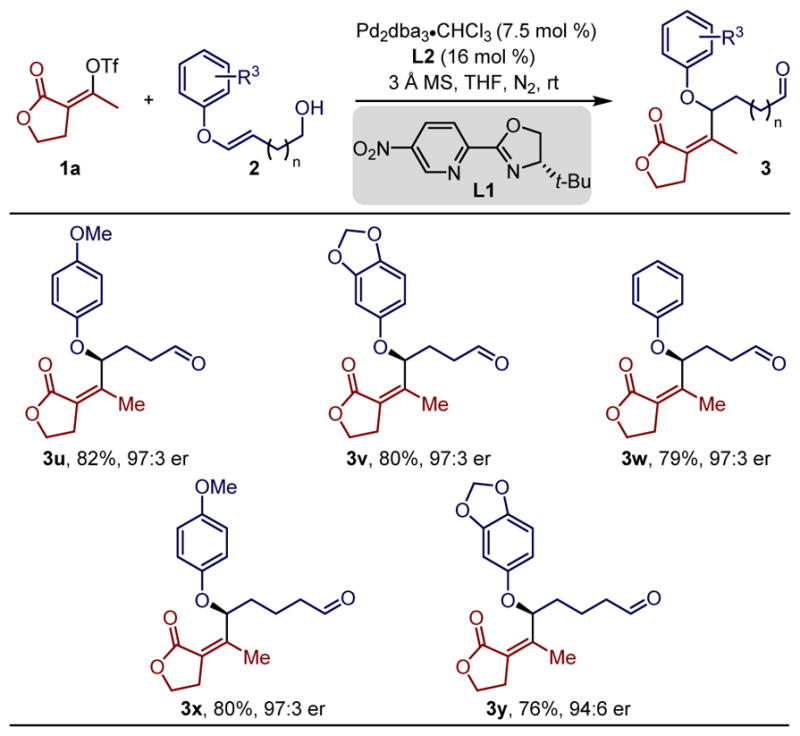
As the redox-relay strategy is unique in establishing remote stereocenters, a range of aryl enol ether substrates having a different number of methylenes between the alkene and the alcohol were investigated (Table 3).13 Significantly, the use of a homoallylic alcohol may allow the isolation of aldehyde products directly, as the degradation via an E1cB process is not possible. Thus, the reaction of the cyclic vinylogous lactone triflate 1a with a p-OMe-substituted aryl enol ether substrate 2u, using slightly higher catalyst loadings provided the γ-alkenylated aldehyde product 3u in 87% yield and 97:3 enantiomeric ratio. Similarly, a sesamol or phenol derived enol ether also provided the remotely alkenylated aldehyde products 3v and 3w in consistently high yield and enantioselectivity. The alkenyl triflate was further installed at a more remote position δ from the aldehyde to form allylic ethers 3x and 3y in good yield and high enantioselectivity.
The use of a cleavable group in the acyclic enol ether substrate has the potential to deliver enantioenriched chiral allylic alcohols. To this end, a one electron oxidative deprotection of the p-methoxyphenyl (PMP) ether was accomplished by treating compound 6 with CAN (Scheme 1a).18 The desired allylic alcohol 6a was obtained in 80% isolated yield with no loss of the stereochemical integrity (Scheme 1). In order to further demonstrate the synthetic applicability of such chiral allylic ethers, we envisioned accessing flavone derivatives containing an alkenyl group at the 2-position. To accomplish this, the β-alkenylated aldehyde 3c generated after the Heck reaction was subjected to a sequential Pinnick oxidation/Friedel-Crafts cyclization19 affording the desired alkenylated flavone product 7 in 78% overall yield and 94:6 enantiomeric ratio, in single purification (Scheme 1b). A slight erosion of the stereochemical integrity was observed and found to be associated with the Pinnick oxidation step (see Supporting Information for details). Such a strategy allows effective incorporation of highly functionalized alkenyl groups into flavonoids, thereby expanding the chemical space, as most reported flavonoids contain an aryl group.20
Scheme 1.
