

Abstract
Septic shock with low cardiac output is very common in children. However, the mechanism underlying myocardial depression is unclear. The role of β3-AR in the development of myocardial depression in sepsis is unknown. In the present study, we generated an adolescent rat model of hypodynamic septic shock induced by lipopolysaccharide (LPS). Neonatal cardiomyocytes were also treated with LPS to mimic myocardial depression in sepsis, which was confirmed via an in vivo left ventricular hemodynamic study, and measurements of contractility and the Ca2+ transient in isolated adolescent and neonatal cardiomyocytes. After 16 h of LPS treatment, cultured neonatal cardiomyocytes showed a diminished Ca2+ transient amplitude associated with an increase in the β3-AR level. With the addition of a β3-AR agonist, the Ca2+ transient in LPS-treated neonatal rat cardiomyocytes gradually decreased over time; such a change was absent in cells treated with nitric oxide synthase (NOS) inhibitors prior to treatment with a β3-AR agonist. In adolescent rats with septic myocardial depression, cardiac function declined as indicated by decreased MAP, dP/dtmax, and dP/dtmix for 6 h after LPS injection; however, the β3-AR level first increased 2 h after LPS treatment and then decreased 6 h after LPS treatment in the absence of exogenous catecholamines. The results indicate that, in vitro, at the cellular level β3-AR may be involved in the development of myocardial depression (Ca2+ transient depression) in sepsis through NOS signaling pathways; however, in vivo, a complicated mechanism for modulating β3-AR may exist.
Keywords: Septic shock, β3-Adrenergic receptor, Myocardial depression, Rat, Ca2+ transient
Introduction
Nowadays, sepsis has become a major public health problem, and is the main cause of death among patients admitted to intensive care units. Notably, septic shock is a subset of sepsis in which underlying circulatory and cellular/metabolic abnormalities are profound enough to substantially increase mortality [1], and is a common cause of death in pediatric intensive care units [2]. Unlike that in adults, “cold shock” with low cardiac output and high peripheral vascular resistance is very common in children. Cardiovascular dysfunction occurs in about 70% of children with severe sepsis [3]; also called septic myocardial depression or septic cardiomyopathy, it increases the mortality of septic shock [4, 5]. However, the underlying mechanism of septic myocardial depression remains unclear.
β-Adrenergic receptors (β-ARs) and their signaling pathways play crucial roles in regulating cardiac function; they are also closely associated with the development of septic myocardial depression. Compared to β1-AR or β2-AR, the expression level of β3-AR is much lower under physiological conditions and increases in pathological conditions [6, 7, 8, 9]. Additionally, β3-AR activation also requires a higher level of catecholamines. Patients with severe sepsis and septic shock have significantly elevated catecholamines in their serum, resulting from the production of endogenous catecholamines, as well as the medical administration of catecholamines that doubles the increase in adrenaline or norepinephrine [10, 11]. Although published in vitro studies suggested that β3-AR activation may contribute to myocardial depression in sepsis [8], its role in the pathogenesis of septic myocardial depression remains unclear.
In the present study, we investigated the dynamic changes of β3-AR expression in conjunction with the development of myocardial depression in an adolescent rat model of septic shock induced by lipopolysaccharide (LPS) and in primary cultured rat cardiomyocytes treated with LPS. We aimed to better understand the effect of β3-AR on the development of myocardial depression, and therefore provide new insights into therapeutic targets of myocardial depression in septic shock.
Materials and Methods
Animal Model of Septic Shock
An adolescent rat model of septic shock induced by intravenous injection of LPS was generated as previously described [12]. Male pathogen-free Wistar rats (Charles River, Beijing, China), 6–8 weeks old, 190–210 g, were first anesthetized by 20% urethane (1 g/kg i.p.). The left femoral artery was cannulated for continuous monitoring of mean arterial blood pressure (MAP) and a PE50 catheter was inserted into the left ventricle through the right common carotid artery in order to monitor left ventricular (LV) hemodynamic changes (Biopac MP150 Biopac Systems, Goleta, CA, USA). The right femoral vein was exposed for the injection of Escherichia coli 055:B5 (L-2880, Sigma-Aldrich, St Louis, MI, USA; 15 mg/kg, 5 mg LPS dissolved in 1 mL 0.9% saline, i.v., 0.1 mL/min). Septic shock was established when MAP decreased to 25–30% of the baseline value, which was about 1.5 h after the injection of LPS. For controls, saline (0.9%, 3 mL/kg, 0.1 mL/min) was injected through the right femoral vein of control rats.
Blood (3–4 mL) was collected from the abdominal aorta at 2, 4, and 6 h after LPS administration and centrifuged (3,000 rpm, 10 min) after sitting at room temperature for 15 min. The supernatant was collected and stored at −80°C for ELISA. The heart was excised within 6 h of LPS administration and stored at −80°C for real-time PCR and immunoblotting.
Adolescent Rat Cardiomyocyte Isolation
Adult rat cardiomyocytes were isolated as previously described [13]. Four hours after an LPS injection, the hearts were isolated from LPS-treated rats, washed, and retroperfused through the aorta using a Langendorff apparatus. The perfusion solution (Tyrode) contained (in mM): 140 NaCl, 5.4 KCl, 1 MgCl2, 1.2 CaCl2, 10 HEPES, and 10 D-Glucose (pH set to 7.4 with NaOH). After 3 min of perfusion with the Ca2+-containing Tyrode, the hearts were further perfused with Ca2+-free Tyrode for 5 min and then subjected to digestion with high-K+ Kraftbrühe (KB) solution containing (in mM): 80 KOH, 40 KCl, 25 KH2PO4 25.0, 3 MgSO4, 50 L-glutamic, 20 taurine, 10 HEPES, 1 EGTA, 10 D-glucose, and 0.03 Ca2+ (pH set to 7.2 with KOH), with the addition of 0.6% collagenase type II and 0.6% BSA. The left ventricles were then removed and chopped into small pieces followed by gentle pipetting in fresh KB solution to completely isolate cardiomyocytes. The resultant, isolated cardiomyocytes were transferred to fresh Tyrode solution and left at room temperature for 1 h. Ca2+ was restored gradually (0.3, 0.6, 1.2 mM Ca2+) over the following period of 30 min. Rats subjected to 4 h of 0.9% saline treatment were used as controls.
