

Abstract
Bone marrow fibrosis is the continuous replacement of blood‐forming cells in the bone marrow with excessive scar tissue, leading to failure of the body to produce blood cells and ultimately to death. Myofibroblasts are fibrosis‐driving cells and are well characterized in solid organ fibrosis, but their role and cellular origin in bone marrow fibrosis have remained obscure. Recent work has demonstrated that Gli1+ and leptin receptor+ mesenchymal stromal cells are progenitors of fibrosis‐causing myofibroblasts in the bone marrow. Genetic ablation or pharmacological inhibition of Gli1+ mesenchymal stromal cells ameliorated fibrosis in mouse models of myelofibrosis. Conditional deletion of the platelet‐derived growth factor (PDGF) receptor‐α (PDGFRA) gene (Pdgfra) and inhibition of PDGFRA by imatinib in leptin receptor+ stromal cells suppressed their expansion and ameliorated bone marrow fibrosis. Understanding the cellular and molecular mechanisms in the haematopoietic stem cell niche that govern the mesenchymal stromal cell‐to‐myofibroblast transition and myofibroblast expansion will be critical to understand the pathogenesis of bone marrow fibrosis in both malignant and non‐malignant conditions, and will guide the development of novel therapeutics. In this review, we summarize recent discoveries of mesenchymal stromal cells as part of the haematopoietic niche and as myofibroblast precursors, and discuss potential therapeutic strategies in the specific targeting of fibrotic transformation in bone marrow fibrosis. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: fibrosis, bone marrow, myofibroblasts, mesenchymal stromal cells, Hedgehog signalling, haematopoietic stem cells, myeloproliferative neoplasms
Bone marrow fibrosis
Bone marrow (BM) fibrosis is the continuous replacement of blood‐forming cells in the BM by excessive scar tissue, leading to failure of the body to produce blood cells and ultimately to death. A number of haematological and non‐haematological disorders are associated with increased BM fibrosis, including myelodysplastic syndromes and myeloproliferative neoplasms (MPNs) 1.
Primary myelofibrosis (PMF) is the prototypical example of progressive development of BM fibrosis and progressive evolution of BM insufficiency. PMF refers to BCR‐ABL1‐negative MPN, and is a clonal disorder of haematopoiesis arising in the haematopoietic stem cell (HSC) 2. The majority of patients with PMF carry mutations that activate JAK–STAT signalling; 60% harbour the JAK2V617F mutation, approximately 30% carry a calreticulin (CALR) mutation, and 8% carry a myeloproliferative leukaemia virus oncogene (MPL) mutation 3, 4, 5, 6, 7, 8. PMF is the most aggressive of the three classic MPNs, and is associated with significantly shortened survival 9, 10. Despite our advanced understanding of the molecular pathogenesis of malignant myeloid diseases, the stepwise pathogenesis of BM fibrosis is poorly understood. Up to just a few months ago, the cells that drive BM fibrosis remained obscure, impeding the cellular and molecular dissection of this process. The recent discovery of Gli1+ and leptin receptor (LepR)+ stromal cells, which are cells of the HSC niche, as fibrosis‐driving cells in the BM opens new avenues for investigation of the molecular and cellular mechanisms of fibrotic transformation. Accumulating evidence suggests that PMF is not only a disease of HSCs and haematopoietic progenitor cells, but is rather a disease of the entire HSC niche. The data presented in this review provide a rationale for dissecting the interaction between HSCs and haematopoietic progenitor cells and their complex microenvironment in more detail, and for evaluating the role of different niche cells in both malignant and non‐malignant diseases leading to BM fibrosis.
Stromal cell populations in the BM microenvironment (the HSC niche)
The emerging picture of the BM niche is that of a complex and multifaceted collection of regulatory cell types, with important roles in bone formation and the maintenance of HSC function and blood production regulation. Several stromal cell populations are implicated in the regulation of haematopoiesis, and recent studies have highlighted the role of specific stromal cell populations in regulating discrete haematopoietic progenitor populations 11, 12, 13. BM mesenchymal stromal cells (MSCs) constitute a rare population of cells found around arterioles and sinusoid vessels, and are in direct contact with endothelial cells; others are located in the endosteal niche adjacent to the bone 14. Early studies of BM MSCs and cells responsible for HSC support implicated mature bone‐forming osteoblasts, which produce granulocyte colony‐stimulating factor, thrombopoietin (TPO), and CXC motif ligand (Cxcl) 12 15. It was further shown that differentiating osteoblastic lineage cells (OBCs) express many factors that are important for HSC function, including Cxcl12, stem cell factor (SCF) (also termed c‐KIT ligand or Steel factor), and angiopoietin‐1 (ANGPT1), as well as extracellular matrix (ECM) proteins such as osteocalcin, collagen 1, and osteopontin (Figure 1) 16, 17. In particular, MCAM/CD146‐expressing subendothelial stromal cells with osteogenic potential (so‐called self‐renewing osteoprogenitor cells) are major producers of ANGPT1, a central molecule of the HSC ‘niche’ involved in vascular remodelling 18, 19. The expansion of osteolineage cells results in increased HSC numbers, whereas their ablation results in HSC loss in the BM and extramedullary haematopoiesis 20, 21. However, their true contribution to HSC maintenance remains controversial, as deletion of Cxcl12 or Scf from mature osteoblasts has no effect on HSCs, probably indicating heterogeneity within the osteolineage cell population. Importantly, although mature osteolineage cells appear to have limited roles in vivo, immature osteolineage cells may have a greater impact on HSC maintenance 22, 23.