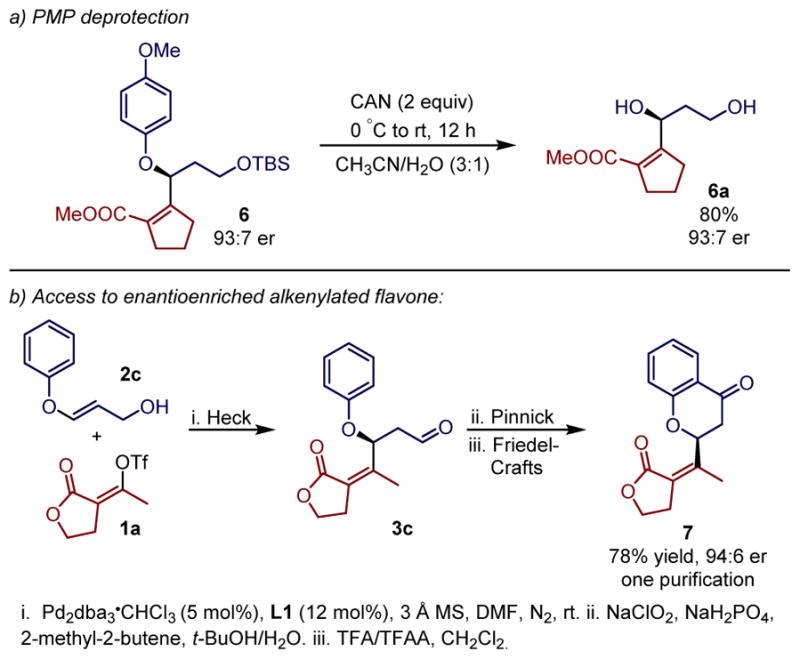
Derivatization of alkenylated products.
In summary, we have developed a unique method for the construction of highly functionalized chiral allylic ethers using a redox-relay Heck strategy. The process utilizes conveniently synthesized aryl enol ether substrates and readily accessible alkenyl triflates to generate chiral allylic ether products in good yield and enantioselectivity. The simple deprotection of the PMP ether provides direct access to chiral allylic alcohols in high enantiomeric ratio. Future work is focused on expanding this approach to other heteroatom containing alkenyl substrates.
Supplementary Material
Acknowledgments
The work was supported by National Institute of Health (NIGMS R01GM063540) and the National Science Foundation REU Program for S.O.S. (CHE-1358740).
Footnotes
Notes
The authors declare no competing financial interests.
Experimental procedures and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (b) Cao B, Park H, Joullié MM. J Am Chem Soc. 2002;124:520. doi: 10.1021/ja017277z. [DOI] [PubMed] [Google Scholar]; (c) Tanaka H, Sawayama AM, Wandless TJ. J Am Chem Soc. 2003;125:6864. doi: 10.1021/ja035429f. [DOI] [PubMed] [Google Scholar]; (d) Mizuguchi E, Achiwa K. Chem Pharm Bull. 1997;45:1209. [Google Scholar]; (e) Pochetti G, Mitro N, Lavecchia A, Gilardi F, Besker N, Scotti E, Aschi M, Re N, Fracchiolla G, Laghezza A, Tortorella P, Montanari R, Novellino E, Mazza F, Crestani M, Loiodice F. J Med Chem. 2010;53:4354. doi: 10.1021/jm9013899. [DOI] [PubMed] [Google Scholar]; (f) Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939. [Google Scholar]; (g) Ishibashi H, Ishihara K, Yamamoto H. J Am Chem Soc. 2004;126:11122. doi: 10.1021/ja0472026. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Wei YF, Luo RS. J Org Chem Res. 2017;5:51. [Google Scholar]
- 3.(a) Trost BM, Tsuji HC, Toste FD. J Am Chem Soc. 2000;122:3534. [Google Scholar]; (b) Trost BM, Toste FD. J Am Chem Soc. 1999;121:4545. [Google Scholar]; (c) Trost BM, Toste FD. J Am Chem Soc. 1998;120:815. [Google Scholar]; (d) Uozumi Y, Kimura M. Tetrahedron: Asymmetry. 2006;17:161. [Google Scholar]; (e) Trost BM, Machacek MR, Tsui HC. J Am Chem Soc. 2005;127:7014. doi: 10.1021/ja050340q. [DOI] [PubMed] [Google Scholar]; (f) Trost BM, Crawley ML. Chem Eur J. 2004;10:2237. doi: 10.1002/chem.200305634. [DOI] [PubMed] [Google Scholar]; (g) Trost BM, Guzner JL, Dirat O, Rhee YH. J Am Chem Soc. 2002;124:10396. doi: 10.1021/ja0205232. [DOI] [PubMed] [Google Scholar]
- 4.(a) Li C, Breit B. Chem Eur J. 2016;22:14655. doi: 10.1002/chem.201603532. [DOI] [PubMed] [Google Scholar]; (b) Lysek R, Borsuk K, Furman B, Kaluza Z, Kazimierski A, Chmielewski M. Curr Med Chem. 2004;11:1813. doi: 10.2174/0929867043364883. [DOI] [PubMed] [Google Scholar]; (c) Evans PA, Leahy DK. J Am Chem Soc. 2000;122:5012. [Google Scholar]