Measurement of Contractility and Ca2+ Transient in Isolated Adolescent Rat Cardiomyocytes
Mechanical properties and intracellular Ca2+ transients were measured simultaneously using an IonOptix Myocam system (IonOptix Inc., Milton, MA, USA). Freshly isolated rat cardiomyocytes were loaded with 5 μM of Fura-2 AM (Molecular Probes, Eugene, OR, USA) at room temperature for 20 min, placed into a perfusion chamber mounted on a microscope stage, and perfused (1.5 mL/min, 37°C) with Tyrode solution containing (in mM): 131 NaCl, 4 KCl, 1.2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, at pH 7.4, with 95% O2. Under a field stimulation of 5 V (1 Hz), cells were excited by UV light (360 and 380 nm, alternately) for Ca2+ transient measurement. The Fura-2 emission at 510 nm was collected with a photomultiplier tube. Only rod-shaped cells with clear edges and a well-defined sarcomere structure and without spontaneous contractions were used for data collection. The kinetics of Ca2+ transients were analyzed in conjunction with myocyte mechanical measurements using the software, IonWizard 6.0 (IonOptix Inc.).
Neonatal Rat Cardiomyocyte Isolation and Culture
One-day-old rats were anesthetized with isoflurane and whole bodies were dipped in 75% ethanol for 5 s. The heart was quickly excised and washed 3 times with PBS. The ventricles were dissected and minced into 1-mm3 pieces followed by a series of digestions in a solution containing 0.06% trypsin and 0.08% collagenase with stirring at 36.8°C. After each digestion, the cell suspension was quickly placed in cell culture medium containing neonatal calf serum and then centrifuged at 800 rpm for 5 min. The resultant cell pellet was resuspended and preplated in a cell culture flask. After sitting in an incubator (37°C, 5% CO2) for 1 h to eliminate fibroblasts, the cardiomyocyte-rich suspension was then plated in 6-well cell culture dishes with a cell intensity of 3 × 105/cm2 for myocyte functional studies and 6 × 105/cm2 for immunoblotting and real-time PCR. Bromodeoxyuridine (0.1 mM) was added to the culture medium to inhibit fibroblast growth. Cells were ready for experiments within 72 h of culture.
Measurement of Ca2+ Transient in Cultured Neonatal Cardiomyocytes
Neonatal rat cardiomyocytes were treated with LPS (1 μg/mL) for 8, 12, and 16 h, respectively. LPS-treated and control cells were loaded with Fluo-4 acetoxymethyl ester (2 μM) for 20 min at room temperature and then placed in a perfusion chamber mounted on a microscope stage. The perfusion solution contained 1.8 mM Ca2+. The intracellular Ca2+ transient was measured by a customized Ca2+ imaging system consisting of an excitation source supplied by an IonOptix Hyperwitch xenon lamp system, which consisted of an excitation filter EX470/40, emission filter EM525/50, dichroic mirror DM495, Olympus IX71 inverted fluorescence microscope with an objective lens (UPLSAPO10X), and an Andor IXon X3 DU897 EMCCD calcium signal acquisition system (binning 4 × 4, sampling frequency 134 Hz). Available myocytes were chosen as a fluorescence region of interest (ROI). The following equation was applied: Catransient = (Fpeak – F0)/(F0 – B), where B represents background fluorescence intensity.
Quantitative Real-Time PCR
Total RNA was isolated from heart tissue or cultured cells according to the manufacturer's instructions supplied with the TRIzol Reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA) and then reverse transcribed with a Thermo First Synthesis Kit. The expression levels of β1-AR, β2-AR, and β3-AR were then determined by real-time PCR. Each targeted cDNA was amplified using SYBR Green via the StepOnePLUS Real-Time PCR system (Applied Biosystems, Waltham, MA, USA). The primers for β1-AR were forward 5′-CCA-TCA-CGC-TGC-CCT-TTC-G-3′ and reverse 5′-CGC-CAC-CAG-TGC-ATG-AGG-AT-3′; for β2-AR they were forward 5′-GCAATTTCTGGTGCGAGTT-3′ and reverse 5′-TGAAGGGCGATGTGATAG-3′; for β3-AR they were forward 5′-TTCCGTCGTCTTCTGTGC-3′ and reverse 5′-CCTGTTGAGCGGTGAGTT-3′. GAPDH was used as an internal control.
Western Blot Analysis
Harvested rat heart tissue and cultured rat cardiomyocytes were homogenized in RIPA buffer. Proteins from the homogenate (30 µg/well) were separated on a one-dimensional 4–10% SDS-polyacrylamide gel and transferred to a polyvinylidene membrane (Bio-Rad Laboratories, Hercules, CA, USA) with a semidry electrotransfer apparatus for 1.5 h at 300 mA. The expression levels of β1-AR, β2-AR, and β3-AR proteins were determined by the monoclonal antibodies: anti-β1-AR (1: 1,000; Abcam, Cambridge, UK), anti-β2-AR (1: 1,000; Abcam), and anti-β3-AR (1: 1,000; Santa Cruz, Dallas, TX, USA), respectively. The immunoblots were visualized by enhanced chemiluminescence (ECL detection kit from GE Healthcare, Chicago, IL, USA).
Statistical Analysis
All experiments were repeated at least 3 times. Data of a normal distribution were presented as the mean ± standard error of the mean (SEM). The Student t test for unpaired data was used to compare between 2 groups. Two-way ANOVA with repeated measures was used to analyze the data of LV function and Ca2+ transient changes with the administration of LPS. The statistical analysis was done using SPSS13.0 software (IBM Corporation, Armonk, NY, USA). p < 0.05 indicated significance.