Figure 1.

The HSC niche in homeostasis: a simplified overview of the HSC niche with a focus on different MSC subsets under homeostatic conditions. Different mesenchymal precursor cells are shown in their localizations in the BM cavity. Relevant secreted and/or cell‐bound factors that regulate HSC functional states are highlighted. EPO, erythropoietin; GFP, green fluorescent protein; NorE, norepinephrine.
The perivascular location of HSCs highlighted the importance of stromal cells in the perivascular area as crucial BM niche components with a significant HSC‐supporting function 24. Within the perivascular region, a large proportion of stromal cells express high levels of Cxcl12, which plays a crucial role in maintaining HSC function. So far, three stromal cell populations characterized by high Cxcl12 expression have been identified: (1) Cxcl12‐abundant reticular (CAR) cells 25, 26; (2) nestin (Nes)+ MSCs (Nes‐MSCs) 27; and (3) leptin receptor (LepR)+ stromal cells (Figure 1) 28, 29. It is likely that there is considerable overlap between these populations. CAR cells are mesenchymal progenitors with adipogenic and osteogenic potential in vitro 26. Importantly, their conditional ablation with diphtheria toxin and the Cxcl12‐regulatory element resulted in a reduction in HSC numbers and long‐term repopulation activity, alongside increased HSC quiescence 26. Nes‐MSCs express the classic mesenchymal‐specific markers platelet‐derived growth factor receptor‐α (PDGFRA/CD140a), CD51, and Sca‐1 30, 31, as well as LepR 32 and the pericyte marker neuron/glial antigen 2 (NG2) 33. Although there is significant functional overlap between MSC populations defined with these markers in terms of multipotency and self‐renewal capacities, their distribution in the BM cavity and their association with the endosteum or blood vessels are distinct 13, 24. NG2+LepR+Nesdim pericyte‐like MSCs are mainly periarteriolar in localization, and are close to the endosteal bone surface. NG2+LepR+Nesbright reticular‐like MSCs are mostly perisinusoidal in localization, and are located more in the central BM cavity, away from the endosteum 33 (Figure 1). Perivascular MSCs are directly involved in maintaining HSCs in the BM niche, as shown by a series of studies investigating the consequence of either Nes‐mediated or Cxcl12‐mediated MSC ablation 26, 27 or Lepr‐mediated or Prx1‐mediated deletion of Scf and Cxcl12 expression in MSCs 22, 23, 29. Nes‐MSCs also express genes that are important for HSC maintenance, such as Cxcl12, Scf, and ANGPT1, although their ablation results in a minor loss of HSCs within the BM 11. Much like CAR cells and Nes‐MSCs, LepR+ stromal cells express high levels of Cxcl12 and SCF, highlighting large overlapping stromal cell populations. Interestingly, CAR cells also express high levels of LepR. Intriguingly, osteoblasts were not targeted by Lepr:Cre in lineage mapping studies, suggesting the presence of a LepR– CAR cell subpopulation with osteogenic potential.
Evidence suggests that dysregulation of the interaction between HSCs and haematopoietic progenitor cells and distinct stromal cell populations, either in malignant haematopoietic diseases or in inflammatory processes, disrupts the tightly regulated process of haematopoiesis in the BM niche and favours a secretory phenotype of stromal cells with decreased haematopoiesis‐supporting capacity.
Stromal cell populations in BM fibrosis
Recent studies using different models of haematopoietic malignancies associated with BM fibrosis were able to mechanistically elucidate how malignant haematopoietic cells and proinflammatory cytokines activate stromal cells in the BM, support their fibrotic and secretory activity, and influence their reduced haematopoiesis‐supporting capacity (Figure 2).
Figure 2.

Stromal cell populations in the HSC niche in BM fibrosis: overview of the HSC niche in BM fibrosis with a focus on the contribution of different MSC populations to disease initiation and progression. Different MSC populations and precursors of myofibroblasts contribute to BM fibrosis by different mechanisms and cytokines, and secreted and/or cell‐bound factors. Dysplastic megakaryocytes are supposed to play a central role in the initiation of BM fibrosis. GFP, green fluorescent protein; ROS, reactive oxygen species.
