

ABSTRACT
Introduction: ACE‐083 is a locally acting follistatin‐based therapeutic that binds myostatin and other muscle regulators and has been shown to increase muscle mass and force in neuromuscular disease mouse models. This first‐in‐human study examined these effects. Methods: In this phase 1, randomized, double‐blind, placebo‐controlled, dose‐ranging study in healthy postmenopausal women, ACE‐083 (50–200 mg) or placebo was administered unilaterally into rectus femoris (RF) or tibialis anterior (TA) muscles as 1 or 2 doses 3 weeks apart. Results: Fifty‐eight postmenopausal women were enrolled, 42 ACE‐083 and 16 placebo. No serious adverse events (AE), dose‐limiting toxicities, or discontinuations resulting from AEs occurred. Maximum (mean ± SD) increases in RF and TA muscle volume were 14.5% ± 4.5% and 8.9% ± 4.7%, respectively. No significant changes in mean muscle strength were observed. Discussion: ACE‐083 was well tolerated and resulted in significant targeted muscle growth. ACE‐083 may have the potential to increase muscle mass in a wide range of neuromuscular disorders. Muscle Nerve 57: 921–926, 2018
Keywords: ACE‐083, muscular dystrophy, muscle volume, myostatin, neuromuscular disease
Abbreviations
- ADA
antidrug antibody
- AE
adverse event
- ECG
electrocardiogram
- GDF
growth differentiation factor
- LC‐MS/MS
liquid chromatography‐mass spectrometry assay
- LLOQ
lower limit of quantitation
- PD
pharmacodynamic
- PK
pharmacokinetic
- RF
rectus femoris
- SAE
serious adverse advent
- TA
tibialis anterior
ACE‐083 is a recombinant fusion protein consisting of a modified form of human follistatin linked to the human immunoglobulin G2 Fc domain. The protein functions as a trap for ligands within the transforming growth factor β superfamily (principally growth differentiation factor [GDF]8 [myostatin], GDF11, and activins A and B) that inhibit skeletal muscle growth and differentiation.1, 2
ACE‐083 is designed to act locally in the muscle into which it is injected by having increased residence time in muscle and rapid inactivation after entering the circulation. It has been shown to increase muscle volume as well as peak tetanic force of the injected muscle in wild‐type mice as well as in mouse models of myogenic and neurogenic neuromuscular diseases, including mdx (Duchenne muscular dystrophy model), superoxide dismutase 1 (amyotrophic lateral sclerosis model), and trembler‐J (Charcot–Marie–Tooth disease model).3, 4, 5, 6 These studies support clinical development of ACE‐083 as a potential treatment for a wide range of neuromuscular disorders, in particular those with focal and/or asymmetric muscle involvement.
The primary objective of this first‐in‐human study of ACE‐083 was to evaluate the safety and tolerability of single and multiple doses of ACE‐083 as a local muscle injection. This study also investigated the magnitude and duration of the pharmacodynamic (PD) effects of ACE‐083 on muscle volume and strength as well as the pharmacokinetics (PK) of ACE‐083 in the circulation.
MATERIALS AND METHODS
Study Design
This study was approved by the Chesapeake Institutional Review Board 6940 Columbia Gateway Drive, Suite 110, Columbia, MD, and conducted in accordance with good clinical practices, the Declaration of Helsinki, and applicable national and local regulations and requirements. All participants provided written informed consent, and the study is registered at clinicaltrials.gov (NCT02257489).
This was a phase 1, randomized, double‐blind, placebo‐controlled, dose‐ranging study conducted at a single study center (Celerion, Lincoln, NE) that enrolled healthy postmenopausal women between October 6, 2014 and April 25, 2016. In total, 7 cohorts were enrolled, each with 8 or 9 randomized participants (6 active and 2 placebo in cohorts 1–5; 6 active and 3 placebo in cohorts 6 and 7).