Results
Depressed Left Ventricular Function in Rats with Septic Shock
LV function with/without LPS treatment was monitored in situ throughout the entire LPS treatment period of 6 h. The LV functional data collected at 0, 2, 4, and 6 h time points are presented in Figure 1. MAP started to decline 1.5 h after the intravenous administration of LPS to rats. Septic shock was defined as once the rats' MAP decrease was about 25–30% of the baseline value, which occurred about 2 h after the LPS injection, and lasted for the subsequent 4 h of the observation period. Controls with a saline injection displayed an unchanged MAP throughout the entire observation period of 6 h (Fig. 1a). Compared to control rats, LPS-treated rats showed a gradual decline in cardiac function as evidenced by significant decreases in heart rate, LV peak rate of a pressure rise, and an LV peak rate of pressure decay, as well as a prolonged relaxation time constant, though the LV end-diastolic pressure remained unaltered (Fig. 1b, c, d, e, f). These data indicate a successful rat model of septic shock with more and more depressed cardiac function when LPS treatment lasted for the 6-h period.
Fig. 1.
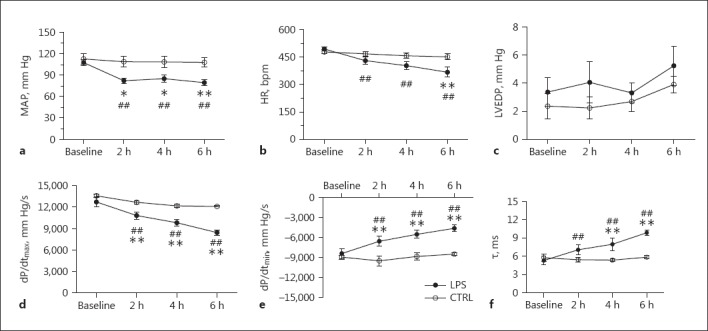
a–f In vivo hemodynamic studies. Septic shock was defined as MAP decreased by 25–30% of its baseline value. Septic shock in rats was induced by intravenous injection of LPS (E. coli 055:B5, 15 mg/kg, 0.1 mL/min) and rats injected (i.v.) with saline were used as controls. LV function was monitored over the entire 6 h of LPS/saline injection. ** p < 0.01, * p < 0.05 versus saline; ## p < 0.01 versus the respective baseline. n = 6.
Unaltered Serum Cardiac Troponin I and Creatine Kinase Isoenzyme in Rats with Septic Shock
Serum cardiac troponin I (cTnI) and creatine kinase isoenzyme (CK-MB) in LPS-treated rats was detected with an ELISA at 2, 4, and 6 h after treatment, and did not show significant changes in comparison to controls (Fig. 2). The results indicate that cell death or tissue damage did not occur during 6 h of LPS treatment in septic rats even though their overall cardiac function was significantly depressed.
Fig. 2.
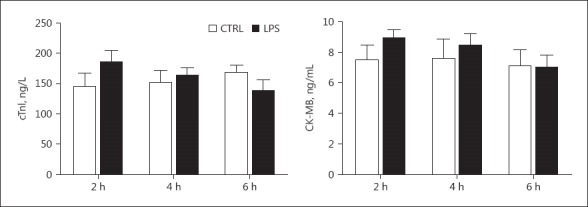
Serum cTnI and CK-MB levels in LPS-treated rats. Blood was collected from the rats at 2, 4, and 6 h after LPS or saline administration. Serum cTnI and CK-MB was detected by ELISA. n = 8.
Depressed Function of Cardiomyocytes Isolated from Rats with Septic Shock
Cardiomyocytes were isolated from the hearts of septic rats euthanized 4 h after LPS treatment. As shown in Figure 3, compared to cardiomyocytes from the control group, cardiomyocytes from septic rats showed significantly lower peak shortening, slower peak rate shortening (+dL/dt), and a peak rate of relengthening (dL/dt). They also showed a longer time to 90% peak shortening (TPS90) and 90% relengthening (TR90), indicating an overall depression of contractile function in cardiomyocytes in septic rats in the absence of neurohumoral factors.
Fig. 3.
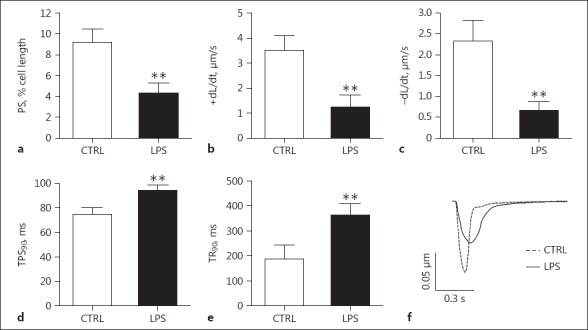
Contractile function of single cardiomyocytes. Cardiomyocytes were isolated from the hearts of the septic rats euthanized at 4 h after LPS treatment. a Peak shortening (PS). b Peak rate of shortening (+dL/dt). c Peak rate of relengthenin (–dL/dt). d Time to 90% peak shortening (TPS90). e Time to 90% of relengthening (TR90). f Representative traces of cellular sarcomere shortening in rat LV myocytes paced at 1 Hz. n = 88 from 5 LPS-treated mice; n = 35 from 5 control mice. ** p < 0.01 compared to the control.
The intracellular Ca2+ transient was also detected in conjunction with cell shortening in isolated cardiomyocytes. In agreement with the contractile data, cardiomyocytes from LPS-treated rats presented significantly lowered baseline and peak Ca2+ concentrations, a decreased amplitude of the Ca2+ transient (delta FFI), prolonged time to 90% Ca2+ decay, and an increased time constant of Ca2+ decay in comparison to controls (Fig. 4). Such data indicated that intracellular Ca2+ handling was negatively altered in cardiomyocytes of rats with septic shock.
Fig. 4.