Two recent studies showed that malignant haematopoietic cells in murine models of MPN 34 and acute myeloid leukaemia (AML) 35 can damage Nes‐MSCs and nerve cells. In these models, the development of haematological malignancy created neuropathic changes in the BM niche, which affected the activity of perivascular MSCs and altered the function of the HSC niche. In particular, they demonstrated that inflammatory mechanisms mediated by the malignant haematopoietic cells affect the dysregulation of Nes‐MSCs. JAK2V617F‐mutated HSCs and haematopoieticprogenitor cells in the murine MPN model led to interleukin (IL)‐1‐mediated damage to nerve fibres and Nes‐MSCs. The subsequent loss of Nes‐MSCs led to an accelerated MPN phenotype through neuropathic changes and to complete disruption of normal haematopoiesis 34. Interestingly, Jak2V617F MPN mice develop BM fibrosis despite a reduction in the size of the MSC compartment with loss of Nes‐MSCs. As in the MPN model, leukaemic cells in an MLL‐AF9 AML transplantation model caused losses of periarteriolar NG2+ cells and nerve fibres, resulting in a sympathetic neuropathy. In this model, an abnormal population of Nes‐MSCs expanded with skewed osteoblastic differentiation and downregulated expression of many HSC retention factors, including Cxcl12 and SCF, but BM fibrosis was not detectable 35. Together, these studies indicate that MPN‐induced and AML‐induced neuropathy promotes the development of a self‐reinforcing malignant niche that favours disease development at the expense of normal HSC maintenance. These studies further demonstrate that Nes‐MSCs are not fibrosis‐driving cells in BM fibrosis, although their reduction and subsequent neuropathy in the BM most likely contribute to dysregulated haematopoiesis and disease progression.
The aberrant differentiation of Nes‐MSC in osteoblasts under leukaemic conditions highlights the role of the endosteal bone in regulation of the BM niche and disease progression. In this context, Schepers et al demonstrated that OBCs, derived from multipotent stromal cells, expand in the presence of malignant haematopoietic cells, resulting in matrix production and trabecular thickening 17. Similarly to the findings of aberrant osteogenic differentiation of Nes‐MSCs in the presence of AML cells, they showed that BCR‐ABL + malignant haematopoietic cells stimulate MSCs to proliferate and adopt an abnormal differentiation programme, resulting in the overproduction of functionally altered OBCs, which accumulate in the BM cavity as inflammatory myelofibrotic cells. Myelofibrotic OBCs also express proinflammatory cytokines (i.e. IL‐1 and tumour necrosis factor‐α), which probably amplify disease development and aberrant myeloid cell production. In summary, these three studies using different models of myeloid malignancies led to the hypothesis that the HSC niche is an oncogenic unit that promotes malignant haematopoiesis at the expense of normal haematopoiesis in terms of a self‐reinforcing niche 17, 36. These studies also highlight the significant role of inflammation in the disturbance of the haematopoietic niche in myeloid malignancies, in particular in the initiation of BM fibrosis (Figure 2).
However, these studies did not identify fibrosis‐driving cells. The identification of cells that drive the development of a fibrotic BM niche with its detrimental consequences for the maintenance of HSCs is a prerequisite for the development of novel targeted therapeutics. Two recent studies using state‐of‐the‐art techniques including genetic fate tracing in vivo, including our own work, provided evidence that LepR+ and Gli1+ cells are key players in the initiation and progression of BM fibrosis 28, 37. Decker et al demonstrated that BM LepR+ mesenchymal stromal lineage cells expand extensively and are fibrogenic in PMF 28. LepR+ MSCs downregulate the expression of key HSC‐supporting factors and upregulate genes associated with fibrosis and osteogenesis, indicating fibrogenic conversion. On the basis of this finding, Decker et al suggested that targeting PDGFRA signalling might be an attractive strategy for treating BM fibrosis. Their data demonstrated that administration of imatinib or conditional deletion of Pdgfra from LepR+ stromal cells suppresses their expansion and ameliorates BM fibrosis (Figure 2). We recently demonstrated that the hedgehog (Hh) transcriptional activator Gli1 marks perivascular MSCs, which contribute substantially to organ fibrosis and constitute a relevant therapeutic target to prevent solid organ dysfunction after injury 38. Gli1+ cells show MSC functional characteristics. The identification of perivascular Gli1+ MSC‐like cells as a major cellular origin of organ fibrosis provided a rationale for studying Gli1+ cells in the BM 38, 39. Periarteriolar Gli1+ cells in the BM have similarities to Nes‐MSCs, but do not express LepR. The majority of Gli1+ cells in the endosteal niche are not associated with glial fibrillary acidic protein+ glia or sympathetic nerve fibres, and only partially express Nes 37. Thus, they might represent a distinct subpopulation of stromal cells in the BM.
Using genetic fate tracing experiments in two murine models of BM fibrosis, we demonstrated that Gli1+ MSCs are fibrosis‐driving cells of the BM (Figure 2). They are recruited from their endosteal and perivascular niche in the presence of mutated haematopoietic cells to become α‐smooth muscle actin (α‐SMA)+ fibrosis‐driving myofibroblasts 37. Importantly, the genetic ablation of Gli1+ cells completely abolishes BM fibrosis and rescues BM failure, providing functional proof that these cells are drivers of the fibrotic transformation. Upon myelofibrotic transformation, Gli1+ cells significantly expand in the BM, in both murine models and patient samples, whereas Nes+ cells decrease in number, suggesting that neuropathic changes lead to dysregulation of the niche accompanied by enhanced expansion and myofibroblast differentiation of Gli1+ MSCs 37.
