

Abstract
A covalently bound flavin cofactor is predominant in the succinate‐ubiquinone oxidoreductase (SQR; Complex II), an essential component of aerobic electron transport, and in the menaquinol‐fumarate oxidoreductase (QFR), the anaerobic counterpart, although it is only present in approximately 10 % of the known flavoenzymes. This work investigates the role of this 8α‐N3‐histidyl linkage between the flavin adenine dinucleotide (FAD) cofactor and the respiratory Complex II. After parameterization with DFT calculations, classical molecular‐dynamics simulations and quantum‐mechanics calculations for Complex II:FAD and Complex II:FADH2, with and without the covalent bond, were performed. It was observed that the covalent bond is essential for the active‐center arrangement of the FADH2/FAD cofactor. Removal of this bond causes a displacement of the isoalloxazine group, which influences interactions with the protein, flavin solvation, and possible proton‐transfer pathways. Specifically, for the noncovalently bound FADH2 cofactor, the N1 atom moves away from the His‐A365 and His‐A254 residues and the N5 atom moves away from the glutamine‐62A residue. Both of the histidine and glutamine residues interact with a chain of water molecules that cross the enzyme, which is most likely involved in proton transfer. Breaking this chain of water molecules could thereby compromise proton transfer across the two active sites of Complex II.
Keywords: density functional calculations, electron transport, enzyme catalysis, flavins, molecular dynamics
Introduction
Flavoenzymes are enzymes that employ flavin cofactors, such as flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN), and are ubiquitously found in Nature because they catalyze a wide range of biological redox reactions. Hundreds of flavoproteins have been reported so far, with a wide range of functions, such as dehydrogenases, monooxygenases, halogenases, and oxidases. Most of these proteins bind the flavin cofactor in a noncovalent manner. However, about 10 % have been shown to contain a covalently bound flavin cofactor with one of five possible bond types that are linked:1 1) at the flavin C8M atom (see Figures 1 and 2); 2) through histidyl and tyrosyl residues; 3) through cysteinyl side chains; 4) at the flavin C6M atom through a cysteinyl side chain; 5) at two positions. Such a flavin:protein covalent bond was first identified in 1955 in the seminal work of Singer and co‐workers on mammalian succinate dehydrogenase.2 Since this report, similar flavin:protein bonds have been identified in different enzymes, and it is now believed that the bond is formed in a self‐catalytic process.1 The actual catalytic function of the bond is still a matter of debate. It has been proposed that the flavin:protein covalent bond could: 1) stabilize the protein structure;1b, 3 2) promote the tight association of different subunits;4 3) prevent the loss of loosely bound flavin cofactors in membrane proteins;1d 4) modulate the redox potential of the flavin microenvironment and facilitate electron‐transfer reactions;3a, 5 5) contribute to substrate binding, as in the case of the cysteinyl linkage.1c
Figure 1.

A) Reactions catalyzed by SQR. B) Possible routes for FAD cofactor oxidation/reduction. UQ=ubiquinone, UQH2=ubiquinol.
Figure 2.

Model systems used for the parameterization of the flavonoid: a) reduced system with a 8α‐N3‐histidyl bond; b) oxidized system with a 8α‐N3‐histidyl bond; c) reduced system without a 8α‐N3‐histidyl bond; d) oxidized system without a 8α‐N3‐histidyl bond.
Regardless of the role of the covalent bond, the binding of the flavin unit is not a requirement for catalysis in most cases. However, the catalytic turnover is more efficient when the flavin cofactor is covalently bound.1a, 5a Interestingly, the FAD:protein covalent bond is present in most of the studied prokaryotic and eukaryotic membrane‐bound succinate‐ubiquinone oxidoreductase (SQR, Complex II) and menaquinol‐fumarate oxidoreductase (QFR) proteins. The exceptions are some soluble fumarate reductases homologous of the Complex II flavoprotein subunit, but these proteins cannot undergo succinate oxidation (i.e., yeast, bacteria from the genus Shewanella, and unicellular parasites).6
Complex II is a component of the aerobic electron‐transport chain, and QFR is a homologue of Complex II in anaerobic respiration. These multifunctional proteins7 normally catalyze enzymatic reactions in vivo in opposite directions. Complex II catalyzes a two‐electron two‐proton transfer between succinate to fumarate (Figure 1 A) and quinone to quinol, whereas QFR catalyzes the reverse reactions. However, QFR when expressed under aerobic conditions can oxidize succinate and reduce the quinone to quinol, whereas SQR acts as a menaquinol‐fumarate reductase under anaerobic conditions.