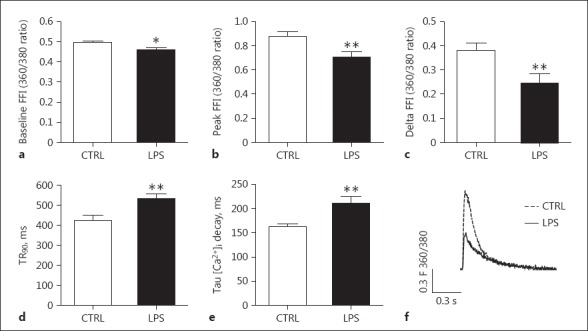
Measurements of intracellular Ca2+ transients in single cardiomyocytes. Cardiomyocytes were isolated from the hearts of the septic rats euthanized at 4 h after LPS treatment. a Baseline Ca2+ concentration (baseline FFI). b Peak Ca2+ concentration during cell shortening (peak FFI). c Amplitude of Ca2+ transient (delta FFI). d Time to 90% Ca2+ decay (TP90). e Time constant of Ca2+ decay (Tau). f Representative traces of cellular calcium transient in rat LV myocytes paced at 1 Hz. n = 88 from 5 LPS-treated mice; n = 35 from 5 control mice. ** p < 0.01 compared to the control.
Altered Ca2+ Transient in Rat Neonatal Cardiomyocytes Treated with LPS
Primarily cultured rat neonatal cardiomyocytes were treated with LPS (1 μg/mL) for 8, 16, and 24 h. Significantly fewer beating cells were present in wells treated with LPS for 24 h than those with a shorter LPS exposure time. Only cells with 8 or 16 h of LPS treatment were used for Ca2+ transient measurements. Compared to cells without LPS treatment, LPS-treated cells decreased the amplitude of the Ca2+ transient and prolonged Ca2+ decay. Such negative changes were only observed in cells exposed to LPS for 16 rather than 8 h. Such data suggest that prolonged LPS exposure depresses the cardiac function of neonatal cardiomyocytes (Fig. 5).
Fig. 5.
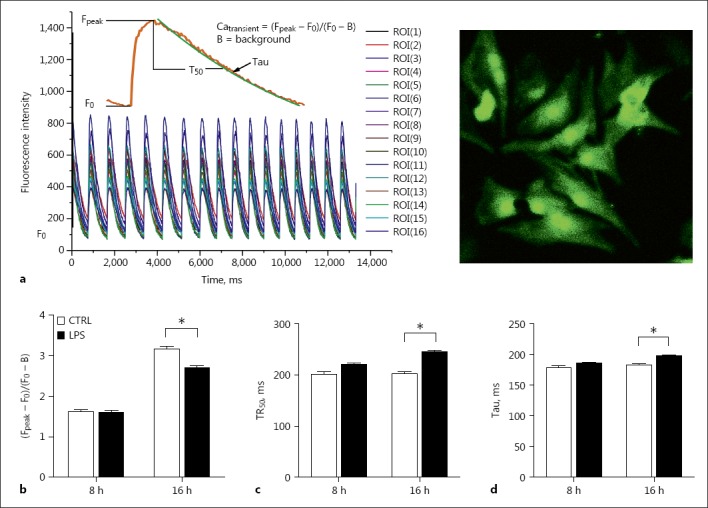
Measurements of intracellular Ca2+ transients in neonatal cardiomyocytes. The cells were exposed to LPS for 8 or 16 h. a A demonstration of Ca2+ transient of multiple cells (left) and a representative view of cultured rat neonatal cardiomyocytes under ×40 magnification (right). b Amplitude of Ca2+ transient (Fpeak – F0/F0 – B). c Time to 50% Ca2+ decay (TR50). d Time constant of Ca2+ decay (Tau). n = 130–150. * p < 0.05 compared to the control.
The Expression Level of β-ARs in Rat Neonatal Cardiomyocytes Treated with LPS
Since neonatal cardiomyocytes showed functional changes after 16 h of LPS treatment, β-AR expression levels were detected at the corresponding time point. As shown in Figure 6a and b, β3-AR expression in LPS-treated cells was increased approximately 2-fold at both mRNA and protein levels, while β1-AR and β2-AR mRNA expressions remained normal.
Fig. 6.
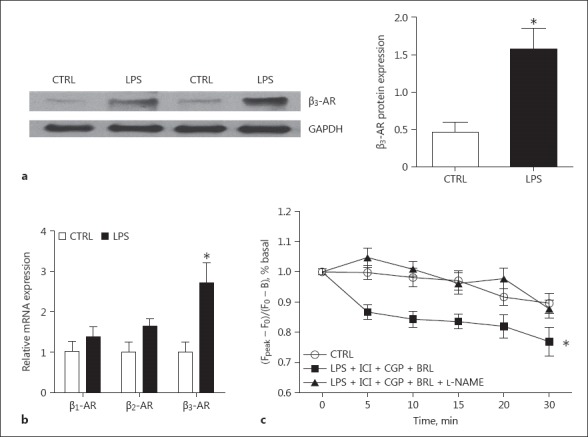
The expression of β-ARs in rat neonatal cardiomyocytes with 16 h of LPS treatment. a The expression level of β3-AR protein detected by immunoblotting. A representative immunoblotting image (left) and the quantitative data (right). n = 4. b The expression levels of β1-, β2-, and β3-AR mRNA. n = 4. c The amplitude of Ca2+ transients in the LPS-treated cardiomyocytes with addition of the following compounds. BRL 37344 (0.1 μM) is a β3-AR agonist. CGP 20712 (0.3 μM) is a specific β1-AR antagonist. ICI 118551 (0.1 μM) is a specific β2-AR antagonist. L-NAME (10 μM) is a NOS inhibitor. The Ca2+ transient was measured at 0, 5, 10, 15, 20, and 30 min after the addition of the β3-AR agonist. n = 40–45. * p < 0.05 versus the control.