- 5.(a) Stanley LM, Bai C, Ueda M, Hartwig JF. J Am Chem Soc. 2010;132:8918. doi: 10.1021/ja103779e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shekhar S, Trantow B, Leitner A, Hartwig JF. J Am Chem Soc. 2006;128:11770. doi: 10.1021/ja0644273. [DOI] [PubMed] [Google Scholar]; (c) Leitner A, Shu C, Hartwig JF. Org Lett. 2005;7:1093. doi: 10.1021/ol050029d. [DOI] [PubMed] [Google Scholar]; (d) Kiener CA, Incarvito C, Hartwig JF. J Am Chem Soc. 2003;125:14272. doi: 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]; (e) Lopez F, Ohmura T, Hartwig JF. J Am Chem Soc. 2003;125:3426. doi: 10.1021/ja029790y. [DOI] [PubMed] [Google Scholar]; (f) Kim D, Reddy S, Singh OV, Lee JS, Kong SB, Han H. Org Lett. 2013;15:512. doi: 10.1021/ol3033237. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kirsch SF, Overman LE. J Am Chem Soc. 2005;127:2866. doi: 10.1021/ja0425583. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kirsch SF, Overman LE, White NS. Org Lett. 2007;9:911. doi: 10.1021/ol070110b. [DOI] [PubMed] [Google Scholar]; (c) Cannon JS, Overman LE. Acc Chem Res. 2016;49:2220. doi: 10.1021/acs.accounts.6b00398. [DOI] [PubMed] [Google Scholar]; (d) Cannon JS, Kirsch SF, Overman L, Sneddon HF. J Am Chem Soc. 2010;132:15192. doi: 10.1021/ja106688j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Werner EW, Mei TS, Burckle AJ, Sigman MS. Science. 2012;338:1455. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patel HH, Sigman MS. J Am Chem Soc. 2015;137:3462. doi: 10.1021/ja5130836. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Patel HH, Sigman MS. J Am Chem Soc. 2016;138:14226. doi: 10.1021/jacs.6b09649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen ZM, Nervig CS, Deluca RJ, Sigman MS. Angew Chem, Int Ed. 2017;56:6651. doi: 10.1002/anie.201703089. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang C, Santiago CB, Crawford JM, Sigman MS. J Am Chem Soc. 2015;137:15668. doi: 10.1021/jacs.5b11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Xu L, Hilton MJ, Zhang X, Norrby PO, Wu YD, Sigman MS, Wiest O. J Am Chem Soc. 2014;136:1960. doi: 10.1021/ja4109616. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hilton MJ, Xu LP, Norrby PO, Wu YD, Wiest O, Sigman MS. J Org Chem. 2014;79:11841. doi: 10.1021/jo501813d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mei TS, Werner EW, Burckle AJ, Sigman MS. J Am Chem Soc. 2013;135:6830. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Ozawa F, Kobatake Y, Hayashi T. Tetrahedron Lett. 1993;34:2505. [Google Scholar]; (b) Kilroy TC, Cozzi PG, Nicole E, Patrick J. Synthesis. 2004;11:1879. [Google Scholar]; (c) Loiseleur O, Hayashi M, Schmees N, Pfaltz A. Synthesis. 