These enzymes are composed of four subunits and contain the following prosthetic groups: FAD, three iron–sulfur clusters, and a heme‐b cofactor. For Complex II, the two larger subunits, namely, the flavoprotein (Fp) and iron–sulfur protein (Ip) subunits, comprise the soluble succinate dehydrogenase domain (present in the mitochondrial matrix). The other two subunits comprise the transmembrane cytochrome bL (CybL) and cytochrome bS (CybS; present at that inner mitochondrial membrane). In most Complex II enzymes, these subunits coordinate a low‐spin hexa‐coordinated heme‐b cofactor with two histidine residues (bis‐His) as axial ligands. The conversion of succinate into fumarate (or the reverse) occurs at the interface of the two domains of the soluble flavoprotein subunit. The ubiquinol and ubiquinone interconversion (or the reverse) occurs in the transmembrane cytochrome b region.8
The reduction of FAD in aqueous solution involves two sequential one‐electron‐ and one‐proton‐transfer reactions. On the other hand, FAD reduction in enzymes can occur by two possible routes: Either through a one‐electron reduction that produces a radical semiquinone or a full two‐electron reduction that yields a hydroquinone directly (all the possible species are depicted in Figure 1 B).
The catalytic mechanism of succinate oxidation is not well described; however, in accordance with the fumarate reduction, the mechanism involves a two‐electron transfer (with hydride transfer followed by the proton). Thus, it is possible that the reaction mechanism could be the same, but in the reverse order. FADH2 should then be reoxidized to FAD and the electrons are transferred through Complex II subunits (through the chain of redox centers: FAD, [Fe2S2], [Fe4S4], and [Fe3S4] clusters) to become involved in the reactant of the second reaction (Figure 1 A).
Previously, we computed the relative energies for the possible spin states of the reduced and oxidized iron–sulfur clusters.9 The [Fe2S2]2+ cluster has a total spin of zero (S=0) and the reduced cluster [Fe2S2]+ has S=1/2 (isoenergetic with S=9/2). The ground state for the [Fe4S4] oxidized cluster was also found to have a spin state of S=0, and the reduced cluster has S=1/2. The [Fe3S4]0 cluster has a spin state of S=2, whereas we found two isoenergetic ground states for the [Fe3S4]+ cluster of S=3/2 and S=5/2. The environment, and more specifically the position and orientation of the amide hydrogen bonds to the sulfur atoms, were shown to affect the stabilization of the reduced clusters. Through the use of these iron–sulfur clusters, the chain of redox centers can extend over 40 Å through Complex II.
Regarding the proton translocation to the ubiquinone molecule, a proton‐uptake pathway entirely made of water molecules that interact with the conserved residues Lys‐230B, Asp‐95C, and Glu‐101C was identified in the native structure of the prokaryotic counterpart (PDB 1NEK).8a, 10 This water channel crosses the membrane anchor and arrives at the ubiquinone binding site.8a It is probable that the eukaryotic complex has also a water channel for proton uptake.
In the QFR Complex, a mutation (to serine (Ser), cysteine (Cys), or tyrosine (Tyr)) of the histidine residue involved in the FAD:protein covalent bond leads to a decrease in the ability to reduce fumarate (>70 %) and a complete loss of succinate dehydrogenase activity. Mutation to arginine leads to a loss of fumarate reductase activity.11 In the case of SQR, a mutation (to serine) of the histidine residue involved in the FAD:protein covalent bond leads to loss of succinate dehydrogenase activity, but the enzyme retained some fumarate reductase activity. All mutants retained the noncovalently bound FAD.12
Therefore, the current evolutionary view is that membrane‐bound enzymes have evolved from a soluble fumarate reductase that contained noncovalent bound FAD and was thus incapable of catalyzing succinate oxidation. Subsequently, the enzyme incorporated iron–sulfur clusters and became associated with the membrane through the transmembrane domains, which provide a site for interaction with ubiquinone. Ultimately, the FAD cofactor has become covalently bound to the protein, thus allowing succinate oxidation.