Importantly, both LepR+ and Gli1+ stromal cells differentiate into myofibroblasts in BM fibrosis, and their differentiation seems to be the common downstream mechanism. Across different organ systems, the majority of investigators agree that myofibroblasts are fibrosis‐driving cells; however, the functional contribution of myofibroblasts in BM fibrosis has remained elusive. Only a few electron‐microscopy studies from the last century and more recent immunohistochemical staining suggested an increase in the number of myofibroblasts in human BM fibrosis 40, 41, 42. The recent genetic fate tracing data studies have shown that both Gli1+ and LepR+ stromal cells are progenitors of myofibroblasts in BM fibrosis. The fact that genetic ablation of Gli1+ cells abolishes BM fibrosis and restores haematopoiesis indicates that Gli1+ MSCs constitute a promising cellular therapeutic target. An open question to be answered in future experiments is which factors and pathways induce the differentiation of multipotent stromal cells into fibrosis‐driving myofibroblasts. Lessons could be learned from mechanisms identified in solid organ fibrosis. Furthermore, it remains elusive how heterogeneous the stromal population is, and whether there is an overlap of Gli1+ and LepR+ cells. Another open question is how the identified cell populations compare with CD146+, SPARC‐expressing cells, which also were shown to react to myeloproliferative stimuli 43, 44. Future single‐cell resolution studies will shed light on this interesting topic.
Similarities to solid organ fibrosis – proinflammatory cytokines and platelets as common mechanisms
Fibrosis as a broad term describes the excess deposition of ECM elements that normally form a complex network of collagens, laminins, entactin, heparan sulphate and other proteoglycans. Fibrosis is often defined as a wound‐healing response and repair of damaged tissues that has got out of control. Damage to tissues can result from various acute or chronic stimuli, including, but not limited to, infections, hypoxia, hypertension, diabetes, autoimmune reactions, and mechanical injury. BM fibrosis is seen in a variety of malignant (e.g. multiple myeloma and myelodysplastic syndrome) and non‐malignant (e.g. chronic autoimmune diseases, infections, and exposure to radiation or toxins) disease states. It is likely that both malignant and non‐malignant diseases activate fibrosis‐driving cells by common downstream mechanisms. PMF is the best‐studied haematopoietic malignancy inducing BM fibrosis, and is often used as the prototypic example of BM fibrosis.
In PMF patients, the haematopoietic phenotype is characterized by a progressive dominance of malignant (clonal) haematopoiesis over normal polyclonal haematopoiesis, the abnormal proliferation of megakaryocytes, the deposition of ECM material within the BM, extensive extramedullary haematopoiesis, abnormal stem cell trafficking, and shortened survival 45, 46, 47, 48. The pathognomonic hyperplasia and dysplasia in the megakaryocyte lineage also results in the excessive production of cytokines and chemokines, such as fibroblast growth factor (FGF), within the BM, and reticulin and collagen fibre deposition in the BM space are additional cardinal signs of disease (Figure 2). This ECM accumulation is supposed to be driven by the extramedullary release of cytokines by the malignant haematopoietic clone and by dysplastic megakaryocytes derived from the malignant clone 49. Growth factors such as PDGF, βFGF and transforming growth factor (TGF)‐β are thought to activate fibrosis‐driving cells, triggering the progressive process of myelofibrosis (MF), and βFGF, vascular endothelial growth factor (VEGF) and IL‐8 can activate endothelial cells that contribute to neoangiogenesis 50. Hence, it is hypothesized that the cross‐talk between HSCs and stromal cells is altered. In this scenario, malignant HSCs release growth factors that condition stromal cells and allow them to gain novel properties. These events subsequently create a pathological microenvironment that allows the malignant clone to develop, leading to imbalances that compromise normal haematopoiesis 45. Therefore, HSC niche deregulation is a key step in myeloproliferative processes.
The common downstream mechanisms in fibrotic transformation in solid organs and in the BM (induced by both malignant and non‐malignant haematopoiesis) appear to be inflammatory programmes and the involvement of megakaryocytes and/or platelets. In solid organ fibrosis, platelets play an important role in the initiation of fibrosis. Platelets are exposed to ECM components, triggering aggregation, clot formation, and haemostasis. Platelet degranulation subsequently promotes vasodilation and increased blood vessel permeability, while also activating myofibroblasts (collagen‐secreting α‐SMA+ fibroblasts) 51. As described above, megakaryocytes also seem to have a driving and initiating role in BM fibrosis, in particular through the secretion of proinflammatory and profibrotic cytokines. In solid organ fibrosis, CXC chemokines play a significant role in the activation and migration of myofibroblasts and fibrocytes. In particular, CXC chemokine receptor (CXC) 4, CC chemokine receptor (CCR) 7 and CCR2 mediate the recruitment of myofibroblasts and, potentially, their differentiation. A central role of platelet‐derived Cxcl4 (also known as platelet factor 4) was demonstrated in solid organs. It was shown that Cxcl4 is secreted not only by activated platelets, but also by plasmacytoid dendritic cells and fibroblasts 52, 53. These studies link megakaryocytes/platelets to proinflammatory programmes and the activation of profibrotic programmes in fibrosis. In MF, studies performed 30 years ago indicated that abnormal megakaryocytes stimulate the proliferation of fibrosis‐driving fibroblasts, partially mediated by Cxcl4 54. Functional and gene expression studies using RNA sequencing recently demonstrated that Cxcl4 also induces migration of Gli1+ stromal cells and their myofibroblast transdifferentiation, thus mediating central cellular processes in BM fibrosis (Figure 2). Significant downregulation of the chemokine stromal‐cell derived factor (SDF)‐1 (Cxcl12) was also observed, in line with previous studies characterizing niche cells in MPN 17, 34. Interestingly, deletion of Cxcl12 in stromal cells was shown to increase the number of circulating platelets 34, 55, and studies in patients with MF have suggested that the BM microenvironment is deprived of active Cxcl12 56. These data led to the hypothesis that Gli1+ stromal cells are activated from their niche by atypical platelets/megakaryocytes (mediated by Cxcl4), which leads to a cascade of myofibroblast transdifferentiation, metabolic reprogramming, and downregulation of Cxcl12. Additional stimuli might be derived from mutant neutrophils that additionally lead to persistent immune stimulation 57.