The Effect of β3-AR Agonist on LPS-Treated Neonatal Cardiomyocytes
In order to test whether β3-AR activation caused myocardial depression in sepsis, a β3-AR agonist, BRL 37344 (0.1 μM), was added to the culture medium after 16 h of LPS treatment. Because of the imperfect selectivity of BRL37344, specific β1- and β2-AR antagonists (CGP 20712, 0.3 μM and ICI 118551, 0.1 μM) were added to block the activation of the 2 receptors. The Ca2+ transient was measured at 0, 5, 10, 15, 20, and 30 min after the addition of the β3-AR agonist. LPS-treated neonatal cardiomyocytes showed a gradual and significant decline in the amplitude of the Ca2+ transient over the time of exposure to the β3-AR agonist (Fig. 6c), while Ca2+ transients in non-LPS-treated cells were not affected by the same treatment (data not shown). The aforementioned negative effect of the β3-AR agonist on LPS-treated cardiomyocytes was totally absent in cells that were treated with a nitric oxide synthase (NOS) inhibitor (L-NAME, 10 μM) 30 min ahead of β3-AR agonist treatment.
The Expression Level of β-ARs in Hearts of Septic Shock Rats
As the rats had developed and maintained septic shock-associated myocardial depression after 2 h of LPS treatment, expression levels of β-ARs in the left ventricle were detected 2, 4, and 6 h after LPS exposure. The expression level of β3-AR protein increased significantly at first at an early stage of septic shock (2 h of LPS treatment) and then decreased to a level significantly lower than the control group after 6 h of LPS treatment (Fig. 7a, b). The expression level of β3-AR mRNA changed over the period of LPS treatment in an identical pattern to its protein level (Fig. 7c). In contrast to β3-AR, the expression of β1- and β2-AR did not show a significant change after LPS treatment at either the mRNA or protein level (Fig. 7b, c).
Fig. 7.
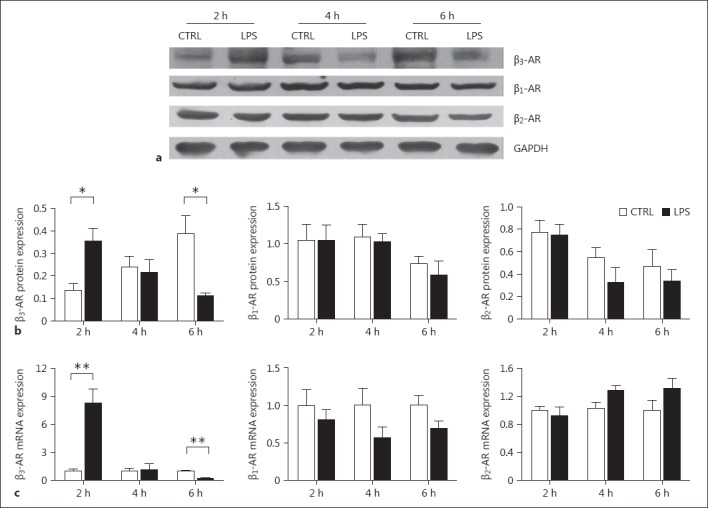
The expression of β-ARs in rats with myocardial depression in septic shock. Heart tissue was harvested from the septic shock rats at 2, 4, and 6 h after LPS treatment. a Representative immunoblotting image of β3-AR expression. b Quantitative data of the expression levels of β1-, β2-, and β3-AR proteins detected by immunoblotting. c Quantitative data of the expression levels of β1-, β2-, and β3-AR mRNA detected by real-time PCR. n = 4. * p < 0.05, ** p < 0.01 versus the control.
Discussion
By using an adolescent rat model of LPS-induced septic shock and isolated adolescent and neonatal rat cardiomyocytes, the present study demonstrated sepsis-associated intrinsic myocardial systolic and diastolic dysfunction both in vivo, at the intact animal level, and in vitro, at the cellular level. Furthermore, for the first time, we studied the expression level of β3-AR when myocardial depression manifested and developed without exogenous catecholamine. In vitro, the increase in β3-AR expression associates with the contractile depression of LPS-treated cultured rat neonatal cardiomyocytes, which involves NOS pathways. However, we did not observe a persistent increasing trend of β3-AR without exogenous catecholamines during the development of septic myocardial depression; the change in β3-AR expression in vivo is likely a protective compensatory mechanism against septic myocardial depression.
Septic shock is a subset of sepsis with circulatory and cellular/metabolic dysfunction associated with a higher risk of mortality. Unlike “warm shock” with high cardiac output and low peripheral vascular resistance in adults, “cold shock” with low cardiac output and high peripheral vascular resistance is very common in children. We used an animal model of septic shock through the intravenous injection of endotoxin, which showed more significant myocardial depression causing low cardiac output and decreased blood pressure, indicating this to be a good animal model for studying childhood septic shock.
Septic shock associated with myocardial dysfunction or septic cardiomyopathy leads to high mortality, yet the underlying mechanisms remain unclear. However, it is hard to measure intrinsic myocardial function in such patients [14]. Studies exist roughly describing septic myocardial depression by using echocardiography and cardiac biomarkers, such as troponin and brain natriuretic peptide [15, 16, 17]. As early as 2 decades ago, Robotham et al. [18] pointed out that the ejection fraction (EF) reflects the net effect of LV contractile function and its responses to afterload; in other words, when a decrease in peripheral arterial resistance lowers afterload, EF may be within the normal range, even with myocardial dysfunction. In septic myocardial depression, peripheral vascular dilation is common; therefore, EF is not an ideal diagnostic criterion for cardiac contractile function. Zaky et al. [15] also pointed out the limitation and insensitivity of using echocardiography to evaluate myocardial function in sepsis. An invasive hemodynamic study is a better way to study intrinsic cardiac contractile function in vivo, as employed in the present study to evaluate myocardial function in vivo in a rat model of LPS-induced sepsis with invasive hemodynamic measurements. Together with in vitro functional studies in isolated adolescent and neonatal cardiomyocytes, our work provides solid evidence of the presence of intrinsic myocardial depression in septic shock induced by LPS.