In this study, we obtained molecular mechanics (MM) parameters for several of the systems involved in these flavoproteins by using DFT calculations and performed classical molecular‐dynamics (MD) simulations of Complex II:FAD and Complex II:FADH2 systems. We performed these operations for the normal histidyl:cofactor covalent bond and a non‐natural histidyl:cofactor noncovalent bond to obtain atomic‐level insight into the importance of the bond. Our results show that the covalently bound histidine residue on its own does not seem to provide the FAD cofactor with new properties. However, this amino acid allows FAD to adopt a different position at the active site, thus engaging in different interactions with the protein, which has consequences in regard to the possible proton and electron pathways across the enzyme.
Results and Discussion
FAD and FADH2 charge parameterization
We performed DFT calculations on the oxidized and two‐electron reduced flavin ring, with and without the 8α‐N3‐histidyl bond (Figure 2). The calculated charges are given in Table S1 (see the Supporting Information). Looking at these small DFT models, the covalently bound histidine residue does not seem to alter the electrostatic properties of the flavin ring significantly, which means that the protein environment must play an important role in the modulation of the redox potential.
FAD and FADH2 interactions with Complex II
For Complex II with the 8α‐N3‐histidyl FAD bond (Complex II:FAD:covalent), the pattern of interactions observed in the MD simulation is quite similar to the pattern found in the crystal structure. This finding gives credence to our parameterization procedure, which we can trust to give reliable interactions within the classical MD simulations. The isoalloxazine ring of riboflavin interacts with the His‐A365, Leu‐A264, and threonine (Thr)‐A225 side chains in Complex II. The Ser‐A414 residue and the backbone atoms of the alanine (Ala)‐A60, Ala‐A61, and Gln‐A62 residues are also close to the ring. The major difference is that Arg‐A409 is close to Arg‐A298 in the crystal structure (distance between the Arg‐A298‐NE and Arg‐A409‐NH2 atoms is 2.96 Å), whereas the distance between these two residues increases according to the MD simulations. In one of the MD simulations, Arg‐A409 moves toward the N1 atom of the flavin ring (the average distance during the last 20 ns of one the simulation replicas is d=3.5(±0.57) Å; see Table S2 in the Supporting Information), whereas in the other MD simulation this residue moves further away and starts to interact with Tyr‐A366 (d=10.25(±0.47) Å; see Table S2 in the Supporting Information). The Arg‐A298 residue also forms a hydrogen bond with Glu‐A267 (d=3.37(±0.81) and 3.08(±0.31) Å; see Table S3 in the Supporting Information). The His‐A365 residue is close to the pteridine‐2,4‐dione moiety in the FAD cofactor and close to His‐A254. The Glu‐A398 residue interacts with a hydroxy group of trihydroxypentane (d=2.49(±0.07) and 2.44(±0.06) Å; see Table S2 in the Supporting Information). There are 2–6 water molecules in the vicinity of the flavin ring.
We observed good outcomes in the MD simulations for the FADH2 cofactor as well. In the simulation of Complex II with the 8α‐N3‐histidyl:FADH2 covalent bond (Complex II:FADH2:covalent), we see a similar pattern of interactions to the pattern described for the Complex II:FAD:covalent system. The most relevant difference is that the Gln‐A62 side chain forms a hydrogen bond with N5 of FADH2 (2.7 Å; Figure 3) and His‐A365 is closer to N5 (3.5 Å) in the Complex II:FADH2:covalent system, whereas Gln‐A62 is rotated and lies far away from the cofactor in the Complex II:FAD:covalent system. Because of the protonation of N5 in FADH2, a new hydrogen‐bonding possibility is present, which benefits the Gln‐A62 residue. Through the rotation of the Gln‐A62 side chain, extra space is created for water molecules, which enables better organization of these molecules in the vicinity of FADH2 relative to FAD. If we look to the complete protein, we can observe that there is a chain of water molecules that connects the FADH2 cofactor to the conserved residue Glu‐B67, which is close to the [Fe2S4] cluster and continues through the molecule (Figure 4). A similar chain of water molecules was observed for the prokaryotic enzyme and was proposed to be the most probable proton‐uptake pathway.8a
Figure 3.