Implications for the therapy of BM fibrosis – treating the deregulated HSC niche
At present, the only curative treatment modality for PMF is allogeneic stem cell transplantation, which is able to restore BM function but carries significant risks of morbidity and mortality, particularly in the older patient population 58, 59. It is hypothesized that myeloid malignancies such as PMF can be worsened by deregulation of the BM microenvironment 60, including MSCs and their progeny.
We will focus here on a discussion of treatment strategies for PMF, as the treatment of BM fibrosis induced by other malignant and non‐malignant processes in the BM is even less well understood and is understudied. The potential therapeutic approaches for PMF can be divided into two categories: (1) eradication of the malignant haematopoietic clone; or (2) targeting BM fibrosis. Currently, no Food and Drug Administration‐approved therapies exist for specific targeting of the fibrotic BM niche, although reversal of BM fibrosis is a clinically achievable goal, as demonstrated in chronic myeloid leukaemia (CML) patients upon BCR/ABL inhibition 61 or in PMF patients upon SCT 62, 63, 64. With the introduction of selective JAK inhibitors over the last 10 years, initial expectations of clonal suppression as measured by elimination of molecular and karyotypic abnormalities were dampened by the observations of persistent clonal haematopoiesis and unaltered MPN BM pathological features. Although JAK inhibitors improve patient constitutional symptoms and reduce splenomegaly, JAK inhibitor monotherapy does not significantly reduce the mutant allele burden in the majority of MPN patients 65, 66, 67.
Increases in the expression of Hh target genes has been observed in granulocytes isolated from MPN patients 68. So far, the exact role of the Hh pathway in PMF and its contribution to BM fibrosis is not fully understood. We have shown that Gli1 expression is significantly increased in stromal cells from MPN patients (Figure 3). Pharmacological targeting with the Gli inhibitor GANT61 inhibits Gli1+ cell expansion and myofibroblast differentiation, and attenuates fibrosis severity 37. These data thus provide a rationale for targeted therapy of Gli proteins in BM fibrosis, as monotherapy or combined therapy with other agents. The efficacy of Gli inhibition in murine BM fibrosis models further suggests that combinations of JAK2 inhibitors with inhibitors of the Hh pathway might provide a novel avenue for targeting stem cell‐derived clonal myeloproliferation (which evades JAK2‐targeted monotherapy). Preclinical and clinical data suggest that Hh pathway inhibitors have therapeutic activity in MF. To date, only Smo inhibitors have been tested in MF, with varying success 69. We hypothesize that Gli proteins in Gli1+ cells can be activated independently of canonical Hh signalling, explaining the mixed response in patients with MF, e.g. by phosphoinositide 3‐kinase–AKT signalling (Figure 3).
Figure 3.
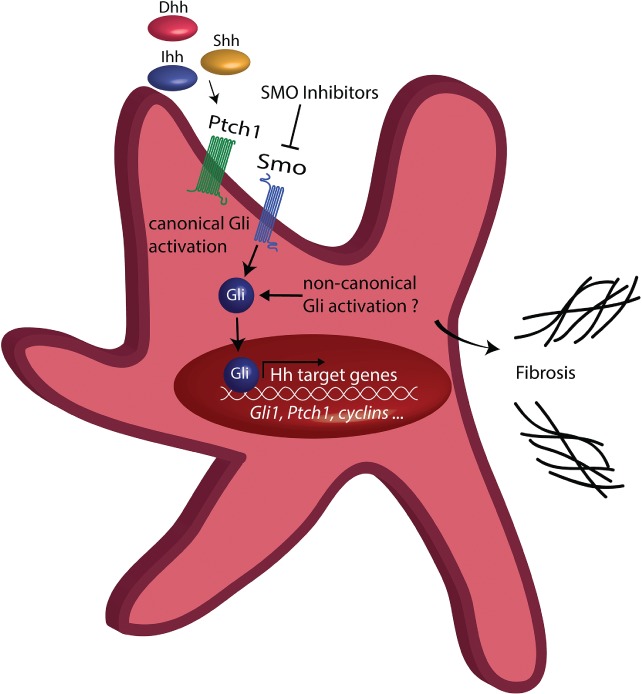
Hh–Gli signalling in myofibroblasts. The canonical Hh pathway is characterized by binding of one of the three ligands desert hedgehog (Dhh), Indian hedgehog (Ihh) or sonic hedgehog (Shh) to the receptor Ptch1, which discontinues its tonic inhibition on the transmembrane protein smoothened (Smo) upon ligand binding. Smo then activates the transcriptional activators of Hh signalling, the so‐called Gli proteins, which will transfer to the nucleus and activate expression of Hh target genes, promoting expansion of myofibroblasts and fibrosis. Non‐canonical Hh signalling can activate Gli proteins directly and independently of the Hh ligands Ptch1 and Smo.