Determination of the starting dosage level of ACE‐083 was based on nonclinical toxicity and pharmacology studies in various animal species. In cohorts 1–3, ACE‐083 (50 mg/ml; Acceleron Pharma, Cambridge, MA) was injected into the rectus femoris (RF) as a single dose (50, 100, or 200 mg per muscle), and in cohorts 4 and 5 it was injected as 2 doses on days 1 and 22 (100 or 200 mg per muscle). In cohorts 6 and 7, ACE‐083 was injected into the tibialis anterior (TA) as 2 doses on days 1 and 22 (100 or 150 mg per muscle). ACE‐083 (or placebo) was administered unilaterally in the right side only. Electromyography guidance was used to administer each dose as 2 or 4 equal‐volume injections (0.5–1.0 ml per injection), depending on the dose, by using a 26‐gauge Myoject needle (Natus, Pleasanton, CA) into predefined muscle locations along the length of the muscle.
Participants were followed for 12 weeks after their final dose. Study visits included physical exams, safety and biomarker assessments, MRI scans, and strength assessments.
Participant Eligibility
Eligible participants were healthy postmenopausal women aged 45–75 years with a body mass index range of 18.5–32 kg/m2. Participants who had received any medication that could affect muscle, including hormone replacement therapy, systemic glucocorticoid therapy, statin medication, insulin, or any other investigational therapy within 3 months prior to study enrollment were excluded. Participants who had received any therapies affecting bleeding risk within 1 week prior to enrollment were also excluded. In addition, because muscle changes were a key endpoint, participants unwilling or unable to maintain their baseline level of physical activity throughout the study were excluded.
Randomization and Blinding
The randomization schedule was computer generated. The pharmacist at the site and a selected research associate were unblinded to the participant treatment assignments. Participants, the investigators, the sponsor, and all other site personnel remained blinded to treatment assignments throughout the study until the database was locked.
Assessments and Endpoints
Safety
Safety end points were assessed at baseline and regularly throughout the study. They included evaluation of adverse events (AE), injection site reactions, clinical laboratory tests (including hematology, chemistry, urinalysis, and biomarkers), electrocardiograms (ECG), vital signs, antidrug antibody (ADA), and physical examinations (see Supp. Info. Table 1). AEs were coded according to the Medical Dictionary for Regulatory Activities v17.1 and were recorded regardless of causality.
A safety review team comprising the principal investigator, medical monitor, and a neuromuscular specialist unaffiliated with either the study or the sponsor met periodically throughout the study and reviewed blinded data from each cohort to make recommendations regarding dose selection, dose escalation, enrollment, and overall safety monitoring. Dose‐limiting toxicity was defined as a serious AE (SAE) possibly or probably related to study drug or an AE, injection‐site reaction, ECG abnormality, laboratory variable abnormality, or vital sign abnormality, possibly or probably related to study drug that was either grade ≥3 (National Cancer Institute‐Common Terminology Criteria for Adverse Events v4.0) or clinically significant in the judgment of the investigator.
Pharmacokinetics
An enzyme‐linked immunosorbent assay with a lower limit of quantitation (LLOQ) of 100 ng/ml was performed on serum from all cohorts; however, detection of inactive breakdown products led to inconclusive results and discontinuation of this assay. A liquid chromatography‐mass spectrometry assay (LC‐MS/MS) with an LLOQ of 10 ng/ml was developed specifically to measure intact ACE‐083 with higher sensitivity. The LC‐MS/MS assay was performed only on serum samples from cohorts 6 and 7 because of limited sample volumes from earlier cohorts. PK samples were collected on days 1, 2, 5, 8, 15, 22, 23, 26, 29, 33, 36, 40, 43, 50, 64, 78, and 92; multiple samples (predose, 3 h postdose, and 6 h postdose) were obtained on days 1 and 22.
Pharmacodynamics
Key PD assessments included MRI of the thigh (RF cohorts) and lower leg (TA cohorts) to measure muscle volume and intramuscular fat. The boundaries of the injected muscle on each cross‐sectional image were identified by a blinded technologist and reviewed and approved by the blinded radiologist assigned to the study. The volume of the injected muscle was calculated by using an automated task developed and validated by the imaging vendor (VirtualScopics, Rochester, NY).
Muscle strength of the knee extensors (RF cohorts) and ankle dorsiflexors (TA cohorts) was measured by both hand‐held dynamometer (microFET2; Hoggan Scientific, Salt Lake City, UT) and fixed system (Biodex System 3 Pro; Biodex Medical Systems, Shirley, NY). Each strength assessment was performed by the same blinded clinical evaluator, as outlined in the clinical evaluator manual. Knee extension was performed with the participant sitting and at 90 ° of flexion; ankle dorsiflexion was performed with the participant supine and the ankle in the neutral state. The participant was instructed to flex as hard as possible for 5 s, 3 times, with 30–60 s rest between each measurement. The best of the 3 attempts was recorded.
Statistical Analysis
No formal a priori power analysis for sample size was conducted. Baseline was defined as the last assessment prior to dosing on study day 1. For each participant, change and percentage change from baseline were calculated for each PD endpoint; treated and contralateral limbs were compared. PD changes in active dosage groups were compared to the pooled placebo group by analysis of covariance. In addition, Dunnett's test was performed to assess trends in muscle volume changes. Safety and PK variables of ACE‐083 were summarized by using descriptive statistics. Fisher's exact test was used to compare the proportions of selected AEs among treatment and muscle groups.
RESULTS
In total, 58 postmenopausal women were enrolled including 42 randomized to ACE‐083 and 16 randomized to placebo. One participant was not evaluated for PD because of missing posttreatment MRI scans (see Supp. Info. Fig. 1 for the study CONSORT diagram). Demographics and baseline characteristics by muscle and treatment group are summarized in Table 1. The median age and range was similar between ACE‐083‐ and placebo‐treated groups; most participants were white and not Hispanic or Latino.
Table 1.
Demographic summary
| RF cohorts 1–5 | TA cohorts 6–7 | Overall | ||||
|---|---|---|---|---|---|---|
| Variable | ACE‐083, n = 30 | Placebo, n = 10 | ACE‐083, n = 12 | Placebo, n = 6 | ACE‐083, n = 42 | Placebo, n = 16 |
| Sex, n (%) | ||||||
| Women | 30 (100) | 10 (100) | 12 (100) | 6 (100) | 42 (100) | 16 (100) |
| Race, n (%) | ||||||
| White | 29 (97) | 10 (100) | 12 (100) | 5 (83) | 41 (98) | 15 (94) |
| Black | 1 (3) | 0 (0) | 0 (0) | 1 (17) | 1 (2) | 1 (6) |
| Ethnicity, n (%) | ||||||
| Not Hispanic or Latino | 29 (97) | 10 (100) | 11 (92) | 6 (100) | 40 (95) | 16 (100) |
| Hispanic or Latino | 1 (3) | 0 (0) | 1 (8) | 0 (0) | 2 (5) | 0 (0) |
| Age, y | ||||||
| Median | 56.0 | 57.5 | 55.5 | 57.5 | 56.0 | 57.5 |
| Minimum, maximum | 45, 70 | 55,73 | 49, 62 | 45,65 | 45, 70 | 45, 73 |
| BMI, kg/m2 | ||||||
| Median | 25.0 | 26.2 | 28.3 | 24.8 | 25.9 | 25.1 |
| Minimum, maximum | 19.2, 31.5 | 22.3, 29.6 | 21.5, 31.6 | 23.8, 31.4 | 19.2, 31.6 | 22.3, 31.4 |
BMI, body mass index; RF, rectus femoris; TA, tibialis anterior; y, years.
Safety and Tolerability
There were no SAEs, dose‐limiting toxicities, grade 3 or higher AEs, or discontinuations resulting from AEs. AEs considered related to treatment and occurring in ≥10% of ACE‐083‐treated participants are shown in Table 2, by muscle and treatment group. AEs involving the injection site accounted for the majority of AEs reported for both ACE‐083‐ and placebo‐treated participants. Injection site pain was the most common AE. Other injection site AEs, reported in approximately 10%–20% of ACE‐083‐treated participants, are described in Table 2. Most of these events were mild in intensity and occurred at the time of or immediately after dosing and were resolved shortly thereafter. There was no statistically significant difference in the incidence of injection site reaction or hemorrhage in the ACE‐083 groups compared with placebo.
Table 2.