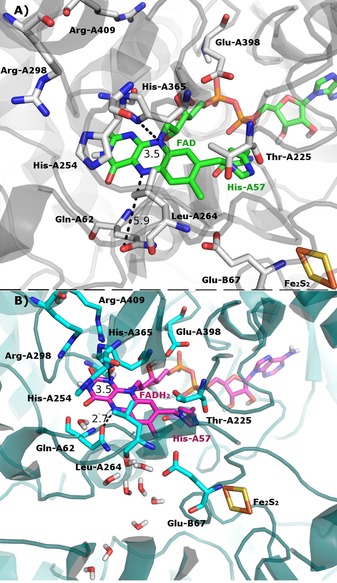
A) Complex II with the 8α‐N3‐ histidyl FAD bond. B) Complex II with the 8α‐N3‐ histidyl FADH2 bond. As it can be seen, there is an ordered chain of water molecules above the isoallxazine ring for the FADH2 system. Arg=arginine, Gln=glutamine, Glu=glutamate, His=histidine, Leu=leucine, Thr=threonine.
Figure 4.

The chain of water molecules that cross the enzyme in Complex II with the 8α‐N3‐ histidyl FADH2 bond system. The Glu‐A267 and Glu‐B67 residues seem to be important for the water‐molecule organization. For simplicity, all hydrogen atoms are not shown, except for the nonpolar hydrogen atom of FADH2 and those of the water molecules.
For Complex II, the water channel goes through most of the protein, and histidine residues close to this channel might also be involved in proton transfer. Another hypothesis, which results from the high proximity of the water channel to the iron–sulfur clusters, is that the oxidation state of the iron–sulfur clusters regulate the protein conformation and consequently the water channel. For other iron–sulfur proteins, it was shown that oxidized metal clusters are poorly solvated relative to reduced metal clusters.13
In solution, the fully reduced FAD and the semiquinone should be mainly deprotonated (i.e., FADH− and FAD.−). However, it has been shown that, in enzymes containing the FAD cofactor, the protein environment modulates the pK a values either through solvent accessibility or by specific interactions with protein residues, which makes the pK a values much higher.14 To the best of our knowledge, the FAD pK a value in Complex II has not been reported. In this enzyme, FAD is reduced by succinate to FADH2 and then should be reoxidized to FAD to reduce ubiquinone to ubiquinol (Figure 1 A).
From the analysis of the structure of the Complex II:FADH2:covalent system, we hypothesized that proton abstraction from the N1 atom should involve transfer to His‐A365. Afterwards, another histidine, His‐A254, is close to His‐A365, and could accept this proton. The His‐A254 residue then interacts with a chain of water molecules. For the second proton transfer, the most probable hypothesis is the direct transfer to the water molecules. In our MD simulations of the fully reduced FADH2 system, we see that the N5 atom forms a hydrogen bond with the Gln‐A62 residue, which can be rearranged in an intermediate structure. The conserved glutamate‐A67 residue, which is close to the [Fe2S4] cluster, also interacts with the putative chain of water molecules. Glutamate can organize the chain of water molecules or can directly participate in the proton transfer.
Covalent‐bond modulation of the interactions between FAD/FADH2 and the SQR enzyme
To better understand how the histidyl:cofactor covalent bond is able to modulate the cofactor:Complex II interactions, we also conducted MD simulations of Complex II:FAD and Complex II:FADH2 systems without the histidyl:cofactor covalent bond (Complex II:FAD:noncovalent; Complex II:FADH2:noncovalent). For the Complex II:FAD:noncovalent system, we observed a rotational and translational movement of the FAD isoalloxazine ring relative to the disposition described above for the MD simulations with the normal histidyl:cofactor covalent bond. The His‐A57 residue does not interact with the C8M atom of FAD, but instead is hydrogen bonded to Glu‐A398 (d(Glu‐A398‐OE2⋅⋅⋅His57‐NE2)=2.99(±0.16) and 3.06(±0.18) Å; Table S3 in the Supporting Information), which seems to explain the observed high mobility of the isoalloxazine ring.