Previous reports have demonstrated the existence of cardiomyocyte ultrastructural damage in sepsis, yet without significant cardiomyocyte death [19]. Recently, Takasu et al. [20] studied myocardial tissue from patients with sepsis and found a lack of significant necrosis, apoptosis, or autophagy compared to control donor hearts. Zaky et al. [15] also suggested that an increased serum troponin level in sepsis is likely due to increased permeability of the cardiomyocyte plasma membrane caused by inflammatory factors rather than cardiac cell death. However, we did not detect any significant change in serum CK-MB or troponin, a specific marker of myocardial damage, in the rat model of septic shock, even after the development of myocardial depression. Therefore, severe myocardial necrosis or damage is not a characteristic of this rat model.
Recently, accumulating evidence has suggested the importance of β3-AR in regulating cardiac function and its potential as a therapeutic target [21, 22]; however, whether or not β3-AR activation correlates with myocardial depression in sepsis remains unclear. Moniotte et al. [8] first reported that cardiac β3-AR is elevated in patients with sepsis. The human β3-AR gene is located on the eighth chromosome and has a 79% similarity to that in the rat, in which the transmembrane region shares the highest homology, while the C-terminus and the third intramembranous region share the lowest [21]. Therefore, we used the rat as the animal model in this study. In contrast to β1/β2-AR that has a positive inotropic effect, β3-AR has a negative inotropic effect on myocardium in humans and several other animal models. β3-AR activation leads to the inhibition of L-type Ca2+ [23]; the NOS inhibitor, L-NAME, can partially block the inhibition [24]. β3-AR activation also decreases the intracellular Na+ concentration via activating the Na+/K+ pump and, in turn, leads to an increase in Ca2+ efflux through the Na+-Ca2+ exchanger, which eventually lowers Ca2+ uptake by the sarcolemma reticulum [25]. However, such a functional change in the Na+/K+ pump leading to myocardial depression was observed in wild-type animals, but not in animal models of heart failure [26].
In the present study, the observed depression in the Ca2+ transient in rat neonatal cardiomyocytes reflects the development of myocardial dysfunction after LPS treatment; at the same time both β3-AR mRNA and protein expression levels increased significantly. Furthermore, the addition of BRL37344, a β3-AR agonist, exaggerated the Ca2+ transient depression over time, in the absence of β1- and β2-AR activation. Such an effect was completely blocked in cells pretreated with an NOS inhibitor, which contrasts with the study by Moniotte et al. [8], where the NOS inhibitor was found to incompletely block the negative inotropic contractility effect of β3-AR activation in culture medium conditioned with LPS-activated macrophage-conditioned medium. These results indicate that β3-AR activation may be involved in the development of myocardial Ca2+ transient dysfunction through NOS signaling in sepsis in vitro.
Our results showed that, without the intervention of exogenous catecholamines, a significant change in blood catecholamine concentration between septic shock and control rats was not observed (data not shown). With the decline in cardiac function and heart rate, the expression level of β3-AR firstly increased and then decreased rather than continuing to increase. A previous study indicated that the activation of β3-AR required more catecholamines than that of β1/β2-AR [27]. Indeed, the concentration of catecholamines increased significantly in patients with severe sepsis; however, such an increase could have both an endogenous and exogenous origin because catecholamines are commonly used to treat patients with severe sepsis or septic shock. Therefore, it is unclear whether β3-AR activation is a risk factor that contributed to septic myocardial depression, although Moniotte et al. [8] detected a significantly high β3-AR level in myocardium from patients who died of severe sepsis. In our septic shock model, myocardial β3-AR increased and then decreased, despite an ever-declining cardiac function. In contrast, β3-AR was slightly increased in the control group without any cardiac dysfunction, which may be related to long-time anesthesia without food or water administration and later operation stresses. A lasting high catecholamine level or stress often triggers a high level of oxygen consumption in the myocardium, Ca2+ overload, and toxin-related myocardium damage. Thus, activation of β3-AR in vivo is more likely to be a compensation mechanism to protect the heart from a high metabolic state during septic shock, which agrees with reported in vivo studies on β3-AR in heart failure and hypertrophic cardiomyopathy models [26, 28, 29]. With the activation of β3-AR, the third generation β-AR blocker, nebivolol, is more effective in inhibiting cardiac remodeling than the β1-AR blocker, metoprolol [30]. The aforementioned studies were performed in chronic disease models while septic myocardial depression is an acute condition. Therefore, β3-AR may play a more complicated role in septic myocardium depression in vivo, although β3-AR activation may be involved in the development of myocardial depression in isolated neonatal cardiomyocytes. Further studies are required to address questions, such as how β3-AR changes and how it affects myocardial function in the administration of exogenous catecholamines during septic shock, and what is the time window for treatment with an β3-AR agonist or antagonist.
In addition to β3-AR, we simultaneously detected changes in cardiac β1-AR and β2-AR at various time points of septic shock and did not find any significant alteration. Early reports indicated that the density of dominant β-AR decreased or stayed normal in sepsis [31, 32]; however, another study found a decrease in stimulatory G protein and an increase in inhibitory G protein [33, 34], all of which may lead to a decrease in intracellular cAMP and thus affect myocardium function.
Our study has several limitations. Unlike isolated adult cardiomyocytes with an elongated shape and clear edge, primary cultured neonatal cardiomyocytes have an irregular shape and thus lack any widely used commercialized measurement of contractile function, so there is no widely accepted method for the accurate measurement of primary cultured neonatal cardiomyocytes. Since cardiac contractile function is coupled with cyclic cytosolic Ca2+ transient change, we indirectly evaluated the contractile function of neonatal cardiomyocytes by measuring Ca2+ transients. Compared to the conventional method, which only measures a cell at a time, our customized system is able to measure multiple cells simultaneously, which improves efficiency and minimizes the variance between each measurement.