Related AEs reported in ≥10% of ACE‐083‐treated participants overall
| RF cohorts 1–5 | TA cohorts 6–7 | Overall | ||||
|---|---|---|---|---|---|---|
| AE preferred term | ACE‐083, n = 30 | Placebo, n = 10 | ACE‐083, n = 12 | Placebo, n = 6 | ACE‐083, n = 42 | Placebo, n = 16 |
| Injection site AEs, n (%) | ||||||
| Injection site pain | 27 (90) | 10 (100) | 11 (92) | 6 (100) | 38 (90) | 16 (100) |
| Injection site discomfort | 4 (13) | 1 (10) | 4 (33) | 3 (50) | 8 (19) | 4 (25) |
| Injection site reaction | 5 (17) | 1 (10) | 1 (8) | 0 (0) | 6 (14) | 1 (6) |
| Injection site hemorrhage | 4 (13) | 0 (0) | 1 (8) | 0 (0) | 5 (12) | 0 (0) |
| Injection site warmth | 1 (3) | 2 (20) | 3 (25) | 1 (17) | 4 (10) | 3 (19) |
| All other AEs, n (%) | ||||||
| Pain in extremity | 6 (20) | 2 (20) | 12 (100) | 5 (83) | 18 (43) | 7 (44) |
| Muscle twitching | 8 (27) | 3 (30) | 0 (0) | 0 (0) | 8 (19) | 3 (19) |
| Muscle tightness | 2 (7) | 1 (10) | 4 (33) | 2 (33) | 6 (14) | 3 (19) |
| Limb discomfort | 3 (10) | 2 (20) | 1 (8) | 0 (0) | 4 (10) | 2 (13) |
| Myalgia | 3 (10) | 0 (0) | 1 (8) | 0 (0) | 4 (10) | 0 (0) |
AE, adverse event; RF, rectus femoris; TA, tibialis anterior.
Noninjection site AEs considered related to treatment are also shown in Table 2. Each had a similar incidence in ACE‐083 and placebo groups. There was no statistically significant difference in the incidence of myalgia between ACE‐083 and placebo. There were statistically significant differences in the incidence of muscle twitching (P = 0.01), pain in extremity (P < 0.001), and muscle tightness (P = 0.02) between the RF and TA cohorts, but there was no significant difference in limb discomfort. There were no notable correlations between AE incidence and increasing dosage or number of injections.
No trends in vital signs, ECG, clinical laboratory tests, or physical examination were noted. Three participants had confirmed positive ADA results, 2 of which occurred at the last time point at low titers (40, 80) and returned to negative. One participant had varying titers of ADA (40–2,560) that decreased to 80 at 12 weeks after the last dose and were not associated with any clinical manifestations; this participant was lost to follow‐up.
Pharmacokinetics
PK results were available for cohorts 6 and 7. Serum ACE‐083 levels were detectable only during the first day after dosing, with most values close to the LLOQ. Only 14 of 252 (5.6%) samples from 6 of 12 (50%) ACE‐083‐treated participants were above the LLOQ; thus, limited PK variables could be estimated (Table 3).
Table 3.
Summary of ACE‐083 LC‐MS/MS PK observations in the TA
| Variable | Cohort 6, 100 mg | Cohort 7, 150 mg | ||
|---|---|---|---|---|
| Dose 1, n = 6 | Dose 2, n = 6 | Dose 1, n = 6 | Dose 2, n = 6 | |
| Participants with at least 1 sample > LLOQ, n (%) | 1 (17) | 0 (0) | 4 (67) | 3 (50) |
| Cmax | ||||
| Minimum (ng/ml) | BLQ | BLQ | BLQ | BLQ |
| Maximum (ng/ml) | 13.3 | BLQ | 25.8 | 37.2 |
| Tmax (postdose) | ||||
| Minimum (h) | 3 | NC | 3 | 3 |
| Maximum (h) | 3 | NC | 24 | 6 |
BLQ, below limit of quantification; Cmax, maximum measured plasma concentration; LC‐MS/MS, liquid chromatography‐mass spectrometry; LLOQ, lower limit of quantitation (10 ng/ml in this assay); NC, not calculated; PK, pharmacokinetic; TA, tibialis anterior; Tmax, time of the maximum measured plasma concentration.