For the Complex II:FADH2:noncovalent system (Figure 5 A), His‐A57 is also now hydrogen bonded to Glu‐A398, instead of interacting with the FADH2 isoalloxazine ring (d(Glu‐A398‐OE2⋅⋅⋅His57‐NE2)=2.97(±0.16) and 3.10(±0.23) Å; Table S3 in the Supporting Information). However, the N5 atom of the isoalloxazine ring is now hydrogen bonded to the backbone of Gln‐A62 for this system, whereas the phenyl ring is involved in a strong T‐shaped stacked interaction with His‐A365. Moreover, Arg‐A409 forms a hydrogen bond with the hydroxy group in Ser‐A414 (d(Arg‐A409‐NH2⋅⋅⋅Ser‐A414‐OG)=3.31(±0.24) and 3.13(±0.53); Table S3 in the Supporting Information). The Ser‐A414 residue also interacts with the O2 atom in FADH2 (d(Ser‐A414‐OG⋅⋅⋅FAD‐O2)=4.03(±0.53) and 3.74(±0.31) Å; Table S2 in the Supporting Information). When looking at water molecules around the structures, we can observe that the 8α‐N3‐histidyl moiety in FADH2 has an average of 2.2 water molecules around the N3 atom and 0.8 molecules around the N1 atom, whereas the noncovalently bound FADH2 only has an average of 1.35 and 0.11 water molecules around the same atoms, respectively.
Figure 5.

Superposition of the Complex II:FADH2 model with and without the covalent bond (purple and yellow, respectively). Two different orientations (i.e., A1 and A2) are shown. As can be seen, the absence of the covalent bond leads to a completely different position of the isoalloxazine ring. The N1 atom is far from the His‐A365 residue, and the N5 atom establishes a hydrogen bond with the backbone of Gln‐A62. There is no continuity with the chain of water molecules close to Glu‐B67, and Leu‐A264 is now in the position previously occupied by Gln‐A62. Without the covalent bond, His‐A57 also moves into a new position.
In summary, the absence of the covalent bond for the Complex II:FADH2:noncovalent interaction leads to the following consequences: 1) His‐A57 moves away from FADH2 and starts to interact with Glu‐A398; 2) the cofactor isoalloxazine ring moves closer to the backbone of Gln‐A62 and the N5 atom moves away from His‐A365, so that there is no connection with the chain of water molecules close to the [Fe2S4] cofactor and Glu‐B67; 3) the covalently bound FADH2 is better solvated than the structure without the covalent bond. Therefore, the MD analysis shows that there seems to be a greater degree of stabilization of the 8α‐N3‐histidyl linkage in the FADH2 cofactor by the protein environment and water molecules and there is better access to the chain of water molecules that connects the two active sites.
QM calculations
QM clusters models were built based on reference structures of the MD simulations of FADH2 covalently and noncovalently bound to the histidine residue. These models correspond to the MD structures with the lowest root‐mean square deviation in relation to the MD average structure. The models were geometry optimized by using the ONIOM method15 with B3LYP and PM6.16. We observed the Arg‐A409, His‐A365, and Ser‐A414 residues are closer to the N1 atom of the flavin unit in the Complex II:FADH2:covalent model. In the Complex II:FADH2:noncovalent model, His‐A365 and Arg‐A409 are substantially further away from N1 and interact with each other. Moreover, there are very few water molecules near the flavin moiety, contrary to the Complex II:FAD H2: covalent model (Figure 6).
Figure 6.

ONIOM QM models of the complexes with FADH2 covalently bound (a) and unbound (b) to the histidine residue. The close‐up image of the cofactors depicts the interactions between the isoalloxazine rings and the protein residues and water molecules. Electrostatic‐potential maps of the isoalloxazine rings with the His‐A57 side chain in the field of the remaining protein and solvent atoms of the model (from −5×10−2 to +5×10−2).
Previously, covalent coupling of a flavin unit had been found to increase the midpoint potential significantly for several enzymes.1a Therefore, it is possible than the conformational changes promoted by the covalent bond lead to better stabilization of the reduced flavin. In fact, when we compare the FAD molecules in the two models we can see that the Complex II:FADH2:covalent establishes interactions with protein side chains (i.e., Gln‐A62 and Arg‐A409) and water molecules, whereas the noncovalent Complex mainly establishes interactions with the protein backbone (Figure 6). Accordingly, in the electrostatic‐potential maps of the isoalloxazine rings, taken from the ONIOM calculations and thus in the field of the remaining protein and solvent molecules, we can observe greater positive charge at the N1 and N5 hydrogen atoms for the Complex II:FADH2:covalent model (Figure 6).