Conclusion
Our present study is an investigation of the role of β3-AR in the development of myocardial depression in hypodynamic septic shock induced by LPS. It demonstrates septic myocardial depression in both the intact animal and in isolated adolescent and neonatal cardiomyocytes treated with LPS. Although β3-AR activation may be involved in the development of myocardial dysfunction through NOS signaling in isolated neonatal cardiomyocytes, a complicated mechanism may exist in vivo of septic shock considering a lack of a persistent increasing trend of β3-AR without exogenous catecholamines during the development of septic myocardial depression. Our findings shed light on an understanding of the role of β3-AR and its related signaling pathways in children with septic myocardial depression, which deserves further exploration.
Statement of Ethics
This study was approved by the Medicine Ethics Committee of Shengjing Hospital of the China Medical University (2017PS232K). The animal experiments conformed to institutional standards.
Conflict of Interest
The authors declare no conflicts of interest.
Funding Sources
This work was funded by the Natural Science Foundation of China (No. 81372039) and Science and Technology Fundation of Shenyang (No. F13-220-9-38)
Acknowledgement
We sincerely thank the expert assistance of the Core Laboratory at China Medical University Shengjing Hospital and the China Academy of Chinese Medical Sciences.
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruth A, McCracken CE, Fortenberry JD, Hall M, Simon HK, Hebbar KB. Pediatric severe sepsis: current trends and outcomes from the Pediatric Health Information Systems database. Pediatr Crit Care Med. 2014;15:828–838. doi: 10.1097/PCC.0000000000000254. [DOI] [PubMed] [Google Scholar]
- 3.Weiss SL, Fitzgerald JC, Pappachan J, Wheeler D, Jaramillo-Bustamante JC, Salloo A, Singhi SC, Erickson S, Roy JA, Bush JL, Nadkarni VM, Thomas NJ. Global epidemiology of pediatric severe sepsis: the Sepsis Prevalence, Outcomes, and Therapies Study. Am J Respir Crit Care Med. 2015;191:1147–1157. doi: 10.1164/rccm.201412-2323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raj S, Killinger JS, Gonzalez JA, Lopez L. Myocardial dysfunction in pediatric septic shock. J Pediatr. 2014;164:72–77. doi: 10.1016/j.jpeds.2013.09.027. e2. [DOI] [PubMed] [Google Scholar]
- 5.Brierley J, Peters MJ. Distinct hemodynamic patterns of septic shock at presentation to pediatric intensive care. Pediatrics. 2008;122:752–759. doi: 10.1542/peds.2007-1979. [DOI] [PubMed] [Google Scholar]
- 6.Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL. Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103:1649–1655. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- 7.Cheng HJ, Zhang ZS, Onishi K, Ukai T, Sane DC, Cheng CP. Upregulation of functional β3-adrenergic receptor in the failing canine myocardium. Circ Res. 2001;89:599–606. doi: 10.1161/hh1901.098042. [DOI] [PubMed] [Google Scholar]
- 8.Moniotte S, Belge C, Sekkali B, Massion PB, Rozec B, Dessy C, Balligand JL. Sepsis is associated with an upregulation of functional β3 adrenoceptors in the myocardium. Eur J Heart Fail. 2007;9:1163–1171. doi: 10.1016/j.ejheart.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Dinçer ÜD, Bidasee KR, Güner Ş, Tay A, Özçelikay AT, Altan VM. The effect of diabetes on expression of β1-, β2-, and β3-adrenoreceptors in rat hearts. Diabetes. 2001;50:455–461. doi: 10.2337/diabetes.50.2.455. [DOI] [PubMed] [Google Scholar]
- 10.Benedict CR, Rose JA. Arterial norepinephrine changes in patients with septic shock. Circ Shock. 1992;38:165–172. [PubMed] [Google Scholar]
- 11.Ostrowski SR, Berg RMG, Windeløv NA, Meyer MAS, Plovsing RR, Møller K, Johansson PI. Coagulopathy, catecholamines, and biomarkers of endothelial damage in experimental human endotoxemia and in patients with severe sepsis: a prospective study. J Crit Care. 2013;28:586–596. doi: 10.1016/j.jcrc.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Fink MP. Animal models of sepsis. Virulence. 2014;5:143–153. doi: 10.4161/viru.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai D, Gao J, Bi X, He H, Shi X, Weng S, Chen Y, Yang Y, Ye Y, Fu G. The Rho kinase inhibitor, fasudil, ameliorates diabetes-induced cardiac dysfunction by improving calcium clearance and actin remodeling. J Mol Med. 2017;95:155–165. doi: 10.1007/s00109-016-1469-1. [DOI] [PubMed] [Google Scholar]
- 14.Repessé X, Charron C, Vieillard-Baron A. Evaluation of left ventricular systolic function revisited in septic shock. Crit Care. 2013;17:164. doi: 10.1186/cc12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaky A, Deem S, Bendjelid K, Treggiari MM. Characterization of cardiac dysfunction in sepsis. Shock. 2014;41:12–24. doi: 10.1097/SHK.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 16.Palmieri V, Innocenti F, Guzzo A, Guerrini E, Vignaroli D, Pini R. Left ventricular systolic longitudinal function as predictor of outcome in patients with sepsis. Circ Cardiovasc Imaging. 2015;8 doi: 10.1161/CIRCIMAGING.115.003865. e003865. [DOI] [PubMed] [Google Scholar]