Pharmacodynamics
Muscle Volume According to MRI
Increases in muscle volume of the injected muscle were observed after administration of ACE‐083 to both the RF and TA muscles, with effects in the contralateral noninjected muscle similar to placebo‐injected muscles. Percentage changes in right (injected) muscle volume for each cohort at 3 and 8 weeks after the last dose are shown in Figures 1 and 2. The maximum (mean ± SD) increase in muscle volume at 3 weeks after the last dose was 14.5% ± 4.5% in the RF (cohort 5, 200 mg × 2 doses) and 8.9% ± 4.7% in the TA (cohort 7, 150 mg × 2 doses). At the highest dose levels in both muscles at 8 weeks after the last dose, increases in muscle volume were observed, but the increases were not statistically significant.
Figure 1.
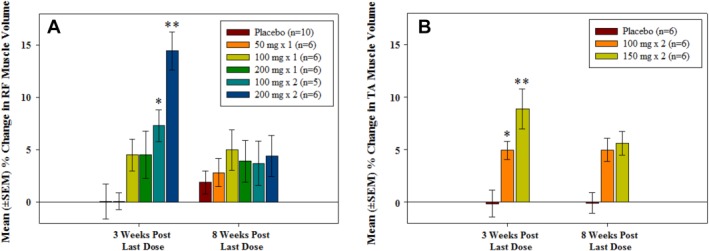
Mean ± SEM percentage change from baseline in muscle volume according to MRI in the right (ACE‐083‐injected) muscle at 3 weeks and 8 weeks after the last dose. (A) Rectus femoris. (B) Tibialis anterior. *P < 0.05, **P < 0.001, Dunnett's test vs. placebo.
Figure 2.
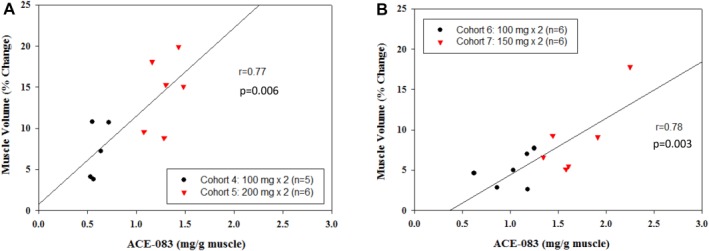
Correlation between right (ACE‐083‐injected) muscle volume increase at 3 weeks after last dose and ACE‐083 dosage per gram muscle (according to MRI for each participant). (A) Rectus femoris cohorts 4 and 5. (B) Tibialis anterior cohorts 6 and 7.
The dose of ACE‐083 relative to the size of the muscle was estimated for each participant by dividing the dose of ACE‐083 administered by the baseline muscle volume of the injected muscle determined by MRI for cohorts 4–7 (Fig. 2). Observed percentage change in muscle volume correlated with the estimated ACE‐083 dose within each muscle (milligrams ACE‐083/gram of muscle), with r = 0.77–0.78. Dose levels > 1.0 mg/g muscle were associated with increases in muscle volume exceeding 5%. No significant changes in intramuscular fat were observed.
Strength
No statistically significant changes in strength of the injected side were observed by either hand‐held dynamometry or fixed system. Although the TA is the primary muscle contributing to dorsiflexion, changes in dorsiflexion strength were not observed. For example, mean percentage changes from baseline for muscle strength fixed system measurements after multiple doses of ACE‐083 (100 mg × 2 and 150 mg × 2) administered in the TA of the right leg showed marked variability in both treated and untreated sides, with mean percentage changes on the treated side ranging from −2.75% to + 5.22%, and no consistent trends were observed compared with placebo.
DISCUSSION
The number of active and placebo‐treated participants in this study is considered appropriate for early‐stage clinical studies and was both feasible and sufficient to achieve the primary objective of the study. Single and multiple doses of ACE‐083 as a local muscle injection into the RF or TA muscle were generally safe and well tolerated. AEs considered related to treatment consisted primarily of events at the injection site occurring in both active‐ and placebo‐treated participants as well as transient pain in extremity, muscle twitching, and muscle tightness. PK data collected in this study support the localized therapeutic effect of ACE‐083 with little to no systemic exposure beyond 24 h postdose.
Muscle volume as assessed by MRI increased with escalating doses of ACE‐083 in the RF and TA compared with placebo‐treated participants. With only 2 doses administered 3 weeks apart, these increases reached maximum changes from baseline at 3 weeks after the last dose that have not previously been seen with other muscle agents. No change in intramuscular fat was observed; however, minimal intramuscular fat was noted on baseline MRI scans.