1997;11:1338. [Google Scholar]; (d) Loiseleur O, Meier P, Pfaltz A. Angew Chem, Int Ed Engl. 1996;35:200. [Google Scholar]; (e) Gilbertson SR, Fu Z. Org Lett. 2001;3:161. doi: 10.1021/ol006747b. [DOI] [PubMed] [Google Scholar]; (f) Gilbertson SR, Denov DG, Rheingold AL. Org Lett. 2000;2:2885. doi: 10.1021/ol006323h. [DOI] [PubMed] [Google Scholar]; (g) Kaukoranta P, Kallstrom K, Andersson PG. Adv Syn Catal. 2007;349:2595. [Google Scholar]; (h) Tietze LF, Thede K, Sannicolo F. Chem Comm. 1999:1811. [Google Scholar]; (i) Hennessy AJ, Malone YM, Guiry PJ. Tetrahedron Lett. 2000;41:2261. [Google Scholar]
- 11.Race NJ, Schwalm CS, Nakamuro T, Sigman MS. J Am Chem Soc. 2016;138:15881. doi: 10.1021/jacs.6b11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Bertelsen S, Dinér P, Johansen RL, Jørgensen KA. J Am Chem Soc. 2007;129:1536. doi: 10.1021/ja068908y. [DOI] [PubMed] [Google Scholar]; (b) Jiang H, Holub N, Jørgensen KA. Proc Natl Acad Sci USA. 2010;107:20630. doi: 10.1073/pnas.0914523107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sundén H, Ibrahem I, Zhao GL, Eriksson L, Córdova A. Chem Eur J. 2007;13:574. doi: 10.1002/chem.200600572. [DOI] [PubMed] [Google Scholar]
- 13.Tellam JP, Kociok-Köhn G, Carbery DR. Org Lett. 2008;10:5199. doi: 10.1021/ol802169j.. For more preparations of other enol ether substrates see: Ma D, Cai Q, Xie X. Synlett. 2005:1767.Kundu D, Maity P, Ranu BC. Org Lett. 2014;16:1040. doi: 10.1021/ol500134p.Wan C, Jones CD, Koenig TM, Pu J, Mitchell D. Tetrahedron Lett. 2003;44:8257.Willis MC, Taylor D, Gillmore AT. Chem Comm. 2003:2222.Nordman G, Buchwald SL. J Am Chem Soc. 2003;125:4978. doi: 10.1021/ja034809y.
- 14.Kuroiwa Y, Matsumura S, Toshima K. Synlett. 2008:2523. [Google Scholar]
- 15.Gritter RJ, Wallace TJ. J Org Chem. 1959;24:1051. [Google Scholar]
- 16.Babinski D, Soltani O, Frantz DE. Org Lett. 2008;10:2901. doi: 10.1021/ol8010002.. For alternative procedures see: Specklin S, Bertus P, Weibel JM, Pale P. J Org Chem. 2008;73:7845. doi: 10.1021/jo8015049.Ritter K. Synthesis. 1993:735.Crisp GT, Meyer AGJ. Org Chem. 1992;57:6972.
- 17.(a) Crouch IT, Neff RK, Frantz DE. J Am Chem Soc. 2013;35:4970. doi: 10.1021/ja401606e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crouch IT, Dreier T, Frantz DE. Angew Chem, Int Ed. 2011;50:6128. doi: 10.1002/anie.201101820. [DOI] [PubMed] [Google Scholar]
- 18.Qin D, Byun HS, Bittman R. J Am Chem Soc. 1999;121:662. [Google Scholar]
- 19.(a) Zhao D, Beiring B, Glorius F. Angew Chem, Int Ed. 2013;52:8454. doi: 10.1002/anie.201302573. [DOI] [PubMed] [Google Scholar]; (b) Vila C, Hornillos V, Fañanás-Mastral M, Feringa BL. Chem Commun. 2013;49:5933. doi: 10.1039/c3cc43105c. [DOI] [PubMed] [Google Scholar]
- 20.Chahar MK, Sharma N, Dobhal MP, Joshi YC. Pharmacog Rev. 2011;5:1. doi: 10.4103/0973-7847.79093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.