Strategies aimed at preventing the development of a profibrotic niche could help to restore normal haematopoiesis and disrupt the malignant self‐reinforcing niche. Several agents are expected to target the microenvironment, including immunomodulatory drugs such as thalidomide and lenalidomide, proteasome inhibitors such as bortezomib, and VEGF‐targeting drugs such as sunitinib and bevacizumab, and have been tested in subsets of PMF patients. However, these clinical trials have had mixed results, and were hampered by severe tolerability issues 70. Considering the importance of inflammatory cytokines in driving fibrotic BM niche remodelling 17 and CML development 71 in murine models, it will be crucial to test the effect of blocking these specific proinflammatory cytokines with various suppressing, neutralizing and/or antagonizing antibodies that are currently available for clinical use. Strategies aimed at either directly protecting nerve cells in the BM or at preserving their interaction with perivascular MSCs 34, 35 might also help to support the maintenance of normal haematopoeisis. The experimental evidence that the clinically approved β3‐adrenergic agonist mirabegron delays disease onset and improves several MPN‐associated features in the Jak2V617F murine MPN model 34 provides a rationale for modulating nerve cells in BM fibrosis.
Summary
BM fibrosis is a reactive secondary process and a cardinal feature of PMF. Identifying Gli1+ and LepR+ cells as the sources of fibrosis‐driving cells was the first crucial step in developing antifibrotic therapies. As we have discussed, there is significant overlap in the stromal cell populations that contribute to HSC maintenance and to the development of haematological malignancies. Importantly, we have identified Gli1+ cells as a stromal cell population that provides an attractive therapeutic target, and have also identified deregulated pathways in these cells that provide novel avenues for future targeted therapies 37, 38.
Author contributions statement
All authors were involved in writing the paper and creating the figures, and gave final approval to the submitted manuscript.
Acknowledgements
This work was supported by the European Haematology Association (EHA; John Goldman Clinical Research grant), a research grant from the MPN foundation (2017 MPNRF/LLS Award), a KWF Kankerbestrijding young investigator grant (11031/2017‐1, Bas Mulder Award; Dutch Cancer Foundation), a Celgene research grant (DEU‐077), and a grant from the European Research Council (ERC) (deFIBER; ERC‐StG 757339), all to RKS, and by grants from the German Research Foundation (KR‐4073/3‐1, SCHN1188/5‐1, and SFB/TRR57), a grant from the European Research Council (ERC‐StG 677448), a START Grant from the RWTH Aachen University (101/15), a Grant of the State of Northrhinewestfalia (Return to NRW), and a grant from the Interdisciplinary Centre for Clinical Research within the faculty of Medicine at the RWTH Aachen University (IZKF O3‐11), all to RK.
No conflicts of interest were declared.
References
- 1. Kuter DJ, Bain B, Mufti G, et al Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol 2007; 139: 351–362. [DOI] [PubMed] [Google Scholar]
- 2. Tefferi A, Thiele J, Orazi A, et al Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007; 110: 1092–1097. [DOI] [PubMed] [Google Scholar]
- 3. Levine RL. JAK‐mutant myeloproliferative neoplasms. Curr Top Microbiol Immunol 2012; 355: 119–133. [DOI] [PubMed] [Google Scholar]
- 4. Klampfl T, Gisslinger H, Harutyunyan AS, et al Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013; 369: 2379–2390. [DOI] [PubMed] [Google Scholar]
- 5. Tefferi A, Guglielmelli P, Lasho TL, et al CALR and ASXL1 mutations‐based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia 2014; 28: 1494–1500. [DOI] [PubMed] [Google Scholar]
- 6. Tefferi A, Lasho TL, Tischer A, et al The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1‐like CALR variants. Blood 2014; 124: 2465–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nangalia J, Massie CE, Baxter EJ, et al Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013; 369: 2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine RL, Gilliland DG. Myeloproliferative disorders. Blood 2008; 112: 2190–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mehta J, Wang H, Iqbal SU, et al Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma 2014; 55: 595–600. [DOI] [PubMed] [Google Scholar]
- 10. Tefferi A. Primary myelofibrosis: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2011; 86: 1017–1026. [DOI] [PubMed] [Google Scholar]
- 11. Anthony BA, Link DC. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol 2014; 35: 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol 2011; 195: 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frenette PS, Pinho S, Lucas D, et al Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping‐stone for regenerative medicine. Annu Rev Immunol 2013; 31: 285–316. [DOI] [PubMed] [Google Scholar]
- 14. El Agha E, Kramann R, Schneider RK, et al Mesenchymal stem cells in fibrotic disease. Cell Stem Cell 2017; 21: 166–177. [DOI] [PubMed] [Google Scholar]
- 15. Calvi LM, Adams GB, Weibrecht KW, et al Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003; 425: 841–846. [DOI] [PubMed] [Google Scholar]
- 16. Winkler IG, Barbier V, Wadley R, et al Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood 2010; 116: 375–385. [DOI] [PubMed] [Google Scholar]
- 17. Schepers K, Pietras EM, Reynaud D, et al Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self‐reinforcing leukemic niche. Cell Stem Cell 2013; 13: 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bianco P, Sacchetti B, Riminucci M. Osteoprogenitors and the hematopoietic microenvironment. Best Pract Res Clin Haematol 2011; 24: 37–47. [DOI] [PubMed] [Google Scholar]
- 19. Sacchetti B, Funari A, Michienzi S, et al Self‐renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007; 131: 324–336. [DOI] [PubMed] [Google Scholar]
- 20. Zhang J, Niu C, Ye L, et al Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003; 425: 836–841. [DOI] [PubMed] [Google Scholar]
- 21. Visnjic D, Kalajzic Z, Rowe DW, et al Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004; 103: 3258–3264. [DOI] [PubMed] [Google Scholar]
- 22. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013; 495: 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greenbaum A, Hsu YM, Day RB, et al CXCL12 in early mesenchymal progenitors is required for haematopoietic stem‐cell maintenance. Nature 2013; 495: 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature 2014; 505: 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sugiyama T, Kohara H, Noda M, et al Maintenance of the hematopoietic stem cell pool by CXCL12–CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006; 25: 977–988. [DOI] [PubMed] [Google Scholar]
- 26. Omatsu Y, Sugiyama T, Kohara H, et al The essential functions of adipo‐osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 2010; 33: 387–399. [DOI] [PubMed] [Google Scholar]
- 27. Mendez‐Ferrer S, Michurina TV, Ferraro F, et al Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010; 466: 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Decker M, Martinez‐Morentin L, Wang G, et al Leptin‐receptor‐expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat Cell Biol 2017; 19: 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ding L, Saunders TL, Enikolopov G, et al Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012; 481: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winkler IG, Sims NA, Pettit AR, et al Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010; 116: 4815–4828. [DOI] [PubMed] [Google Scholar]
- 31. Pinho S, Lacombe J, Hanoun M, et al PDGFRalpha and CD51 mark human nestin+ sphere‐forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med 2013; 210: 1351–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou BO, Yue R, Murphy MM, et al Leptin‐receptor‐expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 2014; 15: 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kunisaki Y, Bruns I, Scheiermann C, et al Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013; 502: 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arranz L, Sanchez‐Aguilera A, Martin‐Perez D, et al Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014; 512: 78–81. [DOI] [PubMed] [Google Scholar]
- 35. Hanoun M, Zhang D, Mizoguchi T, et al Acute myelogenous leukemia‐induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014; 15: 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt T, Kharabi Masouleh B, Loges S, et al Loss or inhibition of stromal‐derived PlGF prolongs survival of mice with imatinib‐resistant Bcr‐Abl1(+) leukemia. Cancer Cell 2011; 19: 740–753. [DOI] [PubMed] [Google Scholar]
- 37. Schneider RK, Mullally A, Dugourd A, et al Gli1+ mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell Stem Cell 2017; 20: 785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kramann R, Schneider RK, DiRocco DP, et al Perivascular Gli1+ progenitors are key contributors to injury‐induced organ fibrosis. Cell Stem Cell 2015; 16: 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schepers K, Campbell TB, Passegue E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell 2015; 16: 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Biagini G, Severi B, Govoni E, et al Stromal cells in primary myelofibrosis: ultrastructural observations. Virchows Arch B Cell Pathol Incl Mol Pathol 1985; 48: 1–8. [DOI] [PubMed] [Google Scholar]