- 17.Masson S, Caironi P, Fanizza C, Carrer S, Caricato A, Fassini P, Vago T, Romero M, Tognoni G, Gattinoni L, Latini R, Albumin Italian Outcome Sepsis Study Investigators Sequential N-terminal pro-B-type natriuretic peptide and high-sensitivity cardiac troponin measurements during albumin replacement in patients with severe sepsis or septic shock. Crit Care Med. 2016;44:707–716. doi: 10.1097/CCM.0000000000001473. [DOI] [PubMed] [Google Scholar]
- 18.Robotham JL, Takata M, Berman M, Harasawa Y. Ejection fraction revisited. Anesthesiology. 1991;74:172–183. doi: 10.1097/00000542-199101000-00026. [DOI] [PubMed] [Google Scholar]
- 19.Zanotti-Cavazzoni SL, Hollenberg SM. Cardiac dysfunction in severe sepsis and septic shock. Curr Opin Crit Care. 2009;15:392–397. doi: 10.1097/MCC.0b013e3283307a4e. [DOI] [PubMed] [Google Scholar]
- 20.Takasu O, Gaut JP, Watanabe E, To K, Fagley RE, Sato B, Jarman S, Efimov IR, Janks DL, Srivastava A, Bhayani SB, Drewry A, Swanson PE, Hotchkiss RS. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–517. doi: 10.1164/rccm.201211-1983OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozec B, Gauthier C. β3-Adrenoceptors in the cardiovascular system: putative roles in human pathologies. Pharmacol Ther. 2006;111:652–673. doi: 10.1016/j.pharmthera.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Balligand J-L. Beta3-adrenoreceptors in cardiovasular diseases: new roles for an “old” receptor. Curr Drug Deliv. 2013;10:64–66. doi: 10.2174/1567201811310010011. [DOI] [PubMed] [Google Scholar]
- 23.Kitamura T, Onishi K, Dohi K, Okinaka T, Isaka N, Nakano T. The negative inotropic effect of beta3-adrenoceptor stimulation in the beating guinea pig heart. J Cardiovasc Pharmacol. 2000;35:786–790. doi: 10.1097/00005344-200005000-00016. [DOI] [PubMed] [Google Scholar]
- 24.Zhang ZS, Cheng HJ, Onishi K, Ohte N, Wannenburg T, Cheng CP. Enhanced inhibition of L-type Ca2+ current by β3-adrenergic stimulation in failing rat heart. J Pharmacol Exp Ther. 2005;315:1203–1211. doi: 10.1124/jpet.105.089672. [DOI] [PubMed] [Google Scholar]
- 25.Audigane L, Kerfant BG, Harchi El A, Lorenzen-Schmidt I, Toumaniantz G, Cantereau A, Potreau D, Charpentier F, Noireaud J, Gauthier C. Rabbit, a relevant model for the study of cardiac β3-adrenoceptors. Exp Physiol. 2009;94:400–411. doi: 10.1113/expphysiol.2008.045179. [DOI] [PubMed] [Google Scholar]
- 26.Bundgaard H, Liu CC, Garcia A, Hamilton EJ, Huang Y, Chia KKM, Hunyor SN, Figtree GA, Rasmussen HH. β3 adrenergic stimulation of the cardiac Na+-K+ pump by reversal of an inhibitory oxidative modification. Circulation. 2010;122:2699–2708. doi: 10.1161/CIRCULATIONAHA.110.964619. [DOI] [PubMed] [Google Scholar]
- 27.Strosberg AD. Structure and function of the β3-adrenergic receptor. Annu Rev Pharmacol Toxicol. 1997;37:421–450. doi: 10.1146/annurev.pharmtox.37.1.421. [DOI] [PubMed] [Google Scholar]
- 28.Moens AL, Leyton-Mange JS, Niu X, Yang R, Cingolani O, Arkenbout EK, Champion HC, Bedja D, Gabrielson KL, Chen J, Xia Y, Hale AB, Channon KM, Halushka MK, Barker N, Wuyts FL, Kaminski PM, Wolin MS, Kass DA, Barouch LA. Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the β3-adrenoreceptor. J Mol Cell Cardiol. 2009;47:576–585. doi: 10.1016/j.yjmcc.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belge C, Hammond J, Dubois-Deruy E, Manoury B, Hamelet J, Beauloye C, Markl A, Pouleur AC, Bertrand L, Esfahani H, Jnaoui K, Götz KR, Nikolaev VO, Vanderper A, Herijgers P, Lobysheva I, Iaccarino G, Hilfiker-Kleiner D, Tavernier G, Langin D, Dessy C, Balligand JL. Enhanced expression of β3-adrenoceptors in cardiac myocytes attenuates neurohormone-induced hypertrophic remodeling through nitric oxide synthase. Circulation. 2014;129:451–462. doi: 10.1161/CIRCULATIONAHA.113.004940. [DOI] [PubMed] [Google Scholar]
- 30.Sorrentino SA, Doerries C, Manes C, Speer T, Dessy C, Lobysheva I, Mohmand W, Akbar R, Bahlmann F, Besler C, Schaefer A, Hilfiker-Kleiner D, Lüscher TF, Balligand JL, Drexler H, Landmesser U. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional β1-blockade. J Am Coll Cardiol. 2011;57:601–611. doi: 10.1016/j.jacc.2010.09.037. [DOI] [PubMed] [Google Scholar]
- 31.Tang C, Liu MS. Initial externalization followed by internalization of beta-adrenergic receptors in rat heart during sepsis. Am J Physiol. 1996;270:R254–R263. doi: 10.1152/ajpregu.1996.270.1.R254. [DOI] [PubMed] [Google Scholar]
- 32.Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc Natl Acad Sci USA. 1989;86:6753–6757. doi: 10.1073/pnas.86.17.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda N, Hattori Y, Akaishi Y, Suzuki Y, Kemmotsu O, Gando S. Impairment of cardiac β-adrenoceptor cellular signaling by decreased expression of Gsα in septic rabbits. Anesthesiology. 2000;93:1465–1473. doi: 10.1097/00000542-200012000-00019. [DOI] [PubMed] [Google Scholar]
- 34.Wu LL, Yang SL, Yang RC, Hsu HK, Hsu C, Dong LW, Liu MS. G protein and adenylate cyclase complex-mediated signal transduction in the rat heart during sepsis. Shock. 2003;19:533–537. doi: 10.1097/01.shk.0000055816.40894.cd. [DOI] [PubMed] [Google Scholar]