Significant changes in strength were not detected. This finding could be related to several factors. First, the participants included in this phase 1 study were healthy individuals with no muscle atrophy or strength deficits at baseline. Second, the RF muscle only accounted for approximately 13% of the total quadriceps muscle volume in these participants, making quantifiable improvements in knee extension strength unlikely. Finally, a maximum of 2 doses was administered; an extended duration of treatment and observation may be required to elicit a measurable improvement in strength.
In future studies with ACE‐083, many of these limitations will be addressed. For example, studies will be longer in treatment duration. In neuromuscular diseases in which atrophy and weakness of specific muscles can have a devastating impact on a patient's daily activities and overall quality of life, substantial increases in muscle volume may be associated with quantifiable improvements in strength and function. In diseases with elevated fat fraction,7, 8 ACE‐083 treatment may reasonably be expected to reduce intramuscular fat.9
The concentrated effect of locally acting ACE‐083 resulted in an amount of muscle growth not previously achieved. For comparison, GDF8 (myostatin)‐directed compounds (e.g., LY2495655, stamulumab [MYO‐029], domagrozumab [PF‐06252616], BMS‐986089) have led to muscle volume increases in the 1%–6% range.7, 8, 9, 10 Studies with muscle volume changes in this range were typically not associated with clinically meaningful changes in strength or function, although some studies did show trends favoring improvement.
A phase 2 trial evaluating bimagrumab, an activin receptor antibody, in patients with sporadic inclusion body myositis (n = 14) showed a mean increase from baseline in lean mass of approximately 5%–6%, which was associated with an approximately 7% increase from baseline in 6‐min walk distance in the treated group.11 However, a phase 3 trial using lower dosage levels (n = 251) showed a mean increase from baseline in lean mass of approximately 2.8% at the highest dosage level and an approximate corresponding mean increase from baseline in 6‐min walk distance of <5%.12, 13 This suggests that the threshold to achieve clinical benefit is likely greater than 5% and that ACE‐083, in comparison to other therapies, has a greater likelihood of achieving this threshold in patients. The ability of ACE‐083 to bind multiple ligands, including GDF8, GDF11, and activins as well as acting locally at high concentrations in the muscle (where these ligands act in largely a paracrine/autocrine manner), may account for the greater increases in muscle volume observed here.14
The promising degree of muscle growth observed in this phase 1 healthy volunteer study supports further evaluation of the impact of ACE‐083 on dystrophic or atrophic muscle. Previous studies in Duchenne muscular dystrophy with a systemic myostatin inhibitor15 have suggested that increases in lean body mass can be achieved in dystrophic muscle, whereas the present study suggests that changes are possible even in healthy muscle, as may be found in primarily neuropathic diseases. Two studies of ACE‐083 are currently ongoing and actively enrolling patients with facioscapulohumeral muscular dystrophy (NCT02927080), and Charcot–Marie–Tooth disease (NCT03124459).
The authors thank Steven A. Greenberg for providing medical review assistance as part of the safety review team; Sharon Wagner (Celerion) and VirtualScopics for study conduct, participant recruitment and treatment, and data acquisition; ICON, Algorithme, and qPharmetra for antidrug antibody and pharmacokinetic data analyses; Bryan Health for facilities and personnel; and Susan Pandya, MD, Ashley Leneus, Amelia Pearsall, Brian Vidal, Xiaosha Zhang, Jade Sun, and Carrie Barron from Acceleron Pharma for trial management, data management, statistical analysis, and writing support.
Ethical Publication Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Supporting Information
Supporting Information
Conflicts of Interest: K. M. Attie, C. E. Glasser, B. Miller, and M. L. Sherman are all employees of Acceleron Pharma. D. Wilson was an employee of Acceleron Pharma at the time of the study. M. R. Gartner was an employee of Celerion at the time of the study.