Conclusion
We have performed MD simulations of Complex II:FAD and Complex II:FADH2 systems with explicit solvent water molecules, not only with the normal histidyl:cofactor covalent bond, but also with a non‐natural histidyl:cofactor noncovalent bond.
Our results have shown that the covalently bound histidine residue on its own does not seem to provide the FAD cofactor with new properties. However, this residue allows the isoalloxazine ring of the cofactor to adopt a strict active‐center disposition, which influences the interactions with the protein, flavin solvation, and possible proton‐transfer pathways because the absence of the bond strongly compromises the interaction of the FADH2 cofactor with two histidine residues (i.e., His‐A365 and His‐A254) and the water molecules in a water chain that connects the two active sites.
Consequently, the protein environments seem to have evolved to modulate water accessibility to the FADH2 cofactor to control flavin stabilization and proton transfer between the two active sites of Complex II.
Computational methods
DFT calculations
The flavin rings, bound or unbound to histidine A57, were geometry optimized with the program ADF.17 All the systems were treated with the exchange‐correlation functionals OPBE.18 Geometry optimizations and energy calculations were performed with the TZP basis set18b and with the Cosmo model19 with a dielectric of value of 4 to represent the protein environment.
Model preparation
Pig SQR structure (pdb code: 1ZOY; 2.4 Å) was used as the starting structure.8b Chains C and D were inserted within a preoptimized 384 2‐oleoyl‐1‐palmitoyl‐sn‐glycero‐3‐phosphoethanolamine (POPE) bilayer. TIP3P water molecules were added to obtain an octrahedral box. The system contained a total of 151 411 atoms. Charges and bonded parameters were obtained for the cofactors when they were absent in the Amber force‐field parameters set. The iron–sulfur cofactors parameters were taken from reference 9. The parameters used for the lipid molecules were the Berger parameters, which were adapted to be used with the Amber program.20 For the systems without the covalent bond, we only modeled the soluble chains due to the large distance between the FAD unit and the transmembrane domains.
MD simulations
MD simulations were performed with the GPU implementation of Amber1421 using the ff99SB parameter set22 for the protein. An initial energy minimization was performed followed by an equilibration of 1000 ps to slowly heat the system from 0 to 300 K. The equilibration was performed in an NVT ensemble by using the Langevin dynamics with small restraints on the protein atoms (10 kcal mol−1). Production simulations were carried out at 300 K in the NPT ensemble using Langevin dynamics with a collision frequency of 1.0 ps−1. Constant‐pressure periodic‐boundary conditions were imposed with an average pressure of 1 atm. Isotropic position scaling was used to maintain pressure with a relaxation time of 2 ps. The time step was set at 2 fs. Shake constraints were applied to all bonds involving hydrogen atoms. The particle mesh Ewald (PME) method was used to calculate electrostatic interactions with a cut‐off distance of 8 Å.23 The total time of each simulation was 50 ns. Two replicas were performed for each simulation. The total combined time of all simulations was 400 ns.
QM calculations
The cluster models were built from the lowest root‐mean square deviation to the average structure from the simulations of FADH2 covalently bound and unbound to the histidine residue. The models contained the isoalloxazine, trihydroxypentane, and diphosphate groups of FADH2, the [Fe2S2] cluster and corresponding cysteinyl ligands, all the protein residues within a radius of 10 Å from the N5 atom in the flavin unit, and the closest water molecules.