- 41. Thiele J, Kuemmel T, Sander C, et al Ultrastructure of bone marrow tissue in so‐called primary (idiopathic) myelofibrosis‐osteomyelosclerosis (agnogenic myeloid metaplasia). I. Abnormalities of megakaryopoiesis and thrombocytes. J Submicrosc Cytol Pathol 1991; 23: 93–107. [PubMed] [Google Scholar]
- 42. Schmitt‐Graeff AH, Nitschke R, Zeiser R. The hematopoietic niche in myeloproliferative neoplasms. Mediators Inflamm 2015; 2015: 347270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tripodo C, Sangaletti S, Guarnotta C, et al Stromal SPARC contributes to the detrimental fibrotic changes associated with myeloproliferation whereas its deficiency favors myeloid cell expansion. Blood 2012; 120: 3541–3554. [DOI] [PubMed] [Google Scholar]
- 44. Tripodo C, Di Bernardo A, Ternullo MP, et al CD146(+) bone marrow osteoprogenitors increase in the advanced stages of primary myelofibrosis. Haematologica 2009; 94: 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Bousse‐Kerdiles MC. Primary myelofibrosis and the ‘bad seeds in bad soil’ concept. Fibrogenesis Tissue Repair 2012; 5: S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rumi E, Cazzola M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017; 129: 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rumi E, Pietra D, Ferretti V, et al JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014; 123: 1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tefferi A, Cervantes F, Mesa R, et al Revised response criteria for myelofibrosis: International Working Group‐Myeloproliferative Neoplasms Research and Treatment (IWG‐MRT) and European LeukemiaNet (ELN) consensus report. Blood 2013; 122: 1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ciurea SO, Merchant D, Mahmud N, et al Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007; 110: 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Le Bousse‐Kerdiles MC, Martyre MC. Dual implication of fibrogenic cytokines in the pathogenesis of fibrosis and myeloproliferation in myeloid metaplasia with myelofibrosis. Ann Hematol 1999; 78: 437–444. [DOI] [PubMed] [Google Scholar]
- 51. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 2007; 117: 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van Bon L, Affandi AJ, Broen J, et al Proteome‐wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med 2014; 370: 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zaldivar MM, Pauels K, von Hundelshausen P, et al CXC chemokine ligand 4 (Cxcl4) is a platelet‐derived mediator of experimental liver fibrosis. Hepatology 2010; 51: 1345–1353. [DOI] [PubMed] [Google Scholar]
- 54. Burstein SA, Malpass TW, Yee E, et al Platelet factor‐4 excretion in myeloproliferative disease: implications for the aetiology of myelofibrosis. Br J Haematol 1984; 57: 383–392. [DOI] [PubMed] [Google Scholar]
- 55. Tzeng YS, Li H, Kang YL, et al Loss of Cxcl12/Sdf‐1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood 2011; 117: 429–439. [DOI] [PubMed] [Google Scholar]
- 56. Migliaccio AR, Martelli F, Verrucci M, et al Altered SDF‐1/CXCR4 axis in patients with primary myelofibrosis and in the Gata1 low mouse model of the disease. Exp Hematol 2008; 36: 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tripodo C, Burocchi A, Piccaluga PP, et al Persistent immune stimulation exacerbates genetically driven myeloproliferative disorders via stromal remodeling. Cancer Res 2017; 77: 3685–3699. [DOI] [PubMed] [Google Scholar]
- 58. Kroger NM, Deeg JH, Olavarria E, et al Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: a consensus process by an EBMT/ELN international working group. Leukemia 2015; 29: 2126–2133. [DOI] [PubMed] [Google Scholar]
- 59. Vannucchi AM. Management of myelofibrosis. Hematology Am Soc Hematol Educ Program 2011; 2011: 222–230. [DOI] [PubMed] [Google Scholar]
- 60. Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood 2009; 114: 1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Thiele J, Kvasnicka HM. Myelofibrosis in chronic myeloproliferative disorders – dynamics and clinical impact. Histol Histopathol 2006; 21: 1367–1378. [DOI] [PubMed] [Google Scholar]
- 62. Daly A, Song K, Nevill T, et al Stem cell transplantation for myelofibrosis: a report from two Canadian centers. Bone Marrow Transplant 2003; 32: 35–40. [DOI] [PubMed] [Google Scholar]
- 63. Kroger N, Giorgino T, Scott BL, et al Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood 2015; 125: 3347–3350; quiz 3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hussein K, Stucki‐Koch A, Alchalby H, et al Cytokine expression pattern in bone marrow microenvironment after allogeneic stem cell transplantation in primary myelofibrosis. Biol Blood Marrow Transplant 2016; 22: 644–650. [DOI] [PubMed] [Google Scholar]
- 65. Verstovsek S, Mesa RA, Gotlib J, et al A double‐blind, placebo‐controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012; 366: 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Harrison C, Kiladjian JJ, Al‐Ali HK, et al JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012; 366: 787–798. [DOI] [PubMed] [Google Scholar]
- 67. Harrison CN, Vannucchi AM, Kiladjian JJ, et al Long‐term findings from COMFORT‐II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016; 30: 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bhagwat N, Keller MD, Rampal RK, et al Improved efficacy of combination of JAK2 and Hedgehog inhibitors in myelofibrosis. Blood 2013; 122(suppl 1): 666.23794067 [Google Scholar]
- 69. Sasaki K, Gotlib JR, Mesa RA, et al Phase II evaluation of IPI‐926, an oral Hedgehog inhibitor, in patients with myelofibrosis. Leuk Lymphoma 2015; 56: 2092–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rambaldi A, Barbui T, Barosi G. From palliation to epigenetic therapy in myelofibrosis. Hematology Am Soc Hematol Educ Program 2008; 2008: 83–91. [DOI] [PubMed] [Google Scholar]
- 71. Reynaud D, Pietras E, Barry‐Holson K, et al IL‐6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 2011; 20: 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]