REFERENCES
- 1. Rodino‐Klapac LR, Haidet AM, Kota J, Handy C, Kaspar BK, Mendell JR. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 2009;39(3):283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walker RG, Poggioli T, Katsimpardi L, Buchanan SM, Oh J, Wattrus S, et al Biochemistry and biology of GDF11 and myostatin: similarities, differences, and questions for future investigation. Circ Res 2016;118(7):1125–1141; discussion 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pearsall RS, Widrick J, Cotton E, Sako D, Liu J, Davies M, et al ACE‐083 increases muscle hypertrophy and strength in C57BL/6 mice. Neuromuscul Disord 2015;25(Suppl 2):S218. [Google Scholar]
- 4. Mulivor A, Sako D, Cannell M, Wallner S, Hevron K, Steeves R, et al A modified cysteine knot ligand trap of the TGFβ superfamily, ACE‐083, increases muscle mass locally in a mouse model of Duchenne muscular dystrophy. Neuromuscul Disord 2014;24(9–10):878. [Google Scholar]
- 5. Pearsall RS, Widrick J, Sako D, Davies M, Heveron K, Castonguay R, et al ACE‐083, a locally‐acting TGF‐β superfamily ligand trap, increases muscle mass and strength in a mouse model of ALS. Poster presented at: MDA Clinical Conference; March 20–23, 2016; Arlington, VA.
- 6.Li J, Cannell M, Suragani RNVS, Pearsall RS, Kumar R. ACE‐083, a locally acting gdf/activin ligand trap, augments dorsiflexor muscle function in a murine model of Charcot–Marie–Tooth (CMT) disease. Poster presented at: Peripheral Nerve Society; July 11, 2017; Sitges‐Barcelona, Spain.
- 7. Janssen BH, Voet NB, Nabuurs CI, Kan HE, de Rooy JW, Geurts AC, et al Distinct disease phases in muscles of facioscapulohumeral dystrophy patients identified by MR detected fat infiltration. PLoS One 2014;9:e85416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morrow JM, Sinclair CD, Fischmann A, Machado PM, Reilly MM, Yousry TA, et al MRI biomarker assessment of neuromuscular disease progression: a prospective observational cohort study. Lancet Neurol 2016;15:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Becker C, Lord SR, Studenski SA, Warden SJ, Fielding RA, Recknor CP, et al Myostatin antibody (LY2495655) in older weak fallers: a proof‐of‐concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol 2015;3(12):948–957. [DOI] [PubMed] [Google Scholar]
- 10. Binks M. Domagrozumab (PF‐06252616): a potential treatment for DMD. Presented at: Parent Project Muscular Dystrophy Connect Conference; June 26–29, 2016; Orlando, FL.
- 11. Jacobsen L. BMS‐986089: an antimyostatin adnectin targeting Duchenne muscular dystrophy. Presented at: Parent Project Muscular Dystrophy Connect Conference; June 26–29, 2016; Orlando, FL.
- 12. Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, et al A phase I/II trial of MYO‐029 in adult subjects with muscular dystrophy. Ann Neurol 2008;63(5):561–571. [DOI] [PubMed] [Google Scholar]
- 13. Amato AA, Sivakumar K, Goyal N, David WS, Salajegheh M, Praestgaard J, et al Treatment of sporadic inclusion body myositis with bimagrumab. Neurology 2014;83(24):2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amato A, Badrising U, Benveniste O, Needham M, Chinoy H, Wu M, et al RESILIENT: a randomized, double‐blind, placebo‐controlled study of bimagrumab in patients with sporadic inclusion body myositis. Arthritis Rheumatol 2016;68(Suppl 10). [Google Scholar]
- 15. U. S. National Library of Medicine . Efficacy and Safety of Bimagrumab/BYM338 at 52 Weeks on Physical Function, Muscle Strength, Mobility in sIBM Patients (RESILIENT). ClinicalTrials.gov. Identifier NCT01925209. Available at: https://clinicaltrials.gov/ct2/show/NCT01925209?term=NCT01925209&rank=1. Updated April 2017. Accessed March 1, 2018.
- 16. Latres E, Mastaitis J, Fury W, Miloscio L, Trejos J, Pangilinan J, et al Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun 2017;8:15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Campbell C, McMillan HJ, Mah JK, Tarnopolsky M, Selby, K , McClure T, et al Myostatin inhibitor ACE‐031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo‐controlled clinical trial. Muscle Nerve 2017;55(4):458–464. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Supporting Information
Supporting Information