The system was divided into layers according to the ONIOM methodology,15 with the high‐level layer comprising the isoalloxazine ring and the covalently bound histidine residue treated with the exchange correlation functional B3LYP and the basis set STO‐3G and 6‐31G* and the remaining atoms with the semiempirical PM6.16 The structures were geometry optimized by using the Gaussian 09 software.24
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The following organizations are thanked for financial support: A.T.P.C. is grateful to the Fundação para a Ciência e Tecnologia (FCT) for the grant IF/01272/2015. AGAUR for fellowship 2010 BP_B00238, the Ministerio de Ciencia e Innovación (MICINN, project number CTQ2011‐25086/BQU), and the DIUE of the Generalitat de Catalunya (project number 2009SGR528 and Xarxa de Referència en Química Teòrica i Computacional). Financial support from MICINN (Ministry of Science and Innovation, Spain) and the FEDER fund (European Fund for Regional Development) was provided by grant UNGI08‐4E‐003. With the support of the Secretary for Universities and Research of the Ministry of Economy and Knowledge of the Government of Catalonia and the Cofund programme of the Marie Curie Actions of the 7th R&D Framework Programme of the European Union. D.F.A.R.D. acknowledges the financial support from INVEST NI RD0314092. The authors are also grateful to the computer resources, technical expertise, and assistance provided by the Barcelona Supercomputing Center—Centro Nacional de Supercomputación.
D. F. A. R. Dourado, M. Swart, A. T. P. Carvalho, Chem. Eur. J. 2018, 24, 5246.
References
- 1.
- 1a. Heuts D. P., Scrutton N. S., McIntire W. S., Fraaije M. W., FEBS J. 2009, 276, 3405–3427; [DOI] [PubMed] [Google Scholar]
- 1b. Heuts D. P., Winter R. T., Damsma G. E., Janssen D. B., Fraaije M. W., Biochem. J. 2008, 413, 175–183; [DOI] [PubMed] [Google Scholar]
- 1c. Huang C. H., Winkler A., Chen C. L., Lai W. L., Tsai Y. C., Macheroux P., Liaw S. H., J. Biol. Chem. 2008, 283, 30990–30996; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Mewies M., McIntire W. S., Scrutton N. S., Protein Sci. 1998, 7, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ackrell B. A., Kearney E. B., Singer T. P., Methods Enzymol. 1978, 53, 466–483. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Caldinelli L., Iametti S., Barbiroli A., Fessas D., Bonomi F., Piubelli L., Molla G., Pollegioni L., Protein Sci. 2008, 17, 409–419; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Heuts D. P., van Hellemond E. W., Janssen D. B., Fraaije M. W., J. Biol. Chem. 2007, 282, 20283–20291; [DOI] [PubMed] [Google Scholar]
- 3c. Nandigama R. K., Edmondson D. E., J. Biol. Chem. 2000, 275, 20527–20532; [DOI] [PubMed] [Google Scholar]
- 3d. Caldinelli L., Iametti S., Barbiroli A., Bonomi F., Fessas D., Molla G., Pilone M. S., Pollegioni L., J. Biol. Chem. 2005, 280, 22572–22581. [DOI] [PubMed] [Google Scholar]
- 4. Eschenbrenner M., Chlumsky L. J., Khanna P., Strasser F., Jorns M. S., Biochemistry 2001, 40, 5352–5367. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Quaye O., Cowins S., Gadda G., J. Biol. Chem. 2009, 284, 16990–16997; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Motteran L., Pilone M. S., Molla G., Ghisla S., Pollegioni L., J. Biol. Chem. 2001, 276, 18024–18030; [DOI] [PubMed] [Google Scholar]
- 5c. Efimov I., Cronin C. N., McIntire W. S., Biochemistry 2001, 40, 2155–2166; [DOI] [PubMed] [Google Scholar]
- 5d. Fraaije M. W., van den Heuvel R. H., van Berkel W. J., Mattevi A., J. Biol. Chem. 1999, 274, 35514–35520. [DOI] [PubMed] [Google Scholar]
- 6. Cecchini G., Annu. Rev. Biochem. 2003, 72, 77–109. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Guest J. R., J. Gen. Microbiol. 1981, 122, 171–179; [DOI] [PubMed] [Google Scholar]
- 7b. Maklashina E., Berthold D. A., Cecchini G., J. Bacteriol. 1998, 180, 5989–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Horsefield R., Yankovskaya V., Sexton G., Whittingham W., Shiomi K., Omura S., Byrne B., Cecchini G., Iwata S., J. Biol. Chem. 2006, 281, 7309–7316; [DOI] [PubMed] [Google Scholar]
- 8b. Sun F., Huo X., Zhai Y., Wang A., Xu J., Su D., Bartlam M., Rao Z., Cell 2005, 121, 1043–1057. [DOI] [PubMed] [Google Scholar]
- 9. Carvalho A. T., Swart M., J. Chem. Inf. Model. 2014, 54, 613–620. [DOI] [PubMed] [Google Scholar]
- 10. Yankovskaya V., Horsefield R., Tornroth S., Luna-Chavez C., Miyoshi H., Leger C., Byrne B., Cecchini G., Iwata S., Science 2003, 299, 700–704. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Doherty M. K., Pealing S. L., Miles C. S., Moysey R., Taylor P., Walkinshaw M. D., Reid G. A., Chapman S. K., Biochemistry 2000, 39, 10695–10701; [DOI] [PubMed] [Google Scholar]
- 11b. Blaut M., Whittaker K., Valdovinos A., Ackrell B. A. C., Gunsalus R. P., Cecchini G., J. Biol. Chem. 1989, 264, 13599–13604. [PubMed] [Google Scholar]
- 12. Robinson K. M., Rothery R. A., Weiner J. H., Lemire B. D., Eur. J. Biochem. 1994, 222, 983–990. [DOI] [PubMed] [Google Scholar]
- 13. Sulpizi M., Raugei S., VandeVondele J., Carloni P., Sprik M., J. Phys. Chem. B 2007, 111, 3969–3976. [DOI] [PubMed] [Google Scholar]
- 14. Bhattacharyya S., Stankovich M. T., Truhlar D. G., Gao J., J. Phys. Chem. A 2007, 111, 5729–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Svensson M., Humbel S., Froese R. D. J., Matsubara T., Sieber S., Morokuma K., J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar]
- 16. Stewart J. J., J. Mol. Model. 2007, 13, 1173–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. te Velde G., Bickelhaupt F. M., Baerends E. J., Fonseca Guerra C., van Gisbergen S. J. A., Snijders J. G., Ziegler T., J. Comput. Chem. 2001, 22, 931–967; [Google Scholar]
- 17b. Fonseca Guerra C., Snijders J. G., te Velde G., Baerends E. J., Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar]
- 18.
- 18a. Hoe W.-M., Cohen A. J., Handy N. C., Chem. Phys. Lett. 2001, 341, 319–328; [Google Scholar]
- 18b. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- 19. Klamt A., Schuurmann G., J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar]
- 20. Cordomí A., Caltabiano G., Pardo L., J. Chem. Theory Comput. 2012, 8, 948–958. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a.Amber, D. A. Case, T. A. Darden, I. T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, University of California, San Francisco, 2008;
- 21b. Götz A. W., Williamson M. J., Xu D., Poole D., Le Grand S., Walker R. C., J. Chem. Theory Comput. 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C., Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Darden T., York D., Pedersen L., J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- 24.Gaussian 09, Revision A.2, M. J. T. Frisch, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Scalmani, G. Barone, V. Mennucci, B. Petersson, G. A. Nakatsuji, H. Caricato, M. Li, X. Hratchian, H. P. Izmaylov, A. F. Bloino, J. Zheng, G. Sonnenberg, J. L. Hada, M. Ehara, M. Toyota, K. Fukuda, R. Hasegawa, J. Ishida, M. Nakajima, T. Honda, Y. Kitao, O. Nakai, H. Vreven, T. Montgomery, J. A. Peralta, Jr., J. E. Ogliaro, F. Bearpark, M. Heyd, J. J. Brothers, E. Kudin, K. N. Staroverov, V. N. Kobayashi, R. Normand, J. Raghavachari, K. Rendell, A. Burant, J. C. Iyengar, S. S. Tomasi, J. Cossi, M. Rega, N. Millam, J. M. Klene, M. Knox, J. E. Cross, J. B. Bakken, V. Adamo, C. Jaramillo, J. Gomperts, R. Stratmann, R. E. Yazyev, O. Austin, A. J. Cammi, R. Pomelli, C. Ochterski, J. W. Martin, R. L. Morokuma, K. Zakrzewski, V. G. Voth, G. A. Salvador, P. Dannenberg, J. J. Dapprich, S. Daniels, A. D. Farkas, Ö. Foresman, J. B. Ortiz, J. V. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary